

Abstract
Dual oxidase 2 is a member of the NADPH oxidase (Nox) gene family that plays a critical role in the biosynthesis of thyroid hormone as well as in the inflammatory response of the upper airway mucosa and in wound healing, presumably through its ability to generate reactive oxygen species, including H2O2. The recently discovered overexpression of Duox2 in gastrointestinal malignancies, as well as our limited understanding of the regulation of Duox2 expression, led us to examine the effect of cytokines and growth factors on Duox2 in human tumor cells. We found that exposure of human pancreatic cancer cells to IFN-γ (but not other agents) produced a profound up-regulation of the expression of Duox2, and its cognate maturation factor DuoxA2, but not other members of the Nox family. Furthermore, increased Duox2/DuoxA2 expression was closely associated with a significant increase in the production of both intracellular reactive oxygen species and extracellular H2O2. Examination of IFN-γ-mediated signaling events demonstrated that in addition to the canonical Jak-Stat1 pathway, IFN-γ activated the p38-MAPK pathway in pancreatic cancer cells, and both played an important role in the induction of Duox2 by IFN-γ. Duox2 up-regulation following IFN-γ exposure is also directly associated with the binding of Stat1 to elements of the Duox2 promoter. Our findings suggest that the pro-inflammatory cytokine IFN-γ initiates a Duox2-mediated reactive oxygen cascade in human pancreatic cancer cells; reactive oxygen species production in this setting could contribute to the pathophysiologic characteristics of these tumors.
Keywords: Cytokine Action, Oxidase, Oxidative Stress, Reactive Oxygen Species (ROS), STAT Transcription Factor, Cancer, Dual Oxidase 2, Dual OxidaseA2, Gastrointestinal Track
Introduction
It has been suggested that pancreatic inflammation accelerates the development and progression of pancreatic cancer, possibly due to cytokine release and generation of reactive oxygen species (ROS)2 (1–4). Recently, pancreatic tumor cells of epithelial origin have been shown to produce appreciably higher concentrations of ROS than their normal cellular counterparts (5). Thus, during recurring episodes of chronic pancreatitis, cytokine-related ROS production could both increase genomic instability, as well as diminish the tumor suppressor function of serine/threonine and tyrosine phosphatases that modulate malignant transformation and cellular proliferation (6). Although at high concentrations ROS can be cytotoxic, physiologic ROS levels play a critical role in the control of tumor cell growth, differentiation, and signal transduction (7). Although ROS are primarily a by-product of aerobic metabolism in mammalian cells, the recently discovered family of epithelial NADPH oxidases (reduced nicotinamide adenine dinucleotide phosphate oxidases (Noxs)), contributes significantly to intracellular ROS production in tumor cells (8, 9).
Dual oxidase 2 (Duox2) belongs to the Nox family of oxidases that is composed of six additional enzymes, including Nox1–5 and dual oxidase 1 (Duox1) (10). Duox2 was originally identified in the thyroid as the H2O2-generating enzyme responsible for thyroid hormone biosynthesis (11). Very recently, Duox2 also has been found in upper airway mucosa, ducts of the salivary gland, and throughout the gastrointestinal tract (7, 12, 13). Duox2 is a membrane glycoprotein with a unique N-terminal heme peroxidase-like domain as well as a membrane-spanning NAD(P)H oxidase domain, with an additional cytosolic segment containing two calcium-binding domains that generate H2O2 in the extracellular milieu (14, 15). The associated dual oxidase maturation factor 2 (DuoxA2), an endoplasmic reticulum-resident protein, is absolutely necessary for post-translational processing and translocation of Duox2 to the plasma membrane to initiate functional enzymatic activity (16). In addition to its role in thyroid hormone biosynthesis, other suggested functions for Duox2 include host defense, promotion of wound healing, and maintenance of airway homeostasis (17–21). Clarification of the contribution of Duox2 to pancreatic cancer could help to identify a novel molecular target for early intervention in this condition.
Interferon-γ (IFN-γ) is a soluble cytokine that plays an important role in innate and adoptive immunity against viral and intracellular bacterial targets (22). IFN-γ also inhibits tumor cell growth by activating distinct cellular signaling programs leading to transcriptional control over a large numbers of genes. It has been documented that IFN-γ can stimulate ROS production in human neutrophils, monocytes, and macrophages through transcriptional up-regulation of the gp91phox and p67phox subunits of the multicomponent NADPH oxidase complex (23). Recent studies have also demonstrated that IFN-γ up-regulates the expression of Duox2 in papilloma virus-immortalized human tracheobronchial epithelial cells (13, 18). In the studies reported here, we have addressed specific biological effects of IFN-γ in the context of Duox2-mediated reactive oxygen production in human pancreatic tumor cells.
IFN-γ binding to its cell surface receptors IFNγR1 and IFNγR2 results in oligomerization of the receptors and activation of the receptor-associated tyrosine kinases, Jak1 and Jak2 (24). Upon phosphorylation by Jak1, Stat1 transduces the signal into transcriptional events. Phosphorylation of the tyrosine 701 moiety of Stat1 enables Stat1 to form homodimers; translocation of the complex to the nucleus and subsequent binding to the GAS promoter element leads to an increased expression of IFN-γ-stimulated genes (25). Besides the tyrosine 701 moiety of Stat1, phosphorylation of Ser727 on Stat1 is also necessary for its full transcriptional activity (26).
Recent studies have shown that IFN-γ signaling also activates several other kinases in addition to the Jaks. For example, IFN-γ engagement with its cell surface receptors can activate phosphoinositide 3-kinase (PI3K) and the mitogen-activated protein kinase (MAPK) cascades (27). Through MAPK, IFN-γ further activates p38 mitogen-activated protein kinase (p38-MAPK) to induce gene transcription. The activation of these signaling pathways can lead to phosphorylation of Stat1 on Ser727, increasing its transcriptional efficacy and the phosphorylation of other substrates, resulting in stimulation of alternate signal transduction cascades that support Stat1-dependent gene transcription (25).
