

Abstract
Background
Pharmacological approaches to inhibit increased leukocyte adhesive interactions in sickle cell disease may represent important strategies for the prevention of vaso-occlusion in patients with this disorder. We investigated, in vitro, the adhesion molecules involved in endothelial-sickle cell disease neutrophil interactions and the effect of simvastatin on sickle cell disease neutrophil adhesion to tumor necrosis factor-α-activated endothelial monolayers (human umbilical vein endothelial cells), and neutrophil chemotaxis.
Design and Methods
Sickle cell disease patients in steady state and not on hydroxyurea were included in the study. Endothelial cells treated, or not, with tumor necrosis factor-α and simvastatin were used for neutrophil adhesion assays. Neutrophils treated with simvastatin were submitted to interleukin 8-stimulated chemotaxis assays.
Results
Sickle cell disease neutrophils showed greater adhesion to endothelial cells than control neutrophils. Adhesion of control neutrophils to endothelial cells was mediated by Mac-1 under basal conditions and by the Mac-1 and LFA-1 integrins under inflammatory conditions. In contrast, adhesion of sickle cell disease neutrophils to endothelium, under both basal and tumor necrosis factor-α-stimulated conditions, was mediated by Mac-1 and LFA-1 integrins and also by VLA-4. Under stimulated inflammatory conditions, simvastatin significantly reduced sickle cell disease neutrophil adhesion, and this effect was reversed by inhibition of nitric oxide synthase. Furthermore, intercellular adhesion molecule-1 expression was significantly abrogated on tumor necrosis factor-α-stimulated endothelium incubated with simvastatin, and statin treatment inhibited the interleukin-8-stimulated migration of both control and sickle cell disease neutrophils.
Conclusions
The integrins Mac-1, LFA-1 and, interestingly, VLA-4 mediate the adhesion of sickle cell disease leukocytes to activated endothelial cell layers, in vitro. Our data indicate that simvastatin may be able to reduce endothelial activation and consequent leukocyte adhesion in this in vitro model; future experiments and clinical trials may determine whether simvastatin therapy could be employed in patients with sickle cell disease, with beneficial effects on vaso-occlusion.
Keywords: inflammation, leukocyte adhesion, simvastatin, sickle cell disease, vaso-occlusion
Introduction
Sickle cell disease (SCD) is characterized by red blood cell sickling and hemolysis; however, inflammatory mechanisms and other types of cells, including leukocytes, also appear to participate in the vaso-occlusive process. Sickle cell crises are often associated with infection. Neutrophil counts are higher in SCD individuals, and polymorphonuclear leukocytosis has been correlated with an increased rate of early death, acute chest syndrome and stroke.1 Data provided by in vitro investigations and in vivo studies of murine models of SCD indicate that the recruitment of large, less deformable, adherent leukocytes to the vascular endothelium, and their interaction with circulating erythrocytes, may impair blood flow and therefore propagate, or even initiate, the vaso-occlusive process.2–4
Neutrophils of SCD individuals are more able to adhere to fibronectin, recombinant intercellular adhesion molecule 1 (ICAM-1) and to endothelial monolayers than are neutrophils from healthy individuals.5,6 A variety of surface adhesion molecules are required for transendothelial migration; the L- and P-selectins are believed to mediate tethering and rolling on the endothelium, while firm adhesion is mediated by the β2 integrins, macrophage 1 (Mac-1; CD11b/CD18) and lymphocyte function associated 1 (LFA-1; CD11a/CD18).7 Expression of Mac-1, an integrin that can bind several extracellular matrix and endothelial proteins, has been shown to be increased on stimulated SCD neutrophils.8,9 Conversely, the very late antigen 4 (VLA-4; CD49d/CD29) integrin is generally thought to be expressed only by eosinophilic leukocytes; however there is evidence to suggest that expression of this adhesion molecule is increased on neutrophils during chronic inflammatory processes.10 Numerous inflammatory markers have been reported to be elevated in the circulation of SCD individuals, including tumor necrosis factor (TNF)-α, C-reactive protein, and interleukins 1β and 8.11–14 Inflammation is hypothesized to contribute to the increased adhesive properties of neutrophils, with the consequent participation of these cells in the vaso-occlusive process.
As such, pharmacological approaches to inhibit increased leukocyte adhesive interactions may represent important strategies for the prevention of SCD vaso-occlusion. Recent reports suggest that statins (HMG-CoA reductase inhibitors) may have clinical applications for the treatment of inflammatory disease states.15 Statins are potent modulators of endothelial cell nitric oxide synthase function and have been shown to upregulate levels of endothelial cell nitric oxide synthase and nitric oxide synthesis.16,17 Statin therapy has been reported to significantly inhibit leukocyte-endothelial cell interactions, independently of any lipid-lowering actions, in normocholesterolemic rats.18 Furthermore, in an experimental SCD mouse model, statin therapy was found to prolong survival following pneumococcal challenge.19
Since leukocyte adhesion to the endothelium may participate in SCD inflammation and, therefore, vaso-occlusion, the first objective of this study was to identify those adhesion molecules involved in endothelial-SCD neutrophil interactions, under in vitro conditions. In addition, we tested the hypothesis that simvastatin may reduce SCD neutrophil adhesion, in vitro, to TNF-α-activated human umbilical vein endothelial cells (HUVEC) and also looked at the effect of this drug on neutrophil chemotaxis, in vitro.
