

Abstract
Insulin potentiates N-methyl-d-aspartate receptors (NMDARs) in neurons and Xenopus oocytes expressing recombinant NMDARs. The present study shows that insulin induced (i) an increase in channel number times open probability (nPo) in outside-out patches excised from Xenopus oocytes, with no change in mean open time, unitary conductance, or reversal potential, indicating an increase in n and/or Po; (ii) an increase in charge transfer during block of NMDA-elicited currents by the open channel blocker MK-801, indicating increased number of functional NMDARs in the cell membrane with no change in Po; and (iii) increased NR1 surface expression, as indicated by Western blot analysis of surface proteins. Botulinum neurotoxin A greatly reduced insulin potentiation, indicating that insertion of new receptors occurs via SNARE-dependent exocytosis. Thus, insulin potentiation occurs via delivery of new channels to the plasma membrane. NMDARs assembled from mutant subunits lacking all known sites of tyrosine and serine/threonine phosphorylation in their carboxyl-terminal tails exhibited robust insulin potentiation, suggesting that insulin potentiation does not require direct phosphorylation of NMDAR subunits. Because insulin and insulin receptors are localized to glutamatergic synapses in the hippocampus, insulin-regulated trafficking of NMDARs may play a role in synaptic transmission and plasticity, including long-term potentiation.
Keywords: insulin receptor tyrosine kinase, regulated exocytosis, Xenopus oocytes
Insulin, insulin receptors, and their substrates are enriched at synapses in hippocampus and cerebral cortex (1, 2), where they are thought to subserve a number of functions including regulation of glucose metabolism, gene expression, and synaptic plasticity (for review see refs. 3 and 4). Insulin promotes axonal growth in cultures of fetal brain cells and is thought to play a role in axonal growth and elongation in brain development (5). Insulin is synthesized by hippocampal neurons in culture and can be released from neurons and brain synaptosomes by depolarization in a Ca2+-dependent manner (6, 7). Insulin inhibits norepinephrine uptake by hippocampal neurons in culture (8) and reduces firing rate of pyramidal neurons in hippocampal slices (9). Insulin potentiates neuronal γ-aminobutyric acid type A receptor activity by recruitment of functional γ-aminobutyric acid type A receptors to postsynaptic structures at CA1 synapses (10) and reduces α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) activity by promoting internalization of AMPARs from synaptic membranes of hippocampal neurons in culture (11, 12). Insulin enhances N-methyl-d-aspartate receptor (NMDAR)-mediated synaptic transmission at CA1 synapses (13) and potentiates activity of recombinant NMDARs expressed in Xenopus oocytes (14).
The insulin receptor is a tyrosine kinase, which, upon stimulation, phosphorylates insulin receptor substrates (IRS-1, IRS-2), leading to activation of diverse signaling pathways and downstream proteins including phosphoinositide 3-kinase, 3-phosphoinositide-dependent kinase, Akt2, and atypical protein kinase C (PKCζ/λ) (15–17). In striated muscle and adipose tissue, insulin stimulation and phosphorylation of IRS proteins promotes increased glucose uptake primarily via translocation of the GLUT4 glucose transporter protein from intracellular storage sites to the plasma membrane (18). Movement of the transporter requires activation of the intrinsic tyrosine kinase of the insulin receptor (19–21). The signaling pathway(s) activated by insulin in neurons to potentiate NMDARs are not yet known.
The present study was undertaken to examine the hypothesis that insulin-induced potentiation of NMDARs expressed in Xenopus oocytes occurs at least in part by rapid recruitment via exocytosis of vesicle-associated channel molecules to the cell surface. Oocytes expressing recombinant receptors provide geometric simplicity and express a homogenous population of receptors, presumably in the absence of PSD-95, which is known to inhibit insulin and PKC potentiation of recombinant NMDARs (22, 23). Moreover, the molecular machinery for protein trafficking is highly conserved from yeast to mammals (24). Experiments involving patch-clamp recording, charge transfer measurements, neurotoxin treatment, and Western analysis of surface proteins demonstrate that insulin delivers new NMDA channels to the plasma membrane by regulated exocytosis, a mechanism that may be relevant to synaptic plasticity.
Materials and Methods
Expression Constructs.
Mouse ɛ1 (corresponding to rat NR2A) cDNA was a gift of M. Mishina (University of Tokyo, Tokyo). Rat NR1–4a (NR1000) cDNA was cloned in this laboratory (25). cDNAs were subcloned into the pBluescript SK(−) vector for oocyte expression. To generate templates for transcription, circular plasmid cDNAs were linearized with BamHI (NR1–4a) or NotI (ɛ1). Capped mRNAs were synthesized as run-off transcripts from linearized plasmid cDNAs with T3 or T7 polymerase (Ambion mMessage mMachine transcription kit, Austin, TX, 2 h at 37°C). Concentration and integrity of mRNAs were assessed after staining with ethidium bromide by direct comparison of sample mRNAs with an RNA standard ladder (GIBCO/BRL).
