

Abstract
Protein binding site residues, especially catalytic residues, play a central role in protein function. Because more than 99% of the ∼12 million protein sequences in the nonredundant protein database have no structural information, it is desirable to develop methods to predict the binding site residues of a protein from its primary sequence. This task is highly challenging, because the binding site residues constitute only a small portion of a protein. However, the binding site residues of a protein are clustered in its functional pocket(s), and their spatial patterns tend to be conserved in evolution. To take advantage of these evolutionary and structural principles, we constructed a database of ∼50,000 templates (called the pocket-containing segment database), each of which includes not only a sequence segment that contains a functional pocket but also the structural attributes of the pocket. To use this database, we designed a template-matching technique, termed residue-matching profiling, and established a criterion for selecting templates for a query sequence. Finally, we developed a probabilistic model for assigning spatial scores to matched residues between the template and query sequence in local alignments using a set of selected scoring matrices and for computing the binding likelihood of each matched residue in the query sequence. From the likelihoods, one can predict the binding site residues in the query sequence. An automated computational pipeline was developed for our method. A performance evaluation shows that our method achieves a 70% precision in predicting binding site residues at 60% sensitivity.
Keywords: functional surface, residue matching technique, spatial template, split pocket
The binding site residues of a protein are essential for the function of the protein. Thus, computational methods have been developed to predict and characterize protein binding sites (1–5). Because the structure of a protein provides much insight into its function, more than 60,000 protein structures have been experimentally determined [in the Protein Data Bank (PDB)] (6). Among them, ∼25,000 are bound structures (7, 8), each of which includes its ligand(s) or interactions with other proteins. Computational methods have been developed to identify the binding site residues in this simplest case (3). For unbound structures, which do not include any ligand, it is more difficult to predict their binding site residues. Currently, the precision of methods for predicting the binding site residues in an unbound structure ranges from 40% to 60% (4). An even more difficult case is for proteins that have only primary sequences and no structural information and no structurally similar homologs. More than 99.5% of the ∼12 million protein sequences in the nonredundant (NR) protein database have no 3D structures. Therefore, it is strongly desirable to be able to predict the binding site residues of a protein from its primary sequence alone. However, difficulties arise from the fact that the binding site residues constitute only a small portion of a protein sequence.
Three strategies have been applied to this challenging task. First, support vector machine (SVM) methods and neural network methods (9, 10) have been used to extract useful information from sequences and structures. These machine-learning methods require training data derived from sequence or structural alignments, sequence profiles, and evolutionary analysis. So far, the prediction precision is only about 50% at 20% sensitivity (11). Second, from experimentally derived structural coordinates of proteins, one may predict the coordinates of a new protein to model the native protein folding and structure from its primary sequence by homology modeling (12). After the theoretical structural coordinates are obtained, predictive tools such as CASTp (13) and SplitPocket (8) can be applied to identify putative binding site residues. For example, Adamian et al. (14) identified the determinants of ρ1 GABAc receptor assembly and channel gating by detecting the binding surfaces of the ligand-gated ion channel located on the transmembrane. Third, a promising strategy is to assemble a library of precomputed structural templates (15–19). Then, for an uncharacterized protein, one can search for putative templates in the library in the hope of matching attributes of the query with those of characterized templates. This approach has proved useful to identify binding site residues of proteins that have structural coordinates. However, the focus was on the applicability of templates to structures but not sequences. For example, one of the active site templates curated by Meng et al. (17) has been used to detect divergent members in the enolase superfamily. These knowledge-based templates require manual extraction from structures and therefore, are difficult to collect in a large-scale manner. One major difficulty is that a large-scale collection requires an automated pipeline to establish a diversified template database for function prediction and characterization in a high-throughput manner. A recent study (5) developed an automated computational method, called signature of local active regions (SOLAR), to construct a basic set of consensus templates of binding surfaces and used the templates to characterize metalloendopeptidase and nicotinamide adenine dinucleotide binding proteins.
