

Abstract
Overexpressed receptors, characteristic of many cancers, have been targeted by various researchers to achieve a more specific treatment for cancer. A common approach is to use the natural ligand for the overexpressed receptor as a cancer-targeting agent which can deliver a chemically or genetically conjugated toxic molecule. However, it has been found that the therapeutic efficacy of such ligand-drug molecular conjugates can be limited, since they naturally follow the intracellular trafficking pathways of the endogenous ligands. Therefore, a thorough understanding of the intracellular trafficking properties of these ligands can lead to novel design criteria for engineering ligands to be more effective drug carriers. This review presents a few commonly used ligand/receptor systems where intracellular trafficking considerations can potentially improve the therapeutic efficacy of the ligand-drug molecular conjugates.
Keywords: Transferrin, Epidermal growth factor, Transforming growth factor-α, Folate, Interleukin-13, Targeted delivery
Introduction
According to the Centers for Disease Control, cancer is currently second only to heart disease as the leading cause of death in the United States with an estimated 569,490 deaths in 2010.38,76 Unfortunately, current standard-of-care treatments still involve highly invasive surgical tumor resections followed by nonspecific radiation and chemotherapy that harm both normal and cancer cells. The issues associated with these treatment modalities have spawned numerous studies, and though a therapy truly specific to cancer has yet to be discovered, much progress has been made to improve the tumor selectivity of treatments in recent years.
Beginning in the early 1980s, researchers began reporting that many receptors were found to have increased expression in cancer cells compared to their normal counterparts. These included growth factor receptors (GFRs), such as epidermal GFR and fibroblast GFR, as well as other receptors such as transferrin, interleukin-13, and folate receptors.53,70,74,80,87 Overexpressed cell-surface receptors have therefore been exploited for several years as cancer-selective targets for therapeutic purposes. By controlling the dosage of receptor-targeted therapeutics, these agents can specifically target cancer cells while minimally affecting normal cells. Such molecular therapeutic approaches include signal transduction inhibitors, molecules to induce an immune response, antibody-drug conjugates (immunotoxins), and natural ligand-drug conjugates. The focus of this review, however, is on natural ligand-drug conjugates that have been studied for their antitumor effects.
Although these molecular conjugates have sometimes been grouped with immunotoxins, ligand-drug conjugates deserve special consideration since they naturally follow the intracellular trafficking pathways of the endogenous ligands. As such, an understanding of the trafficking pathway of the natural ligand can lead to novel approaches for engineering the ligand to be a more effective drug carrier.59,71,72,127,128 Therefore, the purpose of this review is to summarize some of the more extensively investigated ligand-drug molecular conjugates with particular emphasis given to the intracellular trafficking properties of the ligand. Moreover, although ligands have been conjugated to liposomes103and nanoparticles19 to enhance the targeting of these nanoscale drug delivery vehicles, they are not within the scope of this review, since the intracellular trafficking properties of these nanoscale drug carriers can vary from that of the ligand alone due to the large differences in size. In each of the following sections, a brief overview of the ligand/receptor system will be provided, followed by a summary of the associated trafficking pathway as well as notable examples of specific ligand-drug conjugates.
Transferrin
Human serum transferrin (Tf) has been widely investigated for its potential as a targeting agent in cancer therapy. This monomeric glycoprotein is approximately 80 kDa in size and serves the physiological role of sequestering and transporting free iron from systemic circulation to the cells of the body. Each molecule of Tf contains two iron-binding lobes, the N-terminal lobe (N-lobe) and the C-terminal lobe (C-lobe), with each lobe capable of binding a ferric (Fe3+) ion with an equilibrium dissociation constant (K D) of approximately 10−22 M.1,2 This iron-bound Tf, or holo-Tf, can then interact with cells through the surface-localized Tf receptor (TfR), allowing holo-Tf to deliver its iron payload to the cell. Once internalized, these iron atoms are stored within ferritin or used in a variety of biological processes, including cellular growth and proliferation.13
Since cancer cells are characterized by their uncontrolled growth, research has shown that these cells tend to require more iron, and thus, are likely to overexpress TfR. For example, a study by Tsan and coworkers, involving indirect detection of TfRs through immunofluorescence, demonstrated that neoplastic cell lines such as HEp-2 (larynx) and HeLa (cervix) have more TfRs on their surfaces than normal cells such as WI-38 (embryonic lung) and foreskin fibroblasts.15 Moreover, an immunohistochemistry study by Griffin’s group indicated an overexpression of TfRs in malignant brain tissue, including glial tumors and meningiomas, compared to normal brain tissue.87 The differences in expression levels of TfRs among cancer and healthy cells have been exploited by researchers to reduce the nonspecific toxicity of current cancer therapies.
Transferrin Receptor Intracellular Trafficking
The intracellular trafficking pathway of Tf and its receptor (Fig. 1a) is well-characterized and has been reviewed in several journals.67,68,84 As previously mentioned, the pathway begins with the binding of Fe3+ ions to Tf at physiological pH.84 Holo-Tf then binds to TfR on the surface of cells with nanomolar affinity (K D ~ 10−9 M). The Tf/TfR complex is clustered into a clathrin-coated pit which allows the complex to be internalized into an endocytic vesicle by a temperature- and energy-dependent process. The endosome eventually matures and acidifies to a pH of approximately 5.3 due to an ATP-driven proton pump,17 driving the release of iron from both iron-binding lobes of Tf.
Figure 1.
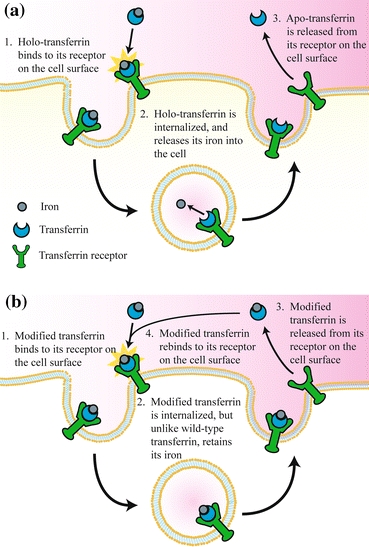
(a) Schematic of the Tf/TfR trafficking pathway. (b) The proposed alternative Tf/TfR trafficking pathway of Tf with decreased endosomal iron release rates.127 Copyright © 2008 Elsevier B. V., with kind permission from Elsevier
The release of iron by Tf is a complicated process involving many steps, a few of which will be briefly discussed here. An important mechanism of endosomal iron release from the N-lobe of holo-Tf involves two lysine residues known as the “dilysine trigger”.21 Though both lysines reside in opposite domains of the N-lobe, they are brought within close proximity in the iron-bound state, where only one of the two lysines is protonated, and therefore positively charged. The iron release “trigger”, which refers to the protonation of the opposite lysine residue upon acidification of the endosome, forces the two domains on each side of Fe3+ apart due to electrostatic repulsions. In the C-lobe, iron release is influenced by a trio of residues coined the “pH-sensitive triad”, the exact mechanism of which remains to be characterized.30 Moreover, iron release is facilitated by the interaction between Tf and TfR at endosomal pH, which explains decreased iron release rates observed for in vitro experiments performed in the absence of the receptor.27
After the release of iron, now in its reduced ferrous (Fe2+) form due to the presence of oxidoreductases in the endosome, Fe2+ enters the cytosol through a divalent cation transporter. Simultaneously, the iron-free Tf/TfR complex is recycled back to the cell surface. Since iron-free Tf (apo-Tf) has a low binding affinity for TfR at the near neutral pH of the cell surface, apo-Tf quickly dissociates from the cell-surface receptor, freeing TfR to bind to new holo-Tf molecules. This entire cycle of the Tf/TfR trafficking pathway typically lasts only 4–5 min.52
While the rapid recycling of Tf contributes to the efficient transport of iron, it also limits the time frame in which the ligand can deliver a conjugated drug. A clear understanding of the intracellular trafficking pathway of Tf is therefore crucial to improving its efficacy as a drug delivery vehicle.58 Through in vitro experiments and mathematical modeling, Murphy and coworkers previously demonstrated that the duration of cellular trafficking of monoclonal anti-transferrin receptor antibodies, or its cellular association, is correlated with the effectiveness of the antibody as a drug delivery vehicle.118,124,125 In other words, an increased duration of the antibody in the cell, i.e., an increased cellular association, would result in greater exposure of the cell to the conjugated drug. Instead of investigating antibodies for TfR, the Kamei laboratory focused on the Tf ligand itself. Specifically, by deriving and analyzing an extensive model for Tf/TfR trafficking, Kamei and coworkers identified a previously unreported design criterion for engineering Tf to enhance its drug delivery efficacy,59,127,128 as discussed in the “Diphtheria Toxin” section.
Transferrin Conjugates
Doxorubicin
Doxorubicin (Adriamycin®) is a widely used anthracycline chemotherapeutic agent in the treatment of leukemia, breast carcinoma, and other solid tumors.55 Doxorubicin manifests its toxicity by inhibiting topoisomerase II and/or intercalating nuclear DNA, thereby inhibiting DNA replication and halting cell growth. However, use of free doxorubicin in cancer treatment often leads to toxic side effects including nephrotoxicity, extravasation, cumulative cardiotoxicity, and myelosuppression. Furthermore, the clinical application of doxorubicin is also limited by the resistance of certain cancer cells to this drug. In recent years, considerable effort has been made to increase the specificity of this anti-neoplastic agent through targeted delivery. Several groups have investigated the potential of Tf as a carrier for doxorubicin by conjugating the two molecules through the formation of a Schiff base, and have seen a significant difference between the efficacy of free doxorubicin and the Tf-doxorubicin conjugate.3,107,126 Additionally, Goldenberg and coworkers demonstrated that Tf-doxorubicin is cytotoxic toward KB nasopharyngeal carcinoma cell lines in vitro despite their cellular resistance toward the unconjugated drug.26 In vivo studies by Micetich and coworkers, in which the drug was delivered to nude mice bearing human mesothelioma tumors, demonstrated a 69% increase in survival for mice treated with Tf-doxorubicin versus 39% for those treated with free doxorubicin, further validating the improved efficacy of the molecular conjugate.107
Diphtheria Toxin
Diphtheria toxin (DT) is a single-chain protein secreted by Corynebacterium diphtheriae that inhibits protein synthesis by inactivation of elongation factor 2 (EF-2), an enzyme integral to this process. This bacterial toxin contains three functional domains: the catalytic EF-2 inactivating domain, the cytosolic translocation domain, and the receptor-binding domain.29 A conjugate of Tf with a mutant of DT, known as CRM107, has exhibited potency against malignant gliomas.115 CRM107 contains a point mutation that significantly inhibits the binding of the drug to its native receptor, a heparin-binding epidermal growth factor-like growth factor precursor, thereby reducing the nonspecific toxicity of Tf-CRM107 (TransMID™).81 Furthermore, in vitro studies in which Tf-CRM107 toxicity was evaluated using a cellular protein synthesis assay indicated that the Tf-CRM107 conjugate is 1000- to 100,000-fold more toxic than free CRM107, illustrating the significant targeting ability of Tf.115 In vivo testing of the efficacy of Tf-CRM107, performed by Youle and Oldfield’s group on solid human gliomas in the flanks of nude mice, further confirmed the increased cytotoxicity exerted by the conjugate. In this study, the injection of Tf-CRM107 every 2 days for a total of four doses led to a significant reduction in tumor size. Conversely, treatment with free CRM107 at the same dosing schedule was 10- to 100-fold less effective in its ability to inhibit tumor growth. The success of Tf-CRM107 eventually led to phase III clinical trials.