Electrophysiology of Recombinant NMDARs Expressed in Xenopus Oocytes.
Adult female Xenopus laevis (Xenopus I, Ann Arbor MI) were maintained in a temperature- and light-controlled environment and treated in accordance with the principles and procedures of the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Selected stage V and VI oocytes from Xenopus were injected with in vitro-transcribed mRNAs (20 ng mRNA/cell; NR1/NR2 = 1:2) (26).
Whole-cell currents were recorded from oocytes (2–6 days after injection) at ambient temperature in the voltage clamp mode as described (26). Currents were elicited by bath application of NMDA with glycine at a holding potential of −60 mV. Oocytes were perfused in Mg2+-free, normal frog Ringer's solution consisting of 116 mM NaCl, 2.0 mM KCl, 1.0 mM CaCl2, 10 mM Hepes, pH 7.2. For insulin receptor activation, oocytes were incubated in insulin (1 μM, 10 min; Sigma).
Single-channel currents were recorded from outside-out patches excised from devitellinized oocytes (2–7 days after injection) as described (27). Single-channel current amplitudes were calculated from means of Gaussian fits to all-point amplitude histograms; open-time durations, from single-channel openings above baseline; nPo was total channel open time divided by recording time. Data are means ± SEMs for 4–12 experiments with oocytes from 2–3 batches of oocytes. Statistical significance was evaluated by Student's t test (SIGMAPLOT 6.0).
Western Blot Analysis of Surface Proteins.
Control and insulin-treated Xenopus oocytes expressing NR1–4a/NR2A (NR1100/NR2A) receptors were screened for NMDA currents in the range of 100 to 300 nA and insulin potentiation of ≈2- to 3-fold. Oocytes were incubated in external recording solution in the absence or presence of insulin (100 μM, 2 min) and washed twice. Surface proteins were biotinylated with the membrane-impermeant reagent sulfosuccinimidyl 2-(biotinamido) ethyl-1,3′dithiopropionate (sulfo-NHS-SS-biotin; Pierce) according to Chen et al. (28). Cell extracts were prepared as described by Hollmann et al. (29). To isolate biotinylated surface proteins from nonsurface proteins, cell extracts were incubated with Neutravidin-linked beads (Pierce; 2 h at 4°C). Bound proteins were eluted from the beads by incubation with SDS/PAGE gel loading buffer containing DTT (which releases biotin from labeled proteins) and subjected to gel electrophoresis with aliquots of total proteins.
Results
Insulin Potentiates NMDAR Currents via Activation of Insulin Receptor Tyrosine Kinase.
To examine the actions of insulin on NMDAR functional activity, we recorded NMDA-elicited whole-cell currents from Xenopus oocytes expressing NR1–4a/NR2A receptors before and after insulin treatment via the bath perfusate. We chose the NR1–4a splice isoform, which has the shortest C-terminal tail, because NR1–4a/NR2A and NR1–4a/NR2B receptors exhibit the highest cell surface expression (30) and highest degree of PKC potentiation (31). Insulin (1 μM, 10 min) markedly increased NMDA-elicited whole-cell currents (Fig. 1A). Insulin potentiation (defined as the ratio of NMDA current amplitude measured after insulin to that before insulin) was 2.8 ± 0.1 (n = 11).
Figure 1.

Insulin potentiates NMDA whole-cell currents. (A) A typical sequence showing NMDA-activated whole-cell currents recorded at Vh = −60 mV from oocytes expressing NR1–4a/NR2A receptors before and after application of insulin (1 μM, 10 min). NMDA, 300 μM; glycine, 10 μM. Insulin potentiated NMDA-elicited currents to 2.8 ± 0.1 (n = 11) times control. (B) The oocytes expressing NR1–4a/NR2A receptors were preincubated at least 1 h in control external solution, containing the nonselective inhibitor of tyrosine kinases, genistein (100 μM). Genistein had no effect on either basal NMDA responses or insulin potentiation. Potentiation was to 2.7 ± 0.2 (n = 3) after preincubation of oocytes in genistein. (C) Tyrphostin A47 (100 μM; 10 min), a selective inhibitor of insulin receptor tyrosine kinases, slightly potentiated the control NMDARs current (to 1.56 ± 0.16 times control), but completely blocked insulin potentiation of NMDA-elicited currents. Insulin potentiation was to 1.02 ± 0.10 times the control NMDA response in the presence of tyrphostin alone. (D) Summary of experiments showing potentiation of NMDA currents by insulin in the absence and presence of genistein and tyrphostin A47.