Because of the proposed growth stimulatory and pro-inflammatory roles of Duox2, we investigated whether pro-inflammatory cytokines or growth factors could affect Duox2 activity. Among several pancreatic cancer lines evaluated, BxPC-3 and AsPC-1 cells are specifically responsive to IFN-γ for the induction of both Duox2 and DuoxA2 mRNA, in a time-dependent manner. Assays for both intra- and extracellular ROS production confirmed that IFN-γ-induced Duox2 and DuoxA2 mRNA expression correlates with Duox2 enzymatic activity in BxPC-3 cells. We hypothesized that Duox2-dependent ROS could play an important role in cancer cell signal transduction, as well as inflammation. Thus, in this article, we examined IFN-γ-mediated signaling events that could regulate Duox2 expression. Our experiments revealed that in addition to the canonical Jak-Stat1 pathway, IFN-γ also activates the p38 MAPK pathway to sustain Ser727 phosphorylation of Stat1. Pharmacological inhibition, as well as genetic manipulation, of p38 MAPK activity abrogated Ser727 phosphorylation of Stat1 and down-regulated Duox2 expression by IFN-γ, suggesting that p38 MAPK is upstream of Stat1. Hence, p38 MAPK activation by IFN-γ appears to play an important role in IFN-γ-induced Duox2 expression in BxPC-3 cells.
In summary, our results suggest that stimulation of the Stat1 pathway by IFN-γ-mediated up-regulation of Duox2 may play an important role in the generation of high local concentrations of extracellular ROS that could contribute to a pro-inflammatory milieu in the pancreas. Considering the essential role ROS play in mediating cell signaling, cell proliferation, survival, and apoptosis, our findings may help to identify a pathway that plays a critical role in the pathologic behavior of human pancreatic tumor cells.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Recombinant human cytokines IFN-γ, IFN-α, IFN-β, TNF-α, IL-6, and growth factors EGF, HGF, and TGF-β were purchased from R & D Systems. Cytokines and growth factors were dissolved in phosphate-buffered saline (PBS) with 1% bovine serum albumin (BSA) and added directly to the culture medium. For untreated control conditions, the same amount of PBS, 1% BSA, without cytokine or growth factor, was added to the medium. Diphenyleneiodonium chloride (catalog number D-2926), cycloheximide (catalog number C-7968), actinomycin D (catalog number A-9415), and human β-actin antibody (catalog number A3853) were purchased from Sigma; ionomycin (catalog number 407952), U0126 (catalog number 662005), SB203580 (catalog number 559389), AG490 (catalog number 658401), and BAPTA-AM (catalog number 196419) were from EMD Chemicals; IRF-1 (sc-497), Stat1 p84/p91 (sc-346), IRF-2 (catalog number sc-101069), and IRF-3 (catalog number sc-33641) antibodies were from Santa Cruz Biotechnology; p-Stat1(Ser727) (catalog number 9177S), Stat3 (catalog number 9132), p-Stat3(Tyr705) (catalog number 9131S), phospho-p38 MAPK(Thr180/Tyr182) (catalog number 9215L), p-Stat1(Tyr701) (catalog number 9167L), p38 MAPK (catalog number 9212), phospho-p44/42 MAPK (Thr202/Tyr204) (catalog number 9101L), and p44/42 MAPK (catalog number 9102) antibodies were from Cell Signaling Technology. Stealth RNAi Negative Control Duplexes (catalog number 12935-100), StealthTM siRNA duplex for Duox2 (catalog number Duox2-HSS121309), Stat1 validated Stealth RNAi DuoPak (catalog number 45-1698), and siRNA for IRF-1 (catalog number s7501) were from Invitrogen. The human Nox1–5 primers including Nox1 (catalog number Hs00246589), Nox2 (catalog number Hs00166163), Nox3 (catalog number Hs00210462), Nox4 (catalog number Hs00276431), Nox5 (catalog number Hs00225846), as well as the primers for human Duox1 (catalog number Hs00213694), Duox2 (catalog number Hs00204187_m1), and DuoxA2 (catalog number Hs01595310_m1), TaqMan Universal PCR mix (catalog number 4364340), human actin primer (catalog number Hs99999903_m1), human Stat1 primer (catalog number Hs00234829_m1), and the human IRF-1 primer (catalog number Hs00971959_m1) were purchased from Applied Biosystems. The QuikChIP Kit (catalog number 3010K) was from Imgenex. Mouse anti-human Duox2 monoclonal antibody S-40 was developed by Creative Biolabs, Port Jefferson Station, NY, using the following procedure. A 53-kDa protein corresponding to a human Duox2 131–540 amino acid fragment was expressed in Escherichia coli. The protein was purified and used as the antigen to produce a monoclonal antibody. Several approaches were used to validate the human Duox2 antibody. These included: (a) transient and stable transfection of HA-tagged human Duox2 into COS-7 and MIA-PaCa cells, with the performance of Western analysis using an antibody raised against the HA tag, as well as our Duox antibody; (b) the use of Duox2 siRNA silencing experiments in IFN-γ-treated BxPC-3 cells; these studies revealed that Duox protein and enzymatic activity corresponded very well with the level of Duox2 mRNA expression; and (c) we also performed immunofluorescence microscopy to demonstrate coincident cellular expression using an antibody against our HA tag and our Duox antibody in MIA-PaCa cells that were transfected with the HA-tagged human Duox2 expression vector. In the course of the experiments to validate this antibody, it was found to cross-react with Duox1. Thus, we have referred to the protein detected by our antibody as “Duox.” 2′,7′-Dichlorodihydrofluorescin diacetate (H2-DCF-DA) was obtained from Molecular Probes (Carlsbad, CA).
Cell Culture
The human pancreatic cancer cell lines BxPC-3 (catalog number CRL-1687), AsPC-1 (catalog number CRL-1682), MIA-PaCa (catalog number CRL-1420), and PANC-1 (catalog number CRL-1469), and two other epithelial cell lines, HeLa (catalog number CCL-2) and HT-29 (catalog number HTB-38), were purchased from ATCC, Manassas, VA. All cell lines were cultured using ATCC recommended medium with 10% FBS. For starvation conditions, cells were grown overnight in the same medium without FBS. We chose to use starvation conditions because although we observed the induction of Duox2 expression by IFN-γ under both serum-containing and serum-starved conditions, after starvation, the induction of Duox2 was stronger. Cells were cultured in a humidified incubator at 37 °C in an atmosphere of 5% CO2 in air.