Design and Methods
Patients
A total of 31 SCD patients, diagnosed as homozygous for HbS (using hemoglobin electrophoresis methods and high pressure liquid chromatography), in steady state and cared for at the Hematology and Hemotherapy Center, participated in this study. The patients’ clinical characteristics are presented in Table 1. Patients were not in crisis, were not on hydroxyurea therapy and had not received blood transfusions in the preceding 3 months. Healthy individuals (aged 23–56 years) were used as controls. Informed written consent was obtained from all patients and controls and the ethics committee of the University of Campinas approved the study.
Table 1.
Clinical details of steady-state SCD patients participating in the study.

Materials
Ham’s F12K, Gibco-Invitrogen (Carlsbad, CA, USA) medium, penicillin, streptomycin, gentamycin, heat-inactivated fetal bovine serum, glutamine, trypsin/EDTA solution, N-nitroso-L-arginine methyl ester (L-NAME) and Ficoll-Paque were obtained from Sigma Chemical (Saint Louis, MO, USA). Simvastatin was from Calbiochem (San Diego, CA, USA) and recombinant TNF-α and interleukin-8 (IL-8) were from R&D Systems (Minneapolis, MN, USA). Tissue culture plates were supplied by Costar (USA). Function-inhibiting monoclonal antibodies, anti-CD11a (clone 38), anti-CD11b (clone ICRF44), anti-CD18 (clone YFC118.3), anti-CD49d (clone HP2/1) and anti-CD29 (clone 12G10) and non-specific control monoclonal antibody were purchased from Serotec Ltd. (Oxford, UK) and antibodies used for flow cytometry, anti-CD54-phycoerythin (ICAM-1, clone HA58) and anti-CD106-fluorescein isothiocyanate (VCAM-1, clone 5110C9) were bought from BD Pharmingen (San Diego, CA, USA).
Isolation of human neutrophils from peripheral blood
Whole blood, collected into heparin-containing vacutainer tubes (BD Biosciences, New Jersey, USA), was placed over two layers of Ficoll-Paque of densities of 1.077 and 1.119 g/L. After separation of monocytes and granulocytes by centrifugation at 700 g for 30 min, the granulocyte layer was washed once in phosphate-buffered saline (PBS; pH 7.4), before lysis of contaminating red cells (10 min, 4°C, lysis buffer; 155 mM NH4Cl, 10 mM KHCO3). Cells were washed once again in RPMI medium before resuspension in RPMI medium. Cells were counted using the Advia Hematology System (Bayer, Tarrytown, NY, USA), cytospun onto slides and a cell differential count performed. Neutrophil suspensions were utilized immediately in assays and only when their purity was greater than 92%; contaminating cells were mainly lymphocytes and eosinophils.
Endothelial cell culture
HUVEC were acquired from the American Type Culture Collection (Manassas, VA, USA) and cultured in 25 cm2 flasks, and 96-well tissue culture plates with Ham’s 12K medium supplemented with endothelial cell growth-stimulating factor, 20 mM HEPES, 100 U/mL penicillin, 100 U/mL streptomycin, 2.5 μg/mL fungisone, 2 mM glutamine and 10% fetal bovine serum. Cells were used after the fourth to sixth passage and cultures were maintained at 37°C under a humidified 5% CO2 room air atmosphere; the medium was replaced every 2 days until confluence (3–5 days).
Pretreatment of human umbilical vein endothelial cells
Confluent HUVEC were treated with 10 ng/mL TNF-α in medium for 3 h before adhesion assays. In the experiments involving treatment with simvastatin, medium containing simvastatin (1 μM, in 0.05% dimethylsulfoxide vehicle) was added to confluent cells, in culture for 4 h before performance of the assays. Control values were obtained by adding 0.05% (v/v) dimethylsulfoxide only. Under all conditions, cell viability was greater than 90%, as judged by trypan blue exclusion.
Neutrophil adhesion assay
HUVEC, grown to confluence in 96-well plates, were pretreated, or not, with simvastatin (1 μM) and/or L-NAME (1 mM) for 4 h in the absence or presence of a 10 ng/mL TNF-α stimulus (3 h). Briefly, neutrophils (50 μL; 2×106 cells/mL) were seeded onto the plate wells and cells were allowed to adhere to HUVEC for 30 min at 37°C in 5% CO2. Following incubation, non-adhered cells were discarded and the wells were washed once with PBS. Ham’s F12k (50 μL) was added to each well and varying concentrations of the original cell suspension (0–100 %) were added to empty wells to form a standard curve. Percentage cell adhesion was calculated by measuring the myeloperoxidase content of each well and comparing it to the standard curve for each individual study subject. In some assays, isolated neutrophils were co-incubated with adhesion molecule-blocking monoclonal antibodies, anti-CD11a, anti-CD11b, anti-CD18 anti-CD49d and negative control IgG during adhesion assays.