In this study, we have developed a method that uses only sequences to identify their binding site residues. Note that the binding site residues of a protein include all substrate binding residues and nonbinding residues in the binding sites of the protein; these include catalytic residues and binding residues. Our approach is based on evolutionary and structural principles and is different from the approaches described above. We note that the binding site residues of a protein are usually clustered in the functional pocket(s) of the protein and possess geometric characteristics (Fig. 1A) and that their spatial patterns tend to be well-conserved in evolution. A key step to use these two principles is to build an extensive database of templates, each of which is a sequence segment that contains a functional pocket (surface). This database is called pocket-containing segment database (PSD) (Fig. 1B). For each template in PSD, we also include geometric, biological, and evolutionary information of the binding site residues. This database takes advantage of the fact that functionally relevant residues are much better conserved than the rest of the protein. To use this database, we design a template-matching technique, termed residue matching profiling (RMP) (Fig. 1C). Because binding surfaces are usually highly conserved in evolution (20, 21), we use their spatial patterns to infer the binding site residues in the query sequence. It works by selecting a good template from PSD and using the binding site residues in the template to match those in a query sequence. Thus, another key step is to establish a criterion for selecting templates for a query sequence; we call this criterion the efficacy of a template. The third important step is to develop a probabilistic model for assessing the binding propensity of each potential binding site residue in the query sequence. Using this model, we assign a spatial score to each pair of matched residues between the template and the query sequence in local alignments using a set of selected scoring matrices, and then, we compute the binding likelihoods of these residues in the query sequence (Fig. 1C). From the likelihoods, we select the putative binding site residues in the query sequence. As will be shown, our approach can effectively identify the binding site residues of a protein from its sequence alone, even when the sequence identity between the query and the selected template is below 30%. This high performance attests to the evolutionary principle that the spatial patterns of functionally important amino acid residues in a protein, such as binding site residues, tend to be well-conserved in evolution. To carry out the above tasks, we develop an automated pipeline.
Fig. 1.

Outline of constructing PSD and conducting RMP. (A) The database SplitPocket (in cyan) contains 48,289 functional surfaces (in green) with their spatial patterns that consist of site-specific binding site residues. (B) Each pocket in SplitPocket is transformed into a PSD template. A template sequence is the shortest subsequence of the primary sequence of interest that starts from the first residue and goes to the last residue of the pocket. The pocket residues in the template sequence are indicated in different colors to signify that the spatial pattern of each pocket residue is transformed to the template sequence by filling in nonpocket residues (in gray). (C) A selected template is repeatedly aligned with the query sequence each time with a scoring matrix in the set of selected scoring matrices. RMP then predicts the binding site residues for the query by generating a binding profile of likelihoods.
Results
Database of Spatial Templates.
As noted above, the binding site residues are usually clustered in functional pockets. Thus, the database of functional pockets that we established previously (i.e., SplitPocket) (8) can be used as a library of binding site residues. The number of binding site residues in a functional pocket in SplitPocket ranges from 5 to 200, with a mean of 30 (Fig. S1A). However, because the binding site residues of a protein are usually dispersed over the primary sequence, it is difficult to align them to any query sequence. Therefore, we transformed SplitPocket into the PSD in which each sequence segment is the shortest subsequence of the primary sequence that contains the functional pocket of interest, starting from the first residue to the last residue of the pocket (Fig. 1B). The functional pocket of a ligand-bound structure is called a split pocket, because it is split by the ligand (3). PSD also contains the spatial attributes of binding site residues of each pocket stored in SplitPocket, including residue composition, relative distances between pocket residues, residue solvent accessible area, and physicochemical features (SI Materials and Methods, Filtering Out the Predicted Residues Located in a Protein Core). Moreover, for the residues with biological annotations, we mapped the annotated coordinates from the feature tables in UniprotKB/Swiss-Prot (22) to the corresponding positions in PSD templates. PSD currently holds 48,289 entries from 24,882 structures. Fig. S1B shows the length distributions of the spatial patterns and templates in PSD; the template length ranges from 7 to 500 residues, with a mean of 211.
