

Abstract
We describe the construction of a soluble protein carrying the N-terminal extracellular domain (ECD) of the α7 subunit of the nicotinic acetylcholine receptor. The approach was to fuse the α7 ECD at the C and N termini of several monomeric and pentameric soluble carrier proteins and to investigate the soluble expression of the product in Escherichia coli. An initial screening of six carrier proteins resulted in the selection of a fusion protein comprising, from the N to the C terminus, the maltose binding protein, a 17-aa linker containing an enterokinase binding site, and the α7 ECD. This protein is soluble upon expression in bacteria and is purified by affinity chromatography. It binds the competitive nicotinic antagonist α-bungarotoxin with 2.5 μM affinity and displays a CD spectrum corresponding to a folded protein. The method might be suitable to produce large quantities of protein for crystallization and immunochemical experiments.
Nicotinic acetylcholine receptors (nAChRs) are members of the superfamily of ligand-gated ion channels that include the glycine, γ-aminobutyric acid type A, GluR-C, and 5HT3 receptors. These proteins, specialized in transduction of chemical signals to electrical response, display significant amino acid sequence similarity, transmembrane topology, and overall secondary, tertiary, and quaternary structure (1).
Despite the large body of experimental data and predictions about their functional organization (2), the three-dimensional structure at atomic resolution is still missing. Too large to be investigated by NMR, these proteins display features that have made many attempts of crystallization unsuccessful. They are, altogether, integral transmembrane proteins, glycosylated, form homopentamers or heteropentamers composed of several types of homologous subunits pseudosymmetrically arranged around an axis perpendicular to the membrane. Furthermore, the nature of the subunit interface appears critical, as it contributes to both the neurotransmitter binding site and the ion channel. Moreover, these ligand-gated ion channels are allosteric proteins, in equilibrium between at least three interconvertible conformations. Finally, these receptors often were found highly difficult to express in large quantities. To date, the best source of nAChR is the electric organ of Torpedo, from which milligram amounts of muscle-type heterooligomeric receptors can be prepared in membrane-bound or detergent-purified forms. The membrane-bound material made possible low-resolution electron microscopy of two-dimensional crystalline arrays of the receptor molecule down to 4.7 Å (3).
The currently accepted transmembrane topology of each subunit consists in a large extracellular N-terminal domain (ECD), which represents about half of the subunit sequence, followed by the transmembrane and cytoplasmic regions (1). The construction of a functional chimera carrying the ECD of the homooligomeric α7 nAChR, fused to the complementary C-terminal domain of the homooligomeric 5HT3 receptor, suggests that both domains may fold autonomously (4). Moreover, the chimera displayed a typical α7 pharmacology, indicating that the extracellular domain contains all of the structural elements contributing to the neurotransmitter binding site.
This work was aimed at developing a technique for overexpressing in bacteria the ECD of the α7 subunit in a ligand-binding conformation. After the observation that proteins could be grafted at the N-terminal end of the full receptor with retention of functionality (5), we explored the expression of the α7 ECD as a fusion with bacterial proteins and a convenient linker.
Experimental Procedures
Oligonucleotides (Table 1) were custom-synthesized by MWG Biotec (Ebersberg, Germany). Protein concentration was estimated by the modified Bradford procedure reported by Read and Northcote (6). SDS/polyacrylamide gel electrophoresis was performed as described by Laemmli (7). Molecular weight standards were supplied by Sigma. CD measurements were performed with a spectropolarimeter JASCO J-715 in 5- or 10-mm quartz cells, and protein solutions (MalE, 16 μg/ml; Mal-ent-α7 1–196, 18 μg/ml) were measured against 25 mM potassium phosphate, pH 7.2, at 20°C.
Table 1.
Oligonucleotides used in this study
| Designation | 5′-sequence-3′ |
|---|---|
| EC-MalE-Rbs-EcoRI | ataatagaattcattaaagaggagaaattaactatgaaaatcgaagaaggtaaactggtaatctgg |
| EC-MalE-1 | catccccgaggttgttgttattgttattgttgttgttgttcgagctcgaattagtctgcgcgtc |
| EC-MalE-NotI-BamHI-2 | tattatggatcctattattatagcggccgctttatcgtcatcatccccgaggttgttgttattgttattg |
| RN-α7-NotI-Vo | ataataatagcggccgctgagttccagaggaggctgtacaaggagctg |
| RN-α7-D196-BamHI | tattatggatccttaatctgggtaaggctctttgcagcactc |
Construction of Expression Plasmid pNCO-MalE.