RNA Extraction, cDNA Synthesis, and Quantitative Real Time PCR Assay
Total RNA was extracted from IFN-γ-treated or untreated BxPC-3 cells using the RNeasy Mini Kit (catalog number 74104 from Qiagen) following the manufacturer's instructions. Two μg of total RNA was used for cDNA synthesis using SuperScript II reverse transcriptase (catalog number 18080-044) and random primers (catalog number 48190-011, Invitrogen) in a 20-μl reaction system, with the following cycles: 25 °C for 5 min, 42 °C for 50 min, 75 °C for 5 min. After reaction, the RT-PCR products were diluted with diethyl pyrocarbonate/H2O to 100 μl for the real time PCR. Real time RT-PCR was performed on 384-well plates in a 20-μl reaction system containing 2 μl of diluted cDNA, 1 μl of primer mixture, 7 μl of H2O, and 10 μl of TaqMan 2× in reaction mixture. The PCR was carried out using the default cycling conditions, and fluorescence was detected with the ABI 7900HT sequence detection system (Applied Biosystems, Foster City, CA). Triplicate determinations were performed for each sample that was used for the real time PCR; the mean value was calculated, and the data in the final figures represents three independent experiments. Relative gene expression was calculated as the ratio of the target gene to the internal reference gene (β-actin) multiplied by 103 based on the Ct values.
Whole Cell Extract (WCE) Preparation and Western Analysis
Cell pellets from BxPC-3 cells treated with or without IFN-γ were lysed with 1× RIPA lysis buffer (catalog number 20–188, Millipore, Temecula, CA) with the addition of a phosphatase inhibitor tablet (Roche Applied Science, catalog number 04-906-837001) and a protease inhibitor tablet (Roche, catalog number 11-836-153001). The protein concentration of the WCE was measured using the BCA protein assay kit (Pierce). WCE was mixed with an equal volume of 2× SDS protein gel loading buffer (catalog number 351-082-661, Quality Biological); 50 μg of WCE was loaded onto a 4–20% Tris glycine gel (Invitrogen, catalog number EC6028), and the proteins were separated and electrophoretically transferred to nitrocellulose membranes using I Blot gel transfer stacks (Invitrogen, catalog number IB 3010-01). The membranes were blocked in 1× TBST buffer with 5% nonfat milk for 1 h at room temperature and then incubated with primary antibody overnight in TBST buffer. Membranes were washed three times in 1× TBST buffer and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature with shaking. The antigen-antibody complex was visualized with SuperSignal West Pico Luminol/Enhancer Solution (catalog number 1856136, Thermo Scientific). To evaluate Duox2 expression, the WCE was mixed with an equal volume of 2× SDS loading buffer without boiling; for other proteins, the mixture of cell extract with loading buffer was boiled for 5 min. As noted by others3 and our laboratory, Duox2 forms an aggregate upon heating of the WCEs; the mobility of the Duox2 protein is much slower with boiling than when the WCEs are not boiled.
Chromatin Immunoprecipitation (ChIP)
ChIP assays to detect the binding of Stat1 on the Duox2 promoter were carried out using the QuikChip Kit (catalog number 30101K, Imgenex, San Diego, CA) following the manufacturer's protocol. In short, starved BxPC-3 cells treated with or without IFN-γ were cross-linked using 1% formaldehyde; the cross-linking was stopped by adding glycine at a final concentration of 125 mm to the medium; then the cells were harvested and resuspended in 1× SDS lysis buffer. The suspended cells were sonicated with 10 pulses of 20 s each, with a 30-s rest on ice between each pulse, using Sonic Dismembrator model 100 from Fisher Scientific to produce 200–1000-bp genomic DNA fragments. For immunoprecipitation, Stat1 antibody or normal rabbit IgG antibody from Santa Cruz were used to pull down the cross-linked DNA protein complex, which was recovered using the QIAquick PCR purification kit (catalog number 28104, Qiagen). The purified DNA was used as a PCR template to amplify the Stat1 target sequence in the human Duox2 promoter. The primer sequences were as follows: 5′-AAACCAGAGTCCCCAAGACC-3′ and 5′-GGAGTGAAGGTGGTGGAAGA-3′. The resulting PCR products were 232 bp in length.
Intracellular ROS Detection by Confocal Microscopy
For intracellular ROS detection by confocal microscopy (28, 29), BxPC-3 cells were seeded in 6-well plates at a density of 1 × 106 cells per well in growth medium overnight. The next day, medium was changed to remove serum, and serum-free medium with different concentrations of IFN-γ was added. Cells were incubated for another 24 h. Following treatment with IFN-γ, the cells were washed twice with phosphate-buffered saline (PBS, pH 7.4), and 10 μm H2-DCF-DA in PBS was applied to live cells for 5 min at 37 °C in the dark on the stage of a confocal microscope. H2-DCF-DA diffuses into cells where the free, non-fluorescent 2′,7′-dichlorodihydrofluorescein in the cytoplasm is oxidized to the green fluorescent moiety, dichlorofluorescein, by intracellular ROS. H2-DCF-DA-loaded cells were rinsed twice in PBS, and then either stimulated with 1 μm ionomycin or exposed to medium for 5 min at 37 °C in the dark. Confocal microscopy was performed using an Olympus Fluoview scanning unit equipped with an argon-krypton laser at 488 nm mounted on an Olympus reflected fluorescence microscope (Olympus, Nagano, Japan). During scanning of H2-DCF-DA-loaded cells, we observed that prolonged (more than 5 s) irradiation with the xenon lamp or slow scanning with the 488-nm laser line causes probe oxidation and an artifactual increase in fluorescence. To avoid this artifact, the cells were initially brought into focus using standard epifluorescence (xenon lamp excitation). Then the epifluorescence was switched off and the specimen was moved beyond the previously illuminated area. A single scan at one focal plane using minimum laser intensity (10%) was obtained, and the image was saved for subsequent measurement of fluorescence intensity with the Olympus proprietary Fluoview software.
Measurement of Intracellular ROS Production by Flow Cytometry
IFN-γ- or solvent-primed BxPC-3 cells (1 × 106) were suspended in 0.5 ml of Krebs buffer containing 7.5 μm of the redox-sensitive dye CM-H2-DCF-DA (Invitrogen, catalog number C6827), and incubated in the dark for 30 min at 37 °C. After 25 min of incubation, either solvent (DMSO) or 1 μm ionomycin was added to the cell suspension, and the incubation was continued at 37 °C for an additional 5 min. The cells were then harvested and resuspended in fresh medium without CM-H2-DCF-DA. Fluorescence was recorded on the FL-1 channel of a FACS Aria flow cytometer (BD Bioscience) and analyzed using Flowjo Software (30).
