

Abstract
Förster resonance energy transfer (FRET) technology has been widely used in biological and biomedical research and is a very powerful tool in elucidating protein interactions in many cellular processes. Ubiquitination and SUMOylation are multi-step cascade reactions, involving multiple enzymes and protein–protein interactions. Here we report the development of dissociation constant (K d) determination for protein–protein interaction and cell-based high-throughput screening (HTS) assay in SUMOylation cascade using FRET technology. These developments are based on steady state and high efficiency of fluorescent energy transfer between CyPet and YPet fused with SUMO1 and Ubc9, respectively. The developments in theoretical and experimental procedures for protein interaction K d determination and cell-based HTS provide novel tools in affinity measurement and protein interaction inhibitor screening. The K d determined by FRET between SUMO1 and Ubc9 is compatible with those determined with other traditional approaches, such as isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR). The FRET-based HTS is pioneer in cell-based HTS. Both K d determination and cell-based HTS, carried out in 384-well plate format, provide powerful tools for large-scale and high-throughput applications.
Keywords: SUMOylation, Förster resonance energy transfer, Kd affinity determination, High-throughput screening
Introduction
Post-translational modification by ubiquitin (Ub) and ubiquitin-like (Ubls) proteins, such as SUMO, plays critical roles in many physiological and pathological processes in eukaryotes, such as signal transductions, genome integrity, protein transport, cell cycles, and tumorigenesis.1,6,10,18,28 SUMO, known as Small Ubiquitin-related MOdifier, has emerged as an important protein modifier in last decade. Composed of ~100 amino acids, SUMO undergoes reversible conjugation to the lysine residues of target proteins (SUMOylation) via the catalysis of various enzymes (Fig. 1). SUMOylation in a target-specific manner can affect a target protein’s intracellular localization, its ability to interact with other proteins or its transcriptional activity.10 SUMO is present in most eukaryotic organisms, suggesting its critical roles in various physiological processes. Although not completely understood yet, SUMOylation of many proteins have been linked to some human diseases including neurodegenerative diseases, viral infection, tumorigenesis, and cancer metastasis.1,3,16,25 (Fig. 1).
Figure 1.
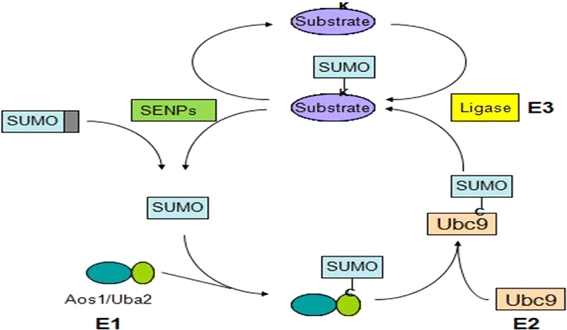
Multiple protein–protein interactions are involved in sumoylation conjugation pathway
Analogous to ubiquitination, conjugation and deconjugation of SUMO require the catalysis of multiple enzymes (Fig. 1). SUMO is synthesized as a precursor protein. Pre-SUMO is then recognized by SUMO-specific peptidases (SENPs) and cleaved into a mature form with a C-terminal Gly–Gly motif.27 The heterodimer Aos1/Uba2, which is the SUMO E1 activating enzyme, then forms a thioester bond with SUMO using the energy from the degradation of ATP.7,12,20 SUMO is further transferred from the E1 enzyme to the active site cysteine of the SUMO E2 conjugating enzyme Ubc9.9,11 With the help of SUMO E3 ligases in vivo, SUMO is finally transferred from Ubc9 to the lysine residue of target proteins.10 SUMOylated proteins can then be recognized by SENPs and SUMO is cleaved off for next cycle of conjugation.27 Protein–protein interactions are crucial for SUMOylation to proceed.
Förster resonance energy transfer (FRET) is very appealing for bioanalysis because of its intrinsic sensitivity to nanoscale changes in D/A separation distance (proportional to r 6). The efficiency of the energy transfer can be determined from either steady-state or time-resolved measurements. This property has been extensively used in biological research to identify protein interactions, monitor intracellular signaling activities in real-time, and survey bioactive molecules by high-throughput screening (HTS).4,8,23 Compared with traditional techniques to identify protein–protein interactions, such as co-immunoprecipitation and yeast two-hybrid assay, FRET offers real-time monitoring and spatial information on molecular interactions in living cells.29
Because of FRET’s high sensitivity and suitability for both spectroscopy and imaging analysis, many efforts have been made to develop it into quantitative assays and HTS assays.2,4,8,23 However, due to the low FRET efficiencies of fluorescent proteins and complexity of fluorescence emissions of donor and acceptor, it has been very challenging process in developing FRET into quantitative assay until recently, an effort utilizing classical ECFP and Venus-YFP to measure K d of SUMO1 and Ubc9 interaction.17 In this study, CFP-tagged SUMO1 and YFP-tagged Ubc9 recombinant proteins were mixed and the fluorescent spectrum was compared with that from CFP and YFP–Ubc9 to derive the FRET-sensitized emission from YFP–Ubc9. The FRET emission intensity was then fitted with YFP–Ubc9 concentration in a hyperbola curve to get the maximum FRET emission intensity, and the bound YFP–Ubc9 concentration were calculated from the FRET emission in a linear relationship. Fitting the bound and free YFP–Ubc9 concentrations with a hyperbola formula resulted in a K d value of 0.59 μM, which is very close to the K d determined by other methods.26
Current FRET-based HTS assays have been mostly limited to in vitro biochemical assay or small-molecule fluorophore in non-protein molecules due to their high quantum yield or high FRET efficiency.4,24 In these assays, organic fluorophores are used to modify proteins of interest in vitro, and compound libraries are added into recombinant protein mixtures. Although in vitro assays based on purified recombinant proteins are easier to handle, they do not accurately replicate the intracellular environment, do not reflect the localization and modification of target proteins in living cells, and cannot determine transport efficiencies of the drug across cell membranes.