The malE gene coding for the maltose binding protein from Escherichia coli (GenBank accession no. AAB59056) was amplified by PCR by using the oligonucleotides EC-MalE-Rbs-EcoRI and EC-MalE-1 as primers (Table 1) and the plasmid pMALC2 (New England Biolabs) as template. The resulting fragment was purified and extended in the 3′ direction by using the primer combination EC-MalE-Rbs-EcoRI and EC-MalE-NotI-BamHI-2 to a final size of 1,227 bp. The resulting fragment was purified and digested with EcoRI and BamHI. The digested fragment was ligated into the plasmid pNCO113 (8), which had been treated with the same restriction enzymes. The resulting plasmid pNCO-MalE was transformed into the E. coli strain XL1-Blue, affording the recombinant strain XL1-pNCO-MalE.
Construction of Expression Plasmid pNCO-Mal-ent-α7 1–196.
The gene coding for the N-terminal domain of the rat α7 receptor (GenBank accession no. AAC33136) was amplified by PCR by using the oligonucleotides RN-α7-NotI-Vo and RN-α7-D196-BamHI as primers (Table 1) and cDNA from rat as template. The resulting 620-bp fragment was purified and digested with NotI and BamHI. The cleaved fragment was ligated into the plasmid pNCO-MalE (this study), which had been treated with the same restriction enzymes. The resulting plasmid pNCO-Mal-ent-α7 1–196 was transformed into the E. coli strain XL1-Blue, yielding the recombinant strain XL1-pNCO-Mal-ent-α7 1–196. DNA was sequenced by the automated dideoxynucleotide method (9) by using a 377 A Prism sequencer from Perkin–Elmer.
Transformation of E. coli XL1-Blue Cells.
E. coli XL1-Blue cells were transformed according to Bullock et al. (10). Transformants were selected on LB agar plates supplemented with ampicillin (150 mg/liter). The constructs were monitored by restriction analysis and DNA sequencing. In the expression plasmids, the genes coding for the fusion proteins are under control of the T5 promotor and the lac operator. Protein expression was induced by the addition of 2 mM isopropyl β-D-thiogalactoside to the lac operator.
Protein Purification.
Recombinant E. coli strains were grown in LB medium containing ampicillin (150 mg/liter) at 37°C with shaking. At an optical density of 0.6, isopropyl β-D-thiogalactoside was added to a final concentration of 2 mM, and incubation was continued for 6 h. The cells were harvested by centrifugation, washed with 0.9% NaCl, and stored at −20°C.
Cells obtained from 500 ml of culture medium were suspended in 35 ml of 20 mM Tris⋅hydrochloride, pH 7.4, containing 200 mM NaCl, 1 mM EDTA (buffer A). The suspension was cooled on ice and subjected to ultrasonic treatment. The suspension was centrifuged, and the supernatant was mixed with 4 ml of amylose agarose (New England Biolabs), which had been equilibrated in buffer A. The suspension was mixed in a 50-ml tube for 1 h at room temperature. It then was placed into an empty chromatography column. In the first step, the column was washed with 40 ml of buffer A. In the second step, the suspension was washed by using 5 ml of 50 mM Tris⋅hydrochloride, pH 7.4 (buffer B). The protein was eluted stepwise with 50 mM Tris⋅hydrochloride, pH 7.4, containing 10 mM maltose (buffer C). The protein was eluted in a volume of about 2 ml.
Binding Experiments.
After purification, the protein was diluted to 0.1 mg/ml in 20 mM Tris⋅hydrochloride, pH 7.4. Aliquots of 100 μl (final volume) were incubated with several concentrations of 125I-labeled α-bungarotoxin (αBgtx, Amersham Pharmacia), with or without unlabeled nicotinic ligands, for 3 h at room temperature. The samples then were filtrated on glass microfiber filters (GF/C, Whatman) incubated with 1% polyethyleneimine, and rinsed twice with PBS buffer containing 0.2% BSA. Filters were counted by using a γ counter. Nonspecific binding was measured by using at least a 50-fold excess of unlabeled αBgtx.