Extracellular H2O2 Measurement Using Amplex Red®
The Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (catalog number A22188 from Invitrogen) was used to detect the extracellular H2O2 released by the IFN-γ activated cells (31). BxPC-3 cells were grown in serum-free medium with or without IFN-γ for 24 h; the cells were then washed twice with 1× PBS, trypsinized, and dispersed thoroughly. Cells were counted to produce a 20-μl cell suspension containing 2 × 104 live BxPC3 cells in 1× KRPG buffer. The cells were mixed with 100 μl of Amplex Red reagent containing 50 μm Amplex Red and 0.1 units/ml of HRP in Krebs-Ringer phosphate glucose (KRPG) buffer with or without 1 μm ionomycin, and incubated at 37 °C for the indicated times. The fluorescence of the oxidized 10-acetyl-3,7-dihydroxyphenoxazine was measured at an excitation wavelength of 530 nm and an emission wavelength of 590 nm using a SpectraMax Multiplate reader (Molecular Devices, Sunnyvale, CA). H2O2 was quantified using an H2O2 standard curve over a 0–2 μm concentration range. Each value in the figures represents a mean of quadruplicate samples.
Statistical Analysis
Results are expressed as the mean ± S.D. from at least triplicate experiments. Statistical differences between mean values of control and treated samples were assessed using Student's t test; p < 0.05 was considered statistically significant. Significance levels were designated as *, p < 0.05; **, p < 0.01, and ***, p < 0.001 throughout.
RESULTS
IFN-γ-induced Duox2 and DuoxA2 Expression
Several human tumor cell lines were exposed to a panel of pro-inflammatory cytokines (IFN-α, IFN-β, IFN-γ, TNF-α, and IL-6) and certain growth factors (EGF, HGF, and TGF-β) to examine the potential of each to regulate Duox2 expression (supplemental Fig. S1). Among the lines tested, BxPC-3 and AsPC-1 human pancreatic cancer cells were responsive to treatment with IFN-γ; the expression of both Duox2 and its cognate maturation factor DuoxA2 (Fig. 1), but not other members of the Nox family of genes (supplemental Fig. S1B), were stimulated; no other cytokine or growth factor produced a substantial effect on Duox2 expression. Analysis by real time PCR revealed that IFN-γ induced both Duox2 and DuoxA2 expression in a dose- and time-dependent manner (Fig. 1, A and B, upper panels, and supplemental Fig. S2A). Using our newly characterized Duox monoclonal antibody that detects both Duox1 and -2, we confirmed that Duox (Duox1 or -2) protein expression correlated well with Duox2 mRNA levels in BxPC-3 and AsPC-1 cells (Fig. 1, A and B, lower panels, and supplemental Fig. S2). Unfortunately, the protein expression of DuoxA2 cannot be assessed currently because DuoxA2 antibodies useful for Western analysis are not available.
FIGURE 1.

IFN-γ induces Duox2 and DuoxA2 expression in BxPC-3 cells in a dose- and time-dependent manner. A, upper panel, represents the dose response for the induction of Duox2 expression by IFN-γ; real time PCR analysis of relative Duox2 and DuoxA2 expression has been normalized to β-actin; error bars indicate the standard deviation from triplicate samples. The lower panel demonstrates expression of the Duox protein by Western analysis in BxPC-3 cells treated with increasing concentrations of IFN-γ for 24 h. Although our antibody recognizes both Duox1 and Duox2, our RNAi experiments (see Fig. 3) strongly suggest that the signals observed from our Western analyses are due to Duox2 protein. B, time course for the induction of Duox2 and DuoxA2 expression by IFN-γ; real time PCR was performed using RNA from BxPC-3 cells treated with 25 ng/ml of IFN-γ for different times, as indicated; error bars represent the standard deviation from triplicate samples. The lower panel shows the change in Duox protein over time following exposure to 25 ng/ml of IFN-γ. ***, p < 0.001.
Both Intracellular ROS and Extracellular H2O2 Generation Were Increased by IFN-γ Priming of BxPC-3 Cells
To determine whether IFN-γ-induced Duox2 and DuoxA2 expression correlated with the functional activity of Duox2, we examined ROS production by BxPC-3 cells exposed to different concentrations of IFN-γ using three different experimental approaches. First, we evaluated intracellular ROS production using the redox-sensitive dye H2-DCFH-DA coupled with confocal microscopy (32). As shown in Fig. 2A, only background levels of fluorescence were demonstrable in cells loaded with H2-DCFH-DA alone or exposed to the calcium ionophore, ionomycin, in addition to the fluorescent dye. BxPC-3 cells treated with 50 ng/ml of IFN-γ for 24 h, on the other hand, demonstrated significantly increased DCF fluorescence, indicative of intracellular ROS production. Intracellular ROS formation increased 3-fold more in cells primed with IFN-γ and subsequently stimulated with ionomycin; furthermore, ROS production increased in an IFN-γ dose-dependent fashion (Fig. 2A, lower panel). These results are in agreement with previous studies that have demonstrated the enhancement of Duox2 activity in the presence of the calcium ionophore, consistent with its EF-hand Ca2+-binding domains (15). To confirm the results obtained with confocal microscopy, flow cytometric quantitation of ROS production was performed, also using the redox-sensitive dye DCFH-DA. These studies clearly demonstrated that exposure to IFN-γ alone for 24 h substantially increased intracellular ROS levels that were enhanced further by ionomycin (Fig. 2B).
FIGURE 2.