Predicting the Binding Site Residues of a Structural Genomics Target.
Proteins targeted by structural genomics projects are proteins of unknown functions. They usually have no known homolog, even when the threshold of sequence identity is set as low as 25%. As an example of the application of our method, we consider Escherichia coli biotin synthesis protein (BIOH) (256 aa; Swiss-Prot P13001), a protein targeted by structural genomics. After matching this query against the spatial templates in PSD, we obtain 65 potential template hits. We arbitrarily select PDB1wpr chain A as our template, because its sequence identity with P13001 is only 22.7%. Although the selected template has a low sequence identity with the query, it is a good template, because it has an efficacy of 2.43, which is considerably higher than the cut-off of 1.25 (Materials and Methods has the definition of efficacy). After determining a local alignment, the structural and evolutionary information of the binding site residues of the template (PDB1wpr.A) are transferred to the matched or similar residues in the query (P13001). For each aligned pair of residues, we assign a score to the predicted residue in the query according to a spatial scoring system for computing a residue matching profile. For example, W22 of the query (P130001) is aligned to F27 of the template (PDB1wpr.A), and the pair W22-F27, a nonperfect match, has a spatial score of 1.32; however, the pair S82-S96, a perfect match, has the highest spatial score of 6. Because we do not know the date of divergence between the query and the template, we sample various scoring matrices to conduct evolutionary matching (Materials and Methods). In addition, by formulating a probabilistic model (Fig. 2) we obtain the 12 predicted residues that have a binding likelihood higher than the cut-off value (Table S1). Table S2 shows a comparison of our RMP method with psi-BLAST (23) and hidden Markov models (HMMER) (24). RMP predicts 12 site-specific residues in P13001, whereas both psi-BLAST and HMMER give continuous subsequences that do not include all of the actual binding site residues (Table S2). In this example, RMP achieves a precision as high as 0.83 with a specificity of 99.2% and sensitivity (recall) of 83%, whereas psi-BLAST has a precision of 0.06 with a specificity of 81.0% and a sensitivity of 25%.
Fig. 2.

Residue-matching profile (RMP). The query protein is P13001 [Escherichia coli biotin synthesis protein (BIOH)], and the template is PDB1wpr.A. (A) The normalized likelihoods of the predicted residues in the query P13001. (The likelihoods are normalized by the sum of the likelihoods.) The x axis shows the sequence coordinate and the amino acid at the position. (B) The same binding profile as in A, but the residues are ranked in the decreasing order of likelihoods. The catalytic triad (S82, D207, and H235) of P13001 is composed of the three residues with the highest likelihoods: 0.123, 0.097, and 0.091.
The BIOH (P13001) structure was solved by the Midwest Center for Structural Genomics, and its function was identified and validated by a panel of enzymatic assays. Fig. 3 shows the predicted binding region with the 12 predicted residues mapped onto the 3D structure of P13001. Among them, 10 of the 12 predicted residues are correct (Fig. 3 A and B). Moreover, the catalytic triad (S82, D207, and H235) is perfectly matched with the three residues on the spatial template of PDB1wpr.A (Fig. 3C). Only two predicted residues (V165 and L166) are false positives, and they are also located on the surface.
Fig. 3.

The binding site residues of E. coli BIOH identified by the technique of RMP. (A) The 12 predicted binding site residues are clustered tightly into a cavity (in green). Among them, S82, D207, and H235 are the catalytic residues (colored in red, yellow, and blue, respectively). (B) V165 and L166 (in brown) are false positives. (C) The side chains of the catalytic residues of BIOH (P13001:PDB1m33; chain A) and those of the hit template (PDB1wpr; chain A) are superimposed.
Importance of Selecting a Good Template.
We use a protein from Bacillus halodurans (Q9K901; 192 residues) as a query to show the importance of having a good template.