Here, we report the theoretical and experimental developments for K d determination of SUMO1 and Ubc9 interaction, and HTS assay development in living cells using an engineered FRET pair, CyPet and YPet. This pair has been showed to have much higher fluorescence quantum yield and FRET efficiency.19 The radiometric FRET signal of CyPet and YPet was 20-fold greater than that of CFP–YFP. This greatly improved dynamic signal range makes CyPet and YPet excellent candidate for both quantitative FRET-based assays and HTS development for inhibitor screening of protein–protein interactions. In contrast to traditional ratiometric criteria of FRET signal, we have differentiated and quantified absolute fluorescence signals contributed by each component, namely donor, acceptor and FRET at the acceptor emission wavelength, which was missed in the traditional ratiometric measurement of FRET. We derived an improved mathematic formula which directly connects the FRET signal-to-K d. The K d of SUMO1 and Ubc9 has been successfully determined using this technique, which fits very well with that determined by other methods, such as surface plasmon resonance (SPR). The same FRET pair, CyPet and YPet, has been used to develop cell-based FRET HTS, which is pioneer in this field. Both K d determination and cell-based HTS were performed in 384-well plate format, which readily allows repeated study and large-scale application, such as genome-wild and industrial applications.
Materials and Methods
DNA Constructs and Protein Expression
The open reading frames of the genes were amplified by PCR and the PCR products were cloned into pCRII-TOPO vector (Invitrogen). After sequencing confirmation, the cDNA encoding CyPet–SUMO1 and YPet–Ubc9 were cloned into pcDNA3.1hygro and pcDNA3.1V5His vectors (Invitrogen), respectively. The cDNAs encoding SUMO1, Ubc9, CyPet–SUMO1 or YPet–Ubc9 were also cloned into pET28(b) vector (Novagen).
BL21(DE3) Escherichia coli (E. coli) cells were transformed with pET28 vectors encoding SUMO1, Ubc9, CyPet–SUMO1 or YPet–Ubc9. The transformed bacteria were inoculated in 2 × YT medium and the expression of polyhistidine-tagged recombinant proteins was induced with 0.1 mM IPTG at 25 °C overnight. The recombinant proteins were purified by Ni2+–NTA agarose beads (QIAGEN) followed by gel filtration HPLC using Superdex75 10/300 GL column on a HPLC purification system (GE Healthcare, ÄKTA™ purifier). YPet–Ubc9 and Ubc9 proteins used for SPR analysis were digested by thrombin to remove the polyhistidine tags prior to purification by HPLC.
FRET Assay and Kd Measurement
Recombinant CyPet–SUMO1 and YPet–Ubc9 proteins were mixed and diluted with phosphate buffered saline (PBS) to a total volume of 30 μL. The final concentration of CyPet–SUMO1 was fixed to 1 μM and the final concentration of YPet–Ubc9 varied from 0 to 4 μM. The mixtures were transferred into a 384-well plate (Falcon) and the fluorescence emission spectrum of each well was measured with a fluorescence multi-well plate reader (Molecular Devices, FlexstationII384). Two excitation wavelengths were used: 414 nm to excite CyPet, and 475 nm to excite YPet. Excited at 414 nm, CyPet has an emission peak at 475 nm (FLDD) (see Fig. 2). With FRET, another emission peak at 530 nm (Emtotal) can be observed which results from the energy transferred from CyPet to YPet. When the mixture is excited at 475 nm, an emission peak at 530 nm (FLAA) can be observed which is from the direct excitation of YPet but not CyPet.
Figure 2.
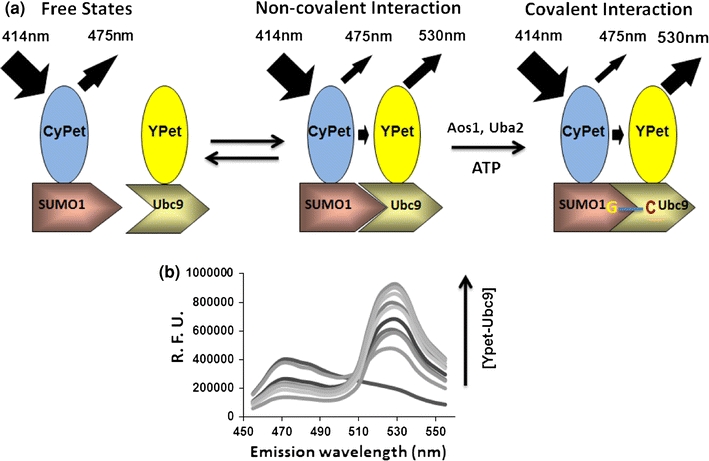
Design and detection of high sensitive FRET-based detection for protein interactions in sumoylation conjugation cascade. (a) The diagram of FRET-based detection of SUMO1 and its E2 ligase, Ubc9, interaction. (b) Emission spectra of protein mixtures with [CyPet–SUMO1] fixed as 1 μM and [YPet–Ubc9] increased from 0 to 7.5 μM. Excitation wavelength is 414 nm
When a mixture of CyPet–SUMO1 and YPet–Ubc9 recombinant proteins was excited at 414 nm, the emission intensity at 530 nm was consisted of three components: the direct emission of CyPet, the sensitized emission of YPet and the direct emission of YPet. Because the sensitized emission from YPet–Ubc9 (EmFRET) is proportional to the amount of YPet–Ubc9 bound to CyPet–SUMO1 ([YPetUbc9]bound), we can convert the concentration of YPet–Ubc9 in both free ([YPetUbc9]free) and bound ([YPetUbc9]bound) forms to functions of EmFRET. Therefore, we can derive the relationship between EmFRET and the total concentration of YPet–Ubc9 ([YPetUbc9]total), and calculate the K d between CyPet–SUMO1 and YPet–Ubc9 based on the algorithm we developed. Data were processed in Prism 5 (GraphPad Software).