Results
Our first attempts consisted in fusing the α7 ECD to carrier proteins naturally assembling into homopentameric structures. The rationale was to improve the folding and assembly of the α7 portion by constraining its pentameric organization. The pentameric carrier proteins were selected on the basis of their high expression level in bacteria and the accessibility of either the N-terminal or C-terminal tails. Using several linkers of different lengths and amino acid compositions, the α7 ECD was fused (i) at the N terminus of the cholera toxin B-subunit (11); (ii) at the N and the C termini of the lumazine synthase from bacteria (12, 13), known to form icosahedral capsids consisting of 12 assembled pentamers (in this case, a wide variety of peptide and protein domains had been attached successfully to both termini without compromising the formation of the icosahedral quaternary structure); and (iii) at the N and C termini of the lumazine synthase from yeast (14), which forms symmetric pentamer structures but no icosahedron. None of these constructs yielded a soluble fusion protein; most of them resulted in the expression of the protein as inclusion bodies.
In parallel, we generated fusion constructs with several monomeric proteins or small peptides. The α7 ECD was fused (i) at the N and C termini of dihydrofolate reductase from E. coli; (ii) at the C terminus of thioredoxin from E. coli; (iii) at the N terminus of a small peptide, called Strep-Tag (15); (iv) at the C terminus of thioredoxin and at the N terminus of the Strep-Tag peptide in a triple construct; (v) at the N and C termini of a 6-histidine peptide; and (vi) at the C terminus of the maltose binding protein (MalE) from E. coli (16). Only the MalE constructs gave soluble protein expression. The constructs selected consist of MalE, followed by three linkers composed of a series of asparagine residues, plus a factor Xa cleavage site (IEGR), an enterokinase cleavage site (DDDDK; Fig. 1), or no cleavage site, followed by three alanines and the coding sequence of the mature rat α7 ECD (residues 1–196) (designated, respectively, Mal-Xa-α7 1–196, Mal-ent-α7 1–196, and Mal-α7 1–196). The α7 sequence was stopped at residue Asp-196 to get the shortest α7 fragment retaining all of the amino acids known to contribute (2) to the agonist/competitive antagonist binding site.
Figure 1.

Design of the fusion proteins between MalE and the extracellular domain of α7 nAChR subunit. The white box corresponds to the MalE protein sequence, black box to the linker segment, dark gray box to the mature rat α7 ECD sequence, and light gray boxes to the α7 transmembrane segments. Dashed lines correspond to the sites where the stop codon was introduced.
Expression of these proteins in XL1-Blue E. coli strains by using the pNCO expression vector (8) was followed by lysis of the cells by sonication in a detergent-free buffer. After centrifugation of this crude material, the supernatant (containing the soluble proteins) and pellets (containing insoluble proteins and inclusion bodies) were analyzed by 10% SDS/PAGE (7). For the three constructs, a significant fraction of the fusion protein was expressed as soluble proteins, with the expected apparent molecular mass of 65 kDa (Fig. 2). However, in the case of Mal-Xa-α7 1–196 and Mal-α7 1–196, the bulk of the protein was found in the insoluble fraction, probably corresponding to inclusion bodies. In the case of Mal-ent-α7 1–196, the amount of soluble protein was much higher and reached the amount of insoluble protein.
Figure 2.
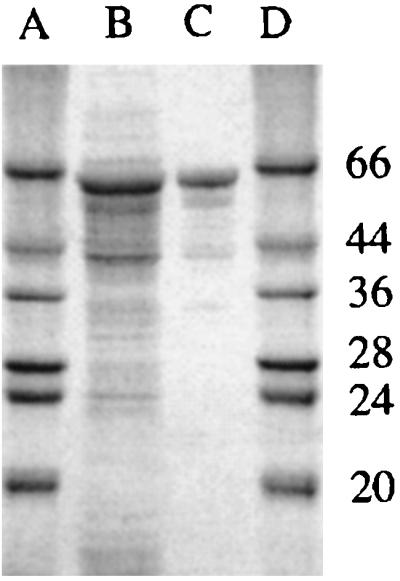
Coomassie-stained SDS/PAGE of the Mal-ent-α7 1–196 fusion protein. Lanes A and D, marker proteins; lane B, crude lysate; lane C, purified fraction of the fusion protein after affinity chromatography.