In vitro neutrophil chemotaxis
Cell migration assays were performed using a 96-well chemotaxis chamber (Chemo Tx; Neuro Probe, Gaithersburg, MD, USA). Twenty-five microliters of cell suspension (4×106 cells/mL in RPMI) were added to the upper compartment of the chamber and separated from the lower chamber, which contained 29 μL of RPMI or IL-8 (100 ng/mL). The upper and lower chambers were separated by a polycarbonate filter (5 μm pore). The chambers were incubated (37°C, 5% CO2) for 120 min. The wells of the upper compartment were emptied by aspiration and then disassembled; cells attached to the upper side of the filter were removed by gentle scraping. To detach adherent neutrophils from the lower surface of the filter, the microtiter plate with attached filter was centrifuged at 1200 rpm for 5 min at room temperature. Plates were then stored frozen overnight before measuring the myeloperoxidase content as described elsewhere.20 The number of migrated neutrophils was calculated by comparing absorbance changes of unknown samples with those of the standard curve, which was formed by measuring the myeloperoxidase values of different neutrophil numbers. For inhibitor incubation, purified neutrophils were pre-incubated with simvastatin (1 μM) before assays for 20 min at 37°C.
Flow cytometry assays
Confluent HUVEC layers were incubated, or not, with simvastatin (1 mM for 4 h) in the absence or presence of a 10 ng/mL TNF-α stimulus (for 3 h). Cells were then washed with PBS (pH 7.4) and detached from 12-well plates with trypsin/EDTA (3 min, 37°C). After washing twice in PBS, cells were incubated with anti-CD54-phycoerythin and anti-CD106-fluorescein isothiocyante monoclonal antibodies (30 min, at room temperature, in the dark; Becton Dickinson, CA). After washing twice with PBS, cell fluorescence (10,000 cells) was determined immediately with a FACScalibur (Becton Dickinson, CA, USA) and analyzed using FACS Diva software. Results are expressed as mean cell fluorescence intensity values compared to those of isotype controls.
Statistical analysis
Results for non-parametric data, comparing control and patient populations, are depicted in graphs as medians and ranges. Differences across groups were determined by the Friedman test (repeated measures) and, when the P value was less than 0.05, specific groups were compared by Dunn’s multiple comparison test. Parametric data (HUVEC cultures) were analyzed by ANOVA (repeated measures), followed by Bonferroni’s test. Statistical significance was established as P values less than 0.05.
Results
Adhesion of control and sickle cell disease neutrophils to non-stimulated and tumor necrosis factor-α-stimulated endothelial cells
Neutrophils from SCD patients showed significantly greater spontaneous adhesion to HUVEC than control neutrophils in static adhesion assays. Pre-treatment of HUVEC with 10 ng/mL TNF-α (3 h, 37°C, 5% CO2) was used to reflect adhesion to activated, inflamed endothelium. Adhesion of control and SCD neutrophils to TNF-α-activated HUVEC was significantly higher than that of adhesion to non-activated HUVEC (Figure 1).
Figure 1.

Adhesion of control and SCD neutrophils to non-stimulated (basal) and TNF-α-stimulated HUVEC. Neutrophils (2x106 cells/mL) from control (n=13) or SCD patients (n=16) were allowed to adhere to HUVEC for 30 min at 37°C, 5% CO2. Results are expressed as percentage of cells adhered (median and range). ***P<0.001, TNF-α-stimulated HUVEC compared to basal; Wilcoxon’s matched pairs test. #P<0.05, Median values differ significantly for SCD and to control cells; Mann-Whitney test.
Effect of adhesion molecule-blocking monoclonal antibodies on control and sickle cell disease neutrophil adhesion to endothelial cells
Adhesion of control neutrophils to HUVEC was significantly inhibited by a CD11b function-blocking monoclonal antibody, but not by monoclonal antibodies against CD11a, the VLA-4-integrin subunit, CD49d, or a non-specific negative control monoclonal antibody (Figure 2A). In contrast, the adhesion of SCD neutrophils to HUVEC was significantly inhibited by the anti-CD11a, the anti-CD11b and anti-CD49d monoclonal antibodies, while a negative control monoclonal antibody did not significantly affect the adhesion of SCD neutrophils (Figure 2B).
Figure 2.

Neutrophil adhesion to HUVEC in the presence of integrin-specific blocking monoclonal antibodies. Neutrophils (2x106 cells/mL) from controls (A, n=6) or from SCD individuals (B, n=8) were allowed to adhere to HUVEC for 30 min at 37°C, 5% CO2, in the presence or absence of integrin-blocking monoclonal antibody, as indicated. *P<0.05; **P<0.01; ***P<0.001, compared to basal adhesion; Friedman’s test, followed by Dunn’s multiple comparison between selected groups and basal adhesion.
Effect of co-incubation with adhesion molecule-blocking monoclonal antibodies on adhesion of control and sickle cell disease neutrophils to tumor necrosis factor-α-stimulated endothelial cells
Under inflammatory conditions, following stimulation of HUVEC with TNF-α (10ng/mL) (3 h, 37°C, 5% CO2), adhesion of control neutrophils to HUVEC was significantly inhibited by anti-CD11a and anti-CD11b monoclonal antibodies (Figure 3A). In contrast, adhesion of SCD neutrophils to HUVEC was significantly inhibited by monoclonal antibodies to CD11a, CD11b and anti-CD49d, but not the negative control monoclonal antibody (Figure 3B).
Figure 3.

Neutrophil adhesion to TNF-α-stimulated HUVEC in the presence of integrin-specific blocking monoclonal antibody. Neutrophils (2x106 cell/mL) from control individuals (A, n=4) or from SCD patients (B, n=5) were allowed to adhere to TNF-α-stimulated HUVEC for 30 min at 37°C, 5% CO2, in the presence or absence of integrin-blocking monoclonal antibody, as indicated. *P<0.05; **P<0.01; ***P<0.001, compared to basal adhesion; Friedman’s test, followed by Dunn’s multiple comparison between selected groups and basal adhesion.