Surface Plasma Resonance Measurement
All analyses of interaction between CyPet–SUMO1 and YPet–Ubc9 or SUMO1 and Ubc9 were performed on BIAcore X100 system equipped with NTA sensor chips (BIAcore AB, Uppsala, Sweden) at a flow rate of 30 μL/min. For immobilization of proteins, the chip was treated with 500 μM NiCl2 for 1 min before 10 μg/mL purified CyPet–SUMO1 or 4 μg/mL purified SUMO1 protein was injected for 180 s and stabilized for 60 s. Then 20–150 μg/mL thrombin-digested YPet-Ubc9 or 5–50 μg/mL thrombin-digested Ubc9 protein was injected for 90 or 120 s and disassociated for 10 min. In order to continuously monitor the non-specific background binding of samples to the NTA surface, YPet–Ubc9 and Ubc9 proteins were injected into a control flow cell without treatment of NiCl2 and CyPet–SUMO1/SUMO1 proteins. All measurements were performed at 25 °C in a buffer containing 50 mM sodium phosphate pH 7.4, 150 mM NaCl and 50 μM EDTA. Data were analyzed with BIAcore X100 evaluation software ver.1.0 (BIAcore).
Mammalian Cell Culture and HTS assay
All cell culture supplies including media, serum, and antibiotics were purchased from Invitrogen (Carlsbad, CA). To generate cell lines which can stably express CyPet–SUMO1 and YPet–Ubc9 proteins, HEK293 cells were transfected with pcDNA3.1hygro–CyPet–SUMO1 using FuGene6 (Roche) in a 6-well plate, diluted to several 10 cm dishes, and selected in DMEM medium supplemented with 150 μg/mL hygromycin. After 2 weeks, cell colonies were transferred and cultured on 96-well plates. In order to determine the expression of CyPet–SUMO1, the fluorescence emission intensities of individual colonies at 475 nm (excited at 414 nm) were scanned using FlexstationII384 (Molecular Devices). The colony having the highest emission intensity was picked up and expanded. This CyPet–SUMO1-expressing HEK293 stable cell line was then transfected with pcDNA3.1V5His–YPet–Ubc9 and selected with 750 μg/mL geneticin for 2 weeks. The fluorescence intensities of cell colonies at 475 and 530 nm (excited at 414 nm) were measured using FlexstationII384 (Molecular Devices), and the colony having the highest Em530/Em475 ratio was picked up and used for subsequent screening assays.
HEK293 cells stably expressing CyPet–SUMO1 and YPet–Ubc9 were diluted to 500 cells per microliter with PBS. Sixty microliter resuspended cells were then aliquoted into each well of 384-well plates (Falcon) using a liquid handler (Molecular Devices, AquaMax DW4). To screen the compound library for inhibitors of SUMO1–Ubc9 interactions, 1 μL 500 μM chemical compound was added to each well using the liquid handling module of FlexstationII384 (Molecular Devices). After incubation at 37 °C for 2 h, they were excited at 414 nm and the fluorescence emission intensities of each well at 475 nm [Em475(Ex414)] and 530 nm [Em530(Ex414)] were measured using FlexstationII384. The emission ratio of Em530(Ex414) to Em475(Ex414) was then calculated for all the wells.
Results
Fluorescence Spectrum Analysis of FRET Signal
Dissociation constant, K d, is an important parameter for characterizing protein–protein interactions. Based on the fact that FRET signal is proportional to the amount of bound FRET pair, we developed a FRET-based technology for K d determination. We believe that FRET-based technology could provide unique advantages compared to traditional techniques for K d determination, such as BIAcore SPR or radiolabeled-ligand binding assay. FRET technology allows real-time monitoring of protein interactions even in a multiple protein complex. The instrumentations for FRET measurement are widely available.15,22 No radioisotope labeling is required, providing environmental-friendly approach. Furthermore, protein concentrations in the system can be accurately determined by fluorescent emission intensity and quantum yield.17
We chose SUMOylation pathway as our model system for this technology development because this pathway involves multiple protein–protein interactions,13 and is also a good potential pathway for chemical inhibitor discovery with our technology (see below). There are two levels of interactions between SUMO1 and it E2 ligase, Ubc9: non-covalent and covalent interactions (Fig. 2a). The non-covalent physical interaction of these two proteins brings two proteins together without any SUMO activation.14 On the other hand, covalent interaction results from an isopeptide bond formation between the C-terminal Gly of SUMO and the Cysteine 93 of Ubc9 after the activation of SUMO peptide by its E1 activating enzyme. To determine the K d of non-covalent interaction of SUMO1 and Ubc9 by FRET approach, we chose a FRET pair, CyPet and YPet, to label SUMO1 and Ubc9 because of their improved FRET efficiency.19 The excitation and emission wavelength of CyPet and YPet are around 414 and 475 nm, 515 and 530 nm, respectively. Specifically, the open reading frames of CyPet and YPet were first ligated into pCRII-TOPO and fused to SUMO1 and Ubc9, respectively. The fusion genes of CyPet–SUMO1 and YPet–Ubc9 were then ligated into pET28 bacterial expression vector. The fusion proteins with poly-histidine and fluorescent tags were then expressed in E. coli and purified by Ni–NTA affinity column followed by HPLC gel filtration purification. To implement FRET-based technology for K d determination, two key issues need to be solved. First, we need to differentiate and quantify fluorescence signal of FRET from other direct fluorescence signals of donor and acceptor at the excitation wavelength. Second, we need to convert fluorescence signals to corresponding concentrations of bound partners, donor and acceptor proteins. We started with the general law of mass action for protein–protein interaction.