To examine the role of tyrosine kinase activation in insulin-induced potentiation of NMDA currents, we applied reagents that target tyrosine kinase activity. Application of the broad-spectrum tyrosine kinase inhibitor genistein (100 μM, 1 h) before and during NMDA application had little or no effect on basal NMDA responses or insulin-induced potentiation of NMDAR responses (Fig. 1B). Insulin potentiation was to 2.7 ± 0.2 times the control NMDA response in the presence of genistein (n = 3; Fig. 1 B and D) vs. 2.8 ± 0.1 in the absence of genistein (n = 11; Fig. 1 A and D). In contrast, the selective insulin receptor inhibitor tyrphostin A47 (100 μM, 10 min) slightly stimulated control NMDA currents (to 1.56 ± 0.16 times the control response; n = 7; Fig. 1C, compare first and second NMDA responses), but completely abolished insulin potentiation of NMDA responses (potentiation to 1.02 ± 0.10 times the control response in tyrphostin A47; n = 7; Fig. 1C, compare second and third responses; Fig. 1D). This finding indicates that insulin-induced potentiation of NMDA currents involves activation of the insulin receptor tyrosine kinase, but does not rule out the involvement of other kinases.
Insulin Increases NMDA Channel nPo but Not Single-Channel Conductance or Mean Open Time in Excised, Outside-Out Patches.
To examine effects of insulin on NMDA channel gating, we recorded channel activity in outside-out patches excised from oocytes expressing NR1–4a/NR2A receptors before and after insulin treatment. We excised patches after insulin treatment, because patch formation can distort the membrane inside the pipette and disrupt exocytosis (32, 51). In control patches, NMDA (100 μM) activated channels with a single channel conductance, γ = 37.3 ± 0.9 pS at ±60 mV (n = 5; Fig. 2 A and C). NMDA channel activity in patches excised after application of insulin (1 μM, 10 min) was markedly potentiated (Fig. 2B). Insulin increased the number of active channels times channel open probability, nPo, by ≈2.3-fold from 0.08 ± 0.02 before insulin (n = 5) to 0.18 ± 0.03 after insulin (n = 5; P < 0.05; Fig. 2D); however, single-channel conductance remained unchanged (γ = 37.2 ± 0.6 pS; n = 5; Fig. 2 B and C). Moreover, insulin slightly, but not significantly, increased the mean duration of openings (Fig. 2E). The distribution of open times was fit by a single exponential consistent with a single open state (τ = 3.36 ± 0.33 msec before insulin; n = 5 vs. τ = 4.04 ± 0.15 msec after insulin; n = 5; P > 0.05).
Figure 2.

Insulin potentiates NMDA single channel activity. (A and B) Representative traces of NMDA-activated channels recorded in outside-out patches excised from control (A) and insulin-treated (B) oocytes expressing NR1–4a/NR2A receptors. Single-channel currents were elicited by application of NMDA (100 μM with 10 μM glycine) directly to the patch and recorded at Vh = −60 mV. (C) Insulin did not change single-channel conductance (γ = 37.3 ± 0.9 pS and γ = 37.2 ± 0.6 pS in control and insulin-treated oocytes, respectively). (D) Quantitation of nP0 measured in outside-out patches from control and insulin-treated oocytes. (E) Distribution of channel mean open times in patches from control (○) and insulin-treated (●) oocytes.
Insulin Increases the Number of Channels in the Plasma Membrane, but Does Not Alter Channel Gating.
The results reported thus far suggest that insulin increases NMDA channel open probability and/or number of active channels at the cell surface. To analyze independently the effects of insulin on number of functional channels in the membrane, N, and channel open probability, Po, we used a modification of the method of Jahr (33) as adapted by Rosenmund et al. (34). This method takes advantage of the essentially irreversible block of NMDA-elicited currents by the open-channel blocker MK-801. NMDA-elicited whole-cell currents were recorded in the continuous presence of MK-801 (5 μM) in control (Fig. 3B Left) and insulin-treated (Fig. 3B Right) oocytes. To determine the number of channels, N, we calculated cumulative charge transfer, Q, which is the total current flow during the time required for complete block by MK-801. N can be calculated from Q, as follows:
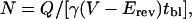 |
where tbl is the time constant for MK-801 block [tbl = 1/(kbl [MK-801])] and kbl = 2.5 × 107 M−1⋅s−1 (33). Insulin does not detectably alter single-channel conductance or mean open time (Fig. 2 C and D) and should not affect kbl. The channel number, N, for control and insulin-treated oocytes was normalized to the NMDA-elicited whole-cell current, which corrects for any differences in levels of expression within and between the two groups. For control oocytes, the mean number of channels per cell, Ncontrol, was 1.01 ± 0.13 × 106 channels per 100 nA. The mean channel density is calculated to be 0.04–0.12 μM−2, assuming a surface area for the oocyte of 3 × 107 μM2 (35) and NMDA-elicited whole-cell currents of 100–300 nA. Insulin increased the number of channels per cell by ≈3.2-fold (n = 6; P < 0.005; Fig. 3E). The increase in channel number per cell accounted for the entire increase in current (Iinsulin/Icontrol = 3.1 ± 0.3; n = 7; Fig. 3 A and D). Po in control and insulin-treated oocytes can be calculated from the near steady-state peak NMDA-elicited current, Icontrol, the single channel current, i, and N, as follows:
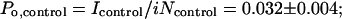 |
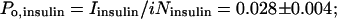 |
from which the ratio, Po, insulin/Po, control = 0.88 (Fig. 3F). These findings indicate that insulin increased the number of active NMDA channels and had no effect on channel open probability.