Effect of simvastatin treatment of endothelial layers on control and sickle cell disease neutrophil adhesion to tumor necrosis factor-α-stimulated and non-stimulated endothelial cells
Treatment of non-stimulated HUVEC cells with simvastatin did not alter the adhesion of either control or SCD neutrophils to HUVEC (Figure 4). In contrast, when HUVEC were pretreated with TNF-α (10 ng/mL), simultaneous pre-treatment of endothelial cells with simvastatin significantly reduced SCD neutrophil adhesion, compared to non-simvastatin-treated HUVEC. In contrast, simvastatin did not alter the adhesion of control neutrophils to TNF-α-stimulated HUVEC (Figure 4).
Figure 4.

Adhesion of control (A) and SCD (B) neutrophils to HUVEC in the presence of simvastatin. Neutrophils from control individuals (n=8) or SCD patients (n=11) were allowed to adhere to HUVEC or to TNF-α-stimulated HUVEC cells in the presence or not of 1 μM simvastatin (SIM) or 1 μM simvastatin and 1 mM L-NAME (SIM+L-N, n=6). ***P<0.001, TNF-α compared to basal adhesion; ••P<0.01 compared to TNF-α alone; ≈≈P<0.01 compared to TNF/SIM; Friedman’s test, followed by Dunn’s multiple comparison.
Importantly, co-incubation of TNF-α-stimulated HUVEC with both simvastatin and the nitric oxide synthase inhibitor, L-NAME (1 mM), reversed the decrease in SCD neutrophil adhesion observed when HUVEC were incubated with simvastatin alone, indicating a role for nitric oxide synthesis in the effect of simvastatin (Figure 4B). L-NAME had no significant effect on neutrophil adhesion to non-TNF-α-stimulated HUVEC, nor to TNF-α-stimulated HUVEC in the absence of simvastatin (P>0.05, data not shown).
Effect of simvastatin treatment on the expression of adhesion molecules on the surface of non-stimulated and tumor necrosis factor-α-stimulated endothelial cells
Flow cytometry assays demonstrated that non-stimulated HUVEC cells had high surface expression of ICAM-1 (Figure 5A); 90.4±0.5% of cells expressed ICAM-1, with a mean fluorescence intensity (MFI) of 13,949±244 units (n=4). Following stimulation of HUVEC with TNF-α (10 ng/mL for 3 h), the surface expression of ICAM-1 was considerably increased to 74,567±6,791 MFI (P<0.001; with 97.4±0.06% cells expressing this adhesion molecule); conversely, when cells were pre-treated with simvastatin (1 μM) for 1 h before the TNF-α stimulation, a significant inhibition of ICAM-1 surface expression was observed (39,352±4,559 MFI; P<0.01; 94.9±0.91%; Figure 5A). In contrast, the level of VCAM-1 expression on resting HUVEC was very low (0.93±0.05%; 125.0±3.4 MFI, n=4). TNF-α stimulation slightly, but significantly, increased VCAM-1 expression on HUVEC (4.23±1.19%; 169.8±10.1 MFI, P<0.01), while pretreatment with simvastatin was able to prevent this TNF-α-induced increase in VCAM-1 (1.08±0.11%; 132.3±5.3 MFI, P<0.01) (Figure 5B).
Figure 5.

Effect of simvastatin on TNF-α-stimulated expression of ICAM-1 (A) and VCAM-1 (B) on HUVEC. HUVEC were stimulated, or not, with TNF-α (10 ng/mL; for 3 h) following their pre-incubation (or not) with 1 μM simvastatin (Sim). Surface ICAM-1 and VCAM-1 expression was determined by flow cytometry using an anti-CD54 phycoerythrin antibody and anti-CD106 fluorescein isothiocyanate, respectively. Data are expressed as mean MFI±SEM (n=4) for ICAM-1 (A) and % positive cells ± SEM (n=4) for VCAM-1 (B). *P<0.05; **P<0.01, ***P<0.001, compared to basal adhesion. ##P<0.01 compared to TNF-α alone. Repeated measures analysis, followed by Bonferroni’s test.
Effects of simvastatin on the chemotaxis of control and sickle cell disease neutrophils
The spontaneous in vitro migration (without the presence of a chemotactic stimulus) of neutrophils from control individuals and SCD patients was similar, although SCD neutrophils demonstrate a tendency, which was not statistically significant, to greater migration (Figure 5). In contrast, when neutrophils were allowed to migrate towards an IL-8 chemotactic stimulus (100 ng/mL), the migration of both control and SCD neutrophils was increased compared to spontaneous migration. Importantly, co-incubation of neutrophils with simvastatin (1 μM) significantly reduced both control and SCD neutrophil chemotaxis towards IL-8 (Figure 6).
Figure 6.