Using Q9K901 as the query to psi-BLAST PDB, we find PDB3cng (a nudix hydrolase from Nitrosomonas europaea) as the best hit. The aligned segment is from positions 57–142 of the PDB3cng.A sequence. The records for PDB3cng.A in PDB, which specify the residues comprising a functional or ligand binding site of the protein, indicate that the residues in the active site (C4, C7, C26, C29, N36, I40, T77, E85, E98, L99, Q110, Y112, F148, R149, and L171) of PDB3cng.A have been experimentally tested. We select the six residues (T77, E85, E98, L99, Q110, and Y112) within the aligned segment to match the binding site residues of Q9K901, but none of them is correctly matched in the psi-BLAST alignment. Thus, PDB3cng.A is not an effective template.
For comparison, from PSD, the template derived from PDB3bhd.A, a human thiamine triphosphatase, is selected, because it has the highest efficacy (3.4) among all potential PSD templates. With this template, we extract the spatial pattern of the 20 binding site residues from its binding site (Fig. S2). Among them, the five annotated residues (E9, K11, Y39, H76, and Y79) are clustered in the active site of the human thiamine triphosphatase. Applying our RMP method (Fig. 4A), we predict 20 putative binding site residues in the query sequence (192 residues); each predicted residue is assigned an RMP score and a likelihood from our probabilistic model (Materials and Methods). We are able to identify 13 well-aligned residues, which are functionally important (true positives in Fig. S2A). In particular, these 13 residues include four of five annotated residues: E8-E9, K10-K11, Y39-Y39, and E77-H76 (the first residue of each pair is from the query). Note that the query and the template have a full-length sequence identity <30%, but their aligned binding site residues show highly similar spatial patterns (Fig. S2B). Fig. 4B shows that the binding site residues of the two proteins are aligned along the diagonal, suggesting evolutionary conservation of the specific patterns of binding site residues.
Fig. 4.

Identifying the putative binding site residues of the query sequence (Q9K901) from B. halodurans. (A) The binding likelihoods of the 20 predicted binding site residues. (B) The diagonalized alignment of the binding site residues between Q9K901 and PBD3bhd.A indicates the high similarity between the spatial patterns of the binding site residues in the two proteins. Functionally important residues (in solid black) have higher binding likelihoods.
The query (Q9K901) protein was targeted by Joint Center for Structural Genomics. Although its structure coordinates have been determined (PDB2gfg), its biochemical function has not been characterized, and its class, architecture, topology, homologous (CATH) superfamily (25) fold signature is not yet assigned. Applying our SplitPocket algorithm for shape analysis (Fig. 5), we identified the actual binding surface in PDB2gfg.A that consists of 20 residues with a similar spatial pattern to that of PDB3bhd.A. Thus, PDB2gfg from B. halodurans and PDB3bhd from humans are homologous but have undergone deep divergence. Our shape analysis indicates that their binding surfaces are highly similar, because 13 of the 20 binding site residues are aligned with a sequence identity of 50% and a root mean square deviation (RMSD) of 2.6 Å at a P value of 10−7 (as opposed to the cut-off P value of 10−4) (1, 3). On the basis of spatial patterns, the significant P value implies that these two binding sites potentially perform similar molecular functions. Thus, their biochemical functions (thiamine triphosphatase; i.e., EC 3.6.1.28 and gene ontology annotations) of PDB3bhd and Q9K901 are likely similar.
Fig. 5.

Shape analysis of the query structure (PDB2gfg.A) and the template (PDB3bhd.A). (A) The actual binding surface of the query. (B) The split pocket (actual binding surface) of the template. (C) The structural alignment is performed based on the superimposition of the two binding surfaces as shown in D by the specific rotation matrix in E, which was computed by the method of fPOP (7). The spatial matching has a RMSD of 2.6 Å at a significant P value of 6.93 × 10−7. (F) In terms of the binding site residues, the sequence identity of the two spatial patterns is as high as 50%, although the full-length sequences of the two proteins have a sequence identity of only ∼34%.