IFN-γ enhances intracellular ROS and extracellular H2O2 production. A, upper panel, interferon-γ induces Duox2-mediated ROS production measured by confocal microscopy. Starved BxPC-3 cells were treated with or without IFN-γ for 24 h. Live staining was performed as described under “Experimental Procedures.” Higher ROS levels were observed in BxPC-3 cells that were pre-treated with IFN-γ followed by stimulation with ionomycin than in those cells treated with IFN-γ or ionomycin alone. Representative confocal images of ROS production (green) or phase-contrast fields (immediately below) are shown. The lower panel provides the quantitation of fluorescent intensities under the different treatment conditions for three separate experiments. B, IFN-γ induces intracellular ROS production as detected by analytical cytometry. Serum was withdrawn from BxPC-3 cells overnight; cells were then treated with or without IFN-γ for 24 h; intracellular ROS production was confirmed using the redox-sensitive dye CM-H2DCFDA. A right-shift in fluorescence intensity indicates increasing amounts of ROS. C, production of extracellular H2O2 after exposure of BxPC-3 cells to IFN-γ. BxPC-3 cells were grown using serum-free RPMI 1640 medium in the presence or absence of IFN-γ for 24 h; after collection, 2 × 104 BxPC-3 cells were mixed with the Amplex Red reagents with or without 1 μm ionomycin for the indicated times. H2O2 levels were calculated using a standard curve of 0–2 μm H2O2. H2O2 production for cells exposed to IFN-γ has been compared with cells treated with solvent alone at each time point. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
In a third approach to defining Duox2 activity after IFN-γ exposure, using the Amplex Red reagent in conjunction with HRP, we found that without ionomycin stimulation, H2O2 steadily accumulated over time; after 3 h of incubation, extracellular H2O2 accumulation was ∼50% higher in IFN-γ-treated tumor cells (p < 0.05). When ionomycin was added to the reaction system, the difference between the H2O2 released by IFN-γ- and solvent-treated cells was substantially higher, increasing continuously over the 3-h incubation period (Fig. 2C, right panel). Importantly, low concentrations (100–200 nm) of the flavoprotein inhibitor diphenyleneiodonium (which produce very modest effects on the electron transport chain) (33) significantly inhibited IFN-γ- and ionomycin-mediated H2O2 production (Fig. 3A, left panel). Furthermore, the calcium chelator BAPTA-AM (10 μm) also significantly decreased extracellular H2O2 production from IFN-γ-primed BxPC-3 cells stimulated with ionomycin (Fig. 3A, right panel). This result further supports the calcium dependence of Duox2 enzymatic activity in BxPC-3 cells.
FIGURE 3.

Duox2 and DuoxA2 are required for IFN-γ-mediated extracellular H2O2 production blocked by the flavin dehydrogenase inhibitor diphenylene iodonium (DPI) or the calcium chelator BAPTA. A, BxPC-3 cells grown in serum-free RPMI 1640 medium were treated with 50 ng/ml of IFN-γ for 22 h and then incubated with or without different concentrations of DPI (left panel) or BAPTA (right panel) for another 2 h; cells were harvested and extracellular H2O2 formation was measured for 1 h in the presence or absence of ionomycin (1 μm). Error bars represent standard deviations from quadruplicate samples. B, Duox2 siRNA abrogates IFN-γ-induced Duox expression and H2O2 production. Upper panel, control or Duox2-specific siRNAs were transiently transfected into BxPC-3 cells; 24 h after transfection, cells were incubated in serum-free medium with or without 25 ng/ml of IFN-γ for a subsequent 24 h. Immediately thereafter, H2O2 production was measured over 1 h as previously described. The lower panel demonstrates the expression of Duox, Stat1, or β-actin by Western analysis for BxPC-3 cells exposed to IFN-γ or solvent, and transfected with Duox2 or control siRNAs as outlined for the measurement of H2O2. C, the left panel demonstrates the effect of control or DuoxA2-specific siRNAs on Duox2 or DuoxA2 expression in BxPC-3 cells treated with IFN-γ (25 ng/ml). Experiments shown in the right panel examine the effect of DuoxA2-specific siRNAs on IFN-γ-induced extracellular H2O2 production. H2O2 formation was determined for 1 h as previously described.
To demonstrate that the IFN-γ-mediated increase in H2O2 production is related to IFN-γ-enhanced Duox2 and DuoxA2 expression, we utilized an RNA interference approach. As shown in the lower panel of Fig. 3B, we successfully inhibited IFN-γ-induced Duox protein expression with a Duox2-specific siRNA; IFN-γ-related H2O2 production was also significantly abrogated by Duox2 siRNA transfection (Fig. 3B, upper panel). To determine whether IFN-γ-induced DuoxA2 expression was an essential component of the IFN-γ-mediated extracellular H2O2 production machinery, two DuoxA2-specific siRNAs or control siRNA were transiently transfected into BxPC-3 cells, and IFN-γ-mediated Duox2 and DuoxA2 RNA expression and extracellular H2O2 generation were analyzed. As demonstrated in the left panel of Fig. 3C, both DuoxA2 siRNAs significantly silenced DuoxA2 expression without producing any effect on Duox2 expression. As expected, both DuoxA2-specific siRNAs also significantly decreased IFN-γ-mediated extracellular H2O2 production as shown in the right panel of Fig. 3C. These results support the accessory role of DuoxA2 in Duox2 function, and reveal that both Duox2 and DuoxA2 are essential for IFN-γ-induced extracellular H2O2 accumulation.
Stat1 Up-regulation Is Required for IFN-γ-induced Duox2 Expression
To examine the molecular mechanism of IFN-γ-induced Duox2 expression, we first studied whether newly synthesized mRNA and protein were required for the observed increase in Duox2 expression in response to IFN-γ. As shown in supplemental Fig. S3A, the inhibitor of transcriptional initiation, actinomycin D, dose dependently blocked IFN-γ-mediated Duox2 induction; the protein synthesis inhibitor cycloheximide demonstrated a similar dose-dependent inhibition of IFN-γ-induced Duox2 expression (supplemental Fig. S3B). These results indicated that IFN-γ-induced Duox2 expression is transcriptionally regulated, but that new protein synthesis is also essential for the induction of Duox2 expression. We then analyzed the kinetics of IFN-γ signaling; IFN-γ induced the phosphorylation of Stat1 on tyrosine 701 within 10 min, and the phosphorylation of tyrosine 701 peaked at 60 min (Fig. 4A). Within 3–6 h after the initiation of IFN-γ exposure, pTyr701-Stat1 began to decrease, although pTyr701-Stat1 was still above baseline levels at 24 h in BxPC-3 cells. The phosphorylation of Ser727 in Stat1 following IFN-γ plateaued within 30–60 min, and activation persisted at this level for 24 h. Total Stat1 levels began to increase 3 h following IFN-γ treatment, and continued to increase for the entire 24-h period of IFN-γ exposure; this pattern of increasing expression mirrors that observed for the induction of Duox2 expression (Fig. 1B). Similar results were obtained using the AsPC-1 cell line (supplemental Fig. S2B). We also examined whether other members of the family of Stat proteins were activated by IFN-γ in BxPC-3 cells; no evidence for activation of any other Stat proteins was found (data not shown). This was not because other Stat members are inactive in these cells; as demonstrated in supplemental Fig. S4, IL-6 was fully capable of activating Stat3 at Tyr705 in the BxPC-3 line without producing a concomitant increase in Duox expression.