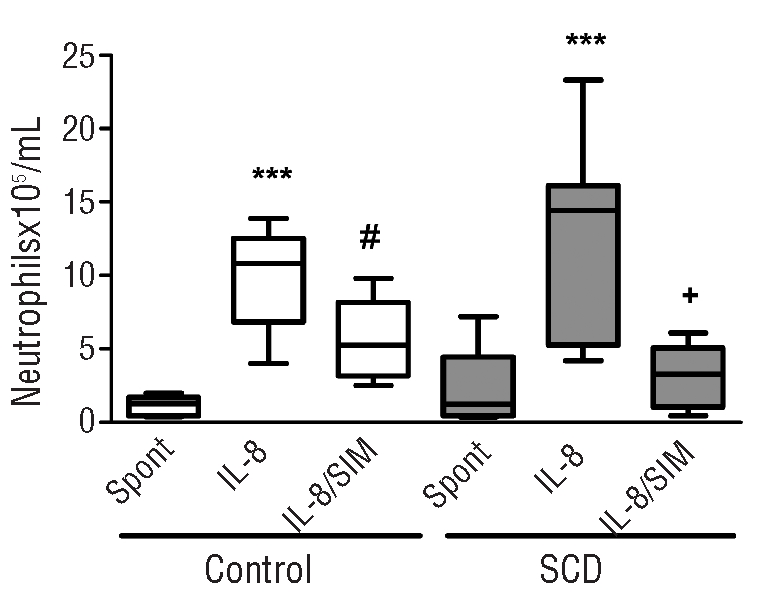
Effect of simvastatin on spontaneous and IL8-stimulated neutrophil chemotaxis. Spontaneous chemotaxis (Spont) and IL-8 (100 ng/mL)-stimulated chemotaxis of neutrophils from control (n=6) and SCD individuals (n=6) was measured following the pre-incubation (or not) of neutrophils with simvastatin (SIM) (1 μM). ***P<0.001, compared with spontaneous chemotaxis; #P<0.05 compared with IL-8-stimulated control neutrophil chemotaxis; +P<0.05, compared with IL-8-stimulated SCD neutrophil chemotaxis; Friedman’s test, followed by Dunn’s multiple comparison.
Discussion
Leukocyte adhesion to the microvascular endothelium and the formation of leukocyte-red blood cell aggregates may make an important contribution to the initiation and propagation of vaso-occlusion in SCD4,21 and, thus, therapeutic options aimed at inhibiting leukocyte adhesive mechanisms could be valuable for preventing vaso-occlusive processes. In vitro studies have demonstrated that SCD neutrophils have an increased capacity to adhere to HUVEC, in vitro, compared to control neutrophils5 and similar studies showed that SCD neutrophils also display augmented adhesion to integrin ligands such as fibronectin and ICAM-1.6 Pre-stimulation of HUVEC with TNF-α, in an assay to mimic inflammatory conditions, resulted in the increased adhesion of both SCD and control leukocytes to stimulated endothelial cells,22 a finding that may be of physiological relevance since increased circulating levels of TNF-α have been reported in a number of SCD populations, both during steady-state14,23 and painful crises.24 Furthermore, plasma TNF-α levels may correlate with bronchial hyperreactivity, lung inflammation and polymorphonuclear leukocyte recruitment to airways.25 As such, TNF-α and other inflammatory molecules may play a role in the endothelial activation that appears central to the inflammatory mechanisms of SCD26 and this activation probably contributes to increase the adhesion of leukocytes to the vascular wall.
While the role of leukocyte-endothelial interactions in vaso-occlusive processes has been clearly indicated by evidence from in vitro and sickle cell mouse models,3,4,9,21,22,27 confirmation of these findings has yet to be found in vivo in humans with SCD. Earlier intravital procedures, performed in the nailfold capillaries of human SCD volunteers, established the participation of red cell-endothelium interactions in red cell entrapment and decreased flow in microcapillaries and the authors of this study further suggested that leukocyte-endothelium adhesive mechanisms may also contribute to such occlusion.28 More recently, computer-assisted intravital microscopy has been used to examine the conjunctival microcirculation of SCD individuals;29,30 however, the studies were restricted to observations of dynamic and morphometric alterations. The difficulty in performing in vivo assays in humans with SCD is a limitation in the field of characterizing the cellular interactions that occur during the vaso-occlusive process, but it is hoped that the development of modern intravital techniques may provide better resolution of the microvascular circulation in humans thus helping to overcome such limitations. We used an in vitro model to study the molecules that may participate in neutrophil-endothelial cell interactions under inflammatory conditions and to investigate the potential that statins (namely, simvastatin) may have to diminish such interactions. Although this in vitro model has some limitations in that it is a static assay and does not account for the presence of other types of cells, endogenous cytokines and other inflammatory mediators, it may be useful for identifying molecules that could represent possible drug targets worthy of future investigation. The possible use of a flow adhesion approach, to afford slightly more physiological conditions to this assay, may be important for confirmation of the findings.
