

Abstract
Bacterial “maintenance of genome stability protein A” (MgsA) and related eukaryotic enzymes play important roles in cellular responses to stalled DNA replication processes. Sequence information identifies MgsA enzymes as members of the clamp loader clade of AAA+ proteins, but structural information defining the family has been limited. Here, the x-ray crystal structure of Escherichia coli MgsA is described, revealing a homotetrameric arrangement for the protein that distinguishes it from other clamp loader clade AAA+ proteins. Each MgsA protomer is composed of three elements as follows: ATP-binding and helical lid domains (conserved among AAA+ proteins) and a tetramerization domain. Although the tetramerization domains bury the greatest amount of surface area in the MgsA oligomer, each of the domains participates in oligomerization to form a highly intertwined quaternary structure. Phosphate is bound at each AAA+ ATP-binding site, but the active sites do not appear to be in a catalytically competent conformation due to displacement of Arg finger residues. E. coli MgsA is also shown to form a complex with the single-stranded DNA-binding protein through co-purification and biochemical studies. MgsA DNA-dependent ATPase activity is inhibited by single-stranded DNA-binding protein. Together, these structural and biochemical observations provide insights into the mechanisms of MgsA family AAA+ proteins.
Keywords: ATPases, Crystal Structure, DNA-Protein Interaction, DNA Recombination, DNA Repair, DNA Replication, AAA+
Introduction
In bacteria, DNA replication forks can stall when they encounter DNA lesions, template strand breaks, DNA-bound proteins, or other impediments. Stalled replication forks occur as often as once per cell generation during normal cell growth and must be repaired (1–5). There are multiple causes of replication fork stalling and/or collapse and a range of recovery mechanisms that likely reflect the variety of DNA/protein structures that are encountered when these events occur. Most of the repair pathways involve recombinational DNA repair and the RecA recombinase (1–6).
MgsA (also called RarA; bacteria), Mgs1 (yeast), and Wrnip1 (mammals) constitute a family of evolutionarily conserved proteins with roles in the recovery of stalled replication processes. Escherichia coli MgsA and its Saccharomyces cerevisiae and Homo sapiens homologs share ∼40% sequence identity and over 55% similarity (7) and are members of the AAA+ (ATPases associated with a variety of cellular activities) family of proteins (8). As implied by their name, AAA+ proteins are involved in diverse cellular pathways, including protein degradation, DNA metabolism, and membrane fusion (9–14). AAA+ proteins contain a conserved 200–250-residue nucleoside triphosphate (NTP)-binding/hydrolysis domain and generally function as oligomeric molecular machines. The NTP-binding sites of AAA+ proteins are typically positioned at the interface of adjacent subunits allowing for intersubunit coupling of NTP binding and hydrolysis events to remodeling of target macromolecules (15, 16).
AAA+ proteins have been classified into various clades based on their sequences and structures (14, 17, 18). Based on these studies, the MgsA/Mgs1/Wrnip1 family is grouped with bacterial and eukaryotic clamp loader proteins in the “clamp loader” AAA+ clade. The clamp loader clade is characterized by a minimal AAA+ fold (14) and a SR(CAT) motif that contains a conserved “Arg finger” residue that is important for NTP hydrolysis (18). The clamp loader proteins load processivity clamps onto primer-template junctions during DNA replication (19–23). Representative clamp loader structures reveal the complexes to be heteropentamers possessing an oligomerization domain C-terminal to the AAA+ module (24, 25). However, insights into the structures of the MgsA family proteins have thus far been lacking.
Although its precise function has remained elusive, genetic experiments suggest E. coli MgsA is important in facilitating the recovery of stalled replication forks (26–28). MgsA function is a prerequisite to RecA protein loading at stalled forks in a temperature-sensitive dnaE mutant (encodes the catalytic subunit of the replicative DNA polymerase III holoenzyme) but not in a temperature-sensitive dnaN mutant (encodes the processivity β clamp) (26). This implies that MgsA is involved in the processing of some stalled replication forks but not others and that this processing may allow, or perhaps promote, RecA loading. Inasmuch as the replisome usually remains intact at the site of a fork when fork stalling occurs in the dnaEts mutant, but not at stalled forks in the dnaNts mutant, it is possible that MgsA somehow facilitates replisome disassembly prior to fork repair. Effects of the dnaE486 mutant, another temperature-sensitive mutant of the catalytic subunit of DNA polymerase III, also suggest a role for MgsA in the processing of stalled forks and maintaining genome stability (28). The dnaE486 mutant has a growth defect and forms filamented cells, indicative of SOS induction, at a semi-restrictive temperature (29, 30). Interestingly, an mgsA deletion suppresses the growth defect of dnaE486 cells but produces a high proportion of anucleate cells, suggesting that MgsA may prevent aberrant DNA replication and allow for RecA filament formation and SOS induction. Similar results have been seen with recA and recQ mutants (28, 31); RecQ helicase is thought to unwind DNA at stalled replication forks generating ssDNA onto which RecA can be loaded. These data indicate that MgsA promotes replication fork repair in some E. coli DNA replisome mutants, possibly through a RecA-dependent pathway. Related to its proposed functions in replication, fluorescently tagged MgsA localizes to the replisome (32, 33), although how this localization is achieved is not known.
Studies of the S. cerevisiae homolog of MgsA, Mgs1, also yield insights into functions of the larger protein family in DNA metabolism. Post-replication repair (PRR) encompasses the error-prone and error-free pathways of repairing stalled replication forks in S. cerevisiae. The Mgs1 protein maintains genome stability by suppressing the error-prone RAD6 PRR pathway in the absence of exogenous DNA damage (34, 35). The RAD6 and RAD52 pathways define two of the major PRR processes in S. cerevisiae. The RAD52 pathway utilizes homologous recombination in an error-free mechanism. Interestingly, when the RAD6 pathway is inhibited by rad6 or rad18 mutations, deleting mgs1 is lethal; however, activation of the RAD52 pathway by overexpressing Rad52 or deleting srs2 suppresses this lethality (34, 35). This suggests that Mgs1 modulates the response to stalled replication forks and may be involved in a novel PRR pathway. Mgs1 also physically interacts with the processivity factor proliferating cell nuclear antigen in yeast (35), which plays a pivotal role in the channeling of stalled forks into the various repair pathways (36–40). Formation of the Mgs1-proliferating cell nuclear antigen interaction could be important in PRR pathway selection.
Data implying roles in replication fork repair for the MgsA protein family extend to humans. H. sapiens Wrnip1, the human MgsA homolog, possesses a ubiquitin-binding zinc finger domain that is involved in its localization to the replisome (41, 42). Treatment of cells with fork-stalling agents causes an increase in the number of Wrnip1 foci suggesting that Wrnip1 is prevalent at stalled replication forks (41). The translesion synthesis DNA polymerases η and κ (43, 44) and ubiquitin ligase RAD18 (41, 45, 46), which are heavily involved in the recovery of stalled replication forks, also possess ubiquitin-binding zinc finger domains and localize to stalled replication forks in a ubiquitin-binding zinc finger-dependent manner. Post-translational modifications, such as ubiquitination and sumoylation of replisome machinery, are known to be important regulatory pathways for DNA replication, recombination, and repair (37, 47). The ubiquitin-binding zinc finger domain of Wrnip1 controls its localization to stalled replication forks with post-translational modifications to replisome components likely functioning as the signal.