FIGURE 4.

An intact JAK-Stat1 signaling pathway and Stat1 up-regulation are essential for IFN-γ-induced Duox2 and DuoxA2 expression. A, signaling pathways involved in the regulation of Duox2 expression in BxPC-3 cells by IFN-γ. BxPC-3 cells were grown in serum-free medium and then exposed to IFN-γ for various times; the expression levels of Stat1, phospho-Stat1 (Ser727 and Tyr701), and IRFs 1–3 were determined subsequently by Western analysis. B, the effect of a 30-min exposure to the JAK inhibitor AG490 on IFN-γ-induced Duox expression and Stat1-related signaling proteins was examined after either 6 (left panel) or 24 h (right panel) of IFN-γ treatment. C, the effect of silencing Stat1 on IFN-γ-induced Duox expression was determined in BxPC-3 cells. Control siRNA and two Stat1-specific siRNAs targeting different domains of human Stat1 were transiently transfected into BxPC-3 cells. Twenty-four hours after transfection, cells were grown for another 24 h in serum-free medium with or without 25 ng/ml of IFN-γ. Western analysis shows the protein expression of Duox, Stat1, and IRF-1 following Stat1 knockdown.
Another important transcription factor, interferon regulatory factor 1 (IRF1), which is downstream of Stat1 and participates in the regulation of IFN-γ signaling (25), is also rapidly induced by IFN-γ, and this induction persists for 24 h (Fig. 4A). On the other hand, expression of the other IRF family members (IRF2 and IRF3) was not induced by IFN-γ (Fig. 4A).
To determine whether the Jak-Stat signaling pathway plays a critical role in IFN-γ-mediated Duox2 expression, we evaluated the effect of a Jak-specific inhibitor (AG-490). As shown in Fig. 4B (right panel), AG-490 (100 μm) strongly inhibited IFN-γ-induced Duox2 expression; AG-490 not only inhibited Tyr701 phosphorylation on Stat1, as expected, but also inhibited total Stat1 expression at both 6- and 24-h time points (Fig. 4B). These observations suggested that IFN-γ-induced Duox2 expression is Stat1 dependent.
To investigate further the role of Stat1 in regulating IFN-γ-mediated Duox2 expression, we performed RNA interference experiments with Stat1-specific siRNAs targeting different domains of human Stat1. Twenty-four h after transfection, serum-free medium with or without 25 ng/ml of IFN-γ was introduced; and BxPC-3 cells were incubated for another 24 h. In the presence of a control siRNA, induction of Duox expression was unchanged (Fig. 4C). In contrast, silencing Stat1 totally abrogated IFN-γ-induced Duox expression (Fig. 4C). This result established that Stat1 is necessary for IFN-γ-induced Duox expression in BxPC-3 cells.
Phosphorylation of Ser727 of Stat1 Is Essential for IFN-γ-mediated Duox2 Expression
Although Jak-Stat signaling pathways have been demonstrated to play a central role in promoting the diverse cellular responses elicited by IFN-γ, some studies have suggested that IFN-γ activates additional signaling networks that can regulate gene expression independent of Stat1 (27). The additional signaling pathways activated by IFN-γ engagement to its cell surface receptors include PI3K/Akt, protein kinase C (PKC) isoforms, calcium/calmodulin kinase II (CaMKII), and MAP kinases (34–36). These signaling pathways either regulate the Ser727 phosphorylation of Stat1 to enhance its transcriptional efficiency or activate other transcription factors leading to a Stat1-independent effect. Using Western analysis, we examined several potential signaling pathways reportedly activated by IFN-γ. PI3K/Akt, PKC, CaMKII, and JNK signaling were not significantly enhanced by IFN-γ in BxPC-3 cells (data not shown). However, two MAPK pathways, p38 and ERK, were significantly activated by IFN-γ, as shown in Fig. 5A. To dissect the possible roles of p38 and ERK activation in the regulation of Duox2 expression by IFN-γ, we evaluated the effects of two MAPK inhibitors, U0126 specific for the ERK pathway, and SB203580 specific for p38 signaling. Starved BxPC-3 cells were pretreated with solvent or the MAPK inhibitors for 30 min, and then stimulated with 25 ng/ml of IFN-γ for 24 h; RNA was extracted and subjected to real time PCR to detect changes in the relative expression levels of Duox2 and DuoxA2. IFN-γ strongly induced both Duox2 and DuoxA2 expression in the presence of solvent (0.1% DMSO); 10 μm U0126 did not attenuate IFN-γ-induced Duox2 or DuoxA2 expression (Fig. 5B). Furthermore, although U0126 blocked the phosphorylation of ERK both at baseline (supplemental Fig. S6) and that produced by IFN-γ, it did not affect Stat1 phosphorylation (Fig. 5E). On the other hand, the p38 MAP kinase inhibitor SB203580 significantly decreased baseline p38 expression and IFN-γ-induced expression of both Duox2 and DuoxA2 (supplemental Fig. S6 and Fig. 5B, left and right panels). SB203580 had no effect on the expression of two other genes induced by IFN-γ in BxPC-3 cells, IRF-1 and Stat1 (supplemental Fig. S5). Western analysis (Fig. 5C) confirmed that SB203580 inhibited IFN-γ-mediated p38 activation at the same time that the Ser727 phosphorylation in Stat1 and Duox2 induction by IFN-γ were suppressed, suggesting that p38 may mediate Ser727 activation in Stat1, and that the phosphorylation of Ser727 is important for IFN-γ-induced Duox2 expression in BxPC-3 cells.
FIGURE 5.