Figure 3.

Insulin delivers new functional NMDA channels to the oocyte surface. (A) Whole-cell recordings were obtained from Xenopus oocytes expressing NR1–4a/NR2A receptors in Ca2+-free (Ba2+) Ringer's solution. Insulin (10 min) potentiated NMDA-elicited currents. Insulin, 1 μM; NMDA, 1 mM; glycine, 50 μM. Vh = −60 mV. (B) Insulin increased the number of functional NMDA channels per cell, N. NMDA currents were recorded in the continuous presence of MK-801 (5 μM) from control (Left) and insulin-treated (Right) oocytes; The NMDA-elicited current increased to a peak value, and then decayed exponentially as channels opened and were blocked by MK-801. The cumulative charge transfer, Q, was obtained by integration of the current trace over time (area indicated by checkerboard pattern). The larger integrated current observed for insulin-treated oocytes indicates increased N. (C) Agonist-evoked currents in B were normalized to the peak current to enable analysis of time constants for decay. The time constant of decay of the NMDA current did not differ for insulin vs. control oocytes, indicating no change channel opening rate, k. (D–H) Quantitation of data in A–C. (D) Ratio of NMDA-elicited currents for insulin-treated vs. control oocytes. (E) Channel number per cell, N, normalized to the initial current, I (to correct for variation in expression levels), for control (●) and insulin-treated oocytes (○). (F) Open probability, Po, for control and insulin-treated oocytes (G and H). Opening rate, kβ, and closing rate, kα, did not differ significantly for control vs. insulin-treated oocytes.
As an independent measure of the effect of insulin on NMDA channel gating, we analyzed the rate of decay of NMDA-elicited current in the presence of MK-801 (Fig. 3C). Provided that kbl[MK-801] >> kα and kβ (the closing and opening rates, respectively), the decay can be described by a single exponential with the rate constant, kβ. Insulin slightly but not significantly reduced kβ (from 2.04 ± 0.15 s−1 for control oocytes to 1.69 ± 0.20 s−1 for insulin-treated oocytes; Fig. 3G). From measurements of Po and kβ, and the relation, Po = kβ/(kα + kβ), we calculated values of kα = 73.6 ± 5.4 s−1 in control oocytes vs. 60.7 ± 7.4 s−1 in insulin-treated oocytes (P > 0.05; Fig. 3H).
These values of kα are substantially less than the ≈250s−1 predicted from the ≈4-ms mean open time of single channels (Fig. 2E). The apparent discrepancy arises because the Po equation applied to macroscopic currents assumes an equilibrium between unliganded and bursting states and will thus yield a kα value corresponding to termination of bursting, rather than of individual openings. kαcalculated in this manner will be overestimated in that Po calculated from N and I is less than the probability that a receptor is liganded. This analysis reveals that insulin does not modulate NMDA channel gating. Moreover, the slow recovery of NMDA-induced responses from MK-801 block indicates that the rates of constitutive exocytosis and endocytosis are relatively slow; thus, a reduction in the rate of endocytosis would account, at most, for a very small component of insulin potentiation (51).
Insulin Promotes Delivery of NMDA Channels to the Cell Membrane via Exocytosis.