To better define the structure and function of the MgsA/Mgs1/Wrnip1 protein family, we have determined the x-ray crystal structure of E. coli MgsA and characterized its biochemical activities. MgsA is distinct among clamp loader-clade AAA+ proteins characterized to date in that it assembles as a homotetramer. MgsA protomers are composed of AAA+ and tetramerization domains, each of which participates in oligomerization to form a highly intertwined quaternary structure. Phosphate is bound at each AAA+ ATP-binding site, but the active sites are in a catalytically incompetent conformation due to displacement of Arg finger residues. MgsA is furthermore shown to form a complex with SSB2 through co-purification and in vitro binding studies. This interaction may help target MgsA to the replication fork in vivo. Together, these studies help define the structural and biochemical mechanisms that underpin MgsA activity.
EXPERIMENTAL PROCEDURES
DNA Substrates
Circular single-stranded DNA was prepared from bacteriophage M13mp18 using previously described methods (48, 49). The concentrations of bacteriophage ssDNA was determined by absorbance at 260 nm, using 36 μg/ml·A260 nm as conversion factor. All DNA concentrations are reported as total nucleotides (micromolar).
Protein Purification
The open reading frame of E. coli mgsA (strain MG1655) was amplified by PCR and subcloned in-frame into NdeI/BamHI-digested pET21A (Novagen). The open reading frame of the resulting plasmid (pEAW354) was sequenced to confirm the sequence integrity. The nuclease-deficient E. coli K12 strain STL2669 (Δ(recA-srlR)306:Tn10 xonA2(sbcB−)), a gift from Susan T. Lovett (Brandeis University), was transformed with pEAW354 and pT7 pol26 (50). Cells were grown in Luria-Bertani medium containing 100 μg/ml ampicillin and 40 μg/ml kanamycin at 37 °C to an A600 nm of ∼0.5. MgsA protein overexpression was induced by the addition of 0.8 mm isopropyl 1-thio-β-d-galactopyranoside and growth at 37 °C for 4 h. All subsequent steps were performed at 4 °C. Pelleted cells were resuspended in sucrose solution (25% (w/v) sucrose, 250 mm Tris-HCl (80% cation), pH 7.7, 7 mm EDTA, 1 μm pepstatin, 1 μm leupeptin, 1 μm E-64) and lysed by addition of lysozyme to a final concentration of 1.6 g/liter and sonication. Insoluble material was removed by centrifugation. MgsA was precipitated from the cell lysate supernatant by the addition of solid NH4(SO4)2 to 35% saturation. In all subsequent steps, 1 mm dithiothreitol (DTT) was added to buffers. The NH4(SO4)2 pellet was resuspended in 20 mm Tris-Cl (80% cation), pH 7.7, 1 mm EDTA, 10% glycerol (R buffer) containing 450 mm NH4(SO4)2 and applied to a butyl-Sepharose (Amersham Biosciences) column. MgsA was eluted using a NH4(SO4)2 linear gradient from 600 to 0 mm in R buffer. MgsA fractions were pooled, dialyzed into P buffer (20 mm phosphate, pH 7.0, 1 mm EDTA and 10% glycerol), and flowed through a ceramic hydroxyapatite (Bio-Rad) column. MgsA in the flow-through was precipitated by adding an equal volume of R buffer (no glycerol) containing 90% saturated NH4(SO4)2, then resuspended in R buffer (<5 ml), and applied onto a HiPrep 16/60 Sephacryl S-300 (Amersham Biosciences) column equilibrated in R buffer supplemented with 1.2 m NaCl. MgsA eluted in three peaks; the third peak contained the most MgsA protein and was pooled. The resulting MgsA protein was >95% pure by gel and was dialyzed into R buffer, aliquoted, and flash-frozen in liquid nitrogen and stored at −80 °C. The purified protein was free of any detectable nuclease activity. The concentration of the MgsA protein was calculated using the native extinction coefficient 5.44 × 104 m−1cm−1. The native extinction coefficient was determined as described previously (51).
To express an R156A MgsA variant, pEAW354 was used as a PCR template with the same upstream primer used in constructing pEAW354, and a downstream primer consisting of bases 521–453 of the mgsA gene. Bases 521–516 include an ScaI restriction enzyme site. The bases ACG at 468–466 coding for the reverse complement of Arg were changed to CGC to code for the reverse complement of Ala. The PCR product was digested with NdeI and ScaI and substituted for the equivalent fragment in pEAW354 to create pEAW662. The presence of the R156A codon change was confirmed by sequencing. The MgsA R156A protein was purified identically to wild-type MgsA.
E. coli SSB (52), SSBΔC8 (53), SSBΔC1 (54), and SSB-mixed (55) proteins were purified as described. The concentration of all SSB protein variants was determined using the extinction coefficient of 2.83 × 104 m−1 cm−1 (56).
MgsA Crystallization and Structure Determination
E. coli MgsA (4 g/liter in R buffer) was crystallized by suspending 1 μl of protein mixed with 1 μl of mother liquor solution (10 mm MES, pH 6.6, 38% ethylene glycol, 16% glycerol) over 1 ml of mother liquor solution in a hanging drop vapor-diffusion experiment. Crystals formed after several weeks at room temperature and were frozen in liquid nitrogen directly from the drop.
Diffraction data were indexed and scaled using HKL2000 (57) and molecular replacement was carried out with Phaser (58) using a structure of the tetramerization domain from H. influenza MgsA as a partial model (Protein Data Bank 3BGE). Phase estimates from molecular replacement were sufficient to produce 2Fo − Fc and Fo − Fc electron density maps that permitted model building of the AAA+ domain through repetitive cycles of manual structure building using COOT (59) and refinement by REFMAC5 (60). Coordinate and structure factor files have been deposited at the Protein Data Bank (code 3PVS).
Analytical Ultracentrifugation
A sample of MgsA was dialyzed overnight against 20 mm Tris-Cl (80% cation), pH 7.7, 150 mm NaCl, 0.1 mm EDTA and then diluted with dialysis buffer to create samples at 3.8, 8.3, and 15 μm. Centrifugation was performed using a Beckman XLA analytical ultracentrifuge, with measurements carried out at 20 °C. Equilibrium data were collected at speeds of 3,600, 4,800, 5,800, 7,800, and 10,000 rpm followed by a final experiment at 5,800 rpm to test for nonreversible aggregation during the experiment. The presence of a nonsedimenting base line was established by a high speed spin to deplete all protein material. Samples were assumed to be at equilibrium when gradients collected 3 h or more apart were superimposable. The density of the buffer was computed as 1.0039 g/ml using the density increment approach (61). The partial specific volume of the protein was computed from the amino acid sequence to be 0.732 ml/g. All data were processed and analyzed using software from Beckman or programs written for Igo Pro (Wavemetrics, Inc, Lake Oswego, OR). Semi-log absorbance data as a function of squared radial distance were globally fitted using models containing one or two molecular species (62). Inclusion of a second species slightly improved the fits. The best fit was generated using a model where the dominant species is tetrameric, with a smaller amount of octamer present.
ATPase Assay
A coupled spectrophotometric enzyme assay (63, 64) was used to measure the ATPase activities of MgsA. The assays were carried out in a Varian Cary 300 dual beam spectrophotometer equipped with a temperature controller and a 12-position cell changer. The cell path lengths are 1 cm and bandpass was 2 nm. The reactions were carried out at 37 °C in 25 mm Tris acetate (80% cation), 1 mm DTT, 3 mm potassium glutamate, 10 mm Mg(OAc)2, 5% (w/v) glycerol, an ATP regeneration system (10 units/ml pyruvate kinase, 2.2 mm phosphoenolpyruvate), a coupling system (3 mm NADH and 10 units/ml lactate dehydrogenase), and the indicated concentrations of MgsA, MgsA R156A, SSB, SSBΔC8, and DNA.
Tandem Affinity Purification
The open reading frame of E. coli mgsA was amplified by PCR and subcloned in-frame into a specialized cloning vector pCN703 to produce an expression vector encoding MgsA with an N-terminal dual affinity tag (includes protein A and calmodulin peptide binding domains separated by a tobacco etch virus protease cleavage site). The open reading frame of the resulting plasmid (pTAP-MgsA) was sequenced to confirm the sequence integrity. E. coli K12 strain MG1655 (DE3) transformed with pTAP-MgsA was grown at 37 °C in 4 liters of Luria-Bertani medium supplemented with 50 μg/ml ampicillin to midlog phase (A600 nm of ∼0.5), induced by the addition of 1 μm isopropyl 1-thio-β-d-galactopyranoside, and grown for an additional 3 h. Cells were harvested by centrifugation, suspended in 50 ml of Nonidet P-40 buffer (6 mm dibasic sodium phosphate, 4 mm monobasic sodium phosphate, 150 mm NaCl, 2 mm EDTA, 50 mm NaF, 4 mg/liter leupeptin, 0.1 mm sodium vanadate, 19.5 mg/liter benzamidine, 8.7 mg/liter phenylmethylsulfonyl fluoride (PMSF), 1% Nonidet P-40 substitute), and lysed by sonication. Soluble lysate was incubated for 1 h at 4 °C with IgG-Sepharose beads (pre-equilibrated in 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% Nonidet P-40); beads were then washed with 3 volumes of equilibration buffer. Tobacco etch virus cleavage buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% Nonidet P-40, 0.5 mm EDTA, 1 mm DTT) with 15 μl of 15 μm tobacco etch virus protease was incubated with the lysate/bead mixture with shaking for ∼12 h at 16 °C. The eluent was incubated with 300 μl of calmodulin affinity resin (Stratagene) and 3 μl of 1 m CaCl2 in calmodulin binding buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% Nonidet P-40, 10 mm magnesium acetate, 1 mm imidazole, 10 mm 2-mercaptoethanol) for 1 h with shaking. Resin was washed with calmodulin binding buffer, and TAP-MgsA was eluted first with a high salt buffer (calmodulin binding buffer adjusted to 1 m NaCl) and then with EGTA elution buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.02% Nonidet P-40, 1 mm magnesium acetate, 10 mm imidazole, 10 mm 2-mercaptoethanol, 20 mm EGTA). High salt and EGTA-eluted protein samples were precipitated with trichloroacetic acid (25% w/v) on ice for 30 min, pelleted by centrifugation, washed twice with ice-cold acetone, and suspended in 30 μl of gel buffer. After SDS-PAGE, individual bands were excised, digested with trypsin, and subjected to MALDI-TOF mass spectrometry for identification of peptides (University of Wisconsin Mass Spectrometry facility).
Surface Plasmon Resonance (SPR)
All SPR measurements were performed on a Biacore 2000 equipped with a sensor chip SA (GE Healthcare). The sensor chip SA was pretreated with three 1-min pulses of 1 m NaCl at 100 μl/min, after which 0.25 pmol (∼300 response units (RU)) of biotinylated dT35 was flowed onto the cell at 5 μl/min in TBS200 (200 mm NaCl, 20 mm Tris, pH 7.5, 0.01% Tween 20, 3 mm EDTA, and 1 mm DTT). The chip was further rinsed with TBS200 at 100 μl/min for 5 min and then flushed with TBS50 (50 mm NaCl, 20 mm Tris-HCl, pH 7.5, 0.01% Tween 20, 3 mm EDT, and 1 mm DTT.) SSB diluted into TBS50 was subsequently loaded to the chip surface at 200 nm (tetramer) until saturated (∼1500 RU) and then rinsed with TBS50 until stable. For SPR measurements with MgsA, various concentrations (50–1500 nm) of MgsA in TBS50 were flowed through the flow cell for 90 s at 30 μl/min and then MgsA was allowed to dissociate for 12 min. Raw RU were background-subtracted against an empty flow cell (∼100 RU at the highest MgsA concentration used), and the maximum RU for each run were plotted against concentration of MgsA and fit to obtain equilibrium binding data using the GraphPad Prism program (version 4.02, SmartDrawNet).
RESULTS
X-ray Crystal Structure of E. coli MgsA
Together with the bacterial and eukaryotic processivity clamp loader proteins, the MgsA family of proteins forms the “clamp loader clade” of AAA+ proteins (reviewed in Refs. 14, 18). Although the structures and direct functions of the clamp loader proteins are well established (24, 25), similar information defining the MgsA family is lacking. A crystallographic approach was therefore taken to better understand how MgsA proteins function.
E. coli MgsA formed crystals that diffracted x-rays to 2.5 Å resolution (Table 1). Experimental phasing experiments with selenomethionine-incorporated protein were not successful due to poor solubility of the derivative proteins. However, the structure of a domain from the Haemophilus influenzae MgsA protein was available for use as a molecular replacement model (Protein Data Bank code 3BGE; unpublished result from the New York SGX Research Center for Structural Genomics available from the RCSB Protein Data Bank). This domain included the C-terminal domain of H. influenzae MgsA and shared 82% identity with the same domain from E. coli MgsA. Molecular replacement produced difference electron density maps that permitted building of the AAA+ domain of E. coli MgsA through several rounds of manual model building and structure refinement.
TABLE 1.
X-ray crystallographic data collection and structure refinement statistics
| Data collection | |
| Space group | P212121 |
| Unit cell | |
| a, b, c | 99.9, 143.7, 163.2 Å |
| α, β, γ | 90°, 90°, 90° |
| Resolution (high resolution shell) | 30.0 to 2.50 Å (2.54 to 2.50 Å) |
| Rsym (high resolution shell)a | 0.073 (0.397) |
| I/σ(I) (high resolution shell) | 24.5 (3.0) |
| Completeness (high resolution shell) | 97.3% (78.8%) |
| Redundancy (high resolution shell) | 7.1 (5.4) |
| Refinement | |
| Resolution | 30.0 to 2.50 Å |
| R/Rfreeb | 0.195/0.266 |
| No. of atoms | |
| Protein | 13,071 |
| Waters/ligands | 476 |
| Average B factors | |
| Protein atoms | 45.9 |
| Waters/ligands | 41.6 |
| Root mean square deviations | |
| Bond lengths | 0.016 Å |
| Bond angles | 1.61° |
| Ramachandran statistics | |
| Residues in core region | 90.8% |
| Residues in allowed region | 9.1% |
| Residues in generously allowed regions | 0.1% |
| Residues in disallowed regions | 0% |
a Rsym = ΣΣj|Ij − 〈I〉|ΣIj, where Ij is the intensity measurement for reflection j, and 〈I〉 is the mean intensity for multiply recorded reflections.
b Rwork/Rfree = Σ‖Fobs| − |Fcalc/|Fobs|, where the working and free R factors are calculated by using the working and free reflection sets, respectively. The free R reflections (5% of the total) were held aside throughout refinement.
The crystal structure of E. coli MgsA revealed three apparent structural domains in the protein as follows: N-terminal ATP-binding (residues ∼22–165) and adjacent helical lid domains (residues ∼166–247 and a short N-terminal helix (14–21)) that are conserved among AAA+ proteins and a C-terminal tetramerization domain (residues ∼251–446) (Fig. 1A). MgsA assembled as a highly intertwined pseudosymmetric tetramer in the crystal in which each protomer physically contacts each of the three remaining subunits in the tetramer (Fig. 1B). The tetramerization domains contribute the bulk of the buried surface area in the tetramer (burying 2069–2168 Å2 of surface area between adjacent molecules) with more modest contributions from the AAA+ domain (413–457 Å2). Overall, an average of 2573 Å2 is buried between pairs of MgsA proteins within the tetramer. This is more buried surface area than in the E. coli clamp loader (PDB code 1JR3) (24), in which 470–1133 and 383–1450 Å2 are buried between the oligomerization and AAA+ domains, respectively. Sedimentation equilibrium analytical ultracentrifugation experiments confirmed that MgsA forms primarily a tetramer in solution. Previous gradient centrifugation studies of the human MgsA homolog, Wrnip1, identified it as a homo-octamer (66). Whether the differences between the apparent quaternary structures of MgsA and Wrnip1 are a result of sequence differences or of differences in the approaches used to examine higher order assemblies is not clear.
FIGURE 1.

Structure of the E. coli MgsA protein. A, ribbon diagram of a single MgsA protomer with ATP-binding domain (orange), lid domain (cyan), and tetramerization domain (green) colored to reflect the domain architecture. Phosphate is modeled in red. B, crystallographic asymmetric unit is shown with one protomer in ribbon form and the three remaining protomers in surface form. C, conservation of sequences in 157 MgsA homologs. Invariant (pink) and highly conserved (magenta) residues are color-coded. The four MgsA protomers of the asymmetric (and biological) unit are shown in four shades of gray. Areas of high conservation are apparent in the active site pocket and as the interface between subunits in the oligomerization domains. D, conservation around the Arg finger (circled in yellow); view is looking down on ATP-binding domain. E, view of a portion of the 2Fo − Fc electron density map contoured to 1.5σ superimposed with the refined MgsA structure.
The conservation of each residue in E. coli MgsA was analyzed across 157 MgsA homologs using the ConSurf program (67). The active site pocket and the surrounding area are extremely conserved, as are the arginine finger and several neighboring residues (Fig. 1, C and D). The final region that is highly conserved is the surface within the tetramerization domains that forms the interface between neighboring subunits. Collectively, the conservation of residues in the active site and the protomer-protomer interface suggest a common mode of ATP hydrolysis and oligomerization throughout MgsA homologs. The interior cavity of the MgsA tetramer is poorly conserved and carries a weak positive charge.
AAA+ and Tetramerization Domains of E. coli MgsA
The N-terminal AAA+ domain includes both the ATP-binding core and a conserved helical lid element that are commonly found among AAA+ proteins. The ATP-binding core domain forms a RecA-like fold in which a central five-stranded parallel β-sheet is sandwiched by α-helices (Fig. 1A). The helical lid element is C-terminal to the ATP-binding core in the AAA+ domain. These first two domains are responsible for NTP binding and hydrolysis in AAA+ proteins (11, 13, 14). The ATP-binding and helical lid domains of MgsA overlay with the related domains from the E. coli clamp loader with root mean square deviations of 2.9 Å over 151 residues and 1.7 Å over 75 residues, respectively, demonstrating the considerable structural conservation within the AAA+ family.
The C-terminal tetramerization domain is composed of several α-helices that pack alongside neighboring subunits to mediate MgsA oligomerization. Overall, the oligomerization domains form a tightly bound collar, and the N-terminal AAA+ domains extend out from the collar in a splayed out arrangement, similar to the architecture seen in the inactive state of the E. coli clamp loader (68). The most striking differences between the structure of MgsA and the clamp loader complexes are that MgsA assembles as a tetramer as opposed to the pentameric arrangement observed for the clamp loaders and that the collar domain of MgsA forms a closed oligomer rather than the open arrangement of the clamp loader complex. Comparison of the MgsA structure to that of the E. coli clamp loader reveals an additional ∼100 residues on the C terminus of MgsA, not present in the clamp loader, that make extensive contact with the neighboring protomer and drive formation of a tetramer (Fig. 2).
FIGURE 2.

Comparison of MgsA to the bacterial clamp loader. A, overlay of the oligomerization domain of the gamma protein of the E. coli clamp loader (Protein Data Bank 1JR3, chain B) (gray) aligned with the oligomerization domain of MgsA demonstrates that MgsA possesses an additional ∼100 C-terminal residues not present in the clamp loader. B, additional ∼100 residues on the C terminus of MgsA (colored in black) wrap around the neighboring protomer to form an extensive interface. C, Arg finger residues (Arg-156) of neighboring protomers are shown as gray sticks demonstrating the positioning relative to the neighboring ATP-binding site (identified by the phosphate ions, shown as red spheres).
MgsA ATPase Active Site Architecture
The ATPase active sites in AAA+ proteins are formed at the interface between neighboring subunits, and the elements that contribute to nucleotide binding and hydrolysis are both well conserved and well characterized for several family members (reviewed in Refs. 13, 18, 69). In the MgsA structure, phosphate ions were found in proximity to all four of the ATP-binding sites (Fig. 1B), suggesting that the protein most likely catalyzes ATP hydrolysis in a manner that is similar to that observed with other AAA+ proteins. The binding sites are essentially identical in all four subunits and are in open positions.
The P loop (or Walker-A motif), located on the loop following β1, is a key ATP-binding and hydrolysis element of AAA+ and related proteins (70). The consensus sequence of the P loop includes a conserved lysine residue that contacts the γ-phosphate of ATP and is essential for ATP hydrolysis in AAA+ proteins (11, 13, 14). The conserved Lys-63 residue in MgsA appears to perform this function, as demonstrated by mutational analysis of the yeast Mgs1 homolog (27). Consistent with this role in ATP binding/hydrolysis, the ϵ-nitrogen of Lys-63 is positioned ∼4 Å from the phosphate ions in each of the MgsA protomers (Fig. 3). Additionally, backbone amide groups from other residues within the MgsA P loop (Gly-60, Thr-61, Gly-62, and Lys-63) are directed toward the phosphate ion, creating a basic pocket that we predict would accommodate the triphosphate of ATP.
FIGURE 3.

MgsA active site architecture. Conserved elements of the MgsA active site are labeled, and key residues are shown as sticks. The distances between the phosphate ion and Lys-63 residue (Walker A) or Arg-209 (sensor II) are shown. W-A, Walker-A; W-B, Walker-B; S-I, sensor-I; S-II, sensor-II.
The metal-binding Walker-B motif is a second integral and well characterized element of ATPases (70). Located at the apex of β3, the MgsA Walker-B motif contains two conserved acidic residues, Asp-113 and Glu-114 (Fig. 3). The R-group of Asp-113 is located ∼6 Å from the active site phosphate, consistent with the placement of the analogous residue in the S. cerevisiae clamp loader structure (25). This distal positioning would allow it to coordinate Mg2+ situated next to the β- and γ-phosphates of ATP (18). The acidic R-group of Glu-114 is also directed toward the phosphate ion and the active site, putting it in position to prime a water molecule for nucleophilic attack (71).
The sensor 1 and 2 elements are additional conserved elements in AAA+ proteins that contact the nucleotide and mediate structural changes throughout the protein in response to hydrolysis (72). The sensor 1 region, located at the apex of β4, sits between the P loop and the Walker-B motif (Fig. 3). Conserved polar residues in this region have been implicated in ATP hydrolysis in AAA+ proteins (73, 74). There are two Thr residues in the sensor 1 region of MgsA; in the crystal structure, the R group of Thr-142 is directed toward the active site, whereas the neighboring Thr-141 is directed away from the active site. The sensor 2 region is located in the α-helical bundle and packs against the P loop (Fig. 3). An Arg in the region, Arg-209 (conserved in clamp loaders (72)), binds the phosphate ion, indicating that it is in a good location for sensing nucleotide binding/hydrolysis and transmitting conformational changes. Sequence changes in this Arg are known to lead to diminished ATP binding (75, 76) in the AAA+ proteins NtrC and PspF and ATP hydrolysis in the AAA+ protein DnaA (77). The effects of mutating the Thr-141 and Arg-209 residues in MgsA have not yet been studied, but the presented structure suggests roles for the residues in ATP binding and/or hydrolysis.
The Arg finger is a highly conserved residue in AAA+ proteins that makes contact with the γ-phosphate of ATP in the neighboring subunit and is an essential element of the active site (69). Interestingly, the Arg finger of MgsA, Arg-156, is positioned ∼40 Å from the active site of the neighboring protomer, suggesting that the presented MgsA structure represents a form of the molecule that is not competent for ATPase activity (Fig. 2C). To confirm that Arg-156 is in fact required for activity, an MgsA variant protein in which the residue was changed to an Ala was purified and tested. The R156A variant is ATPase-deficient (described further below), consistent with the prediction that Arg-156 functions as the Arg finger in MgsA. We hypothesize that ATP binding induces structural changes that positions the Arg finger near the nucleotide of the adjacent protomer and generates a functional active site. Based on results presented below, we further predict that DNA binding assists this conformational rearrangement. This is similar to what has been observed with S. cerevisiae clamp loader protein (25). In that case, the AAA+ modules form tighter interfacial contacts when bound to its cognate processivity clamp and nonhydrolyzable nucleotide, and the active site residues are hypothesized to be optimally positioned for ATP hydrolysis. The two subdomains of AAA+ proteins, the ATP-binding and lid domains, tend to be further apart in the NTP-free state and closer together in the nucleotide-bound state (13). In the S. cerevisiae clamp loader structure, the two subdomains are rotated close together to create a tight nucleotide-binding site. In MgsA, a rotation of the ATP-binding core toward the lid would tighten the interface between the AAA+ modules and may trigger positioning of the Arg finger of one protomer near the active site of the adjacent protomer (Fig. 2C). A similar ATP-dependent interdomain pivoting has been described for the SV40 T-antigen AAA+ protein (15).
MgsA Physically Interacts with SSB
Cellular binding partners that are thought to target eukaryotic Mgs1 and Wrnip1 proteins to sites of DNA replication are known (35, 41, 42), but similar studies have not been carried out with bacterial MgsA. To better understand the functions of MgsA, TAP (78) was used to identify cellular interaction partners of E. coli MgsA. An N-terminal TAP-tagged MgsA protein was expressed in E. coli to facilitate complex formation with its cellular protein partners. A complex including TAP-MgsA and associated proteins was purified by TAP, and the purified components were resolved via SDS-PAGE (Fig. 4). Distinct bands were apparent on the gel, and each was excised and identified by mass spectrometry. Two different proteins were present in the preparations as follows: MgsA and the SSB. The other bands are degradation products of MgsA or SSB. MgsA was also identified in a reciprocal experiment with an N-terminal TAP-tagged SSB but, interestingly, was not identified in an earlier study using a C-terminal TAP-tagged SSB (79). These results show that MgsA and SSB proteins interact in a manner that could require the native SSB C-terminal structure.
FIGURE 4.
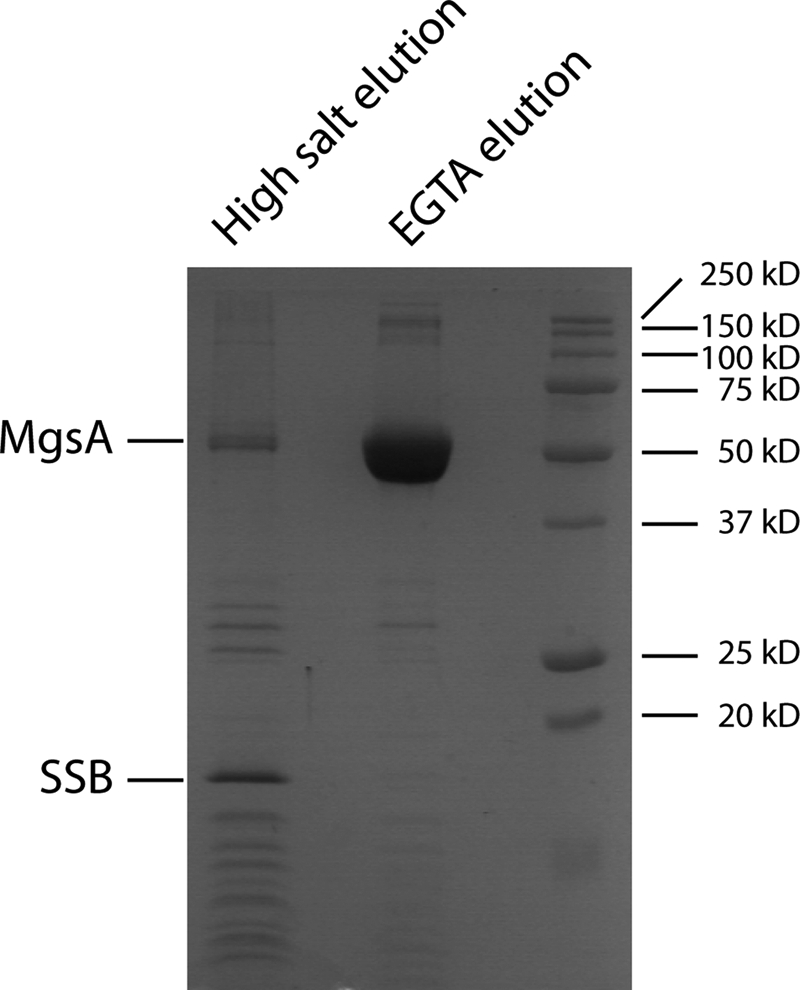
MgsA physically interacts with E. coli SSB in vivo. SDS-PAGE of TAP-MgsA and co-purifying proteins. SSB and MgsA were confirmed by mass spectrometry. Proteins were eluted by a high salt elution followed by an EGTA elution (see under “Experimental Procedures”).
E. coli SSB is composed of two functional domains as follows: an N-terminal DNA-binding domain and a structurally dynamic C-terminal tail ending in a highly conserved amphipathic sequence (SSB-Ct) that mediates interactions with diverse genome maintenance proteins (80). A biochemical approach using SPR was used to examine the interaction between MgsA and SSB interaction and to determine whether this interaction depended upon the SSB-Ct. SSB was flowed over a biotinylated dT35 ssDNA bound to an SPR sensor chip. Under the conditions tested, SSB binding to the ssDNA appears irreversible as evidenced by the stable signal when buffer alone is flowed over the chip. MgsA was subsequently flowed over the SSB/ssDNA substrate, and binding was monitored by the change in response units. Analysis of binding experiments with different MgsA concentration confirmed its interaction with SSB and showed that the MgsA-SSB-ssDNA complex has an apparent dissociation constant (Kd) of 0.36 ± 0.02 μm (Fig. 5). This complex stability is consistent with the Kd range expected from other SSB-protein interactions (80).
FIGURE 5.

MgsA-SSB interaction is largely dependent on the SSB C-terminal tail. Surface plasmon resonance was used to study the MgsA-SSB interaction. Equilibrium response units were plotted versus MgsA concentrations and fitted to hyperbolic binding curves.
SSB variants with altered SSB-Ct sequences were substituted in the SPR assay to test the predicted importance of the SSB-Ct for the MgsA-SSB interaction. The E. coli SSB-Ct sequence (Asp-Phe-Asp-Asp-Asp-Ile-Pro-Phe) is highly conserved among bacteria with the C-terminally most Phe being invariant (80). This conservation reflects the importance of the residues in the SSB-Ct in protein interactions. The variants tested in the MgsA interaction assay were SSBΔC8 (lacks the entire SSB-Ct), SSBΔC1 (lacks the C-terminally most SSB-Ct Phe), and SSB-mixed (final eight residues are rearranged). The most severe binding defect was seen with the SSBΔC8 protein. Substituting SSBΔC8 for wild-type SBB weakened the affinity of MgsA for SSB by ∼20-fold, although the overall change in response units at the highest MgsA concentration was about one-fourth that observed with wild-type SSB. More modest binding defects were seen with SSBΔC1 and SSB-mixed. The SSBΔC1 variant had ∼4-fold weaker binding affinity for MgsA indicating that the C-terminally most residue is important in the MgsA-SSB interaction. Similarly, substituting the SSB-mixed protein weakened the MgsA-SSB interaction by ∼7-fold. Collectively, these data demonstrate the importance of the SSB-Ct for the MgsA-SSB interaction.
MgsA ATPase Activity
The yeast homolog of MgsA, Mgs1, was previously shown to be a DNA-dependent ATPase (27). E. coli MgsA was therefore tested for ATPase activity. In the absence of DNA, MgsA hydrolyzed ATP at a very low level (3.39 ± 0.29 min−1) (Fig. 6A). Addition of M13mp18 circular ssDNA to the reactions strongly stimulated MgsA ATPase activity in a DNA concentration-dependent manner (up to 70.6 ± 4.4 min−1 with 20 μm circular ssDNA) (Fig. 6B). This stimulation of ATPase appeared to level out at a ratio of ∼600–1000 nucleotides of DNA per MgsA tetramer.
FIGURE 6.

MgsA DNA-dependent ATP hydrolysis. A, DNA-independent ATPase activity of MgsA was measured at various MgsA concentrations indicated in the panel. B, MgsA or MgsA R156A (125 nm) was added to reactions with the indicated circular single-stranded DNA concentration. C and D, indicated concentration of E. coli SSB or SSBΔC8 was added to reactions containing 20 μm nucleotides DNA prior to adding 125 nm MgsA.
The effect of SSB on MgsA DNA-dependent ATP hydrolysis was investigated. When SSB was preincubated with the DNA prior to MgsA addition, low concentrations of SSB appeared to have little effect on ATP hydrolysis, whereas higher SSB concentrations (approaching the levels needed to saturate the circular ssDNA) inhibited ATP hydrolysis (Fig. 6C). Supersaturating levels of SSB had little additional inhibitory effects, suggesting that the protein was indirectly inhibiting MgsA by competing for binding to the DNA substrate. Interestingly, when SSBΔC8 was used in the assay, ATP hydrolysis inhibition was apparent at lower SSB concentrations than observed with wild-type SSB (Fig. 6D). This could be due to the higher affinity binding by SSBΔC8 compared with full-length SSB (81) and/or due to the impaired interaction between MgsA and SSBΔC8.
DISCUSSION
Bacterial MgsA and its eukaryotic homologs, Mgs1 and Wrnip1, compose a family of AAA+ enzymes that function in cellular responses to replication fork stalling. How these proteins function and the identity of their substrates are not clear, but their importance in genome maintenance is well documented (27, 28, 34, 35, 82). To better understand the structure and function of MgsA/Mgs1/Wrnip1 family enzymes, we have determined the 2.5-Å resolution x-ray crystal structure of E. coli MgsA. The structure reveals a homotetrameric structure for MgsA that is distinct from other clamp loader clade AAA+ proteins, which function as heteropentamers. The arrangement of the MgsA AAA+ domains does not appear to be competent for catalysis, because the conserved Arg finger elements are not adjacent to the ATPase active sites. This implies that a significant conformational rearrangement takes place upon substrate binding to facilitate formation of an active ATPase site between AAA+ domains. MgsA is shown to have DNA-stimulated ATPase activity and to physically associate with SSB. Interactions between MgsA and SSB appear to be mediated predominantly by the C terminus of SSB, consistent with observations with other SSB-interacting proteins (80). Interaction between MgsA and SSB could help localize MgsA to replication forks, as has been observed in E. coli (32, 33).
The domain architecture of MgsA shares similarity with the clamp loader proteins from bacteria and eukaryotes. In each of these proteins, C-terminal oligomerization domains mediate formation of a collar domain that is decorated with AAA+ domains extending away from the collar. However, the two groups of proteins differ in their oligomeric state (tetramer for MgsA versus pentamer the clamp loaders) in a manner that appears to be dictated by the oligomerization domain. Comparing the structures of MgsA and the E. coli clamp loader shows that more surface area is buried between the subunits in the MgsA than in the clamp loader. Much of this additional area is supplied by an ∼100-residue C-terminal shelf that is present in MgsA but lacking from the clamp loader (Fig. 2A). Nonetheless, the overall similarity among the clamp loader clade proteins suggests that MgsA could possibly perform a clamp loader-like function, loading and/or unloading proteins onto DNA. The precise substrate(s) upon which MgsA acts remains unclear. Efforts to identify an interaction between MgsA and β, the processivity clamp substrate for the bacterial clamp loader, have thus far not identified an interaction (data not shown). Future studies will be required to determine whether MgsA acts upon protein substrates.
AAA+ proteins couple the chemical energy of ATP hydrolysis to the mechanical energy necessary to drive conformational changes within the enzyme that remodel the structures of macromolecular targets (13). A complete understanding of the structural mechanisms governing AAA+ enzyme function therefore requires determination of multiple structural states. Comparing the structure of MgsA presented here to other AAA+ proteins, the arrangement of the AAA+ domains suggests that an inactive conformation of MgsA was captured under our crystallization conditions. However, the local arrangement of the Walker A, Walker B, sensor 1, and sensor 2 motifs in MgsA are all in reasonable positions for ATP binding. This suggests that rearrangement of the AAA+ domains to properly localize the Arg finger elements to the ATP-binding site could be a trigger for ATP hydrolysis. The presented structure provides a physical model for comparison with other AAA+ proteins that could help define the conformational changes that must accompany catalytic activation and defines a structure with which to compare future substrate-bound structures of MgsA. The DNA-stimulated nature of MgsA ATPase activity suggests that association with DNA could be an important molecular cue that induces formation of an active arrangement of AAA+ domains within the MgsA tetramer.
The interaction between MgsA and SSB provides a possible mechanism by which MgsA localizes to sites of DNA replication in cells. Several other genome maintenance proteins utilize interactions with SSB for proper localization and/or biochemical activation (80). Given MgsA's noted cellular localization to sites of replication (32, 33), identification of an interaction between the two proteins is consistent with a similar role for SSB with MgsA. It is not clear whether this interaction functions solely to localize MgsA. Interaction could facilitate MgsA activity or, alternatively, SSB/DNA structures could possibly be substrates upon which MgsA acts. Future experiments will be required to address the importance of the MgsA-SSB interaction on MgsA biochemical activities.
Acknowledgments
We thank Elizabeth A. Wood for construction of the MgsA-expressing plasmid pEAW354 and Darryl R. McCaslin for assistance with the execution and analysis of the analytical ultracentrifugation experiments.
Addendum
While our manuscript was being reviewed, Costes et al. (65) identified a physical interaction between the MgsA and SSB proteins of Bacillus subtilis.
This work was supported, in whole or in part, by National Institutes of Health Grant GM32335 from NIGMS (to M. M. C.) and Grant GM068061 (to J. L. K.).
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (code 3PVS) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
C. Norais and M. M. Cox, unpublished data.
- SSB
- single strand DNA-binding protein
- Ct
- C-terminal tail
- RU
- response unit
- TAP
- tandem-affinity purification
- PRR
- post-replication repair
- SPR
- surface plasmon resonance
- ssDNA
- single-stranded DNA.
REFERENCES
- 1. Cox M. M. (2002) Mutat. Res. 510, 107–120 [DOI] [PubMed] [Google Scholar]
- 2. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., Marians K. J. (2000) Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 3. Kuzminov A. (1999) Microbiol. Mol. Biol. Rev. 63, 751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kowalczykowski S. C. (2000) Trends Biochem. Sci. 25, 156–165 [DOI] [PubMed] [Google Scholar]
- 5. Marians K. J. (2000) Trends Biochem. Sci. 25, 185–189 [DOI] [PubMed] [Google Scholar]
- 6. Michel B., Grompone G., Florès M. J., Bidnenko V. (2004) Proc. Natl. Acad. Sci. U.S.A 101, 12783–12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barre F. X., Søballe B., Michel B., Aroyo M., Robertson M., Sherratt D. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8189–8195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neuwald A. F., Aravind L., Spouge J. L., Koonin E. V. (1999) Genome Res. 9, 27–43 [PubMed] [Google Scholar]
- 9. Davey M. J., Jeruzalmi D., Kuriyan J., O'Donnell M. (2002) Nat. Rev. Mol. Cell Biol. 3, 826–835 [DOI] [PubMed] [Google Scholar]
- 10. Brunger A. T., DeLaBarre B. (2003) FEBS Lett. 555, 126–133 [DOI] [PubMed] [Google Scholar]
- 11. Duderstadt K. E., Berger J. M. (2008) Crit. Rev. Biochem. Mol. Biol. 43, 163–187 [DOI] [PubMed] [Google Scholar]
- 12. Erzberger J. P., Mott M. L., Berger J. M. (2006) Nat. Struct. Mol. Biol. 13, 676–683 [DOI] [PubMed] [Google Scholar]
- 13. Hanson P. I., Whiteheart S. W. (2005) Nat. Rev. Mol. Cell Biol. 6, 519–529 [DOI] [PubMed] [Google Scholar]
- 14. Erzberger J. P., Berger J. M. (2006) Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 [DOI] [PubMed] [Google Scholar]
- 15. Gai D., Zhao R., Li D., Finkielstein C. V., Chen X. S. (2004) Cell 119, 47–60 [DOI] [PubMed] [Google Scholar]
- 16. Wang J., Song J. J., Franklin M. C., Kamtekar S., Im Y. J., Rho S. H., Seong I. S., Lee C. S., Chung C. H., Eom S. H. (2001) Structure 9, 177–184 [DOI] [PubMed] [Google Scholar]
- 17. Ammelburg M., Frickey T., Lupas A. N. (2006) J. Struct. Biol. 156, 2–11 [DOI] [PubMed] [Google Scholar]
- 18. Iyer L. M., Leipe D. D., Koonin E. V., Aravind L. (2004) J. Struct. Biol. 146, 11–31 [DOI] [PubMed] [Google Scholar]
- 19. Bowman G. D., Goedken E. R., Kazmirski S. L., O'Donnell M., Kuriyan J. (2005) FEBS Lett. 579, 863–867 [DOI] [PubMed] [Google Scholar]
- 20. Yao N. Y., Johnson A., Bowman G. D., Kuriyan J., O'Donnell M. (2006) J. Biol. Chem. 281, 17528–17539 [DOI] [PubMed] [Google Scholar]
- 21. Turner J., Hingorani M. M., Kelman Z., O'Donnell M. (1999) EMBO J. 18, 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Donnell M., Kuriyan J. (2006) Curr. Opin. Struct. Biol. 16, 35–41 [DOI] [PubMed] [Google Scholar]
- 23. O'Donnell M. (2006) J. Biol. Chem. 281, 10653–10656 [DOI] [PubMed] [Google Scholar]
- 24. Jeruzalmi D., O'Donnell M., Kuriyan J. (2001) Cell 106, 429–441 [DOI] [PubMed] [Google Scholar]
- 25. Bowman G. D., O'Donnell M., Kuriyan J. (2004) Nature 429, 724–730 [DOI] [PubMed] [Google Scholar]
- 26. Lestini R., Michel B. (2007) EMBO J. 26, 3804–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hishida T., Iwasaki H., Ohno T., Morishita T., Shinagawa H. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8283–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shibata T., Hishida T., Kubota Y., Han Y. W., Iwasaki H., Shinagawa H. (2005) Genes Cells 10, 181–191 [DOI] [PubMed] [Google Scholar]
- 29. Kelman Z., O'Donnell M. (1995) Annu. Rev. Biochem. 64, 171–200 [DOI] [PubMed] [Google Scholar]
- 30. Vandewiele D., Fernández de Henestrosa A. R., Timms A. R., Bridges B. A., Woodgate R. (2002) Mutat. Res. 499, 85–95 [DOI] [PubMed] [Google Scholar]
- 31. Hishida T., Han Y. W., Shibata T., Kubota Y., Ishino Y., Iwasaki H., Shinagawa H. (2004) Genes Dev. 18, 1886–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lau I. F., Filipe S. R., Søballe B., Økstad O. A., Barre F. X., Sherratt D. J. (2003) Mol. Microbiol. 49, 731–743 [DOI] [PubMed] [Google Scholar]
- 33. Sherratt D. J., Soballe B., Barre F. X., Filipe S., Lau I., Massey T., Yates J. (2004) Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hishida T., Ohno T., Iwasaki H., Shinagawa H. (2002) EMBO J. 21, 2019–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hishida T., Ohya T., Kubota Y., Kamada Y., Shinagawa H. (2006) Mol. Cell. Biol. 26, 5509–5517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoege C., Pfander B., Moldovan G. L., Pyrowolakis G., Jentsch S. (2002) Nature 419, 135–141 [DOI] [PubMed] [Google Scholar]
- 37. Watts F. Z. (2006) DNA Repair 5, 399–403 [DOI] [PubMed] [Google Scholar]
- 38. Pfander B., Moldovan G. L., Sacher M., Hoege C., Jentsch S. (2005) Nature 436, 428–433 [DOI] [PubMed] [Google Scholar]
- 39. Papouli E., Chen S., Davies A. A., Huttner D., Krejci L., Sung P., Ulrich H. D. (2005) Mol. Cell 19, 123–133 [DOI] [PubMed] [Google Scholar]
- 40. Maga G., Hubscher U. (2003) J. Cell Sci. 116, 3051–3060 [DOI] [PubMed] [Google Scholar]
- 41. Crosetto N., Bienko M., Hibbert R. G., Perica T., Ambrogio C., Kensche T., Hofmann K., Sixma T. K., Dikic I. (2008) J. Biol. Chem. 283, 35173–35185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bish R. A., Myers M. P. (2007) J. Biol. Chem. 282, 23184–23193 [DOI] [PubMed] [Google Scholar]
- 43. Bienko M., Green C. M., Crosetto N., Rudolf F., Zapart G., Coull B., Kannouche P., Wider G., Peter M., Lehmann A. R., Hofmann K., Dikic I. (2005) Science 310, 1821–1824 [DOI] [PubMed] [Google Scholar]
- 44. Guo C., Tang T. S., Bienko M., Parker J. L., Bielen A. B., Sonoda E., Takeda S., Ulrich H. D., Dikic I., Friedberg E. C. (2006) Mol. Cell. Biol. 26, 8892–8900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M. (2004) EMBO J. 23, 3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakajima S., Lan L., Kanno S., Usami N., Kobayashi K., Mori M., Shiomi T., Yasui A. (2006) J. Biol. Chem. 281, 34687–34695 [DOI] [PubMed] [Google Scholar]
- 47. Haracska L., Torres-Ramos C. A., Johnson R. E., Prakash S., Prakash L. (2004) Mol. Cell. Biol. 24, 4267–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Haruta N., Yu X., Yang S., Egelman E. H., Cox M. M. (2003) J. Biol. Chem. 278, 52710–52723 [DOI] [PubMed] [Google Scholar]
- 49. Neuendorf S. K., Cox M. M. (1986) J. Biol. Chem. 261, 8276–8282 [PubMed] [Google Scholar]
- 50. Mertens N., Remaut E., Fiers W. (1995) Bio/Technology 13, 175–179 [DOI] [PubMed] [Google Scholar]
- 51. Edelhoch H. (1967) Biochemistry 6, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 52. Shan Q., Cox M. M., Inman R. B. (1996) J. Biol. Chem. 271, 5712–5724 [DOI] [PubMed] [Google Scholar]
- 53. Hobbs M. D., Sakai A., Cox M. M. (2007) J. Biol. Chem. 282, 11058–11067 [DOI] [PubMed] [Google Scholar]
- 54. Shereda R. D., Reiter N. J., Butcher S. E., Keck J. L. (2009) J. Mol. Biol. 386, 612–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu D., Keck J. L. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 9169–9174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lohman T. M., Overman L. B. (1985) J. Biol. Chem. 260, 3594–3603 [PubMed] [Google Scholar]
- 57. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 58. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 60. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 61. Laue T. M., Shah B. D. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., Horton J. C. eds) pp. 90–125, Royal Society of Chemistry, Cambridge [Google Scholar]
- 62. Laue T. M. (1995) Methods Enzymol. 259, 61–69 [DOI] [PubMed] [Google Scholar]
- 63. Morrical S. W., Lee J., Cox M. M. (1986) Biochemistry 25, 1482–1494 [DOI] [PubMed] [Google Scholar]
- 64. Lindsley J. E., Cox M. M. (1990) J. Biol. Chem. 265, 9043–9054 [PubMed] [Google Scholar]
- 65. Costes A., Lecointe F., McGovern S., Quevillon-Cheruel S., Polard P. (2010) PLoS Genet. 6, e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tsurimoto T., Shinozaki A., Yano M., Seki M., Enomoto T. (2005) Genes Cells 10, 13–22 [DOI] [PubMed] [Google Scholar]
- 67. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kazmirski S. L., Podobnik M., Weitze T. F., O'Donnell M., Kuriyan J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16750–16755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ogura T., Whiteheart S. W., Wilkinson A. J. (2004) J. Struct. Biol. 146, 106–112 [DOI] [PubMed] [Google Scholar]
- 70. Walker J. E., Saraste M., Runswick M. J., Gay N. J. (1982) EMBO J. 1, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Story R. M., Steitz T. A. (1992) Nature 355, 374–376 [DOI] [PubMed] [Google Scholar]
- 72. Guenther B., Onrust R., Sali A., O'Donnell M., Kuriyan J. (1997) Cell 91, 335–345 [DOI] [PubMed] [Google Scholar]
- 73. Steel G. J., Harley C., Boyd A., Morgan A. (2000) Mol. Biol. Cell 11, 1345–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Karata K., Inagawa T., Wilkinson A. J., Tatsuta T., Ogura T. (1999) J. Biol. Chem. 274, 26225–26232 [DOI] [PubMed] [Google Scholar]
- 75. Lew C. M., Gralla J. D. (2002) J. Biol. Chem. 277, 41517–41524 [DOI] [PubMed] [Google Scholar]
- 76. Rombel I., Peters-Wendisch P., Mesecar A., Thorgeirsson T., Shin Y. K., Kustu S. (1999) J. Bacteriol. 181, 4628–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nishida S., Fujimitsu K., Sekimizu K., Ohmura T., Ueda T., Katayama T. (2002) J. Biol. Chem. 277, 14986–14995 [DOI] [PubMed] [Google Scholar]
- 78. Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. (1999) Nat. Biotechnol. 17, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 79. Butland G., Peregrín-Alvarez J. M., Li J., Yang W., Yang X., Canadien V., Starostine A., Richards D., Beattie B., Krogan N., Davey M., Parkinson J., Greenblatt J., Emili A. (2005) Nature 433, 531–537 [DOI] [PubMed] [Google Scholar]
- 80. Shereda R. D., Kozlov A. G., Lohman T. M., Cox M. M., Keck J. L. (2008) Crit. Rev. Biochem. Mol. Biol. 43, 289–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kozlov A. G., Cox M. M., Lohman T. M. (2010) J. Biol. Chem. 285, 17246–17252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yoshimura A., Seki M., Kanamori M., Tateishi S., Tsurimoto T., Tada S., Enomoto T. (2009) Genes Genet. Syst. 84, 171–178 [DOI] [PubMed] [Google Scholar]