Performance of the Spatial Template Approach.
We evaluated our method on a diverse set of 145 tested sequences using precision recall (PR) curves as described in SI Materials and Methods, Method for Performance Evaluation. The performances at the residue level are obtained for selected spatial templates with a range of sequence identities with the query defined as (i) α (sequence identity ≤ 80%, (ii) β (sequence identity ≤ 60%), and (iii) γ (sequence identity ≤ 35%). The areas under the obtained curves were used to analyze the influence of the spatial template selection criterion (α, β, and γ) on the precision of discovering binding site residues. The curves were obtained by increasing the threshold likelihood value of a binding residue from 0 to 0.25 with a 0.0025 increment. (The likelihoods were normalized by the sum of the likelihoods.) Fig. 6A shows that the method achieves 83%, 80%, and 72% precision at 50% recall (sensitivity) for the α, β, and γ templates, respectively. Overall, the PR-areas under curve (AUC) for α, β, and γ are 0.80, 0.77, and 0.70, respectively. The pair-wise comparisons indicate that the shapes of spatial templates have been highly conserved, because the performance decreases slowly. This is potentially useful when a template has a high efficacy, even if its sequence identity with the query is below 35%. Note that the curve slopes are seen at a steady state. At 60% recall, the spatial template approach achieves a precision >70%. In Fig. 6B, we assessed the accuracy across the range of possible values of threshold for template γ and obtained an optimal likelihood threshold (0.0175) of a binding residue.
Fig. 6.

Performances of selected spatial templates evaluated by PR curves. (A) The AUC for three sets of templates (α, β, and γ). The AUC decreases only 7% when the AUC of the γ-templates is compared with that of the β-templates. (B) The PR curve for the γ-templates is colored according to the spectrum bar of threshold on the right. In Inset, the accuracy of 65% is estimated when the optimal threshold of likelihood is set to 0.0175.
Predicting the catalytic residues of a protein from its primary sequence is an even more challenging task, because the unbalance between the number of true positives and the number of true negatives is even more extreme than in the case of binding site residues. However, as described in SI Materials and Methods, Predictions of Enzyme Catalytic Residues by Selecting Spatial Templates, our RMP method achieves a precision of 57% at a sensitivity of 50% (Fig. S3).
Discussion
Assessing the Spatial Scores for a Residue Matching Profile.
Based on a large-scale analysis of protein binding surfaces (Fig. S1A), the number of binding site residues comprises only ∼10% of a protein. Besides binding site residues, other residues may also be well-conserved in evolution, including the residues that are subject to the constraints for structural stability and protein folding (26, 27). How to separate these two types of residues requires a good strategy when only the primary sequence of the protein is available. Our strategy hinges on establishing a large database of spatial templates and a spatial scoring system. With the scoring system, we are able to generate an RMP to distinguish the residues involved in the function from those involved in stability in a large-scale computation. Moreover, we filter out the residues in a protein core, because they are unlikely involved in any binding reactivity (SI Materials and Methods, Filtering Out the Predicted Residues Located in a Protein Core). In this study, we focused on collecting the binding site residues of proteins to construct an extensive database of spatial templates and developing a criterion for selecting templates and an RMP technique. We found that a template with a high-efficacy value achieves good performance, even when its sequence identity with the query is low. Our study suggests that the likelihood of a binding site residue has an optimal threshold of 0.0175 in the iterative alignments of the evolutionary matching schema (3, 19, 26).
Assessing the Evolutionary Relationships and Structural Folds of PSD Templates.