p38-mediated Stat1Ser727 phosphorylation is necessary for IFN-γ-induced Duox2 and DuoxA2 expression. A, IFN-γ activates p38 and ERK as well as Duox in BxPC-3 cells. Time course demonstrating the effect of IFN-γ treatment on ERK and p38 phosphorylation over 12 h. B, a 30-min pre-treatment with 10 μm SB203580 (p38 MAPK pathway inhibitor), but not with 10 μm U0126 (p42/44 MAPK pathway inhibitor), significantly decreased the enhanced Duox2 (left panel) and DuoxA2 (right panel) mRNA expression that follows 24 h of IFN-γ treatment. C, inhibition of p38 phosphorylation with SB203580 decreases IFN-γ-mediated expression of Duox protein. BxPC-3 cells were grown in serum-free RPMI 1640 medium, and then pretreated with 10 μm SB203580 or an equal volume of DMSO as control for 30 min; the cells were then grown in the presence or absence of the inhibitor for 1 or 24 h with or without 25 ng/ml of IFN-γ, and then analyzed for Stat1-related signaling by Western analysis with 50 μg of whole cell lysate. D, silencing p38 attenuates IFN-γ-induced Stat1 Ser727 phosphorylation and Duox2 expression. BxPC-3 cells were transiently transfected with control or p38-specific siRNA for 24 h. Cells were grown in the absence of serum with or without IFN-γ for another 24 h; 50 μg of cell lysate was then used for Western analysis of the expression of Stat1-related proteins. E, inhibition of the p42/44-MAPK pathway with U0126 does not affect IFN-γ-mediated expression of Duox protein. BxPC-3 cells were grown in serum-free RPMI 1640 medium, and then pretreated with 10 μm U0126 or an equal volume of DMSO as control for 30 min; the cells were then grown in the presence or absence of the inhibitor for 1 or 24 h with or without 25 ng/ml of IFN-γ, and then analyzed for ERK phosphorylation and Stat1-related signaling by Western analysis with 50 μg of whole cell lysate. ***, p < 0.001.
These results are congruent with a recent report indicating that MKK3, p38, and ATF2 are involved in the regulation of Duox2 expression by soluble microbial extracts in CaCo2 human colon cancer cells (20). To confirm the role of p38 kinase in mediating Stat1 Ser727 phosphorylation and Duox2 induction by IFN-γ, human p38-specific siRNA and control siRNA were transiently transfected into BxPC-3 cells; 24 h after transfection, cells were grown in serum-free medium with or without 25 ng/ml of IFN-γ for another 24 h. Cells were then collected, and cell lysates were examined by Western analysis. As shown in Fig. 5D, p38 siRNA suppressed endogenous p38 expression at the same time Stat1 Ser727 phosphorylation and Duox induction by IFN-γ were inhibited.
Stat1 Binds to the Endogenous Human Duox2 Promoter
Because our results strongly suggested that in BxPC-3 cells Stat1 plays a critical role in IFN-γ-induced Duox2 expression, we examined whether Stat1 bound to the human Duox2 promoter, and whether binding of Stat1 to the Duox2 promoter was stimulated by IFN-γ. To address these two questions, we performed chromatin immunoprecipitation assays using BxPC-3 cells treated with or without IFN-γ at different time points. Initially, 3 kb of the human Duox2 promoter was screened to identify putative Stat1 binding sites using the TRANSFAC® search program (37). No canonical Stat1 binding sites were identified; however, several potential binding sites with similarity to the consensus binding site of Stat1 do exist. It has been reported that Stat1 homodimers can bind to some elements of Stat1 target genes with only half GAS sites (38). We focused on one potential Stat1 binding site located between 669 and 676 bp from the transcription start site of the human Duox2 promoter; the sequence is 5′-TTCCTGCAA-3′, which has only one nucleotide changed compared with the conserved GAS element 5′-TTCNNN(G/T)AA-3′. Thus, we designed PCR primers to encompass this site, and performed ChIP assays as shown as in Fig. 6. Control rabbit IgG antibody did not produce a band, indicating that the immunoprecipitation and PCR were specific. Using a Stat1-specific antibody permitted the detection of PCR amplifying products; after 1 h of IFN-γ stimulation, there was substantial Stat1 binding to the endogenous human Duox2 promoter. Signal intensity of PCR products following 24 h of IFN-γ treatment was at least comparable, if not greater, than that from a 1-h exposure to IFN-γ, even though Stat1 phosphorylation on Tyr701 had decreased 24 h following IFN-γ exposure compared with the 1-h time point (Fig. 6).
FIGURE 6.
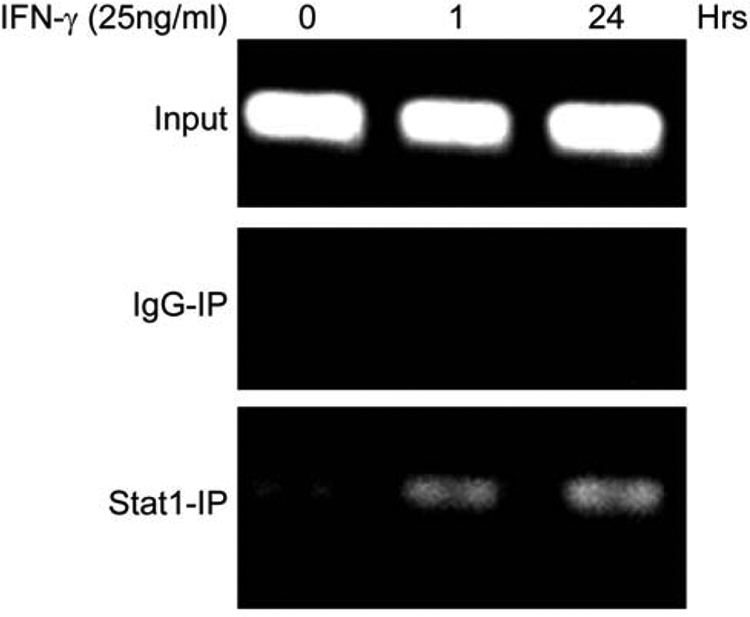
Chromatin immunoprecipitation assay detects Stat1 binding to the Duox2 promoter. Starved BxPC-3 cells were treated with solvent or 25 ng/ml of IFN-γ for 1 or 24 h; cells were then harvested and used for the ChIP assay. Input lanes verifying equal amounts of DNA were used for the initial immunoprecipitation; control IgG- and Stat1-specific antibodies were used to pull down DNA, which was then extracted and used for PCR with primers spanning the potential Stat1 binding site in the Duox2 promoter. The resulting PCR products were separated on a 2% agarose gel and visualized with ethidium bromide staining.