The results reported thus far indicate that insulin increases the number of active NMDA channels at the cell surface, but do not distinguish between insertion of new channels and unmasking of silent channels. To distinguish between these possibilities, we examined the effects of loading oocytes with the light chain of type A botulinum neurotoxin (BoNT A), which is known to cleave SNAP-25 and prevent SNAP-25-dependent exocytosis (36). Microinjection of BoNT A reduced the degree of insulin potentiation of NMDA-elicited currents by about 70% (from 4.7 ± 0.2 for control oocytes; n = 6 to 1.7 ± 0.1 for insulin-treated oocytes; n = 6; P < 0.001 (Fig. 4). No significant change in the resting potential, input resistance, or basal NMDA response was caused by BoNT A (data not illustrated). No effects on insulin potentiation were observed when the vehicle (DTT, 10 mM) was injected. The reduction in insulin potentiation by BoNT A suggests that insulin-induced delivery of NMDA channels to the cell surface is by SNAP-25-dependent exocytosis.
Figure 4.

Insulin delivers new channels to the oocyte surface via regulated exocytosis. (A–C) Microinjection of the light chain of BoNT A (50 ng) into oocytes 5 h before recording reduced insulin potentiation of NMDA-elicited currents by about 70%. (A) Representative NMDA-elicited currents before and after insulin treatment for a control oocyte as in Fig. 1, but in Ca2+-free (Ba2+) Ringer's solution. (B) BoNT A reduced the degree of insulin potentiation. (C) Summary of experiments showing reduction of insulin potentiation by BoNT A.
Insulin Potentiation of NMDARs Does Not Require Direct Tyrosine Phosphorylation of NR1 or the Carboxyl Terminus of NR2A.
Insulin causes tyrosine phosphorylation of the NR2A and NR2B receptors (37). To examine a possible role for direct receptor phosphorylation in insulin-induced potentiation, we used site-directed mutagenesis to remove tyrosine residues. We introduced alanine residues in place of the two tyrosine residues, Y837 and Y865, in the C terminus of NR1–4a; this mutation removes all cytoplasmic tyrosine residues. We replaced Y842 in the C terminus of NR2A with a stop codon; this mutation truncates the C terminus and removes all serine, threonine, and tyrosine residues in that domain (note that two tyrosines are present in the TM1-TM2 intracellular loop of NR2A). Mutant receptors assembled from these subunits exhibited full insulin potentiation. Potentiation was to 3.0 ± 0.7 for NR1–4a(Y837A/Y865A)/NR2ΔA C receptors (n = 7; Fig. 5 B and C) vs. 2.7 ± 0.1 for wild-type NR1–4a/NR2A receptors (n = 7; Fig. 5 A and C). This finding indicates that insulin potentiation does not require direct tyrosine phosphorylation of the C-terminal tails of the NMDAR subunits. The entire increase in the NMDA-elicited whole-cell current for wild-type receptors and the double mutant could be accounted for by an increase in the number of channels (2.8 ± 0.2 for wild-type receptors; n = 5; vs. 3.1 ± 0.6 for mutant receptors; n = 5; Fig. 5D).
Figure 5.

Deletion and truncation mutants of NR1–4a/NR2A receptors exhibit insulin potentiation. Responses of (A) wild-type NR1–4a/NR2A and (B) NR1–4a (Y837A, Y865A)/NR2AΔC receptors before and after insulin treatment and subsequent block by MK-801. Insulin-induced (C) potentiation of NMDA-elicited currents and (D) increase in receptor number (see legends to Figs. 3 and 4) did not differ significantly for wild-type (WT) vs. mutant receptors.
Insulin Treatment Increases Surface Expression of NR1 in Oocytes.
To examine changes in NMDAR subunit surface expression, we performed Western analysis of cell-surface proteins (28). Intact control and insulin-treated oocytes expressing NR1/NR2A receptors were surface-labeled with sulfo-NHS-SS-biotin, and biotinylated surface proteins were separated from nonlabeled intracellular proteins by reaction with Neutravidin beads. Protein samples were subjected to electrophoresis and probed with mAb 54.1 directed to the extracellular loop of the NR1 subunit (ref. 38; Fig. 6A). Analysis of band densities indicated an insulin-induced increase in surface NR1 expression to 2.8 ± 0.3 times that of control; n = 5; P < 0.01 (Fig. 6 B and D), with no change in total cell NR1 protein (Fig. 6C). This finding indicates trafficking of NR1 subunits from the cytoplasm to the cell surface.
Figure 6.

Insulin increases NR1 abundance at the oocyte surface. NR1 surface and total cell expression in control and insulin-treated oocytes, as assessed by Western blot analysis of biotinylated surface protein. (A) Representative Western blot of surface protein from control and insulin-treated oocytes expressing NR1–4a/NR2A receptors probed with anti-NR1 antibody 54.1 (38). Lanes 3, 6, and 12 indicate micrograms of protein in samples of total cell extract before Neutravidin bead extraction loaded on each lane; surface (surf) indicates an aliquot of Neutravidin bead-isolated protein. Insulin increased NR1 abundance in samples of surface protein. (B–D) Quantitative analysis of the effects of insulin on surface expression (B), total cell protein (C), and fractional surface expression (D) of NR1. Mean densities of surface bands were normalized to values for control samples run on the same gels. NR1 subunits expressed at the cell surface increased from 2.0% to 5.3% of total cell NR1; total cell NR1 was unchanged.