Unfortunately, phase III trials were canceled in late 2006 following the results of a conditional power analysis suggesting that its efficacy would not significantly improve upon the standard-of-care treatments for high-grade brain tumors. As mentioned previously in the trafficking section, Tf recycles very rapidly, and this short duration inside the cell probably limits the ability of Tf to deliver its cytotoxic payload. Thus, to identify novel approaches to improving the efficacy of Tf-CRM107, Kamei and coworkers developed a mathematical model of the Tf/TfR trafficking pathway using the principles of mass action kinetics. A sensitivity analysis of the rate constants associated with the various binding events of this model helped determine that, by reducing or inhibiting the iron release rate of Tf within the endosome, its drug delivery efficacy would be significantly improved.59 Specifically, iron would be retained by Tf upon recycling to the cell surface and the conjugate would be reinternalized to participate in another cycle of trafficking due to the preserved high affinity of holo-Tf for TfR, increasing its cellular association (Fig. 1b). The drug delivery efficacy of Tf can therefore be improved, since multiple rounds of trafficking by a single Tf-drug conjugate would increase the probability of delivering the drug. With this design criterion, Kamei and coworkers subsequently engineered two Tf variants with decreased endosomal iron release rates using site-directed mutagenesis.127 These Tf mutants were conjugated to DT and were shown to be more effective than wild-type Tf in delivering DT in vitro to U87 and U251 human glioma cell lines.128 Furthermore, in vivo experiments, in which U87-derived tumors grown on the flanks of mice were treated with the conjugates, validated the improved efficacy of both mutant Tf-DT conjugates compared to their wild-type counterpart. Specifically, mice treated with the mutant Tf-DT conjugates demonstrated near complete tumor regression, while mice treated with the wild-type Tf-DT conjugates only demonstrated a delay in tumor growth under the same experimental conditions (Fig. 2).128 Ongoing studies of these mutant Tf molecules involve their conjugation to CRM107, with the hope that these mutant Tf-CRM107 conjugates would be more effective than TransMID™ in treating malignant gliomas.
Figure 2.
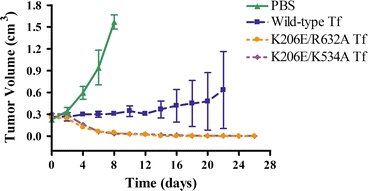
Tumor regression data for nude mice with subcutaneous glioma flank tumors treated with phosphate-buffered saline (PBS) control or DT conjugates of wild-type Tf, or one of two Tf mutants with decreased endosomal iron release rates. The K206E/R632A Tf mutant has a lysine to glutamic acid residue change at position 206 of Tf (in its N-lobe) and an arginine to alanine residue change at position 632 of Tf (in its C-lobe). The K206E/K534A Tf mutant has a lysine to glutamic acid residue change at position 206 of Tf and a lysine to alanine residue change at position 534 of Tf (in its C-lobe).128 Copyright © 2010 American Association for Cancer Research
Epidermal Growth Factor and Transforming Growth Factor-α
Cellular growth and proliferation is tightly regulated by a diverse group of polypeptides known as growth factors (GFs). A review by Moses and coworkers suggests that the fine-tuning of relative proliferation rates necessary for the coordinated growth of cells into different tissues is due to the multiplicity, varying cell type specificity, and coordinated actions of GFs.28 The growth and proliferation of neoplastic cells, on the other hand, are not held to this standard of regulation found in normal cells, since many types of cancers harbor means of escaping cellular growth control by producing autocrine GFs, synthesizing mutant GF receptors, and activating downstream receptor pathways that can altogether bypass the GF requirement.5,44,78 Moreover, studies demonstrate that many tumors overexpress receptors for GFs, favoring the continuous activation of their associated signal transduction pathways leading to enhanced cell motility, proliferation, angiogenesis, and reduced apoptosis.11,53,83,117 This discovery motivated many groups to target these GF receptors as a means of developing cancer-specific therapeutic agents.
This section will focus primarily on the targeting of the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase found to be overexpressed in a variety of cancers. The EGFR-based ligand/receptor system is one of the best characterized cell-signaling pathways, with its inhibition being one of the first examples of targeted therapy leading to suppression of tumor growth in vivo, as extensively reviewed in other journals.11,60,92,101 Moreover, the gene encoding EGFR, ERBB1, has been established as a cellular oncogene, since introducing high levels of this receptor into cultured cells confers the malignant phenotype in the presence of EGFR ligands.60 Though many of the clinically relevant drugs targeting EGFR include monoclonal antibodies and tyrosine kinase inhibitors which effectively downregulate the effects of EGFR signaling,60,74,92 this section will focus on the ability of two commonly used EGFR ligands, epidermal growth factor (EGF) and transforming growth factor-α (TGF-α), to deliver conjugated cytotoxic agents selectively to neoplastic cells.
Epidermal Growth Factor Receptor Intracellular Trafficking
Much of what is known regarding the intracellular signaling and trafficking of EGFR ligand/receptor complexes has been discovered through experimental observation coupled with mathematical modeling.31,104,108 As reviewed by Wiley, Shvartsman, and Lauffenburger, mathematical modeling helped provide important predictions regarding the complex EGFR system that were later tested and refined by the parallel development of powerful molecular, microscopic-imaging-based and biochemical kinetic assays.120 The following information provides some of the key findings regarding the EGFR system utilizing these tools.
There are currently seven known ligands for EGFR, two of which are EGF and TGF-α, each eliciting both distinct and redundant biological activities upon EGFR activation.102 There also exist four receptors in the EGFR family of receptor tyrosine kinases, which include EGFR, erbB-2, erbB-3, and erbB-4. Though some of the other three receptors have the ability to bind their own ligands,116 their importance is in their interaction with EGFR and each other following ligand activation. Altogether, the various EGFR ligands and receptors can modulate the EGFR signal transduction pathway in different cells to perform a variety of biological functions through their specific pairings.
As outlined in Fig. 3, EGFR undergoes a conformational change which allows it to dimerize with erbB-2, erbB-3, or another EGFR upon ligand binding. This interaction triggers phosphorylation of key tyrosine residues in the cytoplasmic tail of EGFR, providing docking sites for proteins containing Src homology 2 and phosphotyrosine binding domains.101 These proteins then initiate several signaling pathways including the mitogen-activated protein kinase kinase (MEK) pathway and the phosphatidylinositol 3 (PI3) kinase/Akt pathway, ultimately leading to activation of the various biological functions previously mentioned.
Figure 3.
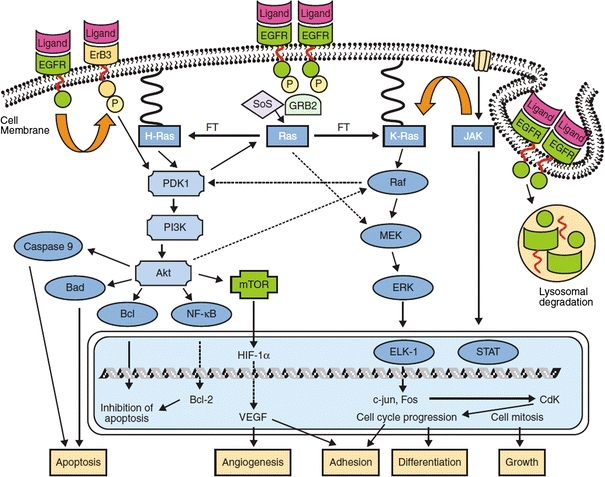
Signal transduction of epidermal growth factor receptor upon ligand activation.112 Copyright © 2005 American Cancer Society, with kind permission from John Wiley and Sons
While this signal can be modulated by the specific ligands and receptors involved in the initial binding event, it can further be modulated by differential intracellular trafficking of the ligand/receptor complex following endocytosis, as evident in the comparison of EGF and TGF-α. Work by the Lauffenburger lab demonstrated that, while EGF-bound EGFR complexes are typically sorted to the lysosome for receptor degradation, TGF-α bound EGFR complexes are sorted to intracellular recycling compartments.24,25,90 This differential trafficking behavior may be due to the fact that while EGF maintains its association to EGFR within the endosomal compartment, TGF-α readily dissociates from EGFR. Since the ligand/receptor interaction is important for maintaining signal transduction, the early dissociation of TGF-α may bias its signaling activity to the cell surface.119 EGF-activated receptors, on the other hand, can continue their signaling far into the intracellular trafficking pathway since the EGF/EGFR complex is maintained intact until just before lysosomal degradation.9
Due to the importance of EGFR overexpression and signaling in the development of the malignant phenotype in many cancers, much of the current generation of cancer-specific therapeutics involve the inhibition of EGFR-associated cell signaling. Currently, the EGFR-specific monoclonal antibody cetuximab (C225, Erbitux™) and the tyrosine kinase inhibitor erlotinib (Tarceva®) are potent inhibitors of EGFR signaling that have obtained Food and Drug Administration (FDA) approval for the treatment of certain cancers.92 However, it has been demonstrated that the benefits from these therapeutic approaches are largely cytostatic rather than cytotoxic, and therefore, researchers are interested in enhancing the antitumor efficacy of these drugs by supplementing them with other therapeutics.60 EGFR ligand-drug conjugates represent one alternative approach that may enhance the efficacy of conventional therapeutics that target EGFR.
EGFR Ligand Conjugates
Ricin
Ricin belongs to a group of plant-derived toxins known as ribosome-inactivating proteins (RIPs) best known for irreversibly damaging ribosomes through their unique RNA N-glycosidase activity and ultimately arresting cellular protein synthesis.4 Recently, Sperti and coworkers revealed that the N-glycosidase activity of RIPs, which is specific to adenine bases, can also damage nuclear DNA. This mode of action, together with protein synthesis inhibition, can induce cellular apoptosis.8 For this reason, RIPs are attractive toxins for the development of cancer-directed therapeutic conjugates. However, type II RIPs, like ricin, contain their own receptor-recognizing domain, which must be removed prior to conjugation to a targeting ligand to obtain cancer-selectivity. For the case of ricin, this truncated variant is known as ricin toxin A. In 1980, Collier and coworkers reported the development of an EGF-ricin toxin A (EGF-RTA) conjugate which demonstrated cytotoxicity at concentrations similar to those required for the biological activity of EGF (10−11–10−9 M) in immortalized 3T3 mouse fibroblast cells.12 This work motivated the Herschman lab to investigate the in vitro therapeutic potential of these conjugates against cancer cell lines such as HeLa and A431 (epidermal) cancer cells.33,106,113 Protein synthesis inhibition assays revealed IC50 values (or the concentrations at which 50% inhibition of protein synthesis occurs) of approximately 10−11 M for both cell lines, consistent with the earlier investigation by the Collier lab. But the most interesting aspect of the HeLa cell study is its comparison of the cytotoxic efficacy of EGF-RTA and an anti-EGFR antibody-ricin toxin A (C225-RTA) immunotoxin.113 The study revealed that, although C225-RTA was able to bind more tightly to EGFR than EGF-RTA, EGF-RTA was internalized to a significantly greater degree, reaching 60% internalization within 15 min while C225-RTA internalization never exceeded 30%. Despite these stark differences in EGFR-mediated cellular association, both agents exhibited a similar concentration-dependent potency in HeLa cells. This result may be due to the added benefit of C225-RTA in blocking the EGFR signal transduction pathway, given the clinical success of C225 as an inhibitor of the EGFR signaling pathway. In contrast, EGF-RTA was simply able to deliver more of its conjugated toxin, suggesting the importance of both EGFR signal inhibition and drug internalization in the development of an effective EGFR-targeted therapeutic. Therefore, combining EGFR ligand-drug conjugates such as EGF-RTA with currently available EGFR inhibiting therapies may significantly improve the efficacy of conventional therapeutics.92
Pseudomonas Exotoxin
Pseudomonas exotoxin (PE) from Pseudomonas aeruginosa is a bacterial toxin similar to DT.82 Like DT, this toxin contains three domains with the same general functions. As in the case of ricin conjugates, its receptor-recognition domain must be inactivated to maintain the integrity of cancer-selectivity upon ligand conjugation. Moreover, its translocation domain must be intact for efficient delivery to the cytosol.110 A truncated variant of PE (PE40) that satisfies both of these conditions is typically used for ligand conjugation. Most interestingly, truncated PE molecules like PE40 have been linked to two different EGFR ligands, EGF and TGF-α (both with M W ~ 6 kDa), which demonstrate different intracellular trafficking dynamics that potentially affect the delivery of the fused toxin. As investigated by the Pastan lab, a recombinant fusion protein of TGF-α and PE40 demonstrated high levels of protein synthesis inhibition against A431 cells which express 3 × 106 EGFR cell−1, moderate levels against KB cells which express 2 × 105 EGFR cell−1, and very low levels against HUT 102 T cell leukemia cells which express less than 103 EGFR cell−1 (Table 1).14,105 A recombinant fusion protein of EGF and a truncated PE (PE∆Ia) similar to PE40, developed by Hwang and coworkers, demonstrated a similar trend in potency against A431, KB, and HUT 102 cells, though their potency was significantly weaker (Table 1).65,69 Moreover, the TGF-α-PE40 recombinant fusion protein, also known as TP-38, has been investigated in phase I/II clinical trials for the treatment of brain and central nervous system tumors with moderate success.97,98 However, further clinical testing was terminated in late 2009 due to safety concerns.
Table 1.
Comparison of 24-h protein synthesis inhibition IC50 values of TGF-α-PE40 and EGF-PE(∆Ia) in a variety of cancer cells with different expression levels of EGFR
Folate
Folate, or vitamin B9, is another natural ligand that has demonstrated great potential as a targeting agent for cancer therapeutics. Unlike Tf, which is a protein, folate exists as a small hydrophilic molecule that is necessary for nucleotide synthesis, and thus cellular survival.70 The vast majority of folate uptake in nearly all cells is due to either a proton-coupled folate transporter or a reduced folate carrier.73,129 The expression of a third carrier, the folate receptor (FR), is typically restricted to normal epithelial tissue but has also been shown to be overexpressed on a large number of epithelial and non-epithelial cancers, with about 1–3 × 106 receptors cell−1.121 Furthermore, while folate-conjugated drugs lack affinity for the two former cell-surface proteins, they demonstrate a very high affinity for FR, an important feature when considering folate as a cancer-targeting agent.
Though FR exists in four known isoforms (α, β, γ, and δ), it is primarily found as either FR-α or FR-β. Of these, FR-α is the most common, present on several types of epithelial tissue including tissues of the fallopian tubes, uterus, epididymis, breast, lung, choroid plexus, and kidney.23 Additionally, cancer cells of the breast, lung, uterus, ovary, kidney, colon, thyroid, and brain have demonstrated overexpression of FR-α compared to their normal counterparts.70 Though the presence of FR-α on normal epithelial tissue is potentially problematic, most of these receptors face the organ’s lumen, and are thus isolated from systemically delivered folate-drug conjugates. Kidney epithelia are an exception, since they come into contact with these conjugates from the blood filtrate. However, FR-α on these epithelial cells participates in a mechanism to prevent folate from being cleared by the body, returning filtered folate-drug conjugates from the lumen of the kidney tubules back to systemic circulation via transcytosis rather than simply internalizing the conjugate. This affords some level of protection to the kidneys.121 Meanwhile, FR-β is only found in activated macrophages and some malignant hematopoietic cell types.121 Thus, folate-conjugated drugs will localize to the kidneys as well as locations of inflammatory immune response in cancer-free patients but, importantly, will also localize to FR-positive tumors in cancer patients. Although this ligand has been more extensively reviewed elsewhere for both targeted cancer therapy and imaging applications,70,95,96,121 this section will cover the basic principles of folate trafficking and examples of its use in cancer therapy.
Folate Receptor Intracellular Trafficking
The trafficking of the folate-drug conjugate with FR begins with the binding of folate to the receptor. It has been estimated that, at physiological pH, folic acid is able to bind to FR-α with picomolar affinity (K D ~ 10−12 M).43 Additionally, drugs linked to the γ-carboxyl group of folate have been shown to have a much higher affinity for FR-α over α-carboxyl-conjugated folate, but the exact K D values for these interactions have not been determined.114
Both FR-α and FR-β are glycosylphosphatidylinositol (GPI)-anchored proteins, meaning that unlike transmembrane proteins, they only contain an extracellular domain that is attached by a chain of sugars and phosphates to a lipid embedded in the outer leaflet of the plasma membrane. Due to their size and shape, these complexes are believed to preferentially reside in lipid rafts.95 Additionally, upon ligand binding, these GPI-anchored proteins have a different endocytic mechanism than many other types of proteins.34 For instance, while the Tf/TfR complex is internalized via clathrin-coated pits, the GPI-anchored FR is primarily internalized through clathrin-independent carriers (CLICs). These ring-shaped tubules form directly from the plasma membrane and are believed to function as a mechanism for the internalization of GPI-anchored proteins.34
Though the mechanism is unclear, these CLICs show an ability to sort different types of proteins and eventually mature into GPI-rich early endosomal compartments (GEECs) which, as the name suggests, contain high concentrations of GPI-anchored proteins like FR. While it remains unclear as to how these processes occur, the GEECs demonstrate an ability to sort some of their contents into early endosomes, shared with Tf/TfR complexes.34,122 These early endosomes are eventually routed to a recycling endosomal compartment (REC) at the perinuclear region, which recycles FR back to the cell surface, often with folate intact.95 At this point, the folate/FR complex can again cycle through this trafficking pathway. After achieving steady-state, it is believed that between 50 and 75% of FR remains within the cell at any given time.95
In designing folate-based cancer therapeutics, Low and coworkers determined that along with understanding the intracellular trafficking pathway of folate, it is necessary to consider the endosomal conditions that will be encountered by folate-based therapeutics. First, to determine whether or not folate encounters reductive environments in its intracellular trafficking pathway, Low and coworkers investigated the intracellular trafficking conditions of folate using a fluorescence resonance energy transfer (FRET) reporter.122 In this case, the reporter (which we will refer to as reporter #1) contained a BODIPY fluorescent probe conjugated to a rhodamine fluorescent probe through a reducible disulfide linkage. Reporter #1 was further conjugated to a folate molecule through a non-reducible amide linkage. Excitation at 488 nm would cause BODIPY to emit light at 520 nm (green) when the disulfide linkage was cleaved, but when intact, the BODIPY emission would be quenched by the nearby rhodamine and result in a final light emission at 595 nm (red). As demonstrated by the confocal images in Fig. 4, it was discovered that the disulfide bond of reporter #1 was reduced very efficiently throughout the entire folate/FR trafficking process.
Figure 4.
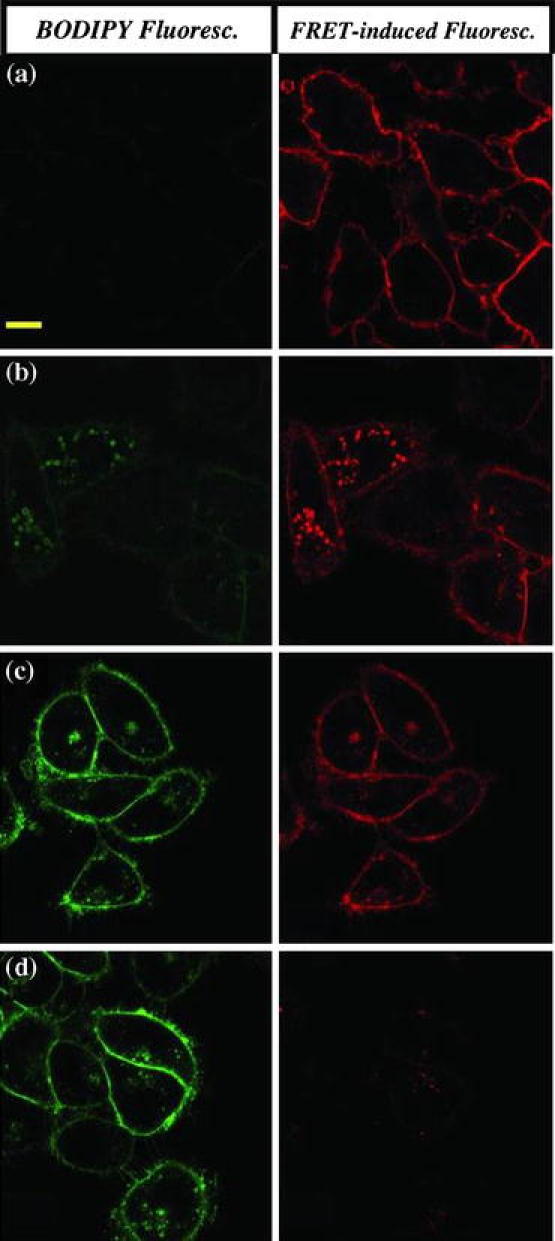
Investigating the reductive nature of intracellular compartments during folate-FR trafficking using FRET. The folate-FRET reporter #1 changes fluorescence from red to green upon disulfide reduction. After a 0.5-h incubation with 100 nM of reporter #1 and a subsequent wash step to remove unbound conjugates, KB cells were imaged with confocal microscopy (a) immediately or after (b) 2, (c) 6, or (d) 12 h of further incubation in culture medium by excitation at 488 nm. Scale bar = 10 μm.122 Copyright © 2006 National Academy of Sciences of the USA
To determine the acidity of the environment encountered by folate/FR complexes, the same group used a similar FRET reporter (which we will refer to as reporter #2) with a BODIPY fluorescent probe and a quenching dabcyl molecule on opposite ends of a pH-sensitive acyl hydrazone linkage. Accordingly, acid-induced cleavage of the quenching dabcyl molecule would result in green fluorescence characteristic of BODIPY.123 As shown in Fig. 5, the acyl hydrazone linkages were cleaved only in the REC after 5.5 h of association with KB cells (Fig. 5c). Through further characterization of this process, Low and coworkers were able to determine that, throughout the folate/FR trafficking pathway, the pH remained above 6.0 with an average pH between 6.6 and 6.8.123 Because folate conjugates encounter an endosomal environment that is more reducing than acidic, a cleavable disulfide linkage, rather than an acid-cleavable linkage, should be used to guarantee effective intracellular drug release.
Figure 5.

Investigating the acidity of intracellular compartments during folate-FR trafficking using FRET. The folate-FRET reporter #2 emits green fluorescence upon acid-induced cleavage. After a 0.5-h incubation with 100 nM of reporter #2 and a subsequent wash step to remove unbound conjugates, KB cells were imaged with confocal microscopy after (a) 1.5, (b) 5.5, or (c) 18.5 h of further incubation in culture medium by excitation at 488 nm.123 Copyright © 2007 American Society for Pharmacology and Experimental Therapeutics
Folate Conjugates
Tubulysin B-H
Tubulysin B is an extremely potent natural peptide produced by certain myxobacteria that destabilizes microtubules and kills cells with IC50 values as low as 8 × 10−10 M.61 Vlahov and coworkers have shown that tubulysin B-H, a tubulysin B molecule that has been functionalized with a hydrazide for conjugation chemistry, retains much of this potency, with IC50 values as low as 2.5 × 10−9 M.61 However, in its free form, tubulysin displays nonspecific toxicity, delaying its further development in cancer treatment. Thus, Vlahov and coworkers have investigated folate conjugates of tubulysin B-H to limit its potency to cancers that overexpress FR. To determine whether the folate-tubulysin B-H conjugate (EC0305) maintained a similar potency as the unconjugated toxin, Vlahov and coworkers performed a series of in vitro studies on KB and RAW 267.4 (macrophage) cells and determined IC50 values to be 1.5 × 10−9 and 2.6 × 10−9 M, respectively.61 Moreover, this conjugate demonstrated an ability to target cancer cells specifically through FR. In vivo studies in mice with established KB dorsal medial tumors intravenously treated with tubulysin B and tubulysin B-H experienced dose-dependent systemic toxicity with no signs of tumor regression when given a maximum of two consecutive doses of 1 μmol drug per kg body weight (μmol kg−1). Conversely, mice treated with EC0305 were found to show partial tumor regression with doses as low as 0.5 μmol kg−1 and complete tumor regression in all tested mice at 1 and 2 μmol kg−1, with no apparent side effects or toxicity. Thus, while native tubulysin B–H was unable to produce an antitumor effect due to its high nonspecific toxicity, EC0305 was able to induce tumor regression with no apparent dose-limiting toxicity.88 Furthermore, the IC50 values of EC0305 were three-fold lower than other similar folate-targeted chemotherapeutics, such as folate-mitomycin C and folate-camptothecin.32,61–64,89,91 Recent studies also demonstrated that EC0305 was more effective against M109 (murine lung) and 4T1-C12 (murine mammary) cancer cells compared to folate-desacetylvinblastine monohydrazide (EC145), a folate-targeted drug that has reached phase II clinical trials.88
Haptens
In contrast to using the folate/FR trafficking pathway to release and deliver a cytotoxic agent into a targeted cell, researchers in the Low laboratory have also utilized folate-hapten conjugates to coat the surface of cancer cells with haptens, i.e., small molecules that induce an immune response when attached to a large carrier molecule.71,72 Although many attempts have been made to generate an immune response to cancer-specific antigens, these attempts have had varying levels of success.7 To address this challenge, the Low laboratory exploited the ability of the folate/FR complex to remain intact throughout its trafficking pathway to coat FR expressing cancer cells with folate-conjugated haptens, which are recognized by endogenous antibodies and lead to antibody-dependent cellular cytotoxicity (ADCC).71 To accomplish this, the Low laboratory created folate-FITC, which was a conjugate between folate and a functionalized fluorescein molecule linked by a short, non-cleavable spacer. Since FITC does not naturally induce an immune response by itself, mice were first inoculated with FITC-carrier conjugates, such as FITC-bovine serum albumin (FITC-BSA) or FITC-keyhole limpet hemocyanin (FITC-KLH), to stimulate mice to develop antibodies against FITC. Next, the folate-FITC conjugates were added along with low levels of interleukin-2 and interferon-α, which help induce ADCC.71 In vivo data showed that mice with M109 tumors were able to demonstrate complete tumor regression and survive significantly longer with no off-target toxicity. Moreover, when mice that had shown tumor remission were reinoculated with M109 tumor cells, they were able to reject the tumor without treatment.72 Investigation into the biological processes of this treatment has confirmed an immunological response with recruitment of natural killer cells and macrophages for tumor clearance, and CD4+ and CD8+ T cells for adaptive immunity.72
Interleukin-13
Interleukin-13 (IL-13) is a natural cytokine in the body produced by activated T-helper type 2 and mast cells that inhibits the production of inflammatory cytokines in monocytes and enhances growth and differentiation of B cells, monocytes, and endothelial cells.37,77 Due to this important role in the immune system as well as its roles in allergy and cancer, IL-13 and its receptors have been extensively reviewed.16,39,130 Through investigations of the IL-13 receptor (IL-13R) composition on various cell lines, the Puri laboratory characterized its complex structure into three types (Fig. 6).37,80 Type I IL-13Rs are made up of IL-13Rα1, IL-13Rα2, and IL-4Rα chains. Type II receptors, on the other hand, only consist of IL-13Rα1 and IL-4Rα chains. Type III receptors are composed of IL-13Rα1, IL-4Rα, and IL-2Rγc chains. Depending on the type of receptor activated by IL-13, the IL-13/IL-13R complex can either initiate cellular signaling through the activation of Janus kinases and the signal transduction activator of transcription (STAT) protein (type II and III)79 or internalize with an undetermined degree of signal transduction (type I), although the purpose of this latter function has yet to be fully elucidated.48 Additionally, it was discovered that only the interaction between IL-13 and the type I IL-13R was able to induce internalization due to the presence of the receptor’s unique IL-13Rα2 chain.48
Figure 6.
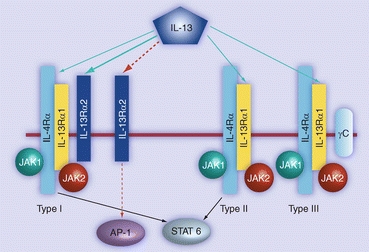
Signal transduction of the three types of interleukin-13 receptors upon ligand activation.42 Copyright © 2009 Future Medicine Ltd
In a study by Puri and coworkers, the type I receptors were found to be overexpressed on a variety of cancers including renal cell carcinoma, glioma, and ovarian adenocarcinomas.36,40,50,51,80 In another study, Puri’s group investigated type I IL-13R expression in 25 brain tumor explants, as well as normal human astrocyte cultures and normal brain tissue. RT-PCR analysis showed expression of IL-13Rα2 in 72% of all brain tumor samples and 82% of glioblastoma multiforme cases; moreover, it was shown that the IL-13Rα2 chain was expressed almost exclusively among brain tumor explants rather than normal cells and tissue.41 Due to its overexpression in cancer cells and ability to internalize, the type I IL-13R has been exploited as a target to deliver therapeutics specifically to cancers without the risk of toxicity to normal tissue. However, the reason for its overexpression in cancer is unknown.42
Interleukin-4 (IL-4) is a cytokine that shares common receptor subunits with IL-13, and thus common signaling pathways.109 Like IL-13, IL-4 is also commonly overexpressed in many types of cancers and has been used as a targeting agent for cancer treatment. Though this section will focus on IL-13 and its receptors, more information on IL-4 and its role in cancer drug delivery can be found in reviews published by the Puri laboratory.45,85
Interleukin-13 Receptor Intracellular Trafficking
In most cells, IL-13 first binds to an IL-13Rα1 chain (K D ~ 3 × 10−8 M) through interactions involving charged lysine, arginine, and glutamic acid residues on the αD helix of IL-13. This interaction recruits an IL-4Rα chain to bind to the IL-13/IL-13Rα1 complex (K D ~ 1.5 × 10−7 M) at residues on IL-13’s αA and αB helices, completing the formation of type II IL-13R. Once IL-13 has been bound to both receptor chains, its affinity greatly increases (K D ~ 3–4 × 10−11 M), indicating cooperative binding.54,75
This type of binding initiates cell signaling for the activation of transcription factors. The cytoplasmic regions of both IL-13Rα1 and IL-4Rα chains found in type II receptors interact with various tyrosine kinases, ultimately interacting with PI3 kinase (involved in cellular proliferation) and STAT6 (involved in altering gene expression).49 In cells that express type III IL-13R, signaling similar to type II receptors is believed to take place; however, kinetic studies have shown reduced specificity and a decrease in the rate of IL-13 binding to IL-13Rα1 in the presence of the IL-2Rγc chain in type III IL-13R.57 Endocytosis, however, is only induced by the additional binding of IL-13 to the IL-13Rα2 chain, found only in type I IL-13R. This endocytic behavior of type I IL-13R was determined by studies demonstrating large concentrations of IL-13Rα2 found within intracellular vesicles. Moreover, IL-13 has a much higher affinity for IL-13Rα2 than IL-13Rα1, effectively inhibiting cell signaling in exchange for endocytosis of IL-13 in cells that express IL-13Rα2.22
Closer examination of the IL-13Rα2 chain has revealed that it exists in three compartments: intracellular vesicles, the plasma membrane, and extracellularly in a soluble form (Fig. 7).18 In most cells, IL-13Rα2 is predominantly located in intracellular vesicles; however, the IL-13Rα2 chain can quickly be mobilized to the cell surface upon inflammation, accounting for the higher expression of IL-13R in many inflammatory states associated with allergy and parasitic infection. Due to its short cytoplasmic tail which lacks signaling motifs, membrane-bound IL-13Rα2 has been hypothesized to act primarily as decoy receptors that inhibit IL-13 signaling by competitive binding particularly due to its higher affinity for IL-13 over IL-13Rα1. Its soluble form, a product of either alternative splicing of the IL-13Rα2 mRNA or cleavage of the membrane-bound IL-13Rα2,109 can sequester IL-13 before it has a chance to bind type II IL-13R and also inhibit signal transduction. Coupled with its propensity to induce internalization of IL-13/type I IL-13R complexes, IL-13Rα2 plays an important role in IL-13 signal regulation that involves an unidentified intracellular trafficking process. As demonstrated by the previously discussed ligand/receptor systems, an understanding of this process can help to improve the therapeutic efficacy of current generation IL-13-conjugated cancer therapeutics, some of which are discussed in the next section.
Figure 7.
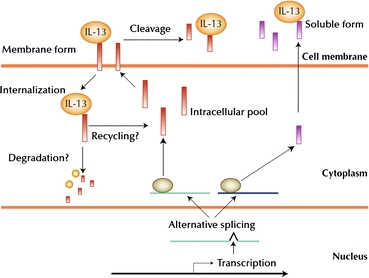
Alternative splicing of the two IL-13Rα2 isoforms, their distribution into extracellular, cell surface, and intracellular compartments, and their interaction with IL-13. Upon IL-13 binding to the membrane-bound IL-13Rα2, the complex is internalized. The intracellular trafficking pathway of this process is yet unknown, though evidence of continuous cycling between cell surface and intracellular pools has been shown.109 Copyright © 2007 Current Medicine Group LLC, with kind permission from Springer Science + Business Media
Interleukin-13 Conjugates
Diphtheria Toxin
DT has been fused to IL-13 as a vehicle for targeted delivery to cancer cells. For example, the Vallera laboratory developed a recombinant fusion protein made up of IL-13 and a truncated variant of DT (DAB389), lacking its receptor-recognition domain, called DTIL13.66 In vitro studies of DTIL13 on glial cell lines have demonstrated that its cytotoxicity increases with an increase in the density of IL-13R expressed on the cell. In vivo testing by Vallera’s group, however, showed mixed results. In the first set of trials, testing in a U373 xenograft mouse tumor model resulted in tumor regression of all mice treated intratumorally with doses of 1 and 10 μg. In the second trial, five more mice were given 10 μg doses. While reduction of tumor size was initially observed in all mice, two became completely cured while others relapsed. This suggests a possible mechanism of resistance to the drug for yet an unknown reason. Further testing was carried out in intracranial brain tumor models in mice.93 Intratumoral administration of DTIL13 indicated that no toxic side effects occurred for 1 μg injections of the drug, whereas loss of body weight was observed at higher doses. Overall, survivability was increased by 35% in mice treated with the 1 μg dose. Furthermore, Vallera and coworkers developed a new drug called DTAT13, which combines the components of DTIL13 with an N-terminal fragment of the urokinase-type plasminogen activator (uPa), allowing it to target cancer cells overexpressing the uPa receptor as well.94 This formulation has shown greater efficacy than both DTIL13 and DTAT, which is DTAT13 without IL-13. Pharmacokinetic studies demonstrated that the presence of both ligands does not impede the binding of the other and allowed faster internalization and killing of cancer cells than DTIL13 or DTAT. DTAT13 was also observed to have a longer in vivo half-life, giving it more time to associate with tumors. Furthermore, the Vallera laboratory studied a similar bispecific recombinant fusion protein which targeted both IL-13R and EGFR with similar results.111
Pseudomonas Exotoxin
Similar to EGF and TGF-α, IL-13 has also been conjugated to PE. For example, the Puri group developed a mutant form of PE (PE38QQR) similar to the PE40 variant used in the TGF-α conjugates.20 The fusion of this mutant to IL-13 led to the development of IL13-PE38QQR, also known as cintredekin besudotox, which was used to target cells overexpressing type I IL-13R. Though this molecule has demonstrated an ability to inhibit protein synthesis, it has also shown the capability of inducing caspase-mediated apoptosis by damaging mitochondria.47 Through in vitro studies of multiple glioblastoma cell lines expressing IL-13R at different densities, Puri and coworkers were able to achieve IC50 values that were less than 2 × 10−11 M, which were still high enough doses to completely kill cells in some cases, indicating high sensitivity to the recombinant fusion protein.35,46 Subsequently, in vivo testing performed in mice with established subcutaneous U251 xenografts showed that increasing the dosage of the drug impeded tumor growth while imparting minimal effect on non-cancerous cells when administered intravenously, intraperitoneally, and intratumorally, with intratumoral administration yielding the best results. However, due to the drug’s short in vivo half-life, frequent administration was necessary to enhance its efficacy and achieve complete regression. This led to the implementation of a convection-enhanced delivery system, as used in the delivery of other therapeutics such as Tf-CRM107, in which a catheter placed into the tumor employs a pressure gradient to deliver the drug.6 Because large biomolecules such as proteins have low diffusion coefficients, convection-enhanced delivery has been effective in increasing the drug’s entry into the tissue by supplementing diffusion with convection. This has allowed for the drug to be administered to a larger volume of the tumor compared to the volume achieved by a simple intratumoral injection.
Although early clinical testing was successful and cintredekin besudotox showed promise, progress stalled during phase III clinical trials due to adverse events in patients, including aphasia and hemiparesis, as well as other nervous system, vascular, and psychiatric disorders, indicating that specificity for target cells was not as strong as initially believed.56 As a solution, Lowenstein and Castro’s group developed an inducible adenoviral vector encoding both (i) a mutated form of human IL-13, which binds more tightly to the IL-13Rα2 chain of type I IL-13R, fused to PE (mhIL-13-PE) and (ii) a mutant form of human IL-4 (mhIL-4), which inhibits potential binding of mhIL-13-PE to normal brain cells by binding directly to the physiological IL-4R and IL-13R (without interacting with the IL-13Rα2 chain).10 In addition, because this adenoviral vector is regulated by a tetracycline-responsive promoter, mhIL-13-PE can be continuously expressed in the presence of tetracycline, significantly increasing its in vivo half-life. Implementation of the vector in vitro showed loss of cell function in over 70% of IN859 and U251 glioma cell lines, while showing no toxicity against control COS-7 monkey kidney cells which do not express IL-13Rα2. Furthermore, in vivo testing in an intracranial U251 xenograft mouse tumor model resulted in 70% long-term survival with no effects on brain structure and function or overt toxic effects.
Concluding Remarks
Natural ligand-drug molecular conjugates for cancer have been extensively studied over the past several years. Though these conjugates have demonstrated some preclinical and clinical successes, not many have obtained FDA approval for the treatment of cancer. The general lack of FDA-approved ligand-drug conjugates may be attributed to the natural properties of the targeting ligand, which may limit its efficacy as a drug delivery vehicle. Although natural ligands can effectively target cancer cells via their cell-surface receptors, once bound, ligand-drug conjugates are at the mercy of the ligand’s physiological trafficking pathway. For instance, the rapid recycling rate of Tf, though highly efficient for the delivery of iron, often restricts its ability to deliver a conjugated drug to a single 4–5 min intracellular pass.59 In the case of EGF-drug conjugates, the much slower lysosomal degradation pathway helps to increase the association of the conjugates with cancer cells. However, the continued propagation of the EGF signal transduction pathway due to the increased cellular association may counteract the therapeutic efficacy of the drug.9,108,119 Lysosomal degradation may also negatively affect protein toxins used in EGF-drug conjugates, such as ricin and PE, which would be subjected to the proteolytic enzymes found in lysosomes.
Through experimentation and mathematical modeling, a more thorough understanding of the intracellular trafficking properties of various ligands has been achieved. Some research groups have capitalized on this understanding to find innovative approaches to manipulating these trafficking properties for the advancement of ligand-based cancer therapeutics. To address the issue of the short residence time of Tf inside cells, i.e., its limited cellular association, the Kamei laboratory discovered a novel design criterion whereby its cellular association, and therefore its drug delivery efficacy, can greatly be enhanced by manipulation of its binding interaction with iron.59,127 In another study, Low and coworkers developed folate-hapten conjugates for cell-surface decoration to elicit an immune attack against cancer, since it had been shown that folate/FR complexes remain intact throughout their trafficking pathway.71,72 These examples demonstrate that consideration of intracellular trafficking properties, and not merely cell-surface binding events, can result in the engineering of effective targeting agents to cancers.
Consideration of cell-level kinetics has the potential for improving success rates in clinical trials, much like compartment models of the body that have been developed and studied for several years to aid in the design of drugs and their dosing. Although a qualitative mechanistic understanding of cell-level kinetics can help in engineering drugs, mathematical modeling of these phenomena can lead to more specific design criteria as noted by Rao, Lauffenburger, and Wittrup.86 These cell-level kinetic models have also been seamlessly integrated with compartment models, since the cells of interest can be used to represent one of the compartments. Sarkar and Lauffenburger derived such a model for the cytokine granulocyte colony-stimulating factor,99 and in an earlier study, also performed molecular modeling to identify mutations in granulocyte colony-stimulating factor that would satisfy cell-level predictions that could lead to enhanced potency.100 This approach of quantitatively bridging molecular, cellular and whole body length scales can be extended to natural ligand-drug molecular conjugates for cancer, and we are excited about this evolving multi-scale approach to rational drug conjugate design.
Acknowledgment
The authors would like to thank the support of the UCSF Brain Tumor Specialized Program of Research Excellence NIH P50CA097257.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Aisen P, Leibman A, Zweier J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J. Biol. Chem. 1978;253:1930–1937. [PubMed] [Google Scholar]
- 2.Baker EN, Baker HM, Kidd RD. Lactoferrin and transferrin: functional variations on a common structural framework. Biochem. Cell Biol. 2002;80:27–34. doi: 10.1139/o01-153. [DOI] [PubMed] [Google Scholar]
- 3.Barabas K, Sizensky JA, Faulk WP. Transferrin conjugates of adriamycin are cytotoxic without intercalating nuclear DNA. J. Biol. Chem. 1992;267:9437–9442. [PubMed] [Google Scholar]
- 4.Barbieri L, Battelli MG, Stirpe F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta. 1993;1154:237–282. doi: 10.1016/0304-4157(93)90002-6. [DOI] [PubMed] [Google Scholar]
- 5.Barnes D, Sato G. Serum-free cell culture: a unifying approach. Cell. 1980;22:649–655. doi: 10.1016/0092-8674(80)90540-1. [DOI] [PubMed] [Google Scholar]
- 6.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. USA. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. Failure of cancer vaccines: the significant limitations of this approach to immunotherapy. Anticancer Res. 2000;20:2665–2676. [PubMed] [Google Scholar]
- 8.Brigotti M, Alfieri R, Sestili P, Bonelli M, Petronini PG, Guidarelli A, Barbieri L, Stirpe F, Sperti S. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. Faseb J. 2002;16:365–372. doi: 10.1096/fj.01-0521com. [DOI] [PubMed] [Google Scholar]
- 9.Burke P, Schooler K, Wiley HS. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol. Biol. Cell. 2001;12:1897–1910. doi: 10.1091/mbc.12.6.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Candolfi M, Xiong W, Yagiz K, Liu C, Muhammad AK, Puntel M, Foulad D, Zadmehr A, Ahlzadeh GE, Kroeger KM, Tesarfreund M, Lee S, Debinski W, Sareen D, Svendsen CN, Rodriguez R, Lowenstein PR, Castro MG. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proc. Natl Acad. Sci. USA. 2010;107:20021–20026. doi: 10.1073/pnas.1008261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castillo L, Etienne-Grimaldi MC, Fischel JL, Formento P, Magne N, Milano G. Pharmacological background of EGFR targeting. Ann. Oncol. 2004;15:1007–1012. doi: 10.1093/annonc/mdh257. [DOI] [PubMed] [Google Scholar]
- 12.Cawley DB, Herschman HR, Gilliland DG, Collier RJ. Epidermal growth factor-toxin A chain conjugates: EGF-ricin A is a potent toxin while EGF-diphtheria fragment A is nontoxic. Cell. 1980;22:563–570. doi: 10.1016/0092-8674(80)90366-9. [DOI] [PubMed] [Google Scholar]
- 13.Cazzola M, Bergamaschi G, Dezza L, Arosio P. Manipulations of cellular iron metabolism for modulating normal and malignant cell proliferation: achievements and prospects. Blood. 1990;75:1903–1919. [PubMed] [Google Scholar]
- 14.Chaudhary VK, FitzGerald DJ, Adhya S, Pastan I. Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc. Natl. Acad. Sci. USA. 1987;84:4538–4542. doi: 10.1073/pnas.84.13.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen DC, Newman B, Turkall RM, Tsan MF. Transferrin receptors and gallium-67 uptake in vitro. Eur. J. Nucl. Med. 1982;7:536–540. doi: 10.1007/BF00571645. [DOI] [PubMed] [Google Scholar]
- 16.Chomarat P, Banchereau J. Interleukin-4 and interleukin-13: their similarities and discrepancies. Int. Rev. Immunol. 1998;17:1–52. doi: 10.3109/08830189809084486. [DOI] [PubMed] [Google Scholar]
- 17.Ciechanover A, Schwartz AL, Dautry-Varsat A, Lodish HF. Kinetics of internalization and recycling of transferrin and the transferrin receptor in a human hepatoma cell line. Effect of lysosomotropic agents. J. Biol. Chem. 1983;258:9681–9689. [PubMed] [Google Scholar]
- 18.Daines MO, Tabata Y, Walker BA, Chen W, Warrier MR, Basu S, Hershey GK. Level of expression of IL-13R alpha 2 impacts receptor distribution and IL-13 signaling. J. Immunol. 2006;176:7495–7501. doi: 10.4049/jimmunol.176.12.7495. [DOI] [PubMed] [Google Scholar]
- 19.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Debinski W, Obiri NI, Pastan I, Puri RK. A novel chimeric protein composed of interleukin 13 and Pseudomonas exotoxin is highly cytotoxic to human carcinoma cells expressing receptors for interleukin 13 and interleukin 4. J. Biol. Chem. 1995;270:16775–16780. doi: 10.1074/jbc.270.28.16775. [DOI] [PubMed] [Google Scholar]
- 21.Dewan JC, Mikami B, Hirose M, Sacchettini JC. Structural evidence for a pH-sensitive dilysine trigger in the hen ovotransferrin N-lobe: implications for transferrin iron release. Biochemistry. 1993;32:11963–11968. doi: 10.1021/bi00096a004. [DOI] [PubMed] [Google Scholar]
- 22.Donaldson DD, Whitters MJ, Fitz LJ, Neben TY, Finnerty H, Henderson SL, O’Hara RM, Jr, Beier DR, Turner KJ, Wood CR, Collins M. The murine IL-13 receptor alpha 2: molecular cloning, characterization, and comparison with murine IL-13 receptor alpha 1. J. Immunol. 1998;161:2317–2324. [PubMed] [Google Scholar]
- 23.Elnakat H, Ratnam M. Distribution, functionality and gene regulation of folate receptor isoforms: implications in targeted therapy. Adv. Drug Deliv. Rev. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 24.French AR, Sudlow GP, Wiley HS, Lauffenburger DA. Postendocytic trafficking of epidermal growth factor-receptor complexes is mediated through saturable and specific endosomal interactions. J. Biol. Chem. 1994;269:15749–15755. [PubMed] [Google Scholar]
- 25.French AR, Tadaki DK, Niyogi SK, Lauffenburger DA. Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J. Biol. Chem. 1995;270:4334–4340. doi: 10.1074/jbc.270.9.4334. [DOI] [PubMed] [Google Scholar]
- 26.Fritzer M, Szekeres T, Szuts V, Jarayam HN, Goldenberg H. Cytotoxic effects of a doxorubicin-transferrin conjugate in multidrug-resistant KB cells. Biochem. Pharmacol. 1996;51:489–493. doi: 10.1016/0006-2952(95)02225-2. [DOI] [PubMed] [Google Scholar]
- 27.Giannetti AM, Halbrooks PJ, Mason AB, Vogt TM, Enns CA, Bjorkman PJ. The molecular mechanism for receptor-stimulated iron release from the plasma iron transport protein transferrin. Structure. 2005;13:1613–1623. doi: 10.1016/j.str.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 28.Goustin AS, Leof EB, Shipley GD, Moses HL. Growth factors and cancer. Cancer Res. 1986;46:1015–1029. [PubMed] [Google Scholar]
- 29.Greenfield L, Johnson VG, Youle RJ. Mutations in diphtheria toxin separate binding from entry and amplify immunotoxin selectivity. Science. 1987;238:536–539. doi: 10.1126/science.3498987. [DOI] [PubMed] [Google Scholar]
- 30.Halbrooks PJ, Giannetti AM, Klein JS, Bjorkman PJ, Larouche JR, Smith VC, MacGillivray RT, Everse SJ, Mason AB. Composition of pH-sensitive triad in C-lobe of human serum transferrin. Comparison to sequences of ovotransferrin and lactoferrin provides insight into functional differences in iron release. Biochemistry. 2005;44:15451–15460. doi: 10.1021/bi0518693. [DOI] [PubMed] [Google Scholar]
- 31.Haugh JM, Wells A, Lauffenburger DA. Mathematical modeling of epidermal growth factor receptor signaling through the phospholipase C pathway: mechanistic insights and predictions for molecular interventions. Biotechnol. Bioeng. 2000;70:225–238. [PubMed] [Google Scholar]
- 32.Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg. Med. Chem. Lett. 2006;16:5350–5355. doi: 10.1016/j.bmcl.2006.07.076. [DOI] [PubMed] [Google Scholar]
- 33.Herschman HR. The role of binding ligand in toxic hybrid proteins: a comparison of EGF-ricin, EGF-ricin A-chain, and ricin. Biochem. Biophys. Res. Commun. 1984;124:551–557. doi: 10.1016/0006-291x(84)91589-4. [DOI] [PubMed] [Google Scholar]
- 34.Howes MT, Kirkham M, Riches J, Cortese K, Walser PJ, Simpson F, Hill MM, Jones A, Lundmark R, Lindsay MR, Hernandez-Deviez DJ, Hadzic G, McCluskey A, Bashir R, Liu L, Pilch P, McMahon H, Robinson PJ, Hancock JF, Mayor S, Parton RG. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J. Cell Biol. 2010;190:675–691. doi: 10.1083/jcb.201002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Husain SR, Joshi BH, Puri RK. Interleukin-13 receptor as a unique target for anti-glioblastoma therapy. Int. J. Cancer. 2001;92:168–175. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1182>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 36.Husain SR, Puri RK. Interleukin-13 fusion cytotoxin as a potent targeted agent for AIDS-Kaposi’s sarcoma xenograft. Blood. 2000;95:3506–3513. [PubMed] [Google Scholar]
- 37.Husain SR, Puri RK. Interleukin-13 receptor-directed cytotoxin for malignant glioma therapy: from bench to bedside. J. Neurooncol. 2003;65:37–48. doi: 10.1023/a:1026242432647. [DOI] [PubMed] [Google Scholar]
- 38.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J. Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 39.Joshi BH, Hogaboam C, Dover P, Husain SR, Puri RK. Role of interleukin-13 in cancer, pulmonary fibrosis, and other T(H)2-type diseases. Vitam. Horm. 2006;74:479–504. doi: 10.1016/S0083-6729(06)74019-5. [DOI] [PubMed] [Google Scholar]
- 40.Joshi BH, Kawakami K, Leland P, Puri RK. Heterogeneity in interleukin-13 receptor expression and subunit structure in squamous cell carcinoma of head and neck: differential sensitivity to chimeric fusion proteins comprised of interleukin-13 and a mutated form of Pseudomonas exotoxin. Clin. Cancer Res. 2002;8:1948–1956. [PubMed] [Google Scholar]
- 41.Joshi BH, Plautz GE, Puri RK. Interleukin-13 receptor alpha chain: a novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000;60:1168–1172. [PubMed] [Google Scholar]
- 42.Joshi BH, Puri RK. IL-13 receptor-alpha2: a novel target for cancer therapy. Immunotherapy. 2009;1:321–327. doi: 10.2217/imt.09.8. [DOI] [PubMed] [Google Scholar]
- 43.Kamen BA, Smith AK. A review of folate receptor alpha cycling and 5-methyltetrahydrofolate accumulation with an emphasis on cell models in vitro. Adv. Drug Deliv. Rev. 2004;56:1085–1097. doi: 10.1016/j.addr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Kaplan PL, Anderson M, Ozanne B. Transforming growth factor(s) production enables cells to grow in the absence of serum: an autocrine system. Proc. Natl. Acad. Sci. USA. 1982;79:485–489. doi: 10.1073/pnas.79.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawakami K, Kawakami M, Puri RK. Overexpressed cell surface interleukin-4 receptor molecules can be successfully targeted for antitumor cytotoxin therapy. Crit. Rev. Immunol. 2001;21:299–310. [PubMed] [Google Scholar]