DISCUSSION
In these experiments, we have demonstrated that the proinflammatory cytokine IFN-γ specifically up-regulates expression of the Nox family member Duox2, as well as its critical maturation factor DuoxA2, in human pancreatic cancer cell lines BxPC-3 and AsPC-1, and that the regulation of DUOX2 is Stat1-dependent. These results are of interest because recent studies have suggested that one consequence of the chronic inflammatory state often accompanying pancreatic adenocarcinoma is the generation of high local concentrations of ROS that could be generated by cytokine-, growth factor-, or extracellular matrix-related activation of epithelial NADPH oxidases (39, 40). PANC-1 and MIA PaCa-2 human pancreatic cancer cell lines express the Nox4 isoform, which is responsible for generation of the ROS that play an important role in survival signaling and apoptosis in these cells (41). Our laboratory has recently examined the expression of Nox family members in human tumors and tumor cell lines (9). We found that Duox2 was highly expressed in colorectal cancers. That finding is supported by the observation that pre-malignant adenomatous lesions of the large intestine demonstrate a 10-fold increase in DUOX2 expression when compared with adjacent non-malignant tissues from the same patients (42). Finally, we have recently conducted an immunohistochemical evaluation of Duox2 expression using tumor tissue microarrays; in that investigation, we found increased Duox2 levels, compared with non-malignant control tissues, in adenocarcinomas of the stomach, colon, and pancreas.4 Thus, members of the Nox family of membrane oxidases, including Duox2, may play a role in the pathophysiology of carcinomas of the gastrointestinal tract, including pancreatic cancer, particularly in the setting of chronic inflammation.
Because Nox-mediated ROS have been demonstrated to enhance genetic instability (43), we were particularly interested in quantitating ROS production following IFN-γ treatment of BxPC-3 cells. We found that Duox2/DuoxA2-dependent intracellular ROS and extracellular H2O2 production was significantly increased in IFN-γ-treated cells (Figs. 2, B and C, and 3, B and C) grown in Ca2+-containing medium in the absence of ionomycin. Although ionomycin enhanced Duox2-dependent ROS formation further, these results suggest that exposure of BxPC-3 cells to IFN-γ leads to a fully functional Duox2-DuoxA2 complex wherein Duox2-dependent ROS could play a direct role in the establishment of cellular redox homeostasis. Our results are consistent with those of Linderholm and colleagues (44) who recently found that up-regulation of Duox2 by all-trans-retinoic acid in respiratory tract epithelium is associated with a significant increase in apical H2O2 production. Furthermore, our experiments support the possibility that up-regulation of DUOX2, leading to high levels of functional Duox2 expression, could contribute to a pro-angiogenic extracellular milieu that would favor both tumor growth and leukocyte infiltration (2, 21, 45).
Other than its calcium dependence (as shown in Fig. 3A), there is only a modest current understanding of the regulation of Duox2 expression. In certain human lung cancer cell lines and in lung cancer surgical specimens, Duox2 appears to be under epigenetic regulation; hypermethylation of the Duox2 promoter in lung cancers, compared with adjacent normal lung tissue taken at the time of surgery, appears to be common (46). In the current study, we found that actinomycin D, a transcriptional inhibitor, as well as cycloheximide, an inhibitor of translation, both blocked IFN-γ-induced Duox2 expression (supplemental Fig. S3). The fact that new protein synthesis, as well as transcription, was required for IFN-γ-mediated Duox2 expression was surprising; identification of the specific factors in the Duox2 transcriptome that mediate the up-regulation by IFN-γ will require further investigation.
The effect of IFN-γ on Duox2 expression was first demonstrated using human tracheo-bronchial epithelial cells (13, 18); from a wide variety of cytokines examined in that system, only IFN-γ and viral infection produced an effect on Duox2. These results suggest that Duox2 plays a critical role in host defense against infectious agents in the lung (47, 48). However, the mechanism by which IFN-γ increases Duox2 expression has, until recently, not been examined (49). To understand the IFN-γ-induced signaling events that result in enhanced Duox2 expression, we investigated the roles played by the Jak-Stat1 and p38-MAPK pathways in these events. Our results strongly suggest that Jak and p38-MAPK play pivotal roles in the activation of Stat1, which, in turn, appears to be essential for full IFN-γ-inducible Duox2 expression as well as production of H2O2 in pancreatic cancer cells.
Stat1 is known to play a major role in the signaling cascade initiated by IFN-γ. However, a recent study reported that IFN-γ-induced Duox2 expression did not utilize the common Stat1 signaling pathway in the respiratory tract epithelial cell line, HBE1 (49). Although HBE1 cells activated the Jak-Stat pathway and increased Duox2 expression >20-fold following IFN-γ stimulation, nonspecific Jak inhibition did not affect Duox2 expression, whereas at the same time the expression of a different IFN-γ-sensitive gene, CXCL10, was significantly decreased. On the other hand, we have provided evidence in this study that the Jak-Stat1 signaling pathway is essential for IFN-γ-mediated Duox2 as well as DuoxA2 expression in BxPC-3 cells. The specific cell or tissue context responsible for the divergence of these observations is unclear.
In BxPC-3 pancreatic carcinoma cells, up-regulation of Stat1 as well as its phosphorylation on both Tyr701 and Ser727 play an important role in Duox2 induction by IFN-γ. The Jak-specific small molecule inhibitor AG-490 not only inhibited Tyr701 phosphorylation of Stat1, it also inhibited total Stat1 up-regulation as well as Duox2 induction by IFN-γ. Direct evidence for the role of the Jak-Stat pathway was provided through the use of Stat1-specific siRNA; this siRNA not only silenced IFN-γ-induced total Stat1 expression, but also completely inhibited IFN-γ-induced Duox2 expression. Of importance, we found that no other member of the Stat family of proteins was activated in IFN-γ-treated BxPC-3 cells; furthermore, our results with IL-6, a known activator of Stat3 (50) (supplemental Fig. S4), suggested that Stat3 was not involved in IFN-γ-mediated Duox2 up-regulation. However, the contribution of total Stat1 versus phosphorylation of Tyr701 or Ser727 of Stat1 in the up-regulation of Duox2 by IFN-γ cannot be fully explained by our studies. Although Tyr701 phosphorylation of Stat1 is an early event in IFN-γ-treated BxPC-3 cells, which suggests that phosphorylation of Tyr701 may be essential for nuclear localization, we also found that Tyr701 phosphorylation of Stat1 decreases at later time points following IFN-γ stimulation. This suggests that Stat1 may form stable homodimers that could bind more efficiently to DNA sequences harboring half- and full-GAS sites on the Duox2 promoter.
IFN-γ activates several signaling cascades, in addition to the Jak-Stat pathway, including Akt, p38-MAPK, NF-κB, p42/44-MAPK, and JNK. