

Abstract
The phytochrome family of sensory photoreceptors interacts with phytochrome interacting factors (PIFs), repressors of photomorphogenesis, in response to environmental light signals and induces rapid phosphorylation and degradation of PIFs to promote photomorphogenesis. However, the kinase that phosphorylates PIFs is still unknown. Here we show that CK2 directly phosphorylates PIF1 at multiple sites. α1 and α2 subunits individually phosphorylated PIF1 weakly in vitro. However, each of four β subunits strongly stimulated phosphorylation of PIF1 by α1 or α2. Mapping of the phosphorylation sites identified seven Ser/Thr residues scattered throughout PIF1. Ser/Thr to Ala scanning mutations at all seven sites eliminated CK2-mediated phosphorylation of PIF1 in vitro. Moreover, the rate of degradation of the Ser/Thr to Ala mutant PIF1 was significantly reduced compared with wild-type PIF1 in transgenic plants. In addition, hypocotyl lengths of the mutant PIF1 transgenic plants were much longer than the wild-type PIF1 transgenic plants under light, suggesting that the mutant PIF1 is suppressing photomorphogenesis. Taken together, these data suggest that CK2-mediated phosphorylation enhances the light-induced degradation of PIF1 to promote photomorphogenesis.
Keywords: Helix-Loop-Helix Transcription Factors, Posttranslational Modification, Protein Degradation, Protein Phosphorylation, Ubiquitination, CK2, bHLH Factor, Phosphorylation, Photomorphogenesis, Proteasomal Degradation
Introduction
Because light is an essential environmental signal for modulating plant growth and development throughout the life cycle, plants have several different classes of photoreceptors sensing and responding to major bandwidths of the light spectrum (1, 2). Of these photoreceptors, the phytochrome (phy)3 family (which consists of five members, phyA to phyE, in Arabidopsis) senses and responds to a broad spectrum (red, far-red, and blue) of light signals and pleiotropically regulates plant growth from seed germination to flowering time (3–5). Within a cell, phys are synthesized as an inactive Pr conformer that resides in the cytosol. Exposure to red (R) light triggers a conformation shift to a biologically active Pfr form, which migrates into the nucleus and initiates phy signaling (6). The active Pfr form can be reverted back to the inactive Pr form by exposure to far-red (FR) light. The active Pfr form interacts with multiple signaling partners within the nucleus (7) and controls the expression of a large number (10–30% of the genome) of genes to regulate photomorphogenesis (8–10).
One of the ways phys exert this strong effect on gene expression is by differentially regulating the stability of positively and negatively acting transcription factors functioning in light signaling pathways (11, 12). For example, the positively acting transcription factors (e.g. HY5, HFR1, LAF1, and possibly others) are degraded in the dark and stabilized under red, far-red, and blue light conditions. By contrast, the negatively acting transcription factors (e.g. the phytochrome interacting factors (PIFs)) are stable in the dark and degrade in response to light. The net effect of this stabilization and destabilization of transcription factors shapes the transcriptome that regulates photomorphogenesis.
PIFs have been shown to play central roles in phy signaling pathways (13–15). They belong to the bHLH class of transcription factors and are encoded by six family members in Arabidopsis (PIF1 and PIF3–7) (13, 14, 16). PIFs physically interact with the Pfr forms of phys with differential affinity for various phys. PIFs bind to G-box DNA sequence motifs and regulate gene expression (17–22). Genetic and photobiological experiments showed that PIFs function as negative regulators of photomorphogenesis largely by modulating the level of phyB under red light (23–25). Moreover, quadruple (pifQ) mutant seedlings of pif1, pif3, pif4, and pif5 are constitutively photomorphogenic, suggesting that PIFs repress photomorphogenesis in the dark (24, 26, 27). This morphological phenotype is reflected at the gene expression level, where a majority of the light-regulated genes are constitutively expressed in the pifQ mutant in the dark (27, 28). In wild-type seedlings, light signals perceived by phys promote degradation of PIFs through the ubiquitin/26 S proteasomal pathway to derepress gene expression and promote photomorphogenesis (13, 15, 29).
Although the mechanisms of dark-induced degradation of positively acting transcription factors are well understood (12), the light-induced degradation of PIFs is only beginning to be understood. All PIFs except PIF7 are rapidly phosphorylated and ubiquitinated in response to light in vivo prior to their degradation (26, 30, 31). The mutant PIF1 and PIF3 that fail to interact with the Pfr forms of phyA and phyB do not undergo light-dependent phosphorylation, suggesting that phy interaction is necessary for the light-dependent phosphorylation and subsequent degradation of PIFs. However, the putative kinase(s) and the putative E3 ligase(s) responsible for phosphorylation of PIFs and recognition of the phosphorylated forms for subsequent ubiquitination and degradation are still unknown.
CK2 (formerly known as casein kinase II), a ubiquitous Ser/Thr kinase, has been implicated in regulating light signaling and circadian rhythms in Arabidopsis (32–38). The CK2 holoenzyme consists of two catalytic α and two regulatory β subunits. The Arabidopsis genome has four α subunit and four β subunit genes (39). Various members of both the α and β subunit families have been shown to be localized in the cytoplasm, nucleus, and also in chloroplasts (39). A CK2-like activity has been shown to phosphorylate HY5, a positively acting component in a phy signaling pathway (34). In addition, CK2 phosphorylates HFR1, a positively acting HLH transcription factor functioning under FR and blue light pathways (35). Phosphorylation of both HY5 and HFR1 has been shown to stabilize these transcription factors (34, 35). Because CK2-mediated phosphorylation stabilizes the positively acting transcription factors involved in light signaling in Arabidopsis, we reasoned that CK2 might also phosphorylate the negatively acting transcription factors (e.g. PIFs) and regulate their stability/function in Arabidopsis. Here we show that CK2 phosphorylates PIF1 at multiple sites in vitro. In addition, CK2-mediated phosphorylation promotes rapid light-induced degradation of PIF1 to fine-tune photomorphogenesis.
EXPERIMENTAL PROCEDURES
Protein Expression and Kinase Assays
The PIF1 ORF was cloned in-frame with the 6× His tag in the pET21d vector and verified by sequencing. The vector was transformed into an Arctic DE3 cell for protein expression and purification using the His tag as described (40, 41). CK2 phosphorylation assays were performed as described (40, 41). Briefly, 20 μl of kinase assay mixtures contained 50 mm Hepes-potassium hydroxide (pH 7.6), 5 mm MgCl2, 2.4 mm DTT, 0.2 mm γ-[32P]ATP (∼250 cpm/pmol), 100 mm KCl, ∼1 pmol CK2, and ∼10–20 pmol PIF1. The reaction was incubated at 30 °C for 30 min and terminated by the addition of 4× SDS loading buffer. Samples were boiled 3 min and separated on 10% SDS-PAGE gels. The gels were dried and exposed to a phosphorImager.
Mapping CK2 Phosphorylation Sites in PIF1
The identification of CK2 phosphorylation sites in PIF1 was essentially performed as described (41). Briefly, the phosphopeptides were enriched using an iron-based affinity resin (Phos-Select, Sigma), followed by analysis on a MALDI-TOF/TOF mass spectrometer (4700 proteomics analyzer, ABSciex, Foster City, CA). Phosphopeptides were identified in the MS by mass shift using the MASCOT (Matrix Science) search algorithm, and the MS/MS were acquired for potential phosphopeptides. These were manually interpreted to confirm the presence of strong neutral loss of phosphoric acid ions (MH+ − 98) and to assign the most probable location of the phosphorylation site based on shifts in b and y fragment ions.
Plant Growth Conditions and Phenotypic Analyses
Plants were grown in Metro-Mix 200 soil (Sun Gro Horticulture, Bellevue, WA) under constant light at 24 °C ± 0.5 °C. Monochromatic R and FR light sources and the spectroradiometer (model EPP2000, StellarNet Inc., Tampa, FL) used to measure fluence rates were as described (42). Seeds were surface-sterilized and plated on Murashige-Skoog growth medium containing 0.9% agar without sucrose as described (42). After stratification at 4 °C in the dark, seeds were exposed to 3 h of white light at room temperature to induce germination before placing them in the dark for an additional 21 h. The plates were then either placed in the dark or under specific wavelengths of light for an additional 3 days. For quantitation of hypocotyl lengths, a digital photograph was taken, and at least 30 seedlings were measured using the publicly available software ImageJ. The experiments were repeated at least three times.
Construction of Plasmids and Generation of Transgenic Plants
The PIF1 ORF was amplified by PCR and cloned into the pENTRY vector (Invitrogen) and recombined with pNTAPa (43) to produce 35S:TAP-PIF1. A 1.6-kb PIF1 promoter fragment was amplified with primers containing restriction sites (supplemental Table S1) and was cloned into the pPZP121 vector. A 3.5-kb fragment containing the TAP-PIF1 from the 35S:TAP-PIF1 construct was cloned into pPZP121-pPIF1 to generate pPIF1:TAP-PIF1 in the pPZP121 background. The specific amino acid mutations in full-length PIF1 were introduced using a site-directed mutagenesis kit in the pENTRY vector background (Stratagene, La Jolla, CA). The 35S:LUC-PIF1 (LP) line is as described (42). The Ser-464–466 to Ala and Ser-459–461 to Ala mutations were created in the LUC-PIF1 background using the site-directed mutagenesis kit (Stratagene) and transformed into the pif1 mutant. The CK2 β1 and β2 subunits were amplified by PCR and cloned into the pENTRY vector and subsequently recombined with the pB7FWG2 vector to produce the 35S:CK2β1/2-GFP fusion constructs (44). All constructs were verified by sequencing. The resulting vectors were transformed into Agrobacterium strain GV3101 by electroporation and transformed into the wild-type Col-0 background using the floral dip method. Transgenic plants were selected on appropriate antibiotics, and homozygous lines were selected from single insert lines.
Protein Extraction and Western Blotting
Protein extraction and Western blotting were essentially performed as described (4, 26). Briefly, 4-day-old dark-grown seedlings were either kept in the dark or exposed to pulses of R or FR light followed by incubation in the dark for various times as indicated on each figure before protein extraction. To detect TAP-PIF1 and LUC-PIF1 proteins in transgenic plants, boiling denaturing buffer (100 mm MOPS (pH 7.6), 5% SDS, 10% glycerol, 4 mm EDTA, 40 mm β-mercaptoethanol, 1× protease inhibitor mixture (Hoffmann-La Roche) was added at a 1:3 (w/v) ratio before grinding. PMSF (2 mm) was also added during extraction. To detect native PIF1 in wild-type plants, about 0.2 g of tissue was ground in 1 ml of extraction buffer (100 mm Tris-HCl (pH 6.8), 20% glycerol, 5% SDS, 80 μm MG132, 20 mm DTT, 1 mm bromphenol blue, 2 mm PMSF, and 1× protease inhibitor mixture (Hoffmann-La Roche)) and boiled for 2 min. Total proteins were separated on 8% SDS-PAGE gels, blotted onto a PVDF membrane, and probed with anti-PIF1, anti-myc, or anti-RPT5 antibodies. Membranes were developed with the SuperSignal West Pico chemiluminescent substrate kit (Pierce) and visualized on an x-ray film. The intensity of the PIF1 and the control bands from each blot were quantified using ImageJ software, and the PIF1 values were divided by the control values to make a ratio for each sample. The dark control for each sample is set to 1 from these ratios, and the relative values of the other samples are calculated based on dark values. These relative values are shown in each figure under the blots.
RESULTS
PIF1 Is Phosphorylated by CK2 in Vitro
Sequence analyses using three software packages (Scansite 2.0, NetPhosK 1.0, and KinasePhos 2.0) revealed that PIF1 has multiple putative CK2 phosphorylation sites (supplemental Table S2). We purified recombinant His-tagged PIF1, two CK2 α (α1 and α2) subunits, and four β (β1–4) subunits and performed in vitro kinase assays as described (40). Results show that PIF1 is weakly phosphorylated by the α1 subunit of CK2. However, addition of the β1 subunit strongly stimulated phosphorylation of PIF1 (Fig. 1A). Fig. 1B shows that phosphorylation of PIF1 was inhibited in proportion to the concentration of heparin, a specific inhibitor of CK2 activity (35). This experiment showed that CK2, and not a contaminating kinase, phosphorylates PIF1 in vitro. Taken together, these data strongly suggest that PIF1 is a bona fide substrate for Arabidopsis CK2 kinase in vitro.
FIGURE 1.

CK2 phosphorylates PIF1 in vitro. A, phosphorylation of PIF1 by CK2 is enhanced by the β subunit. The autoradiogram shows that HIS-PIF1 was strongly phosphorylated by a recombinant CK2 holoenzyme in vitro. CK2 phosphorylation assays were performed in 20 μl of kinase assay mixtures that contained 50 mm Hepes-KOH (pH 7.6), 5 mm MgCl2, 2.4 mm DTT, 100 mm KCl, 0.2 mm γ-[32P]ATP (∼250 cpm/pmol), ∼1 pmol CK2 α or αβ holoenzyme, and ∼10–20 pmol PIF1. The reaction was incubated at 30 °C for 30 min and terminated by the addition of 4× SDS loading buffer. Samples were boiled for 3 min and separated on 10% SDS-PAGE gels. The gels were dried and exposed to a phosphorImager. B, autoradiogram showing that heparin effectively inhibited HIS-PIF1 phosphorylation by CK2 in a dosage-dependent manner. The kinase assays were performed as described in A. The asterisk indicates a nonspecific band. C, different subunit combinations of CK2 differentially phosphorylate PIF1 in vitro. The kinase assays were performed as described in A. Statistical analyses for significant differences are shown in supplemental Table S3.
Effect of Different Subunit Composition on the Phosphorylation of PIF1
Because the Arabidopsis genome encodes four α and four β subunits of CK2 (39), we investigated whether different CK2 α and β subunit combinations have differential phosphorylation activity toward PIF1 as described (40, 41). Results show that all the αβ holoenzyme combinations had stronger activity than either of the two α subunits alone. However, among all the holoenzyme combinations, the α1β2, α2β3, and α2β4 combinations showed stronger activities toward PIF1 compared with other combinations (Fig. 1C and supplemental Table S3), suggesting that various holoenzyme combinations have differential activities toward PIF1. Relatively weak phosphorylation by the catalytic α subunit and strong stimulation by the regulatory β subunit might provide plants with enhanced regulatory power to modulate PIF1 activity in vivo. Moreover, CK2 subunit genes are differentially expressed in different tissues (39), suggesting that PIF1 might also be regulated in a tissue-specific manner.
Mapping of the Phosphorylation Sites in PIF1
To map the CK2 phosphorylation sites in PIF1, we performed in vitro kinase assays using cold ATP and the α1β1 holoenzyme. We then mapped the phosphorylation sites using mass spectrometry as described (41) and identified six (Thr-10, Thr-197, Ser-202, Ser-464, Ser-465, and Ser-469) phosphorylation sites scattered throughout the PIF1 polypeptide with a cluster at the C-terminal end (Fig. 2A, top panel). Some of the sites were identified with higher confidence than others. For example, the MS/MS of 1–27 and 463–489 are the best, with 196–213 moderate to weak, and 468–489 weak (Fig. 2A, bottom panel, and supplemental Fig. S1). Because we wanted to eliminate all the CK2 phosphorylation sites, we performed site-directed mutagenesis to change the six Ser/Thr sites to Ala, including the high- and low-confidence ones, for investigating the biological functions of PIF1. We then purified recombinant mutant PIF1, named PIF1-6M. In vitro kinase assays using either the α1 or α1β1 holoenzyme showed that a majority of the phosphorylation of the mutant PIF1-6M is eliminated, whereas the wild-type PIF1 is strongly phosphorylated by the holoenzyme (Fig. 2B). Because PIF1-6M is still phosphorylated by CK2 and the S466A mutation in the S464–465A (two CK2 sites mapped in this study, Fig. 2A) background displayed a strong reduction in the light-induced degradation of PIF1 (Fig. 4), we reasoned that Ser-466 might also be a CK2 site, as predicted by the ScanSite 2.0 and KinasePhos software packages (supplemental Table S2). We created the S466A mutation in the PIF1-6M background and named it PIF1-7M. The in vitro kinase assay showed that phosphorylation by CK2 is completely eliminated in PIF1-7M, whereas PIF1-6M is weakly phosphorylated by CK2 (Fig. 2C). As expected, WT PIF1 showed strong phosphorylation by CK2. Taken together, these data suggest that we have successfully mapped all the CK2 phosphorylation sites in PIF1.
FIGURE 2.

PIF1 is phosphorylated by CK2 at multiple sites. A, diagram showing the CK2 phosphorylation sites identified in PIF1 (top panel). Identification of phosphorylation sites in PIF1 by MALDI mass spectrometry (bottom panel). MS/MS from the phosphopeptide 463 VSSSKESEDHGNHTTGAAALEHHHHHH 489 is shown. The spectrum is dominated by the neutral loss of 98-Da phosphoric acid that is characteristic of phosphopeptides. Fragments are observed from the C-terminal charge retention y ions up to y24, all without phosphorylation, indicating that the phosphorylation site is localized at Ser-464 and/or Ser-465. B, autoradiogram showing phosphorylation of wild-type HIS-PIF1 and HIS-PIF1-6M mutant proteins by purified recombinant CK2. In vitro phosphorylation of mutant PIF1 by CK2 is severely reduced. C, autoradiogram showing phosphorylation of wild-type HIS-PIF1, HIS-PIF1- 6M, and HIS-PIF1-7M mutant proteins by the recombinant CK2 α1β1 holoenzyme. In vitro phosphorylation of HIS-PIF1-7M by CK2 is completely eliminated.
FIGURE 4.

Ser-464–466 are necessary for the rapid light-induced degradation of PIF1. A, light-induced phosphorylation and degradation of a PIF1 containing either Ser-459–461 to Ala (top panel) or Ser-464–466 to Ala (bottom panel) compared with wild-type LUC-PIF1. The rate of light-induced degradation of LUC-PIF1(S464–466A) is strongly reduced compared with wild-type LUC-PIF1 (bottom panel). The asterisk indicates a cross-reacting band. The numbers under the protein gel blots show the relative PIF1 level in the wild-type LUC-PIF1, LUC-PIF1(S459–461A), and LUC-PIF1(S464–466A) transgenic lines. The PIF1 level in each dark samples is set as 1. B, LUC-PIF1(S464–466A) promotes hypocotyl growth under red light. Photographs of seedlings of various genotypes grown in the dark or under R light (7 μmol m−2s−1) for 4 days. Scale bars = 5 mm. A second allele of LUC-PIF1(S464–466A)#33 displaying a similar long hypocotyl phenotype under red light is shown in supplemental Fig. S4. C, bar graph showing the mean hypocotyl lengths of various genotypes as indicated. Seedlings were grown as described in B. Error bars represent mean ± S.E. (n ≥ 30). *, p < 0.05.
Reduced Degradation of TAP-PIF1-6M Compared with Wild-type TAP-PIF1 under R Light
Previously, phosphorylation of HFR1 and HY5 by CK2 has been shown to stabilize these transcription factors from ubiquitin-mediated degradation (34, 35). To investigate the effect of CK2-mediated phosphorylation on the stability and in vivo function of PF1, we generated homozygous transgenic plants expressing four single mutants (T10A, S202A, S464A, and S465A) and PIF1-6M as TAP fusion proteins using the endogenous PIF1 promoter (supplemental Fig. S2). All four single mutants were phosphorylated and degraded in response to a pulse of R light (data not shown). However, the rate of degradation of TAP-PIF1-6M is significantly slower than that of wild-type TAP-PIF1 after a pulse of R light (Fig. 3A, two independent transgenic lines are shown in the top and bottom panels). Moreover, the light-induced phosphorylation is still present in TAP-PIF1-6M, similar to the wild-type TAP-PIF1, as indicated by the characteristic band shift under light conditions observed previously (26). To experimentally verify that these band shifts represent phosphorylated forms of PIF1, we performed alkaline phosphatase treatment as performed previously (26). Results show that the slow-mobility forms of PIF1 under light are largely converted to fast-mobility forms of PIF1, similar to the dark samples after alkaline phosphatase treatment (supplemental Fig. S3). These data suggest that, in response to light, either CK2 phosphorylates PIF1 at different sites than the ones mutated, or another light-specific kinase may phosphorylate PIF1 at different locations than the CK2 phosphorylation sites.
FIGURE 3.

CK2-mediated phosphorylation of PIF1 is necessary for rapid light-induced degradation of PIF1 in vivo. A, light-induced phosphorylation and degradation of a PIF1 containing six CK2 phosphorylation sites mutated (TAP-PIF1-6M) compared with wild-type TAP-PIF1. Two independent transgenic lines were compared with the corresponding wild-type TAP-PIF1 controls (top and bottom panels). α-RPT5 blots are shown as loading control. Numbers under the protein gel blots show the relative PIF1 level in the wild-type TAP-PIF1 and TAP-PIF1-6M transgenic lines. The PIF1 level in each dark samples is set as 1. B, TAP-PIF1-6M promotes hypocotyl growth under red light. Photographs of seedlings of various genotypes grown in the dark or under R light (7 μmol m−2s−1) for 4 days. Scale bars = 5 mm. Protein levels for various independent transgenic lines are shown (supplemental Fig. S2). C, bar graph showing the mean hypocotyl lengths for various genotypes as indicated. Seedlings were grown as described in B. Error bars represent mean ± S.E. (n ≥ 30). *, p < 0.05.
To determine the roles of TAP-PIF1-6M in phy signaling, we investigated the photomorphogenic phenotypes under R light. Results show that the hypocotyl lengths of all the transgenic TAP-PIF1-6M seedlings were longer compared with the wild-type TAP-PIF1 transgenic seedlings under R light conditions (Figs. 3, B and C), suggesting that TAP-PIF1-6M is functioning as a negative regulator of phy signaling, similar to the wild-type TAP-PIF1. The increased stability of TAP-PIF1-6M under light promotes hypocotyl elongation.
Ser-464–466 Are Necessary for Rapid Light-induced Degradation of PIF1
Previously, we have shown that both the N- and C-terminal regions of PIF1 are necessary for light-induced degradation of PIF1 (26). The N-terminal region contains the active phyA (APA) and active phyB (APB) binding domains necessary for phytochrome interactions. However, the molecular determinants present at the C-terminal region in PIF1 are still unknown. To map the C-terminal amino acids necessary for light-induced phosphorylation and degradation, we mutated two clusters of three serine residues at the C-terminal region of PIF1 (Ser-459–461 and Ser-464–466) to Ala separately. The mutant PIF1s was fused to luciferase (LUC) and expressed using a constitutive (CaMV35S) promoter in the pif1 background. Homozygous transgenic plants were selected and assayed for PIF1 stability and photomorphogenic phenotypes. Results show that the rate of degradation of the mutant LUC-PIF1(S459–461A) was similar to that of wild-type LUC-PIF1 under R light (Fig. 4A, top). Strikingly, the rate of degradation of the mutant LUC-PIF1(S464–466A) was strongly reduced compared with wild-type LUC-PIF1, despite the presence of the light-induced phosphorylation in this mutant PIF1 (Fig. 4A, bottom panel). Consistent with these data, the hypocotyl lengths of the mutant LUC-PIF1(S464–466A) were much longer than the wild-type LUC-PIF1, whereas the hypocotyl lengths of the mutant LUC-PIF1(S459–461A) were largely similar to that of the wild-type LUC-PIF1 (Fig. 4, B and C, and supplemental Fig. S4). We have also included the LUC-PIF1-3M transgenic line as a control. LUC-PIF1-3M has reduced affinity for both phyA and phyB because of three mutations (G47A, L95A, and N144A) at the APA and APB domains (26). LUC-PIF1(S464–466A) transgenic plants showed a similar phenotype compared with that of the LUC-PIF1-3M transgenic lines (Fig. 4, B and C). These data strongly suggest that Ser-464–466 are necessary for the rapid light-induced degradation of PIF1.
Ser-464 and Ser-465 were identified as CK2 phosphorylation sites by MS/MS mapping (Fig. 2A). Site-directed mutagenesis showed that Ser-466 is also a CK2 site, as the residual phosphorylation observed in PIF1-6M is eliminated in PIF1-7M (Fig. 2C). Because Ser-464–466 to Ala scanning mutations showed similar but stronger phenotypes compared with PIF1-6M (Figs. 3 and 4), these data suggest that Ser-464–466 play a major role in regulating PIF1 stability. Conversely, it is likely that the other CK2 sites (e.g. Thr-10, Thr-197, Ser-202, and Ser-469) play minor roles, if any, in regulating PIF1 stability.
The Light-induced Degradation of PIF1 Is Enhanced in CK2 β Subunit Overexpression Lines
The α and β subunits of CK2 are each encoded by four genes in Arabidopsis (39), and single and higher-order mutants in all the subunits are not available. In addition, the dominant-negative CK2 α mutant showed severe developmental defects (45). PIF1 degradation is unaffected in the ck2β3 single-mutant background under both R and FR light conditions (supplemental Fig. S5, A and B). We overexpressed CK2 β1 and β2 subunits and selected homozygous transgenic plants (supplemental Fig. S6). We then examined the rate of degradation of native PIF1 in these lines under R and FR light conditions. Results show that the rate of degradation of native PIF1 is enhanced under both R and FR light conditions in the CK2 β1 and β2 overexpression lines compared with wild-type seedlings (Fig. 5A, top and bottom panels).
FIGURE 5.

Degradation of PIF1 is faster in CK2 β1 and CK2 β2 overexpression lines under red light and far-red light. A, protein gel blots showing native PIF1 levels in wild-type and two independent CK2 β subunit overexpression lines. Four-day-old dark-grown seedlings were exposed to red pulse (Rp) (top panel) or far-red pulse (FRp) (bottom panel) at the indicated fluences and then incubated in the dark for the times indicated before harvesting for protein extraction. The numbers under the protein gel blots show the relative PIF1 level in the wild-type and CK2 β subunit overexpression lines. The PIF1 level in the wild-type Col-O dark sample is set as 1. α-RPT5 blots are shown as loading control. B, CK2 β1 and CK2 β2 overexpression lines are hypersensitive to FR light compared with the wild-type control. Photographs of seedlings of various genotypes grown in the dark or under FR light (0.4 μmol m−2s−1) for 4 days are shown. Scale bars = 5 mm. C, bar graph showing the mean hypocotyl lengths of various genotypes as indicated. Seedlings were grown as described in C. Error bars represent mean ± S.E. (n ≥ 30). *, p < 0.05.
We examined seedling deetiolation phenotypes for these transgenic plants compared with the wild type. Results showed that the CK2 β1 and β2 overexpression lines were modestly hypersensitive to FR light compared with wild-type seedlings (Fig. 5, B and C). Because CK2 regulates the circadian clock (33, 36, 37), we also examined the circadian clock phenotypes by performing leaf movement assays. Results showed that only CK2 β1 overexpression line 20 has a shorter period compared with wild-type and the other transgenic plants (supplemental Fig. S7). CK2 β1 line 20 has the strongest overexpression of the β1 subunit compared with other transgenic plants (supplemental Fig. S6). It is possible that strong overexpression is necessary to robustly regulate seedling deetiolation and circadian clock phenotypes. Taken together, these data suggest that CK2 may regulate the stability/activity of PIF1 and other factors in vivo and modulate deetiolation and the circadian clock in Arabidopsis.
DISCUSSION
Previously, it was shown that PIF1 and PIF3–6 were phosphorylated in response to light before being degraded through the 26 S proteasomal pathway (13, 15, 26). Interactions with both phyA and phyB are necessary for the light-induced phosphorylation and degradation under all three light conditions (4, 13, 15, 26). This study provides biochemical evidence that PIF1 is a substrate for Arabidopsis CK2, a ubiquitous Ser/Thr kinase present in all organisms. Phosphorylation by CK2 appears to be necessary for the rapid light-induced degradation of PIF1.
Several lines of evidence suggest that PIF1 is a bona fide substrate for Arabidopsis CK2. First, in silico studies predicted that PIF1 has multiple CK2 phosphorylation sites (supplemental Table S2). Second, in vitro kinase assays demonstrated that PIF1 is strongly phosphorylated by purified Arabidopsis CK2 α and β subunit holoenzyme combinations (Fig. 1A). Third, CK2-mediated phosphorylation of PIF1 is inhibited by a specific inhibitor, heparin, in a concentration-dependent manner (Fig. 1B). Fourth, mapping of the phosphorylation sites identified seven phosphorylation sites scattered throughout the PIF1 polypeptide with a cluster of sites at the C-terminal end (Fig. 2, A–C, and supplemental Fig. S1). Fifth, CK2-mediated phosphorylation enhances the rapid light-induced degradation of PIF1 (Figs. 3 and 4). Sixth, overexpression of CK2 β subunits accelerates the light-induced degradation of native PIF1 in vivo (Fig. 5A). These data provide strong evidence that PIF1 is a substrate for Arabidopsis CK2 and that CK2 phosphorylation specifically plays a role in the light-induced degradation of PIF1 to promote photomorphogenesis. However, the above data do not show that PIF1 is a substrate for CK2 in vivo. Further experiments are necessary to demonstrate whether CK2 phosphorylates PIF1 in vivo.
Previously, CK2 substrates have been identified in plants. These include translation initiation factors (e.g. eIF2α, eIF2β, eIF3c, eIF4B, and eIF5) (40, 41), a chromatin remodeling enzyme (histone deacetylase 2B) (40), circadian clock components (e.g. CCA1 and LHY) (33, 36, 37, 46), HMBG proteins from maize and Arabidopsis (47), abscisic acid responsive protein Rab17 in maize (48), and positively acting transcription factors (e.g. HY5 and HFR1) involved in light signaling pathways (34, 35). However, phosphorylation by CK2 has been shown to either stabilize or modulate the activity of these factors (34, 35, 40). In contrast, our data show that phosphorylation by CK2 promotes the light-induced degradation of PIF1 through the ubiquitin/26 S proteasomal pathway (Fig. 3A). This is similar to the posttranslational regulation of mammalian IκBα and promyelocytic leukemia proteins, where CK2-mediated phosphorylation enhanced their polyubiquitination and proteasomal degradation (49, 50). Similar to PIF1, CK2 phosphorylates a cluster of sites at the C terminus of IκBα, and this phosphorylation is UV-light inducible (50). Therefore, CK2-mediated stabilization and destabilization of proteins might represent an evolutionarily conserved mechanism.
Although our data provide strong evidence that CK2 promotes the light-induced degradation of PIF1 in vivo, PIF1-6M (which lacks the majority of the CK2 phosphorylation sites) and the PIF1(S464–466A) mutants are still robustly phosphorylated in response to light in vivo as observed previously for wild-type PIF1 (Figs. 3A and 4A and supplemental Fig. S3) (26). These data suggest that either CK2 phosphorylates PIF1 in response to light at different sites than the ones identified and mutated, or a separate light-specific kinase may phosphorylate PIF1 at different Ser/Thr residues under light. Because phy interaction is necessary for the light-induced phosphorylation and degradation of PIFs (12, 13, 26) and because phyA has been shown to have Ser/Thr kinase activity (51), it is possible that phys might directly phosphorylate PIFs in response to light. However, convincing in vivo evidence for the phyA kinase hypothesis is still lacking. Therefore, these data suggest that phosphorylation of PIF1 by CK2 and phosphorylation by either phys directly or a phy-associated kinase is necessary for the rapid light-induced degradation of PIF1. Further work is needed to identify the additional phosphorylation sites by either phytochromes or a phytochrome-associated kinase.
In addition, the mechanism of CK2-mediated enhanced degradation of PIF1 is still unknown. It is possible that phosphorylation of PIF1 by CK2 enhances the affinity of PIF1 for phys, as phy interaction has been shown to be necessary for rapid degradation of PIF1 (26). However, this is unlikely, as the isolated APA and APB domains, which are located within the first 150 amino acids, are both necessary and sufficient for physical interaction with phyA and phyB, respectively (Fig. 2A) (26, 52). Although PIF1 has one CK2 phosphorylation site near the APB sequence (Thr-10) (Fig. 2A), the results show that the CK2 sites at the C-terminal end (Ser-464, Ser-465 and Ser-466) play the major role in PIF1 degradation compared with other CK2 sites (Figs. 2, 3A, and 4A). An alternative hypothesis is that CK2-mediated phosphorylation of PIF1 at the C terminus enhances the interaction of PIF1 with substrate recognition factors responsible for polyubiquitination and subsequent degradation (e.g. F-box proteins in Skp-Cullin-F-box (SCF) complex). This is more likely because enhancement of proteasomal degradation of multiple factors by signal-induced phosphorylation has been demonstrated (50, 53). This possibility is also consistent with our previous data that isolated N- (1–150 amino acids) and C-terminal (151–478 amino acids) regions are not phosphorylated and degraded in response to light (26). Conversely, both N- and C-terminal regions are necessary for the light-induced degradation of PIF1 in vivo (26). Identification of factors responsible for recognition and polyubiquitination of PIF1 will help distinguish these possibilities.
In summary, our data and those of others show that light stabilizes positively acting transcription factors (e.g. HY5, LAF1, and HFR1) (34, 35, 54) and induces proteasomal degradation of negatively acting transcription factors (e.g. PIFs) to promote photomorphogenesis (Fig. 6). CK2 phosphorylates both positively and negatively acting transcription factors involved in light signaling. However, the significance of CK2 phosphorylation is opposite for these two classes of transcription factors. CK2-mediated phosphorylation enhances the stability of the positively acting factors, whereas it decreases the stability of the negatively acting factors (e.g. PIF1 and possibly other PIFs). This contrasting but apparently synergistic effect is expected to promote robust photomorphogenesis. However, our phenotypic analyses showed that the CK2 β subunit overexpression lines have modest seedling deetiolation and circadian phenotypes (Figs. 5, B and C, and supplemental Fig. S7). Moreover, a stable PIF1 is expected to promote hypocotyl growth as observed (Figs. 3 and 4) (26), whereas a CK2 dominant-negative mutant displayed a short hypocotyl phenotype in the dark (45). These apparent contradictions might be explained in part by the fact that CK2-mediated phosphorylation of HY5 reduces the DNA binding ability of HY5 (34). In addition, all organisms have multiple CK2 substrates in vivo that may function in multiple pathways. More than 300 substrates have been identified for CK2 in an animal system (55). Therefore, the phenotypes of the CK2 mutants and overexpression lines will reflect the net effect of CK2 on these diverse pathways that regulate plant growth and development. Identification and characterization of these substrates will shed light on how CK2 optimizes photomorphogenesis.
FIGURE 6.
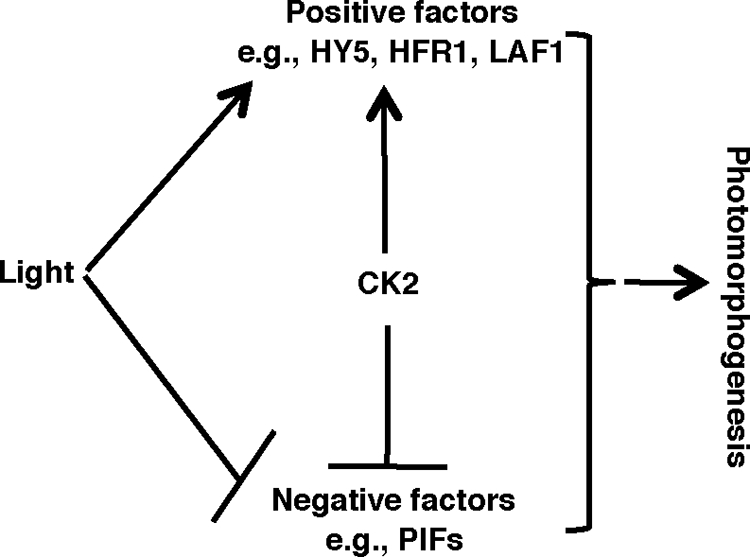
Model of CK2 function in regulating photomorphogenesis. Light induces rapid degradation of the negative regulators (e.g. PIFs) and stabilization of the positive regulators (e.g. HY5, HFR1, and LAF1) to promote photomorphogenesis. CK2 phosphorylates both the negative regulators and the positive regulators. However, CK2-mediated phosphorylation stabilizes the positive regulators and destabilizes the negative regulators in response to light to promote photomorphogenesis.
Acknowledgments
We thank Tuyen Cao and Jimmy Wang for technical assistance, and Jacqulyn Tolson for phosphopeptide enrichment.
This work was supported, in whole or in part, by National Institutes of Health Grant GM-23167 (to E. M. T.) and NIEHS National Institutes of Health Grant P30ES007784 (to M. D. P.). This work was also supported by Department of Energy Grant DE-FG02-04ER15575, National Science Foundation Grant MCB0214996, and Welch Foundation Grant F1339 (to K. S. B.); and by National Science Foundation Grants IOS-0822811 and IOS-0849287 (to E. H.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7, Tables S1–S3, experimental procedures, and references.
- phy
- phytochrome
- R
- red
- FR
- far-red
- PIF
- phytochrome interacting factor
- MOPS
- 4-morpholinepropanesulfonic acid
- LUC
- luciferase
- APA
- active phyA binding domain
- APB
- active phyB binding domain.
REFERENCES
- 1. Rockwell N. C., Su Y. S., Lagarias J. C. (2006) Annu. Rev. Plant Biol. 57, 837–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bae G., Choi G. (2008) Annu. Rev. Plant Biol. 59, 281–311 [DOI] [PubMed] [Google Scholar]
- 3. Franklin K. A., Quail P. H. (2010) J. Exp. Bot. 61, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Castillon A., Shen H., Huq E. (2009) Genetics 182, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schaefer E., Nagy F. (2006) Photomorphogenesis in Plants and Bacteria, 3rd Ed., Springer, Dordrecht, The Netherlands [Google Scholar]
- 6. Fankhauser C., Chen M. (2008) Trends Plant Sci. 13, 596–601 [DOI] [PubMed] [Google Scholar]
- 7. Huq E., Quail P. H. (2005) in Handbook of Photosensory Receptors (Briggs W. R., Spudich J. L. eds) pp. 151–170, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 8. Ma L., Li J., Qu L., Hager J., Chen Z., Zhao H., Deng X. W. (2001) Plant Cell 13, 2589–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quail P. H. (2007) J. Integr. Plant Biol. 49, 11–20 [Google Scholar]
- 10. Jiao Y., Lau O. S., Deng X. W. (2007) Nat. Rev. Genet. 8, 217–230 [DOI] [PubMed] [Google Scholar]
- 11. Huq E. (2006) Trends Plant Sci. 11, 4–7 [DOI] [PubMed] [Google Scholar]
- 12. Henriques R., Jang I. C., Chua N. H. (2009) Curr. Opin. Plant Biol. 12, 49–56 [DOI] [PubMed] [Google Scholar]
- 13. Castillon A., Shen H., Huq E. (2007) Trends Plant Sci. 12, 514–521 [DOI] [PubMed] [Google Scholar]
- 14. Duek P. D., Fankhauser C. (2005) Trends Plant Sci. 10, 51–54 [DOI] [PubMed] [Google Scholar]
- 15. Leivar P., Quail P. H. (2011) Trends Plant Sci. 16, 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toledo-Ortiz G., Huq E., Quail P. H. (2003) Plant Cell 15, 1749–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martínez-García J. F., Huq E., Quail P. H. (2000) Science 288, 859–863 [DOI] [PubMed] [Google Scholar]
- 18. Oh E., Kang H., Yamaguchi S., Park J., Lee D., Kamiya Y., Choi G. (2009) Plant Cell 21, 403–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moon J., Zhu L., Shen H., Huq E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 9433–9438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oh E., Yamaguchi S., Hu J., Yusuke J., Jung B., Paik I., Lee H. S., Sun T. P., Kamiya Y., Choi G. (2007) Plant Cell 19, 1192–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hornitschek P., Lorrain S., Zoete V., Michielin O., Fankhauser C. (2009) EMBO J. 28, 3893–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Toledo-Ortiz G., Huq E., Rodríguez-Concepción M. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 11626–11631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leivar P., Monte E., Al-Sady B., Carle C., Storer A., Alonso J. M., Ecker J. R., Quail P. H. (2008) Plant Cell 20, 337–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leivar P., Monte E., Oka Y., Liu T., Carle C., Castillon A., Huq E., Quail P. H. (2008) Curr. Biol. 18, 1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jang I. C., Henriques R., Seo H. S., Nagatani A., Chua N. H. (2010) Plant Cell 22, 2370–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen H., Zhu L., Castillon A., Majee M., Downie B., Huq E. (2008) Plant Cell 20, 1586–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shin J., Kim K., Kang H., Zulfugarov I. S., Bae G., Lee C. H., Lee D., Choi G. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 7660–7665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leivar P., Tepperman J. M., Monte E., Calderon R. H., Liu T. L., Quail P. H. (2009) Plant Cell 21, 3535–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen M., Galvão R. M., Li M., Burger B., Bugea J., Bolado J., Chory J. (2010) Cell 141, 1230–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Al-Sady B., Ni W., Kircher S., Schäfer E., Quail P. H. (2006) Mol. Cell 23, 439–446 [DOI] [PubMed] [Google Scholar]
- 31. Shen Y., Khanna R., Carle C. M., Quail P. H. (2007) Plant Physiol. 145, 1043–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee Y., Lloyd A. M., Roux S. J. (1999) Plant Physiol. 119, 989–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sugano S., Andronis C., Ong M. S., Green R. M., Tobin E. M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 12362–12366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hardtke C. S., Gohda K., Osterlund M. T., Oyama T., Okada K., Deng X. W. (2000) EMBO J. 19, 4997–5006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park H. J., Ding L., Dai M., Lin R., Wang H. (2008) J. Biol. Chem. 283, 23264–23273 [DOI] [PubMed] [Google Scholar]
- 36. Daniel X., Sugano S., Tobin E. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 3292–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Portolés S., Más P. (2007) Plant J. 51, 966–977 [DOI] [PubMed] [Google Scholar]
- 38. Andronis C., Barak S., Knowles S. M., Sugano S., Tobin E. M. (2008) Mol. Plant 1, 58–67 [DOI] [PubMed] [Google Scholar]
- 39. Salinas P., Fuentes D., Vidal E., Jordana X., Echeverria M., Holuigue L. (2006) Plant Cell Physiol. 47, 1295–1308 [DOI] [PubMed] [Google Scholar]
- 40. Dennis M. D., Browning K. S. (2009) J. Biol. Chem. 284, 20602–20614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dennis M. D., Person M. D., Browning K. S. (2009) J. Biol. Chem. 284, 20615–20628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen H., Moon J., Huq E. (2005) Plant J. 44, 1023–1035 [DOI] [PubMed] [Google Scholar]
- 43. Rubio V., Shen Y., Saijo Y., Liu Y., Gusmaroli G., Dinesh-Kumar S. P., Deng X. W. (2005) Plant J. 41, 767–778 [DOI] [PubMed] [Google Scholar]
- 44. Karimi M., Meyer B. D., Hilson P. (2005) Trends Plant Sci. 10, 103–105 [DOI] [PubMed] [Google Scholar]
- 45. Moreno-Romero J., Espunya M. C., Platara M., Ariño J., Martínez M. C. (2008) Plant J. 55, 118–130 [DOI] [PubMed] [Google Scholar]
- 46. Sugano S., Andronis C., Green R. M., Wang Z. Y., Tobin E. M. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 11020–11025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stemmer C., Leeming D. J., Franssen L., Grimm R., Grasser K. D. (2003) Biochem. 42, 3503–3508 [DOI] [PubMed] [Google Scholar]
- 48. Riera M., Figueras M., López C., Goday A., Pagès M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9879–9884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scaglioni P. P., Yung T. M., Cai L. F., Erdjument-Bromage H., Kaufman A. J., Singh B., Teruya-Feldstein J., Tempst P., Pandolfi P. P. (2006) Cell 126, 269–283 [DOI] [PubMed] [Google Scholar]
- 50. Kato T., Jr., Delhase M., Hoffmann A., Karin M. (2003) Mol. Cell 12, 829–839 [DOI] [PubMed] [Google Scholar]
- 51. Yeh K. C., Lagarias J. C. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13976–13981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khanna R., Huq E., Kikis E. A., Al-Sady B., Lanzatella C., Quail P. H. (2004) Plant Cell 16, 3033–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karin M., Ben-Neriah Y. (2000) Annu. Rev. Immunol. 18, 621–663 [DOI] [PubMed] [Google Scholar]
- 54. Seo H. S., Yang J. Y., Ishikawa M., Bolle C., Ballesteros M. L., Chua N. H. (2003) Nature 423, 995–999 [DOI] [PubMed] [Google Scholar]
- 55. Meggio F., Pinna L. A. (2003) FASEB J. 17, 349–368 [DOI] [PubMed] [Google Scholar]