- 46.Kawakami M, Kawakami K, Puri RK. Apoptotic pathways of cell death induced by an interleukin-13 receptor-targeted recombinant cytotoxin in head and neck cancer cells. Cancer Immunol. Immunother. 2002;50:691–700. doi: 10.1007/s00262-001-0242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawakami M, Kawakami K, Puri RK. Tumor regression mechanisms by IL-13 receptor-targeted cancer therapy involve apoptotic pathways. Int. J. Cancer. 2003;103:45–52. doi: 10.1002/ijc.10778. [DOI] [PubMed] [Google Scholar]
- 48.Kawakami K, Taguchi J, Murata T, Puri RK. The interleukin-13 receptor alpha2 chain: an essential component for binding and internalization but not for interleukin-13-induced signal transduction through the STAT6 pathway. Blood. 2001;97:2673–2679. doi: 10.1182/blood.v97.9.2673. [DOI] [PubMed] [Google Scholar]
- 49.Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science. 2003;300:1527–1528. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- 50.Kioi M, Kawakami M, Shimamura T, Husain SR, Puri RK. Interleukin-13 receptor alpha2 chain: a potential biomarker and molecular target for ovarian cancer therapy. Cancer. 2006;107:1407–1418. doi: 10.1002/cncr.22134. [DOI] [PubMed] [Google Scholar]
- 51.Kioi M, Shimamura T, Nakashima H, Hirota M, Tohnai I, Husain SR, Puri RK. IL-13 cytotoxin has potent antitumor activity and synergizes with paclitaxel in a mouse model of oral squamous cell carcinoma. Int. J. Cancer. 2009;124:1440–1448. doi: 10.1002/ijc.24067. [DOI] [PubMed] [Google Scholar]
- 52.Klausner RD, Van Renswoude J, Ashwell G, Kempf C, Schechter AN, Dean A, Bridges KR. Receptor-mediated endocytosis of transferrin in K562 cells. J. Biol. Chem. 1983;258:4715–4724. [PubMed] [Google Scholar]
- 53.Kobrin MS, Yamanaka Y, Friess H, Lopez ME, Korc M. Aberrant expression of type I fibroblast growth factor receptor in human pancreatic adenocarcinomas. Cancer Res. 1993;53:4741–4744. [PubMed] [Google Scholar]
- 54.Kraich M, Klein M, Patino E, Harrer H, Nickel J, Sebald W, Mueller TD. A modular interface of IL-4 allows for scalable affinity without affecting specificity for the IL-4 receptor. BMC Biol. 2006;4:13. doi: 10.1186/1741-7007-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kratz F, Beyer U, Roth T, Tarasova N, Collery P, Lechenault F, Cazabat A, Schumacher P, Unger C, Falken U. Transferrin conjugates of doxorubicin: synthesis, characterization, cellular uptake, and in vitro efficacy. J. Pharm. Sci. 1998;87:338–346. doi: 10.1021/js970246a. [DOI] [PubMed] [Google Scholar]
- 56.Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, Shaffrey M, Ram Z, Piepmeier J, Prados M, Croteau D, Pedain C, Leland P, Husain SR, Joshi BH, Puri RK. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12:871–881. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuznetsov VA, Puri RK. Kinetic analysis of high affinity forms of interleukin (IL)-13 receptors: suppression of IL-13 binding by IL-2 receptor gamma chain. Biophys. J. 1999;77:154–172. doi: 10.1016/S0006-3495(99)76879-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lao BJ, Kamei DT. Improving therapeutic properties of protein drugs through alteration of intracellular trafficking pathways. Biotechnol. Prog. 2008;24:2–7. doi: 10.1021/bp070080b. [DOI] [PubMed] [Google Scholar]
- 59.Lao BJ, Tsai WL, Mashayekhi F, Pham EA, Mason AB, Kamei DT. Inhibition of transferrin iron release increases in vitro drug carrier efficacy. J. Control Release. 2007;117:403–412. doi: 10.1016/j.jconrel.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laskin JJ, Sandler AB. Epidermal growth factor receptor: a promising target in solid tumours. Cancer Treat. Rev. 2004;30:1–17. doi: 10.1016/j.ctrv.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 61.Leamon CP, Reddy JA, Vetzel M, Dorton R, Westrick E, Parker N, Wang Y, Vlahov I. Folate targeting enables durable and specific antitumor responses from a therapeutically null tubulysin B analogue. Cancer Res. 2008;68:9839–9844. doi: 10.1158/0008-5472.CAN-08-2341. [DOI] [PubMed] [Google Scholar]
- 62.Leamon CP, Reddy JA, Vlahov IR, Vetzel M, Parker N, Nicoson JS, Xu LC, Westrick E. Synthesis and biological evaluation of EC72: a new folate-targeted chemotherapeutic. Bioconjug. Chem. 2005;16:803–811. doi: 10.1021/bc049709b. [DOI] [PubMed] [Google Scholar]
- 63.Leamon CP, Reddy JA, Vlahov IR, Westrick E, Dawson A, Dorton R, Vetzel M, Santhapuram HK, Wang Y. Preclinical antitumor activity of a novel folate-targeted dual drug conjugate. Mol. Pharm. 2007;4:659–667. doi: 10.1021/mp070049c. [DOI] [PubMed] [Google Scholar]
- 64.Leamon CP, Reddy JA, Vlahov IR, Westrick E, Parker N, Nicoson JS, Vetzel M. Comparative preclinical activity of the folate-targeted Vinca alkaloid conjugates EC140 and EC145. Int. J. Cancer. 2007;121:1585–1592. doi: 10.1002/ijc.22853. [DOI] [PubMed] [Google Scholar]
- 65.Lee CH, Lee EC, Tsai ST, Kung HJ, Liu YC, Hwang J. An EGF-pseudomonas exotoxin A recombinant protein with a deletion in toxin binding domain specifically kills EGF receptor bearing cells. Protein Eng. 1993;6:433–440. doi: 10.1093/protein/6.4.433. [DOI] [PubMed] [Google Scholar]
- 66.Li C, Hall WA, Jin N, Todhunter DA, Panoskaltsis-Mortari A, Vallera DA. Targeting glioblastoma multiforme with an IL-13/diphtheria toxin fusion protein in vitro and in vivo in nude mice. Protein Eng. 2002;15:419–427. doi: 10.1093/protein/15.5.419. [DOI] [PubMed] [Google Scholar]
- 67.Li H, Qian ZM. Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 2002;22:225–250. doi: 10.1002/med.10008. [DOI] [PubMed] [Google Scholar]
- 68.Li H, Sun H, Qian ZM. The role of the transferrin-transferrin-receptor system in drug delivery and targeting. Trends Pharmacol. Sci. 2002;23:206–209. doi: 10.1016/s0165-6147(02)01989-2. [DOI] [PubMed] [Google Scholar]
- 69.Liao CW, Hseu TH, Hwang J. A target-specific chimeric toxin composed of epidermal growth factor and Pseudomonas exotoxin A with a deletion in its toxin-binding domain. Appl. Microbiol. Biotechnol. 1995;43:498–507. doi: 10.1007/BF00218456. [DOI] [PubMed] [Google Scholar]
- 70.Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 2009;13:256–262. doi: 10.1016/j.cbpa.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 71.Lu Y, Low PS. Folate targeting of haptens to cancer cell surfaces mediates immunotherapy of syngeneic murine tumors. Cancer Immunol. Immunother. 2002;51:153–162. doi: 10.1007/s00262-002-0266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu Y, Sega E, Low PS. Folate receptor-targeted immunotherapy: induction of humoral and cellular immunity against hapten-decorated cancer cells. Int. J. Cancer. 2005;116:710–719. doi: 10.1002/ijc.21126. [DOI] [PubMed] [Google Scholar]
- 73.Matherly LH, Hou Z, Deng Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007;26:111–128. doi: 10.1007/s10555-007-9046-2. [DOI] [PubMed] [Google Scholar]
- 74.Mendelsohn J. Targeting the epidermal growth factor receptor for cancer therapy. J. Clin. Oncol. 2002;20:1S–13S. [PubMed] [Google Scholar]
- 75.Miloux B, Laurent P, Bonnin O, Lupker J, Caput D, Vita N, Ferrara P. Cloning of the human IL-13R alpha1 chain and reconstitution with the IL4R alpha of a functional IL-4/IL-13 receptor complex. FEBS Lett. 1997;401:163–166. doi: 10.1016/s0014-5793(96)01462-7. [DOI] [PubMed] [Google Scholar]
- 76.Minino, A. M., J. Xu, K. D. Kochanek, and B. Tejada-Vera. Death in the United States, 2007. NCHS Data Brief 1–8, 2009. [PubMed]
- 77.Minty A, Chalon P, Derocq JM, Dumont X, Guillemot JC, Kaghad M, Labit C, Leplatois P, Liauzun P, Miloux B, et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature. 1993;362:248–250. doi: 10.1038/362248a0. [DOI] [PubMed] [Google Scholar]
- 78.Moses HL, Proper JA, Volkenant ME, Wells DJ, Getz MJ. Mechanism of growth arrest of chemically transformed cells in culture. Cancer Res. 1978;38:2807–2812. [PubMed] [Google Scholar]
- 79.Murata T, Noguchi PD, Puri RK. IL-13 induces phosphorylation and activation of JAK2 Janus kinase in human colon carcinoma cell lines: similarities between IL-4 and IL-13 signaling. J. Immunol. 1996;156:2972–2978. [PubMed] [Google Scholar]
- 80.Murata T, Obiri NI, Debinski W, Puri RK. Structure of IL-13 receptor: analysis of subunit composition in cancer and immune cells. Biochem. Biophys. Res. Commun. 1997;238:90–94. doi: 10.1006/bbrc.1997.7248. [DOI] [PubMed] [Google Scholar]
- 81.Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell. 1992;69:1051–1061. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- 82.Pastan I, FitzGerald D. Pseudomonas exotoxin: chimeric toxins. J. Biol. Chem. 1989;264:15157–15160. [PubMed] [Google Scholar]
- 83.Pietras K, Sjoblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–443. doi: 10.1016/s1535-6108(03)00089-8. [DOI] [PubMed] [Google Scholar]
- 84.Ponka P, Lok CN. The transferrin receptor: role in health and disease. Int. J. Biochem. Cell Biol. 1999;31:1111–1137. doi: 10.1016/s1357-2725(99)00070-9. [DOI] [PubMed] [Google Scholar]
- 85.Puri RK. Development of a recombinant interleukin-4-Pseudomonas exotoxin for therapy of glioblastoma. Toxicol. Pathol. 1999;27:53–57. doi: 10.1177/019262339902700111. [DOI] [PubMed] [Google Scholar]
- 86.Rao BM, Lauffenburger DA, Wittrup KD. Integrating cell-level kinetic modeling into the design of engineered protein therapeutics. Nat. Biotechnol. 2005;23:191–194. doi: 10.1038/nbt1064. [DOI] [PubMed] [Google Scholar]
- 87.Recht L, Torres CO, Smith TW, Raso V, Griffin TW. Transferrin receptor in normal and neoplastic brain tissue: implications for brain-tumor immunotherapy. J. Neurosurg. 1990;72:941–945. doi: 10.3171/jns.1990.72.6.0941. [DOI] [PubMed] [Google Scholar]
- 88.Reddy JA, Dorton R, Dawson A, Vetzel M, Parker N, Nicoson JS, Westrick E, Klein PJ, Wang Y, Vlahov IR, Leamon CP. In vivo structural activity and optimization studies of folate-tubulysin conjugates. Mol. Pharm. 2009;6:1518–1525. doi: 10.1021/mp900086w. [DOI] [PubMed] [Google Scholar]