A large-scale sequence comparison of PSD templates reveals conservation in local regions, even when the overall identity between two sequences is low. Interestingly, the selected templates consist of ∼21% of the CATH fold domains that are indeed associated with functions. Note that not all classified fold domains are directly involved in biochemical reactions. These spatial templates yield clues of conserved domains that are often adopted by proteins as modules to fulfill diverse biological functions. For example, by sequence comparison, a total of 430 fold domains in PSD is identified and mapped into a subset of the CATH (2,178 categories in version 3.2) homologous superfamilies. Essentially, they are functional substructures across the fold space. The PSD templates with structure coordinates are proteins that can be used for substructural predictions and designing a binding region. Especially for a query protein with structural coordinates, the prediction of its binding site residues becomes much easier if an appropriate template can be found in PSD. For example, the query protein Q9K901 has structural coordinates (PDB2gfg). From PSD, we find the template PDB3bhd.A, and using footprinting Pockets Of Proteins (fPOP) (7) to conduct a surface comparison (alignment), we identify all of the 20 binding site residues of Q9K901 (Fig. 5).
Inferring Function by PSD Templates.
Each of the PSD templates contains the spatial pattern of a functional surface, which includes structural, functional, and evolutionary information about the protein, such as the spatial distances between binding site residues. The conservation of a spatial pattern is largely caused by functional constraints and allows us to infer the binding site residues of a protein from those of a remote homolog (Fig. 4B). Moreover, comparing the spatial patterns of two proteins allows one to perform functional inference (19) in a reliable manner.
Materials and Methods
The materials used are described in SI Materials and Methods, Data Used for Performance Evaluation. Some methods are also described in SI Materials and Methods.
Defining Spatial Matching Scores and Evaluating Templates.
Let m be the number of residues in template T and denote  by
by 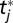 if tj is a binding site residue. Let n be the number of residues in query sequence Q. Let B be a 20 × 20 scoring matrix (see below), and for two amino acids μ and b, let
if tj is a binding site residue. Let n be the number of residues in query sequence Q. Let B be a 20 × 20 scoring matrix (see below), and for two amino acids μ and b, let  be their log-odd score. In the alignment between query sequence Q and template T, assume that ri ∈ Q is aligned with tj ∈ T. Then, for tj = μ, residues ri ∈ Q are classified into four categories
be their log-odd score. In the alignment between query sequence Q and template T, assume that ri ∈ Q is aligned with tj ∈ T. Then, for tj = μ, residues ri ∈ Q are classified into four categories  , with the following spatial matching scores
, with the following spatial matching scores 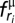 (Eq. 1):
(Eq. 1):
 |
where 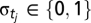 is a biological annotation of the amino acid residue tj in template T from UniprotKB/Swiss-Prot and PDB,
is a biological annotation of the amino acid residue tj in template T from UniprotKB/Swiss-Prot and PDB, 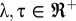 are two scaling parameters to be estimated, and
are two scaling parameters to be estimated, and 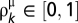 is defined as follows (Eq. 2):
is defined as follows (Eq. 2):
 |
where ‖ ‖ means the number of elements in the set,  , and
, and  for
for 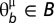 .
.
The value of 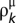 is directly computed from the scoring matrix B, whereas parameters λ and τ can be estimated from the input of tested sequences (see below). Note that we set
is directly computed from the scoring matrix B, whereas parameters λ and τ can be estimated from the input of tested sequences (see below). Note that we set 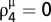 ; that is, when a residue in Q is aligned to a nonbinding residue, its score is 0, because we are only interested in binding site residues. In this approach, we can evaluate the quality of a template using spatial matching scores. Thus, we define the efficacy of template T as (Eq. 3)
; that is, when a residue in Q is aligned to a nonbinding residue, its score is 0, because we are only interested in binding site residues. In this approach, we can evaluate the quality of a template using spatial matching scores. Thus, we define the efficacy of template T as (Eq. 3)
 |
where  is the set of aligned residue pairs (i.e., the set of residues in the query sequence that are matched to binding site residues in T). A high efficacy value means that a high percentage of the binding site residues in the template are matched to the residues in the query sequence.
is the set of aligned residue pairs (i.e., the set of residues in the query sequence that are matched to binding site residues in T). A high efficacy value means that a high percentage of the binding site residues in the template are matched to the residues in the query sequence.