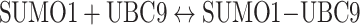 |
The K d for this pair can be calculated as following
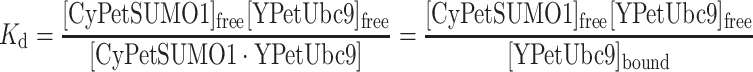 |
1 |
We then differentiate the fluorescent signals at 530 nm into three fractions: FRET emission (EmFRET), YPet direct emission, and CyPet direct emission when excited at 414 nm (Eq. 2; Fig. 3a),
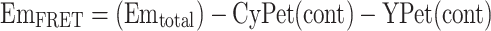 |
2 |
where CyPet(cont) is the fluorescence signal contribution of donor, YPet(cont) is the fluorescence signal contribution of acceptor.
Figure 3.
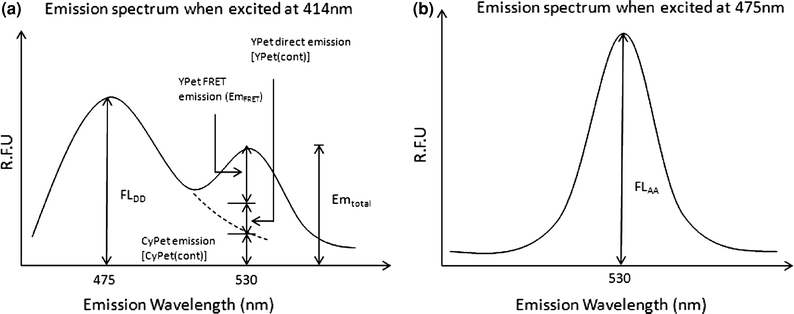
Spectrum analysis of FRET signals. (a) Dissection of emission spectra from mixture of CyPet–SUMO1 and YPet–Ubc9. (b) Emission spectrum of YPet equations used to determine sensitized FRET emission, to convert sensitized emission intensity to bound concentration, to calculate the concentration of free YPet–Ubc9 and to fit bound concentration against free concentration to get the K d value
We then speculated and found out that the direct emission of donor, CyPet, at 530 nm is proportional to its emission at 475 nm when excited at 414 nm with a ratio factor of x, while the direct emission of acceptor, YPet, at 530 nm is proportional to its emission at 530 nm when excited at 475 nm with a ratio factor of y (Eq. 3; Fig. 3b).
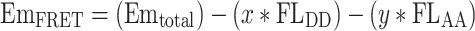 |
3 |
where FLDD is fluorescence signal of donor when excited at donor wavelength (414 nm), and FLAA is fluorescence signal of acceptor when excited at acceptor wavelength (475 nm). From a series measurement of different concentrations of CyPet alone or YPet alone, we determined x = 0.378 and y = 0.026.
Kd Determination of the Non-covalent SUMO1 and Ubc9 Interaction by FRET
In order to determine the K d between SUMO1 and Ubc9, we need to convert the protein concentrations to functions of FRET signal intensity and derive its relationship with K d. Because the total concentration of CyPet–SUMO1 in our assay is fixed, the formula of K d (Eq. 1) can be expressed as:
 |
4 |
Equation 4 can be converted to:
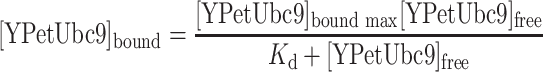 |
5 |
Because the FRET signal intensity (EmFRET) is proportional to the concentration of bound YPetUbc9 in the mixture with all other parameters fixed, [YPetUbc9]bound in each condition is then determined by the sensitized emission intensity in a linear relationship:
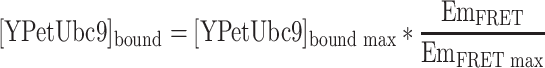 |
6 |
Combining Eqs. (5) and (6) yields:
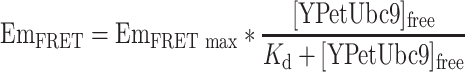 |
7 |
If we set the total concentration of CyPet–SUMO1 to a constant a and the concentration of total and free YPet–Ubc9 to variables X and Y, respectively, we can convert the concentration of bound YPet–Ubc9 and free CyPet–SUMO1 to:
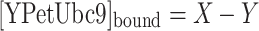 |
8 |
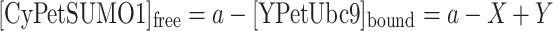 |
9 |
Based on the definition of K d and Eqs. (8) and (9), we can derive
 |
We then solved the above equation as following,
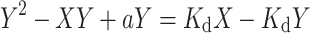 |
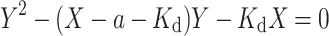 |
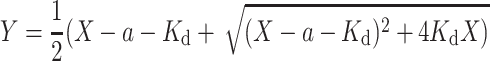 |
10 |
Combining Eqs. (7) and (10) yields:
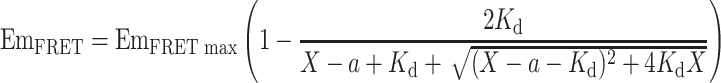 |
11 |
Therefore, by fitting the dataset of EmFRET and total YPet–Ubc9 concentration (X) with Eq. (11) we will be able to derive the value of maximum EmFRET (EmFRETmax) and K d.
Before we measured the FRET signals between CyPet–SUMO1 and YPet–Ubc9, we first characterized potential interaction interference of tags used in our assay, poly-His, CyPet and YPet because it was reported that CyPet and YPet may have a weak dimerization activity by themselves. We would like to know any potential interaction affinity from these tags. A dose-dependent FRET signal increase was observed for CyPet–SUMO1 and YPet–Ubc9, while as control pairs all with poly-His tag, CyPet and YPet, CyPet and YPet–Ubc9, or CyPet–SUMO1 and YPet did not show any significant FRET signals within the experimental concentration (Fig. 4a). These results suggest that, in our experimental condition, these tags would not form a dimmer by themselves and therefore generate interference for K d measurement of SUMO1 and Ubc9.
Figure 4.
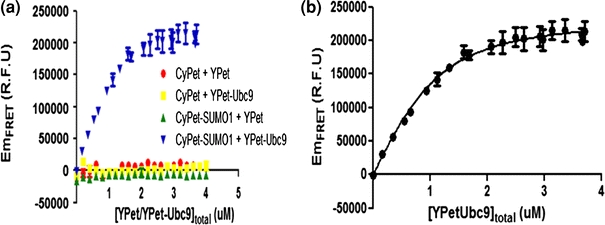
Determination of interaction affinity K d between CyPet–SUMO1 and YPet–Ubc9 using FRET technology. (a) Interaction specificity and quantitation among pairs. (b) Plot of FRET-sensitized YPet–Ubc9 emission vs. the total YPet–Ubc9 concentration
In our K d determination experiment, we fixed the CyPet–SUMO1 concentration to 1 μM and increased the concentration of YPet–Ubc9 from 0 to 4 μM in a total volume of 30 μL. The fluorescence emission spectra of the mixture were then determined by FlexstationII384 under excitation wavelength of 414 nm (Fig. 2b) or 475 nm (data not shown). After the FRET emission intensity under each condition was calculated, the dataset of EmFRET and [YPetUbc9]total were fitted in Eq. (11) by least-square fitting with constant a fixed to 1 (Fig. 4b). We got an EmFRETmax of (2.38 ± 0.05) × 105 RFU and K d of 0.33 ± 0.04 μM, which is comparable to the previously published results from Martin et al. 17 (0.59 ± 0.09 μM) and Tatham et al. 26 (0.25 ± 0.07 μM).
Kd Determination of the Non-covalent SUMO1 and Ubc9 Interaction by SPR
To validate our results of K d measurement from the FRET assay, we then measured the interaction affinity of CyPet–SUMO1 and YPet–Ubc9 by SPR. His-tagged CyPet–SUMO1 was expressed in bacterial cells and purified using Ni–NTA agarose beads. After dialysis, His-tagged CyPet–SUMO1 was immobilized onto a SPR NTA sensor chip. YPet–Ubc9 was obtained by cleaving the His-tagged YPet–Ubc9 on Ni beads directly by thrombin digestion. The cleaved YPet–Ubc9 was then further purified by HPLC. Non-specific binding of YPet–Ubc9 to the NTA chip was subtracted as described in “Materials and Methods” section. The binding kinetics analysis showed the binding response of the bound CyPet–SUMO1 to injections of a series of different concentrations of YPet–Ubc9 (Fig. 5a). CyPet–SUMO1 bound with moderate kinetics to YPet–Ubc9, with a calculated K d of 0.35 μM. This K d is consistent with the K d of 0.33 ± 0.04 μM determined by FRET assay described above.
Figure 5.
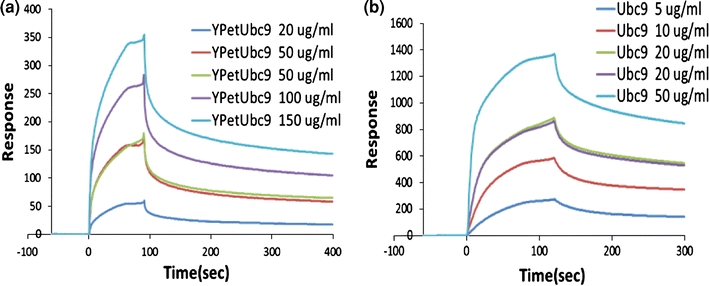
Determination of interaction affinity K d by surface plasma resonance measurement. (a) Determination of K d between CyPet–SUMO1 and YPet–Ubc9 using BIAcore, resulting in K d = 0.35 μM. (b) Determination of K d between SUMO1 and Ubc9 using BIAcore, resulting in K d = 0.10 μM
To further analyze the possible interference of fluorescence tag to the interaction, we performed a control experiment in SPR. The interaction of SUMO1 and Ubc9 by themselves was analyzed. Similar to above experiment, His-tagged SUMO1 was immobilized onto NTA sensor chip. Cleaved Ubc9 was purified by HPLC. Bound SUMO1 shows binding response to a series of injected different Ubc9 concentrations in a similar kinetics as fluorescent protein-tagged fusion proteins (Fig. 5b). The K d of SUMO1 and Ubc9 interaction was calculated as 0.1 μM, which is comparable with that of fluorescent protein-tagged SUMO1 and Ubc9.