Our data indicate that the adhesion of control neutrophils to endothelial cells, in vitro, is mediated mainly by the Mac-1 integrin (CD11b/18) with a contribution from the LFA-1 integrin (CD11a/18), under inflammatory conditions. In contrast, the adhesion of SCD neutrophils to endothelium (under both basal and TNF-α-stimulated conditions), at least in vitro, appears to be mediated by the Mac-1 and LFA-1 integrins and also by VLA-4 (CD49d/CD29), an integrin expressed at low levels on neutrophils during certain inflammatory conditions.31 Previous studies from our laboratory have indicated that, under experimental conditions similar to those used herein, neither Mac-1 (CD11b/CD18) nor LFA-1 (CD11a/CD18) surface expression is significantly altered on non-stimulated SCD neutrophils.6,9 Integrins, including Mac-1 and LFA-1,32,33 are known to mediate adhesive interactions via conformational changes that result in increased ligand affinity and data consistently indicate that increases in integrin affinity, rather than significant changes in surface protein expression, bring about the observed increase in adhesive properties of SCD neutrophils. The Mac-1/LFA-1 integrin ligand, ICAM-1, was found to be highly expressed on resting endothelial cells and further increased by TNF-α, suggesting that this adhesion molecule may be the major ligand for neutrophil adhesion on HUVEC. Interestingly, we found evidence for a role of the β1 integrin, VLA-4 (CD49d/CD29), in SCD neutrophil adhesion to endothelium. Previous data from individuals of the same SCD population indicated a low level of expression of CD49d on the surface of SCD neutrophils.6 Involvement of this β1 integrin has been implicated in the recruitment of neutrophils during chronic inflammation,10,34 and it can be postulated that the inflammatory state that is associated with SCD may stimulate an increased function of this adhesion molecule on neutrophils. Both the resting and TNF-α-stimulated endothelial cells used in this study were found to express extremely low levels of VCAM-1, a primary ligand for the VLA-4 integrin. However, HUVEC are known to express other VLA-4 ligands, such as fibronectin (found in association with the cell surface) and Lu/BCAM, particularly after TNF-α stimulation.35–37 It is, therefore, possible that the VLA-4-mediated interactions of SCD neutrophils observed in this study are mediated by these adhesion molecules, although future studies are necessary to clarify this point.
Treatment of HUVEC with simvastatin appears to protect the endothelium from an inflammatory stimulus, leading to a significant reduction in the adhesion of SCD neutrophils to TNF-α–stimulated HUVEC. Recent studies suggest that statins have anti-inflammatory properties in the atherosclerotic plaque.38 The mechanisms involved in inhibiting inflammation have been related to the inhibition of the expression of endothelial adhesion molecules, such as ICAM-1.39 Accordingly, the surface presentation of ICAM-1 on TNF-α-stimulated endothelial cells was found to be significantly diminished by simvastatin pretreatment under the conditions utilized in our assays. Interestingly, we demonstrated that, under TNF-α-stimulated inflammatory conditions, simvastatin significantly reduced SCD neutrophil adhesion, compared to non-simvastatin-treated HUVEC; in contrast, simvastatin did not alter the adhesion of control neutrophils to HUVEC. These results indicate that, under marked inflammatory conditions, simvastatin may reduce the ability of endothelial cells to interact with leukocytes. This effect may be partly explained by some of the properties of statins, which appear to be able to restore endothelial function, and increase the production of endothelial nitric oxide, by endothelial stimulation and sub-regulation of endothelial nitric oxide synthase.40 Importantly, co-incubation of TNF-α-stimulated HUVEC with both simvastatin and the nitric oxide synthase inhibitor, L-NAME, reversed the decrease in SCD neutrophil adhesion to HUVEC observed for simvastatin alone, indicating that the stimulation of nitric oxide synthase in HUVEC may play a role in the protective effects of simvastatin on the endothelial layer. A preliminary study investigating the effect of short-term simvastatin administration on markers of vascular dysfunction in patients with SCD, related increased nitric oxide availability and anti-inflammatory effects in treated individuals.41
Finally, chemotaxis assays indicated a tendency (P>0.05) towards higher migratory properties in SCD neutrophils, compared to control neutrophils, as previously reported.42 Interestingly, IL-8-stimulated migration of both control and SCD neutrophils was significantly inhibited by pre-treatment of the neutrophils with simvastatin. These data are of interest, since they indicate that simvastatin may also have an anti-inflammatory effect on the neutrophils themselves and not just on the endothelium. The chemotactic properties of neutrophils are central to their inflammatory response function and inhibition of neutrophil chemotaxis by simvastatin indicates another potential beneficial effect of this drug in SCD.38,43
In summary, our data illustrate that the Mac-1 and LFA-1 integrins and, interestingly, VLA-4 may mediate the adhesion of leukocytes to activated endothelial cell layers, at least in vitro. Our data indicate that simvastatin reduces endothelial activation and consequent leukocyte adhesion in this in vitro model; future experiments and clinical trials are required to determine whether simvastatin therapy and other anti-inflammatory approaches could be employed in patients with SCD, with beneficial effects on vaso-occlusion.
Footnotes
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
Funding: this study was supported by FAPESP and CNPq, Brazil. The Hematology and Hemotherapy Center, UNI-CAMP forms part of the National Institute of Blood, Brazil (INCT de Sangue –CNPq/MCT/FAPESP).