Parameter Estimation and the Criterion for Selecting Templates.
The parameters λ and τ are related to biological annotation and evolutionary conservation. Here, we empirically obtain λ ≈ 2 and τ ≈ 4 (Fig. S4) by evaluating the performance of the predicted residues of 100 sequences randomly selected from PSD, with geometric measurements and experimental annotations of binding site residues from UniprotKB/Swiss-Prot. The method of the PR curve described in SI Materials and Methods, Method for Performance Evaluation is used to assess the final prediction. We repeat the process until the prediction performance reaches a steady state.
The selection of templates from PSD for producing RMPs can have a significant effect on the prediction. Spatial scores are used to determine the quality of a template by how well the binding site residues on a template are aligned with those on a query sequence. The majority of the templates have the number of binding site residues ranging from 10 to 50 (3) (Fig. S1A). Therefore, an alignment length of a template T that has ≥50 residues and an associated efficacy of ≥1.25 means that most of the aligned pairs are similar residues. In particular, the efficacy for a template usually has a higher value when the first and last residues are similar or perfect matches to the query sequence. Therefore, these two residues can be used to screen templates in PSD. If the efficacy of a template is ≥1.25, it is qualified as a template. A template with an efficacy ≥2.5 is considered efficacious. In this way, we sample all potential templates in PSD. We apply the above criterion to screen potential templates in PSD.
Matching a Query Sequence Against PSD to Select Templates.
We match the query sequence Q against the templates in the PSD by screening sequence alignments with the Smith–Waterman algorithm (28). We gather the hits with their Smith–Waterman scores and rank the selected hits with their efficacy values. By evaluating the efficacy of each hit template, we can rapidly select useful templates. Within an optimal local alignment between a query and a hit template, the hit template is used to transfer its spatial patterns to the corresponding positions in the primary sequence of the query protein. Iteratively, we collect such hit templates to create an RMP based on the following probabilistic model.
Computing the Binding Likelihood of a Site-Specific Residue.
To compute the binding propensity of a residue ri ∈ Q, i = {1, … , n}, we formulate a probabilistic model for computing the binding likelihood of ri. In the context of protein sequence, the method looks for matched or similar binding site residues and attempts to enumerate all possible combinatory compositions of a binding shape similar to the template. Under the assumption that each unit evolutionary time interval is equally likely, the probability  for residue ri to be a specific binding residue μ is calculated as (Eq. 4)
for residue ri to be a specific binding residue μ is calculated as (Eq. 4)
 |
where Bw ∈ S are the similarity scoring matrices, Ch ∈ G are the gap penalties in the Smith–Waterman algorithm, and Tz ∈ H are the selected templates.
Because it is difficult to know how long ago the query sequence separated from the selected template, we use a set of scoring matrices (S) that represent various degrees of sequence divergence (3, 19, 26). The degree of divergence, however, is measured by the rate of residue substitution in a 20 × 20 matrix. Specifically, we select BLOSUM45, BLOSUM50, BLOSUM62, BLOSUM80, and BLOSUM90 (29) as the set of scoring matrices (S). Using these scoring matrices, we perform template evaluation and the matching between a targeted sequence and a selected template. Then, we use each of them to locate putative binding resides in a query sequence by matching their spatial pattern to that of the binding site residues in the template. For all selected templates, we conduct iterative alignments to assess the potential binding site residues of the query sequence using the above scoring matrices. We thereby obtain the binding likelihoods of site-specific positions of functionally important residues for the query sequence. This approach, called residue matching profiling (RMP), is carried out in a fully automated pipeline, and the method can be used to predict the binding regions of a novel protein.
Supplementary Material
Acknowledgments
We thank Dr. Herbert Edelsbrunner for helpful discussions and Dr. Jie Liang for the Volbl package. This study was supported by National Institutes of Health Grant GM30998.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1102210108/-/DCSupplemental.