Development of Cell-Based HTS Assay
Although FRET assay has been widely used in basic research, it has not been robustly developed into HTS format, especially in cell-based HTS because of the challenging requirements of high signal-to-noise ratio and low variation.4,24 To explore whether this FRET pair can be developed into HTS platform, we went further to test whether this FRET assay is suitable for HTS format or not. In general, a large number of compounds (>100,000 compounds) and various robots normally involved in HTS require reproducible, low variation and cost-effective biological assays. For the FRET-based screening platform, the ideal assay would use cell lines stably expressing fluorescence donor and acceptor pairs in which FRET signals are strong enough to be detected by HTS instruments, such as plate readers.
Establish Stable Cell Lines Expressing Cypet–SUMO1 and Ypet–Ubc9 for Cell-Based FRET Screening
To achieve high reproducibility, low variation and low cost for HTS, a stable cell line expressing both CyPet–SUMO1 and YPet–Ubc9 was established (“Materials and Methods” section). Briefly, we generated the stable cell line in a sequential manner. We first introduced CyPet–SUMO1 into HEK293 cells and selected cell lines with high level of CyPet expression. Then we introduced the second partner, YPet–Ubc9. After the second selection, individual stable colonies were picked up and their FRET signals were examined (Fig. 6a). We also generated control cell lines expressing only CyPet–SUMO1 alone and with other YPet pair, such as YPet–Aos1 for HTS and follow-up assays. When the cells were excited at 414 nm, we routinely got emission ratio (Em530/Em475) of stable cell lines at around 0.8–1.2 (Fig. 6b), while as that of the control cell line (CyPet–SUMO1) was 0.4–0.6. Typical spectrums of stable cell line and control cells were shown in Fig. 6b when exited at 414 nm and in Fig. 6c when exited at 475 nm. To further validate the results, we also carried out the FRET assay with fluorescence microscopy imaging measurements. The cells stably expressing CyPet–SUMO1/YPet–Ubc9 showed fluorescence emission in the yellow channel when excited at 442 nm, while the control cells stably expressing CyPet–SUMO1 showed little fluorescence emission in the yellow channel when excited at the same wavelength (Fig. 6d).
Figure 6.
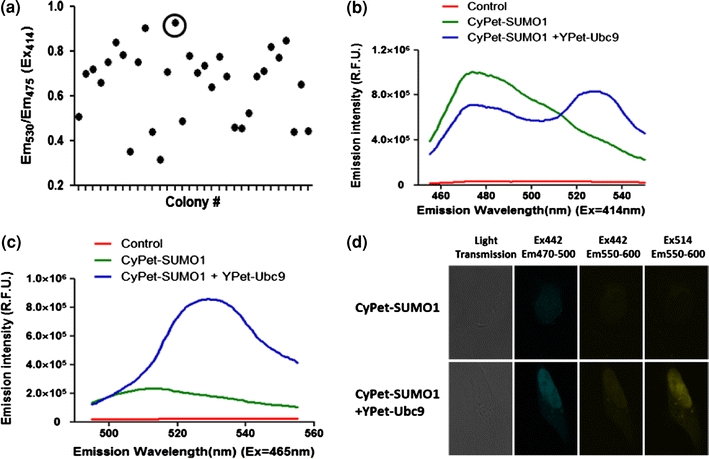
Characterizations of stable cell line for FRET assay. (a) FRET characterization of individual stable cell lines. (b) Fluorescence spectrum of control cells, single transfected stable cell line, and double transfected stable cell line when excited at 414 nm. (c) Fluorescence spectrum of control cells, single transfected stable cell line, and double transfected stable cell line when excited at 465 nm. (d) Fluorescence image analysis of stable cell lines
To estimate the dynamic range of FRET signal changes of the stable cell line in oder to estimate the screening criteria, we performed SUMO1/Ubc9 disruption assays using detergent and/or urea, which could disrupt protein–protein interactions. To get a full range of FRET signals with no interactions of the FRET pair, we examined the FRET spectrum of stable cells in PBS or lysed cells using lysis buffer containing 200 mM NaCl, 10 mM Tris with 1.0% Triton X100, or lysis buffer plus 1 M urea. The FRET signal was decreased from 1.2 to 0.6 by the lysis buffers or lysis buffer plus urea (Fig. 7a). Even with 1 M urea, the FRET signal did not decrease any further, indicating that all the interactions of CyPet–SUMO1/YPet–Ubc9 had been fully disrupted. The dynamic range of FRET signal from 1.2 to 0.6 reflects from the full interaction to no interaction of CyPet–SUMO1 and YPet–Ubc9, which will be used as criteria for the screening hit pickup (Fig. 7b and see below for Pilot screening).
Figure 7.
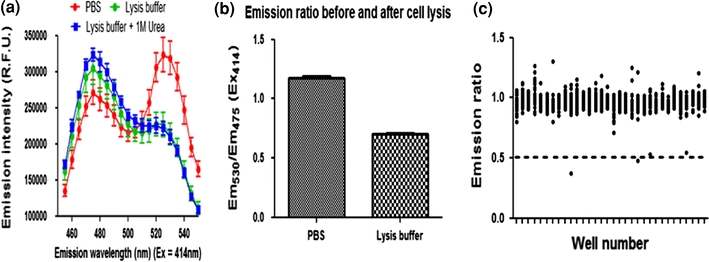
Reversible interaction of SUMO1 and Ubc9 in living cells. (a) FRET assays of stable HEK293 cell line containing CyPet–SUMO1 and YPet–Ubc9 after different treatments with or without protein interaction disrupting reagents, such as Triton-100 or Urea. (b) Plot of FRET ratio (Em530/Em475) of cells treated with or without disrupting buffers as in (a). (c) Positive hit selection of seven hundred screened compounds
Assay Development in the 384-Well Plate Format and Pilot Screening
A highly predictable and reproducible assay is required for HTS and miniaturization is a critical step in the HTS development.24 We thus attempted to develop this FRET-based Ubc9 inhibitor screening into a consistent and high signal-to-noise assay. In principle, a hit should generate a signal in an assay with 95–99% confidence depending on the screens. That means that a hit should be picked by its signal at least beyond three SDs. To achieve high-quality HTS, the ratiometric FRET signal (R = Em530nm/Em475nm) was used as screening signal and the coefficient of variation (CV) of signals in 384-well plate format was used as a variation assessment. We tested different numbers of cells/well and measured the above parameters for different times on different days. Consistent results were obtained. The ratiometric FRET signal (R = Em530nm/Em475nm) of the stable cell line expressing CyPet–SUMO1/YPet–Ubc9 was 1.14 ± 0.04. The signal of the control stable cell line expressing CyPet–SUMO1 was 0.54 ± 0.03. The CVs of all the samples were less than 10%, which met the criteria of HTS requirement, 15%. These data indicate that signal-to-noise is quite separated with tight variations. These results suggest that screening inhibitor(s) of SUMO1–Ubc9 interaction in 384-well plates is feasible for HTS.
To validate our FRET-based HTS platform, we conducted a small-scale pilot screening with a fraction of compound collections available to us. We screened 2000 compounds and found the mean of Em530/Em475 to be 0.95 and standard deviation (SD) to be 0.15. After we filtered out five right side outliers (Em530/Em475 > 3.0), the FRET signals of most compounds were centered at mean. The cut-off of primary hits was set up below 3 × SD, which the ratio of Em530/Em475 is 0.5. A total of 136 hits were picked up and the hit rate was 1.13%. After filtered out those compounds (97 compounds) with self-fluorescence, 39 compounds were kept for further validation and characterization. These data are compatible to a typical HTS hit rate, indicating that this assay platform is feasible for HTS.
Discussion
The high efficient FRET pair, CyPet and YPet, has provided suitable tools for both K d measurements and HTS assay development in living cells. We have demonstrated a robust approach for K d measurement in solution using FRET assay in steady state. The K d from FRET assay has been validated by other traditional methods, such as SPR. Another group also demonstrated the application of FRET assay using classical CFP and YFP for K d measurement. We have derived a significantly different and improved theoretical and experimental procedure to derive K d from fluorescent signal by characterizing the quantitative contributions of donor and acceptor alone and FRET signal. This novel mathematic algorithm of FRET analysis and simplified experimental method enable us to measure K d using one formula and derive more accurate K d because of the elimination of a step for the estimation of maximum bound protein in the middle step. This novel method not only saves the experimental steps but also provide more rigorous mathematic process than the multi-step approach. The data derived from our approach fits into the same category of data determined by other approach and are very tight to each others.17,26 This has demonstrated that interaction affinity measurement by FRET assay is not only feasible but also accurate. This novel approach would provide a very convenient way for K d measurement.
Although FRET assay has been widely used in protein–protein interaction assay using in vitro and spectroscopic method, it has not been developed into a robust HTS technology.4,19,24 Our FRET assay targets protein–protein interactions in vivo. The cell-based HTS will serve as homogenous cell-based assay and will not necessarily reflect a direct physical interaction of two proteins, but its cellular interaction activity which could be mediated by other partner(s). The compounds need to permeate the mammalian cell membrane in order to show activity in this cell-based assay. Therefore, there will be some discrepancy of compound behavior between in vitro assay and in cell-based assay, which is a very challenging task in pharmaceutical industry. In addition, the screening compounds need to be stable in cell culture medium and in cytoplasm, which provide another level testing for compounds to be effective inside cells.
The strategy using fluorescent proteins as fluorescent marker for bound and free partner measurements can be a general method for determining interaction affinities of protein–protein interactions and HTS technology.5,21,29 The approach can be used to measure affinity involving multiple proteins. There are several advantages of FRET-based K d measurement and HTS in living cells over other methods. First, this approach is environmentally friendly, which only requires DNA constructions without radioisotope labeling. Second, the fluorescent-tagged proteins are in aqueous phase, which is mostly close to their natural environment in cells. Third, the fluorescence intensity can be determined by general fluorescence spectroscopy or fluorescence plate reader, which is widely available. Finally, cell-based FRET screening of protein–protein interaction inhibitors is a new screening approach, which not only provides robustness of screening but also evaluate properties of chemical compounds for stability and membrane permeability at the same time. Therefore, the high sensitive FRET-based assays could be a powerful approach in developing genome-wide protein interaction affinity determination and inhibitor screenings of protein–protein interaction.