References
- 1.Okpala I. The intriguing contribution of white blood cells to sickle cell disease - a red cell disorder. Blood Rev. 2004;18(1):65–73. doi: 10.1016/s0268-960x(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 2.Chiang EY, Frenette PS. Sickle cell vaso-occlusion. Hematol Oncol Clin North Am. 2005;19(5):771–84. doi: 10.1016/j.hoc.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Brittain JE, Knoll CM, Ataga KI, Orringer EP, Parise LV. Fibronectin bridges monocytes and reticulocytes via integrin alpha4beta1. Br J Haematol. 2008;141(6):872–81. doi: 10.1111/j.1365-2141.2008.07056.x. [DOI] [PubMed] [Google Scholar]
- 4.Chaar V, Picot J, Renaud O, Bartolucci P, Nzouakou R, Bachir D, et al. Aggregation of mononuclear and red blood cells through α4β1-Lu/BCAM interaction in sickle cell disease. Haematologica. 2010;95(11):1841–8. doi: 10.3324/haematol.2010.026294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fadlon E, Vordermeier S, Pearson TC, Mire-Sluis AR, Dumonde DC, Phillips J, et al. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998;91(1):266–74. [PubMed] [Google Scholar]
- 6.Canalli AA, Franco-Penteado CF, Saad STO, Conran N, Costa FF. Increased adhesive properties of neutrophils in sickle cell disease may be reversed by pharmacological nitric oxide donation. Haematologica. 2008;93(4):605–9. doi: 10.3324/haematol.12119. [DOI] [PubMed] [Google Scholar]
- 7.Chesnutt BC, Smith DF, Raffler NA, Smith ML, White EJ, Ley K. Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation. 2006;13(2):99–109. doi: 10.1080/10739680500466376. [DOI] [PubMed] [Google Scholar]
- 8.Lum AF, Wun T, Staunton D, Simon SI. Inflammatory potential of neutrophils detected in sickle cell disease. Am J Hematol. 2004;76(2):126–33. doi: 10.1002/ajh.20059. [DOI] [PubMed] [Google Scholar]
- 9.Assis A, Conran N, Canalli AA, Lorand-Metze I, Saad ST, Costa FF. Effect of cytokines and chemokines on sickle neutrophil adhesion to fibronectin. Acta Haematol. 2005;113(2):130–6. doi: 10.1159/000083451. [DOI] [PubMed] [Google Scholar]
- 10.Burns JA, Issekutz TB, Yagita H, Issekutz AC. The alpha 4 beta 1 (very late antigen (VLA)-4, CD49d/CD29) and alpha 5 beta 1 (VLA-5, CD49e/CD29) integrins mediate beta 2 (CD11/CD18) integrin-independent neutrophil recruitment to endotoxin-induced lung inflammation. J Immunol. 2001;166(7):4644–9. doi: 10.4049/jimmunol.166.7.4644. [DOI] [PubMed] [Google Scholar]
- 11.Croizat H. Circulating cytokines in sickle-cell patients during steady-state. Br J Haematol. 1994;87(3):592–7. doi: 10.1111/j.1365-2141.1994.tb08318.x. [DOI] [PubMed] [Google Scholar]
- 12.Francis RB, Jr, Haywood LJ. Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. J Natl Med Assoc. 1992;84(7):611–5. [PMC free article] [PubMed] [Google Scholar]
- 13.Malave I, Perdomo Y, Escalona E, Rodriguez E, Anchustegui M, Malave H, et al. Levels of tumor necrosis factor alpha/cachectin (TNF alpha) in sera from patients with sickle cell disease. Acta Haematol. 1993;90(4):172–6. doi: 10.1159/000204452. [DOI] [PubMed] [Google Scholar]
- 14.Lanaro C, Franco-Penteado CF, Albuqueque DM, Saad ST, Conran N, Costa FF. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J Leukoc Biol. 2009;85(2):235–42. doi: 10.1189/jlb.0708445. [DOI] [PubMed] [Google Scholar]
- 15.Shovman O, Levy Y, Gilburd B, Shoenfeld Y. Antiinflammatory and immunomodulatory properties of statins. Immunol Res. 2002;25(3):271–85. doi: 10.1385/IR:25:3:271. [DOI] [PubMed] [Google Scholar]
- 16.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272(50):31725–9. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 17.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97(12):1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 18.Pruefer D, Scalia R, Lefer AM. Simvastatin inhibits leukocyte-endothelial cell interactions and protects against inflammatory processes in normocholesterolemic rats. Arterioscler Thromb Vasc Biol. 1999;19 (12):2894–900. doi: 10.1161/01.atv.19.12.2894. [DOI] [PubMed] [Google Scholar]
- 19.Rosch JW, Boyd AR, Hinojosa E, Pestina T, Hu Y, Persons DA, et al. Statins protect against fulminant pneumococcal infection and cytolysin toxicity in a mouse model of sickle cell disease. J Clin Invest. 2010;120 (2):627–35. doi: 10.1172/JCI39843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradley PP, Priebat DA, Christensen RD, Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78(3):206–9. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 21.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Nat Acad Sci USA. 2002;99(5):3047–51. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finnegan EM, Turhan A, Golan DE, Barabino GA. Adherent leukocytes capture sickle erythrocytes in an in vitro flow model of vaso-occlusion. Am J Hematol. 2007;82(4):266–75. doi: 10.1002/ajh.20819. [DOI] [PubMed] [Google Scholar]
- 23.Tavakkoli F, Nahavandi M, Wyche MQ, Perlin E. Plasma levels of TNF-alpha in sickle cell patients receiving hydroxyurea. Hematology. 2004;9(1):61–4. doi: 10.1080/1024533032000158869. [DOI] [PubMed] [Google Scholar]
- 24.Pathare A, Al Kindi S, Alnaqdy AA, Daar S, Knox-Macaulay H, Dennison D. Cytokine profile of sickle cell disease in Oman. Am J Hematol. 2004;77(4):323–8. doi: 10.1002/ajh.20196. [DOI] [PubMed] [Google Scholar]
- 25.Kips JC, Tavernier J, Pauwels RA. Tumor necrosis factor causes bronchial hyperresponsiveness in rats. Am Rev Respir Dis. 1992;145(2 Pt 1):332–6. doi: 10.1164/ajrccm/145.2_Pt_1.332. [DOI] [PubMed] [Google Scholar]
- 26.Conran N, Costa FF. Hemoglobin disorders and endothelial cell interactions. Clin Biochem. 2009;42(18):1824–38. doi: 10.1016/j.clinbiochem.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Kasschau MR, Barabino GA, Bridges KR, Golan DE. Adhesion of sickle neutrophils and erythrocytes to fibronectin. Blood. 1996;87(2):771–80. [PubMed] [Google Scholar]
- 28.Lipowsky HH, Sheikh NU, Katz DM. Intravital microscopy of capillary hemodynamics in sickle cell disease. J Clin Invest. 1987;80(1):117–27. doi: 10.1172/JCI113036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung AT, Chen PC, Larkin EC, Duong PL, Ramanujam S, Tablin F, et al. Microvascular abnormalities in sickle cell disease: a computer-assisted intravital microscopy study. Blood. 2002;99(11):3999–4005. doi: 10.1182/blood.v99.11.3999. [DOI] [PubMed] [Google Scholar]
- 30.Cheung AT, Miller JW, Craig SM, To PL, Lin X, Samarron SL, et al. Comparison of real-time microvascular abnormalities in pediatric and adult sickle cell anemia patients. Am J Hematol. 2010;85(11):899–901. doi: 10.1002/ajh.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao JX, Issekutz AC. The beta 1 integrin, very late activation antigen-4 on human neutrophils can contribute to neutrophil migration through connective tissue fibroblast barriers. Immunology. 1997;90(3):448–54. doi: 10.1111/j.1365-2567.1997.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eniola-Adefeso O, Huang RB, Smith CW. Kinetics of LFA-1 mediated adhesion of human neutrophils to ICAM-1--role of Eselectin signaling post-activation. Ann Biomed Eng. 2009;37(4):737–48. doi: 10.1007/s10439-009-9647-8. [DOI] [PubMed] [Google Scholar]
- 33.Hogg N, Leitinger B. Shape and shift changes related to the function of leukocyte integrins LFA-1 and Mac-1. J Leukoc Biol. 2001;69(6):893–8. [PubMed] [Google Scholar]
- 34.Issekutz AC, Nakazato S, Issekutz TB. Differential roles of VLA-4(CD49d/CD29) and LFA-1(CD11a/CD18) integrins and E-and P-selectin during developing and established active or adoptively transferred adjuvant arthritis in the rat. Immunol Cell Biol. 2003;81(5):397–408. doi: 10.1046/j.1440-1711.2003.01187.x. [DOI] [PubMed] [Google Scholar]
- 35.Pellegatta F, Radaelli A, Ferrero E, Toninelli E, Vidal MJ, Chierchia SL, et al. Inducible nitric oxide synthase modulates fibronectin production in the EA.hy926 cell line and cultured human umbilical vein endothelial cells. J Cardiovasc Pharmacol. 1994;24(6):1014–9. doi: 10.1097/00005344-199424060-00023. [DOI] [PubMed] [Google Scholar]
- 36.Daramola OA, Heyderman RS, Klein NJ, Shennan GI, Levin M. Detection of fibronectin expression by human endothelial cells using a enzyme-linked immunosorbent assay (ELISA): enzymatic degradation by activated plasminogen. J Immunol Methods. 1997;202(1):67–75. doi: 10.1016/s0022-1759(96)00237-2. [DOI] [PubMed] [Google Scholar]
- 37.El Nemer W, Gauthier E, Wautier MP, Rahuel C, Gane P, Galacteros F, et al. Role of Lu/BCAM in abnormal adhesion of sickle red blood cells to vascular endothelium. Transfus Clin Biol. 2008;15(1–2):29–33. doi: 10.1016/j.tracli.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 38.Kawakami A, Tanaka A, Nakajima K, Shimokado K, Yoshida M. Atorvastatin attenuates remnant lipoprotein-induced monocyte adhesion to vascular endothelium under flow conditions. Circ Res. 2002;91(3):263–71. doi: 10.1161/01.res.0000028454.42385.8b. [DOI] [PubMed] [Google Scholar]
- 39.Alber HF, Frick M, Sussenbacher A, Dorler J, Dichtl W, Stocker EM, et al. Effect of atorvastatin on peripheral endothelial function and systemic inflammatory markers in patients with stable coronary artery disease. Wien Med Wochenschr. 2007;157(3–4):73–8. doi: 10.1007/s10354-007-0377-y. [DOI] [PubMed] [Google Scholar]
- 40.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6(9):1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoppe CC, Stewart KR, Higa A, Larkin S, Liao JK, Styles LA, et al. The effect of short-term simvastatin on markers of vascular dysfunction in patients with sickle cell disease (SCD) Blood. 2009;114:Abs260. [Google Scholar]
- 42.Canalli AA, Franco-Penteado CF, Traina F, Saad ST, Costa FF, Conran N. Role for cAMP-protein kinase A signalling in augmented neutrophil adhesion and chemotaxis in sickle cell disease. Eur J Haematol. 2007;79(4):330–7. doi: 10.1111/j.1600-0609.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 43.Martin-Ventura JL, Blanco-Colio LM, Gomez-Hernandez A, Munoz-Garcia B, Vega M, Serrano J, et al. Intensive treatment with atorvastatin reduces inflammation in mononuclear cells and human atherosclerotic lesions in one month. Stroke. 2005;36(8):1796–800. doi: 10.1161/01.STR.0000174289.34110.b0. [DOI] [PubMed] [Google Scholar]