References
- 1.Binkowski TA, Adamian L, Liang J. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J Mol Biol. 2003;332:505–526. doi: 10.1016/s0022-2836(03)00882-9. [DOI] [PubMed] [Google Scholar]
- 2.Xie L, Bourne PE. A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites. BMC Bioinformatics. 2007;8(Suppl 4):S9. doi: 10.1186/1471-2105-8-S4-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tseng YY, Li WH. Identification of protein functional surfaces by the concept of a split pocket. Proteins. 2009;76:959–976. doi: 10.1002/prot.22402. [DOI] [PubMed] [Google Scholar]
- 4.Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLOS Comput Biol. 2009;5:e1000585. doi: 10.1371/journal.pcbi.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dundas J, Adamian L, Liang J. Structural signatures of enzyme binding pockets from order-independent surface alignment: A study of metalloendopeptidase and NAD binding proteins. J Mol Biol. 2011;406:713–729. doi: 10.1016/j.jmb.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman HM, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tseng YY, Chen ZJ, Li WH. fPOP: Footprinting functional pockets of proteins by comparative spatial patterns. Nucleic Acids Res. 2010;38:D288–D295. doi: 10.1093/nar/gkp900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tseng YY, Dupree C, Chen ZJ, Li WH. SplitPocket: Identification of protein functional surfaces and characterization of their spatial patterns. Nucleic Acids Res. 2009;37:W384–W389. doi: 10.1093/nar/gkp308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutteridge A, Bartlett GJ, Thornton JM. Using a neural network and spatial clustering to predict the location of active sites in enzymes. J Mol Biol. 2003;330:719–734. doi: 10.1016/s0022-2836(03)00515-1. [DOI] [PubMed] [Google Scholar]
- 10.Petrova NV, Wu CH. Prediction of catalytic residues using Support Vector Machine with selected protein sequence and structural properties. BMC Bioinformatics. 2006;7:312. doi: 10.1186/1471-2105-7-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer JD, Mayer CE, Söding J. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics. 2008;24:613–620. doi: 10.1093/bioinformatics/btm626. [DOI] [PubMed] [Google Scholar]
- 12.Eswar N, et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;50:2.9.1–2.9.31. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 13.Dundas J, et al. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adamian L, et al. Structural model of rho1 GABA(C) receptor based on evolutionary analysis: Testing of predicted protein-protein interactions involved in receptor assembly and function. Protein Sci. 2009;18:2371–2383. doi: 10.1002/pro.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binkowski TA, Freeman P, Liang J. pvSOAR: Detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res. 2004;32:W555–W558. doi: 10.1093/nar/gkh390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boeckmann B, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng EC, Polacco BJ, Babbitt PC. Superfamily active site templates. Proteins. 2004;55:962–976. doi: 10.1002/prot.20099. [DOI] [PubMed] [Google Scholar]
- 18.Torrance JW, Bartlett GJ, Porter CT, Thornton JM. Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J Mol Biol. 2005;347:565–581. doi: 10.1016/j.jmb.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 19.Tseng YY, Dundas J, Liang J. Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns. J Mol Biol. 2009;387:451–464. doi: 10.1016/j.jmb.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glaser F, et al. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- 21.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc Natl Acad Sci USA. 2008;105:5441–5446. doi: 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu CH, et al. The Universal Protein Resource (UniProt): An expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschul SF, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 25.Orengo CA, et al. CATH—a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 26.Tseng YY, Liang J. Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: A Bayesian Monte Carlo approach. Mol Biol Evol. 2006;23:421–436. doi: 10.1093/molbev/msj048. [DOI] [PubMed] [Google Scholar]
- 27.Tourasse NJ, Li WH. Selective constraints, amino acid composition, and the rate of protein evolution. Mol Biol Evol. 2000;17:656–664. doi: 10.1093/oxfordjournals.molbev.a026344. [DOI] [PubMed] [Google Scholar]
- 28.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 29.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.