- 89.Reddy JA, Dorton R, Westrick E, Dawson A, Smith T, Xu LC, Vetzel M, Kleindl P, Vlahov IR, Leamon CP. Preclinical evaluation of EC145, a folate-vinca alkaloid conjugate. Cancer Res. 2007;67:4434–4442. doi: 10.1158/0008-5472.CAN-07-0033. [DOI] [PubMed] [Google Scholar]
- 90.Reddy CC, Wells A, Lauffenburger DA. Comparative mitogenic potencies of EGF and TGF alpha and their dependence on receptor-limitation versus ligand-limitation. Med. Biol. Eng. Comput. 1998;36:499–507. doi: 10.1007/BF02523222. [DOI] [PubMed] [Google Scholar]
- 91.Reddy JA, Westrick E, Santhapuram HK, Howard SJ, Miller ML, Vetzel M, Vlahov I, Chari RV, Goldmacher VS, Leamon CP. Folate receptor-specific antitumor activity of EC131, a folate-maytansinoid conjugate. Cancer Res. 2007;67:6376–6382. doi: 10.1158/0008-5472.CAN-06-3894. [DOI] [PubMed] [Google Scholar]
- 92.Rocha-Lima CM, Soares HP, Raez LE, Singal R. EGFR targeting of solid tumors. Cancer Control. 2007;14:295–304. doi: 10.1177/107327480701400313. [DOI] [PubMed] [Google Scholar]
- 93.Rustamzadeh E, Hall WA, Todhunter DA, Low WC, Liu H, Panoskaltsis-Mortari A, Vallera DA. Intracranial therapy of glioblastoma with the fusion protein DTIL13 in immunodeficient mice. Int. J. Cancer. 2006;118:2594–2601. doi: 10.1002/ijc.21647. [DOI] [PubMed] [Google Scholar]
- 94.Rustamzadeh E, Vallera DA, Todhunter DA, Low WC, Panoskaltsis-Mortari A, Hall WA. Immunotoxin pharmacokinetics: a comparison of the anti-glioblastoma bi-specific fusion protein (DTAT13) to DTAT and DTIL13. J. Neurooncol. 2006;77:257–266. doi: 10.1007/s11060-005-9051-7. [DOI] [PubMed] [Google Scholar]
- 95.Sabharanjak S, Mayor S. Folate receptor endocytosis and trafficking. Adv. Drug Deliv. Rev. 2004;56:1099–1109. doi: 10.1016/j.addr.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 96.Salazar MD, Ratnam M. The folate receptor: what does it promise in tissue-targeted therapeutics? Cancer Metastasis Rev. 2007;26:141–152. doi: 10.1007/s10555-007-9048-0. [DOI] [PubMed] [Google Scholar]
- 97.Sampson JH, Akabani G, Archer GE, Berger MS, Coleman RE, Friedman AH, Friedman HS, Greer K, Herndon JE, 2nd, Kunwar S, McLendon RE, Paolino A, Petry NA, Provenzale JM, Reardon DA, Wong TZ, Zalutsky MR, Pastan I, Bigner DD. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol. 2008;10:320–329. doi: 10.1215/15228517-2008-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sampson JH, Akabani G, Archer GE, Bigner DD, Berger MS, Friedman AH, Friedman HS, Herndon JE, 2nd, Kunwar S, Marcus S, McLendon RE, Paolino A, Penne K, Provenzale J, Quinn J, Reardon DA, Rich J, Stenzel T, Tourt-Uhlig S, Wikstrand C, Wong T, Williams R, Yuan F, Zalutsky MR, Pastan I. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neurooncol. 2003;65:27–35. doi: 10.1023/a:1026290315809. [DOI] [PubMed] [Google Scholar]
- 99.Sarkar CA, Lauffenburger DA. Cell-level pharmacokinetic model of granulocyte colony-stimulating factor: implications for ligand lifetime and potency in vivo. Mol. Pharmacol. 2003;63:147–158. doi: 10.1124/mol.63.1.147. [DOI] [PubMed] [Google Scholar]
- 100.Sarkar CA, Lowenhaupt K, Horan T, Boone TC, Tidor B, Lauffenburger DA. Rational cytokine design for increased lifetime and enhanced potency using pH-activated “histidine switching”. Nat. Biotechnol. 2002;20:908–913. doi: 10.1038/nbt725. [DOI] [PubMed] [Google Scholar]
- 101.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin. Cancer Res. 2006;12:5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 102.Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J. Cell. Physiol. 2009;218:460–466. doi: 10.1002/jcp.21635. [DOI] [PubMed] [Google Scholar]
- 103.Shmeeda H, Mak L, Tzemach D, Astrahan P, Tarshish M, Gabizon A. Intracellular uptake and intracavitary targeting of folate-conjugated liposomes in a mouse lymphoma model with up-regulated folate receptors. Mol. Cancer Ther. 2006;5:818–824. doi: 10.1158/1535-7163.MCT-05-0543. [DOI] [PubMed] [Google Scholar]
- 104.Shvartsman SY, Muratov CB, Lauffenburger DA. Modeling and computational analysis of EGF receptor-mediated cell communication in Drosophila oogenesis. Development. 2002;129:2577–2589. doi: 10.1242/dev.129.11.2577. [DOI] [PubMed] [Google Scholar]
- 105.Siegall CB, Xu YH, Chaudhary VK, Adhya S, Fitzgerald D, Pastan I. Cytotoxic activities of a fusion protein comprised of TGF alpha and Pseudomonas exotoxin. Faseb J. 1989;3:2647–2652. doi: 10.1096/fasebj.3.14.2556314. [DOI] [PubMed] [Google Scholar]
- 106.Simpson DL, Cawley DB, Herschman HR. Killing of cultured hepatocytes by conjugates of asialofetuin and EGF linked to the A chains of ricin or diphtheria toxin. Cell. 1982;29:469–473. doi: 10.1016/0092-8674(82)90163-5. [DOI] [PubMed] [Google Scholar]
- 107.Singh M, Atwal H, Micetich R. Transferrin directed delivery of adriamycin to human cells. Anticancer Res. 1998;18:1423–1427. [PubMed] [Google Scholar]
- 108.Starbuck C, Lauffenburger DA. Mathematical model for the effects of epidermal growth factor receptor trafficking dynamics on fibroblast proliferation responses. Biotechnol. Prog. 1992;8:132–143. doi: 10.1021/bp00014a007. [DOI] [PubMed] [Google Scholar]
- 109.Tabata Y, Hershey GK. IL-13 receptor isoforms: breaking through the complexity. Curr. Allergy Asthma Rep. 2007;7:338–345. doi: 10.1007/s11882-007-0051-x. [DOI] [PubMed] [Google Scholar]
- 110.Theuer CP, FitzGerald D, Pastan I. A recombinant form of Pseudomonas exotoxin directed at the epidermal growth factor receptor that is cytotoxic without requiring proteolytic processing. J. Biol. Chem. 1992;267:16872–16877. [PubMed] [Google Scholar]
- 111.Vallera DA, Stish BJ, Shu Y, Chen H, Saluja A, Buchsbaum DJ, Vickers SM. Genetically designing a more potent antipancreatic cancer agent by simultaneously co-targeting human IL13 and EGF receptors in a mouse xenograft model. Gut. 2008;57:634–641. doi: 10.1136/gut.2007.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Venook AP. Epidermal growth factor receptor-targeted treatment for advanced colorectal carcinoma. Cancer. 2005;103:2435–2446. doi: 10.1002/cncr.21123. [DOI] [PubMed] [Google Scholar]
- 113.Vollmar AM, Banker DE, Mendelsohn J, Herschman HR. Toxicity of ligand and antibody-directed ricin A-chain conjugates recognizing the epidermal growth factor receptor. J. Cell. Physiol. 1987;131:418–425. doi: 10.1002/jcp.1041310314. [DOI] [PubMed] [Google Scholar]
- 114.Wang S, Lee RJ, Mathias CJ, Green MA, Low PS. Synthesis, purification, and tumor cell uptake of 67 Ga-deferoxamine–folate, a potential radiopharmaceutical for tumor imaging. Bioconjug. Chem. 1996;7:56–62. doi: 10.1021/bc9500709. [DOI] [PubMed] [Google Scholar]
- 115.Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003;65:3–13. doi: 10.1023/a:1026246500788. [DOI] [PubMed] [Google Scholar]
- 116.Wells A. EGF receptor. Int. J. Biochem. Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 117.Wells A, Kassis J, Solava J, Turner T, Lauffenburger DA. Growth factor-induced cell motility in tumor invasion. Acta Oncol. 2002;41:124–130. doi: 10.1080/028418602753669481. [DOI] [PubMed] [Google Scholar]
- 118.Wenning LA, Yazdi PT, Murphy RM. Quantitative analysis of protein synthesis inhibition and recovery in CRM107 immunotoxin-treated HeLa cells. Biotechnol. Bioeng. 1998;57:484–496. doi: 10.1002/(sici)1097-0290(19980220)57:4<484::aid-bit13>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 119.Wiley HS, Burke PM. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2:12–18. doi: 10.1034/j.1600-0854.2001.020103.x. [DOI] [PubMed] [Google Scholar]
- 120.Wiley HS, Shvartsman SY, Lauffenburger DA. Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol. 2003;13:43–50. doi: 10.1016/s0962-8924(02)00009-0. [DOI] [PubMed] [Google Scholar]
- 121.Xia W, Low PS. Folate-targeted therapies for cancer. J. Med. Chem. 2010;53:6811–6824. doi: 10.1021/jm100509v. [DOI] [PubMed] [Google Scholar]
- 122.Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl. Acad. Sci. USA. 2006;103:13872–13877. doi: 10.1073/pnas.0601455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Characterization of the pH of folate receptor-containing endosomes and the rate of hydrolysis of internalized acid-labile folate-drug conjugates. J. Pharmacol. Exp. Ther. 2007;321:462–468. doi: 10.1124/jpet.106.117648. [DOI] [PubMed] [Google Scholar]
- 124.Yazdi PT, Murphy RM. Quantitative analysis of protein synthesis inhibition by transferrin-toxin conjugates. Cancer Res. 1994;54:6387–6394. [PubMed] [Google Scholar]
- 125.Yazdi PT, Wenning LA, Murphy RM. Influence of cellular trafficking on protein synthesis inhibition of immunotoxins directed against the transferrin receptor. Cancer Res. 1995;55:3763–3771. [PubMed] [Google Scholar]
- 126.Yeh CJ, Faulk WP. Killing of human tumor cells in culture with adriamycin conjugates of human transferrin. Clin. Immunol. Immunopathol. 1984;32:1–11. doi: 10.1016/0090-1229(84)90037-0. [DOI] [PubMed] [Google Scholar]
- 127.Yoon DJ, Chu DS, Ng CW, Pham EA, Mason AB, Hudson DM, Smith VC, MacGillivray RT, Kamei DT. Genetically engineering transferrin to improve its in vitro ability to deliver cytotoxins. J. Control Release. 2009;133:178–184. doi: 10.1016/j.jconrel.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yoon DJ, Kwan BH, Chao FC, Nicolaides TP, Phillips JJ, Lam GY, Mason AB, Weiss WA, Kamei DT. Intratumoral therapy of glioblastoma multiforme using genetically engineered transferrin for drug delivery. Cancer Res. 2010;70:4520–4527. doi: 10.1158/0008-5472.CAN-09-4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao R, Min SH, Wang Y, Campanella E, Low PS, Goldman ID. A role for the proton-coupled folate transporter (PCFT-SLC46A1) in folate receptor-mediated endocytosis. J. Biol. Chem. 2009;284:4267–4274. doi: 10.1074/jbc.M807665200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zurawski G, de Vries JE. Interleukin 13, an interleukin 4-like cytokine that acts on monocytes and B cells, but not on T cells. Immunol. Today. 1994;15:19–26. doi: 10.1016/0167-5699(94)90021-3. [DOI] [PubMed] [Google Scholar]