Discussion
The present study demonstrates that insulin-induced potentiation of NMDAR activity occurs via delivery of new channel molecules to the cell surface by regulated exocytosis. Patch-clamp recording from outside-out patches excised from Xenopus oocytes indicate that insulin increases channel number times open probability (nPo), with no change in mean open time, unitary conductance, or reversal potential. The constancy of unitary conductance and charge transfer measurements of NMDA-elicited whole-cell currents during block by the open channel blocker MK-801 indicate that insulin increases the number of functional NMDARs in the cell membrane with no change in Po. Block of insulin potentiation by BoNT A indicates that insertion of new receptors occurs via a SNAP-25 mediated form of SNARE-dependent exocytosis. Western blot analysis of surface proteins provides direct evidence for delivery by insulin of new NR1 subunits to the cell surface. Recent studies involving electrophysiology and immunolabeling indicate that activation of PKC also induces recruitment of NMDARs to the cell surface of oocytes and hippocampal neurons in culture (51).
Experiments involving truncation and deletion mutants reveal the unexpected finding that NMDARs assembled from subunits lacking all known sites of tyrosine phosphorylation in their C-terminal domains can exhibit insulin potentiation. These results suggest that insulin-induced potentiation of NMDA receptor activity does not occur by direct phosphorylation of the C-terminal tails of the receptor protein, but rather of associated targeting, anchoring, or signaling protein(s). Together, these findings are consistent with a mechanism whereby insulin, acting through the insulin receptor tyrosine kinase, phosphorylates one or more protein(s) involved in receptor signaling or trafficking.
Insulin acts via the insulin receptor tyrosine kinase to initiate a signaling cascade involving phosphoinositide 3-kinase, phosphoinositide-dependent kinase, Akt2, and PKC (17, 39). The finding that insulin potentiation of NMDAR activity is blocked by the selective PKC inhibitor calphostin C supports a pathway involving PKC (14). One potential target of PKC is SNAP-25, which can be directly phosphorylated by PKC in an activity-dependent manner (40, 41). A model consistent with these findings is that insulin, acting via its receptor, regulates membrane fusion events of NMDAR-containing vesicles by PKC-mediated phosphorylation of SNAP-25. The association of SNAP-25 or its binding partners with NMDARs, however, is as yet unclear. That insulin potentiates NMDARs via activation of the insulin receptor is indicated by our finding that tyrphostin A47 blocks insulin potentiation. Specificity of insulin action on NMDARs is indicated by the observation that receptors containing NR2C show little or no insulin potentiation and that coexpression of NMDARs with PSD-95 differentially affects insulin potentiation of NR1/NR2A vs. NR1/NR2B receptors (13, 22).
Findings from the present study suggest a mechanism whereby insulin could regulate cell surface expression of NMDARs at the postsynaptic membrane, thereby modulating neuronal excitability. Turnover studies of neuronal NMDARs indicate that there is a large intracellular pool of NR1 subunits; thus NR1 subunits are present in large numbers in dendritic shafts and spines, where they rapidly assemble with NR2 subunits and insert in the membrane (42). Our finding of insulin-induced regulation of NMDAR trafficking in oocytes are in contrast to that of Lin et al. (12) that insulin promotes endocytosis of AMPARs, but does not affect NMDAR trafficking in hippocampal neurons. Rapid removal of AMPARs and γ-aminobutyric acid type A receptors from the plasma membrane under conditions of normal synaptic transmission (10–12, 43–45) and synaptic plasticity (46) is well established.
There is evidence that membrane fusion events contribute to NMDAR-dependent long-term potentiation (47), but the relative contribution of NMDARs and AMPARs is controversial (48). Although activity-dependent targeting of NMDARs to postsynaptic sites occurs on the order of many hours or days (49), experience-dependent insertion of new NMDARs into the membrane in the visual cortex can be achieved within 1–2 h and may underlie rapid modification in synaptic strength (50). Findings from the present study suggest that insulin, acting via its receptor induces phosphorylation of one or more proteins involved in receptor trafficking and targeting, thereby increasing NMDAR density at synapses. Given that insulin, insulin receptors, and NMDARs are widely expressed throughout the central nervous system, regulation by insulin of NMDA channel activity provides a potentially important way to modulate excitatory synaptic transmission.
Acknowledgments
This work was supported by National Institutes of Health Grants NS 20752 and NS 31282 (to R.S.Z.). M.V.L.B. is the Sylvia and Robert S. Olnick Professor of Neuroscience.
Abbreviations
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- BoNT
botulinum neurotoxin
- NMDA
N-methyl-d-aspartate
- NMDARs
NMDA receptors
- PKC
protein kinase C
References
- 1.Folli F, Ghidella S, Bonfanti L, Kahn C R, Merighi A. Mol Neurobiol. 1996;13:155–183. doi: 10.1007/BF02740639. [DOI] [PubMed] [Google Scholar]
- 2.Abbott M A, Wells D G, Fallon J R. J Neurosci. 1999;19:7300–7308. doi: 10.1523/JNEUROSCI.19-17-07300.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nystrom F H, Quon M J. Cell Signalling. 1999;11:563–574. doi: 10.1016/s0898-6568(99)00025-x. [DOI] [PubMed] [Google Scholar]
- 4.Nawa H, Saito M, Nagano T. Crit Rev Neurobiol. 1997;11:91–100. doi: 10.1615/critrevneurobiol.v11.i1.50. [DOI] [PubMed] [Google Scholar]
- 5.Schechter R, Beju D, Gaffney T, Schaefer F, Whetsell L. Brain Res. 1996;736:16–27. doi: 10.1016/0006-8993(96)00664-6. [DOI] [PubMed] [Google Scholar]
- 6.Clarke D W, Mudd L, Boyd F T, Jr, Fields M, Raizada M K. J Neurochem. 1986;47:831–836. doi: 10.1111/j.1471-4159.1986.tb00686.x. [DOI] [PubMed] [Google Scholar]
- 7.Wei L T, Matsumoto H, Rhoads D E. J Neurochem. 1990;54:1661–1665. doi: 10.1111/j.1471-4159.1990.tb01219.x. [DOI] [PubMed] [Google Scholar]
- 8.Boyd F T, Jr, Clarke D W, Muther T F, Raizada M K. J Biol Chem. 1985;260:15880–15884. [PubMed] [Google Scholar]
- 9.Palovcik R A, Phillips M I, Kappy M S, Raizada M K. Brain Res. 1984;309:187–191. doi: 10.1016/0006-8993(84)91028-x. [DOI] [PubMed] [Google Scholar]
- 10.Wan Q, Xiong Z G, Man H Y, Ackerley C A, Braunton J, Lu W Y, Becker L E, MacDonald J F, Wang Y T. Nature (London) 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- 11.Beattie E C, Carroll R C, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka R C. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- 12.Lin J W, Ju W, Foster K, Lee S H, Ahmadian G, Wyszynski M, Wang Y T, Sheng M. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Brown J C, III, Webster W W, Morrisett R A, Monaghan D T. Neurosci Lett. 1995;192:5–8. doi: 10.1016/0304-3940(95)11593-l. [DOI] [PubMed] [Google Scholar]
- 14.Liao G Y, Leonard J P. J Neurochem. 1999;73:1510–1519. doi: 10.1046/j.1471-4159.1999.0731510.x. [DOI] [PubMed] [Google Scholar]
- 15.Shepherd P R, Withers D J, Siddle K. Biochem J. 1998;333:471–490. doi: 10.1042/bj3330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White M F, Yenush L. Curr Top Microbiol Immunol. 1998;228:179–208. doi: 10.1007/978-3-642-80481-6_8. [DOI] [PubMed] [Google Scholar]
- 17.Whitehead J P, Clark S F, Urso B, James D E. Curr Opin Cell Biol. 2000;12:222–228. doi: 10.1016/s0955-0674(99)00079-4. [DOI] [PubMed] [Google Scholar]
- 18.Olson A L, Pessin J E. Annu Rev Nutr. 1996;16:235–256. doi: 10.1146/annurev.nu.16.070196.001315. [DOI] [PubMed] [Google Scholar]
- 19.White M F, Kahn C R. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- 20.Czech M P, Corvera S. J Biol Chem. 1999;274:1865–1868. doi: 10.1074/jbc.274.4.1865. [DOI] [PubMed] [Google Scholar]
- 21.Cheatham B, Kahn C R. Endocr Rev. 1995;16:117–142. doi: 10.1210/edrv-16-2-117. [DOI] [PubMed] [Google Scholar]
- 22.Liao G Y, Kreitzer M A, Sweetman B J, Leonard J P. J Neurochem. 2000;75:282–287. doi: 10.1046/j.1471-4159.2000.0750282.x. [DOI] [PubMed] [Google Scholar]
- 23.Yamada Y, Chochi Y, Takamiya K, Sobue K, Inui M. J Biol Chem. 1999;274:6647–6652. doi: 10.1074/jbc.274.10.6647. [DOI] [PubMed] [Google Scholar]
- 24.Bennett M K, Scheller R H. Proc Natl Acad Sci USA. 1993;90:2559–2563. doi: 10.1073/pnas.90.7.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durand G M, Gregor P, Zheng X, Bennett M V L, Uhl G R, Zukin R S. Proc Natl Acad Sci USA. 1992;89:9359–9363. doi: 10.1073/pnas.89.19.9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng X, Zhang L, Wang A P, Bennett M V L, Zukin R S. J Neurosci. 1997;17:8676–8686. doi: 10.1523/JNEUROSCI.17-22-08676.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Araneda R C, Lan J Y, Zheng X, Zukin R S, Bennett M V L. Biophys J. 1999;76:2899–2911. doi: 10.1016/S0006-3495(99)77445-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen N, Luo T, Raymond L A. J Neurosci. 1999;19:6844–6854. doi: 10.1523/JNEUROSCI.19-16-06844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollmann M, Maron C, Heinemann S. Neuron. 1994;13:1331–1343. doi: 10.1016/0896-6273(94)90419-7. [DOI] [PubMed] [Google Scholar]
- 30.Okabe S, Miwa A, Okado H. J Neurosci. 1999;19:7781–7792. doi: 10.1523/JNEUROSCI.19-18-07781.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanson G T, Kamboj S K, Cull-Candy S G. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao Y, Ferrer-Montiel A V, Montal M, Tsien R Y. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- 33.Jahr C E. Science. 1992;255:470–472. doi: 10.1126/science.1346477. [DOI] [PubMed] [Google Scholar]
- 34.Rosenmund C, Feltz A, Westbrook G L. J Neurosci. 1995;15:2788–2795. doi: 10.1523/JNEUROSCI.15-04-02788.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zampighi G A, Loo D D, Kreman M, Eskandari S, Wright E M. J Gen Physiol. 1999;113:507–524. doi: 10.1085/jgp.113.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montecucco C, Schiavo G. Q Rev Biophys. 1995;28:423–472. doi: 10.1017/s0033583500003292. [DOI] [PubMed] [Google Scholar]
- 37.Christie J M, Wenthold R J, Monaghan D T. J Neurochem. 1999;72:1523–1528. doi: 10.1046/j.1471-4159.1999.721523.x. [DOI] [PubMed] [Google Scholar]
- 38.Gazzaley A H, Weiland N G, McEwen B S, Morrison J H. J Neurosci. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng X, Zhang L, Wang A P, Bennett M V L, Zukin R S. Proc Natl Acad Sci USA. 1999;96:15262–15267. doi: 10.1073/pnas.96.26.15262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Genoud S, Pralong W, Riederer B M, Eder L, Catsicas S, Muller D. J Neurochem. 1999;72:1699–1706. doi: 10.1046/j.1471-4159.1999.721699.x. [DOI] [PubMed] [Google Scholar]
- 41.Shimazaki Y, Nishiki T, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. J Biol Chem. 1996;271:14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- 42.Huh K H, Wenthold R J. J Biol Chem. 1999;274:151–157. doi: 10.1074/jbc.274.1.151. [DOI] [PubMed] [Google Scholar]
- 43.Nishimune A, Isaac J T, Molnar E, Noel J, Nash S R, Tagaya M, Collingridge G L, Nakanishi S, Henley J M. Neuron. 1998;21:87–97. doi: 10.1016/s0896-6273(00)80517-6. [DOI] [PubMed] [Google Scholar]
- 44.Osten P, Srivastava S, Inman G J, Vilim F S, Khatri L, Lee L M, States B A, Einheber S, Milner T A, Hanson P I, et al. Neuron. 1998;21:99–110. doi: 10.1016/s0896-6273(00)80518-8. [DOI] [PubMed] [Google Scholar]
- 45.Song I, Kamboj S, Xia J, Dong H, Liao D, Huganir R L. Neuron. 1998;21:393–400. doi: 10.1016/s0896-6273(00)80548-6. [DOI] [PubMed] [Google Scholar]
- 46.Carroll R C, Lissin D V, von Zastrow M, Nicoll R A, Malenka R C. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- 47.Lledo P M, Zhang X, Sudhof T C, Malenka R C, Nicoll R A. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- 48.Malenka R C, Nicoll R A. Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 49.Rao A, Craig A M. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 50.Quinlan E M, Philpot B D, Huganir R L, Bear M F. Nat Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- 51.Lan, J.-Y., Skeberdis, V. A., Jover, T., Grooms, S. Y., Lin, Y., Araneda, R., Zheng, X., Bennett, M. V. L. & Zukin, R. S. (2001) Nat. Neurosci., in press. [DOI] [PubMed]