Acknowledgments
We are very grateful to Prof. Victor G.J. Rodgers for the valuable advices on nonlinear regression of K d measurement. We would like to thank Elizabeth Gillard for careful comments on our manuscript. We thank Prof. Xuemei Chen for allowing us to use HPLC system. We also thank Glenn Hicks for screening compound selection and David Carter for helps in compound transfer and fluorescence microscope at Institute Integrative Genome Biology at UCR. We thank all the members in Liao’s group for very close collaborative work and helps for the work. This work was supported by the National Institutes of Health Grant AI076504 to J.L.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Alarcon-Vargas D, Ronai Z. SUMO in cancer–wrestlers wanted. Cancer Biol. Ther. 2002;1(3):237–242. doi: 10.4161/cbt.74. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Puhl HL, 3rd, Ikeda SR. Estimating protein–protein interaction affinity in living cells using quantitative Forster resonance energy transfer measurements. J. Biomed. Opt. 2007;12(5):054011. doi: 10.1117/1.2799171. [DOI] [PubMed] [Google Scholar]
- 3.Cheng J, et al. SENP1 enhances androgen receptor-dependent transcription through desumoylation of histone deacetylase 1. Mol. Cell. Biol. 2004;24(13):6021–6028. doi: 10.1128/MCB.24.13.6021-6028.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dams G, et al. A time-resolved fluorescence assay to identify small-molecule inhibitors of HIV-1 fusion. J. Biomol. Screen. 2007;12(6):865–874. doi: 10.1177/1087057107304645. [DOI] [PubMed] [Google Scholar]
- 5.Giepmans BN, et al. The fluorescent toolbox for assessing protein location and function. Science. 2006;312(5771):217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 6.Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004;18(17):2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 7.Gong L, et al. Molecular cloning and characterization of human AOS1 and UBA2, components of the sentrin-activating enzyme complex. FEBS Lett. 1999;448(1):185–189. doi: 10.1016/S0014-5793(99)00367-1. [DOI] [PubMed] [Google Scholar]
- 8.Gordon GW, et al. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998;74(5):2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang DT, et al. Basis for a ubiquitin-like protein thioester switch toggling E1–E2 affinity. Nature. 2007;445(7126):394–398. doi: 10.1038/nature05490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson ES. Protein modification by SUMO. Annu. Rev. Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 11.Johnson ES, Blobel G. Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J. Biol. Chem. 1997;272(43):26799–26802. doi: 10.1074/jbc.272.43.26799. [DOI] [PubMed] [Google Scholar]
- 12.Johnson ES, et al. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997;16(18):5509–5519. doi: 10.1093/emboj/16.18.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knipscheer P, Sixma TK. Protein–protein interactions regulate Ubl conjugation. Curr. Opin. Struct. Biol. 2007;17(6):665–673. doi: 10.1016/j.sbi.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Knipscheer P, et al. Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 2007;26(11):2797–2807. doi: 10.1038/sj.emboj.7601711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li IT, Pham E, Truong K. Protein biosensors based on the principle of fluorescence resonance energy transfer for monitoring cellular dynamics. Biotechnol. Lett. 2006;28(24):1971–1982. doi: 10.1007/s10529-006-9193-5. [DOI] [PubMed] [Google Scholar]
- 16.Liu B, et al. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 2004;5(9):891–898. doi: 10.1038/ni1104. [DOI] [PubMed] [Google Scholar]
- 17.Martin SF, et al. Quantitative analysis of multi-protein interactions using FRET: application to the SUMO pathway. Protein Sci. 2008;17(4):777–784. doi: 10.1110/ps.073369608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller S, et al. SUMO, ubiquitin’s mysterious cousin. Nat. Rev. Mol. Cell Biol. 2001;2(3):202–210. doi: 10.1038/35056591. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005;23(3):355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 20.Olsen SK, et al. Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature. 2010;463(7283):906–912. doi: 10.1038/nature08765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piston DW, Kremers GJ. Fluorescent protein FRET: the good the bad and the ugly. Trends Biochem. Sci. 2007;32(9):407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Sapsford KE, Berti L, Medintz IL. Materials for fluorescence resonance energy transfer analysis: beyond traditional donor–acceptor combinations. Angew. Chem. Int. Ed. Engl. 2006;45(28):4562–4589. doi: 10.1002/anie.200503873. [DOI] [PubMed] [Google Scholar]
- 23.Saucerman JJ, et al. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc. Natl Acad. Sci. U.S.A. 2006;103(34):12923–12928. doi: 10.1073/pnas.0600137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solly K, et al. Miniaturization and HTS of a FRET-based membrane potential assay for K(ir) channel inhibitors. Assay Drug Dev. Technol. 2008;6(2):225–234. doi: 10.1089/adt.2008.123. [DOI] [PubMed] [Google Scholar]
- 25.Steffan JS, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304(5667):100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 26.Tatham MH, et al. Role of an N-terminal site of Ubc9 in SUMO-1, -2, and -3 binding and conjugation. Biochemistry. 2003;42(33):9959–9969. doi: 10.1021/bi0345283. [DOI] [PubMed] [Google Scholar]
- 27.Yeh ET. SUMOylation and De-SUMOylation: wrestling with life’s processes. J. Biol. Chem. 2009;284(13):8223–8227. doi: 10.1074/jbc.R800050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeh ET, Gong L, Kamitani T. Ubiquitin-like proteins: new wines in new bottles. Gene. 2000;248(1–2):1–14. doi: 10.1016/S0378-1119(00)00139-6. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, et al. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002;3(12):906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]