Loading the crude soluble supernatants on an amylose resin resulted in the purification of the three proteins to more than 90% purity, as judged by SDS/PAGE (Fig. 2). The level of expression after this one-step purification was typically 1–5 mg/liter of culture for Mal-ent-α7 1–196 and only 0.1–0.3 mg for Mal-Xa-α7 1–196 and Mal-α7 1–196. These observations indicate that (i) the maltose binding protein is properly folded within the fusion construct, as it binds amylose resin and maltose, and (ii) the nature of the linker is critical for the solubility of the fusion proteins in this expression system. We thus studied the properties of the Mal-ent-α7 1–196, which gave the highest yields of soluble expression.
CD spectra were recorded to roughly estimate the secondary structure of Mal-ent-α7 1–196. Spectra corresponding to Mal-ent-α7 1–196 and MalE are shown in Fig. 3. The CD spectrum of MalE points to a correct folding of the protein with a minimum at about 210–230 nm and a maximum at about 190–200 nm, corresponding to 41% α-helix and 16% β-sheet, in agreement with published spectra. Mal-ent-α7 1–196 gave a similar CD spectrum. Because MalE represents two-thirds of the total molecule, these data confirm that the MalE portion is correctly folded within Mal-ent-α7 1–196, but they are not informative about the folding of the α7 portion.
Figure 3.

CD spectra of MalE (dashed line) and Mal-ent-α7 1–196 (solid line) in 25 mM K/Na-phosphate, pH 7.2.
To investigate whether the α7 moiety of the fusion construct was folded in a manner close to the native conformation, we studied the ability of the fusion constructs to bind αBgtx, which is known to display a high affinity (1.9 nM) for the native homooligomeric receptor (17). Equilibrium binding measurements were performed by using a rapid filtration assay in a buffer containing the minimum concentration of salts that were found to inhibit toxin binding, as previously observed for a soluble α1 ECD (18). Fig. 4 shows that 125I-αBgtx significantly binds to Mal-ent-α7. The specificity of the binding was demonstrated by the competition with unlabeled αBgtx. The affinity of αBgtx was measured by performing a saturation curve, by using isotopic dilution to reach micromolar concentrations. Fig. 5 shows that the αBgtx binding was saturable, and the Scatchard analysis gave a Kd of 2.5 ± 0.3 μM. The specific activity of the sample was 4.8 ± 0.5 nmol of αBgtx binding sites per mg of protein (measured by Bradford analysis). This value corresponds to 32% of the total protein content but is likely underestimated because some material is lost during the filtration procedure.
Figure 4.

Binding of 125I-αBgtx on MalE-ent-α7 1–196 (first bar), and in the presence of various nicotinic competitors. Values represent the mean of three experiments performed in duplicate.
Figure 5.

Typical Scatchard experiment for the binding of 125I-αBgtx on Mal-ent-α7 1–196.
These data demonstrate that Mal-ent-α7 1–196 retained the ability to bind αBgtx with, however, a 3rd order of magnitude decrease in affinity. A large decrease in affinity was also observed for nicotine, D-tubocurarine and methyllicaconitine, which were found to compete with toxin binding only at very high concentrations (Fig. 4), whereas they display, respectively, Kd of 1.3 μM, 7.3 μM, and in the subnanomolar range for the closely related chick α7 homooligomeric receptor expressed in Xenopus oocyte (17, 19). These results also contrast with those obtained with the soluble chicken α7(1–208)-mAb142 tag construct expressed in small quantities in the Xenopus oocyte, which was reported to bind αBgtx and nicotine with Kd values, respectively, of 0.4 nM and 90 nM (20). Because this α7(1–208) construct contains the α7 sequence of the entire N-terminal domain, we constructed an additional fusion protein where α7 was extended until the first transmembrane segment, Mal-ent-α7 1–209 (Fig. 1). Its expression level, solubility, and binding affinity for αBgtx were found similar to that of Mal-ent-α7 1–196.
We investigated the quaternary organization of Mal-ent-α7 1–196 by centrifugation on 5–20% sucrose gradients. Under conditions where the detergent-solubilized Torpedo receptor is found in the middle fractions of the gradient, the Mal-ent-α7 1–196 was found in all fractions of the bottom half of the gradient, indicating the presence of pentameric and higher molecular weight complexes (data not shown). Yet, Centricon concentration yields solutions with 3 mg of protein per ml without precipitation. We also found that enzymatic cleavage of the protein with enterokinase followed by centrifugation results in recovery of the MalE fragment but not of the α7 fragment, indicating that MalE is required to maintain α7 in a soluble state.
Discussion
During the past few years, several attempts to express ECD were reported. Upon small-scale expression in eukaryotic systems, both the ECD of the α1, β1, γ, and δ subunits of the muscle-type receptor, which consists of heterooligomers of stoichiometry (α1)2β1γδ (21, 22), and the ECD of the α7 subunits, which forms homooligomers (20), were generated as soluble proteins. These ECD were found to associate in a manner similar to that of the full-length receptor, the coexpression α1, β1, γ, and δ subunits being required to form pentamers (22), whereas the α7 ECD gave multimeric entities whose velocity sedimentation profile was consistent with a pentameric structure (20). In addition, the soluble α7 ECD multimers display a typical α7 pharmacology. These observations provided direct demonstration that the ECDs represent autonomous folding units assembling in a native-like manner with a high affinity for the neurotransmitter.
High amounts of ECD were produced in the bacteria by overexpression of the ECD of the α1 subunit as inclusion bodies followed by renaturation procedures (23, 24). In the best case, the ECD were generated in a monomeric state with an affinity for αBgtx of 4 nM, which is 100 times lower that of the native oligomeric receptor. Approximately half of the ECD of α1 (fragment 143–210) also was generated as a soluble αBgtx binding protein (18).
In this paper, we describe the expression as a soluble protein of the ECD of α7 in milligram quantities. Because the native and ECD α7 but not the α1 subunit is able to form homooligomers in vivo and in vitro, the expression of the α7 ECD is expected to result in multimeric entities, as observed upon expression in the Xenopus oocyte (20). We chose to express the α7 ECD as a fusion protein and selected constructs that are expressed as soluble proteins in bacteria. This approach has several advantages, in particular in comparison with the insoluble expression-renaturation procedure currently used. First, it allows for rapid screening of numerous constructs with different fusion proteins and linkers. In this context, we found that the level of soluble expression was altered by subtle changes in the amino acid sequence, especially in the linker region. Second, it allows to recover the protein directly in solution. Indeed, our tests of cleavage between the α7 and MalE portions suggest that expressing α7 as a fusion protein is required to get it soluble. Finally, the MalE portion simplifies the purification procedure.
Several observations suggest a native-like folding of the Mal-ent-α7 1–196 protein: the protein is soluble, the MalE portion binds amylose resin and maltose, the α7 portion binds α-bungarotoxin, and the CD spectra indicates a correct folding of the MalE part. One puzzling observation, however, is the occurrence of high molecular weight multimeric forms, which indicates that the protein assembles in both pentamers and higher molecular weight complexes. One possibility could be that the α7 portions do not interact with each other with the correct geometry, leading to supramolecular entities of more than five protein copies.
Differences between the binding properties of the Mal-ent-α7 1–196 protein and the native α7 receptor oligomers also were observed with, in particular, a 3rd order of magnitude decrease in αBgtx affinity and a large reduction in nicotine and D-tubocurarine affinity. It is well established that the binding site is located at the subunit interfaces in the native protein, and a slight alteration of α7–α7 interfaces, either caused by the presence of the large MalE protein fused at the N-terminal or by the occurrence of multimeric forms of α7, might cause an alteration of the binding properties. Also, the isolated ECD is not expected to undergo the conformational transitions occurring in the fully assembled receptor.
We have thus successfully expressed as a soluble protein the ECD of the α7 subunit of nAChR. Further studies should help elucidate the quaternary structure of the Mal-ent-α7 1–196 and generate a protein suitable for crystallization experiments.
Acknowledgments
We thank Brigitte Vulliez-Le-Normand for useful comments and technical assistance. This work was supported by grants from the European Economic Community Biotech Program, Association de la Recherche contre le Cancer, Fondation Gilbert Lagrue, Association Française contre les Myopathies, and Collége de France.
Abbreviations
- ECD
extracellular domain
- nAChR
nicotinic acetylcholine receptor
- αBgtx
α-bungarotoxin
References
- 1.Le Novere N, Corringer P J, Changeux J P. Biophys J. 1999;76:2329–2345. doi: 10.1016/S0006-3495(99)77390-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corringer P J, Le Novere N, Changeux J P. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 3.Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. J Mol Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- 4.Eisele J L, Bertrand S, Galzi J L, Devillers-Thiery A, Changeux J P, Bertrand D. Nature (London) 1993;366:479–483. doi: 10.1038/366479a0. [DOI] [PubMed] [Google Scholar]
- 5.David-Watine B, Shorte S L, Fucile S, de Saint Jan D, Korn H, Bregestovski P. Neuropharmacology. 1999;38:785–792. doi: 10.1016/s0028-3908(99)00015-5. [DOI] [PubMed] [Google Scholar]
- 6.Read S M, Northcote D H. Anal Biochem. 1981;116:53–64. doi: 10.1016/0003-2697(81)90321-3. [DOI] [PubMed] [Google Scholar]
- 7.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 8.Stüber D, Matile H, Garotta G. In: Immunological Methods. Lefkovits I, Pernis P, editors. Vol. 4. Orlando, FL: Academic; 1990. pp. 121–152. [Google Scholar]
- 9.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bullock W O, Fernandez J M, Short J M. BioTechniques. 1987;5:376–379. [Google Scholar]
- 11.Dertzbaugh M T, Peterson D L, Macrina F L. Infect Immun. 1990;58:70–79. doi: 10.1128/iai.58.1.70-79.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ladenstein R, Schneider M, Huber R, Bartunik H D, Wilson K, Schott K, Bacher A. J Mol Biol. 1988;203:1045–1070. doi: 10.1016/0022-2836(88)90128-3. [DOI] [PubMed] [Google Scholar]
- 13.Ritsert K, Huber R, Turk D, Ladenstein R, Schmidt-Base K, Bacher A. J Mol Biol. 1995;253:151–167. doi: 10.1006/jmbi.1995.0542. [DOI] [PubMed] [Google Scholar]
- 14.Meining W, Mortl S, Fischer M, Cushman M, Bacher A, Ladenstein R. J Mol Biol. 2000;299:181–197. doi: 10.1006/jmbi.2000.3742. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt T G, Skerra A. Protein Eng. 1993;6:109–122. doi: 10.1093/protein/6.1.109. [DOI] [PubMed] [Google Scholar]
- 16.Maina C V, Riggs P D, Grandea A G, Slatko B E, Moran L S, Tagliamonte J A, McReynolds L A, Guan C D. Gene. 1988;74:365–373. doi: 10.1016/0378-1119(88)90170-9. [DOI] [PubMed] [Google Scholar]
- 17.Anand R, Peng X, Ballesta J, Lindstrom J. Mol Pharmacol. 1993;44:1046–1050. [PubMed] [Google Scholar]
- 18.Grant M A, Gentile L N, Shi Q L, Pellegrini M, Hawrot E. Biochemistry. 1999;38:10730–10742. doi: 10.1021/bi983007q. [DOI] [PubMed] [Google Scholar]
- 19.Palma E, Bertrand S, Binzoni T, Bertrand D. J Physiol (London) 1996;491:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wells G B, Anand R, Wang F, Lindstrom J. J Biol Chem. 1998;273:964–973. doi: 10.1074/jbc.273.2.964. [DOI] [PubMed] [Google Scholar]
- 21.West A P, Jr, Bjorkman P J, Dougherty D A, Lester H A. J Biol Chem. 1997;272:25468–25473. doi: 10.1074/jbc.272.41.25468. [DOI] [PubMed] [Google Scholar]
- 22.Tierney M L, Unwin N. J Mol Biol. 2000;303:185–196. doi: 10.1006/jmbi.2000.4137. [DOI] [PubMed] [Google Scholar]
- 23.Schrattenholz A, Pfeiffer S, Pejovic V, Rudolph R, Godovac-Zimmermann J, Maelicke A. J Biol Chem. 1998;273:32393–32399. doi: 10.1074/jbc.273.49.32393. [DOI] [PubMed] [Google Scholar]
- 24.Alexeev T, Krivoshein A, Shevalier A, Kudelina I, Telyakova O, Vincent A, Utkin Y, Hucho F, Tsetlin V. Eur J Biochem. 1999;259:310–319. doi: 10.1046/j.1432-1327.1999.00041.x. [DOI] [PubMed] [Google Scholar]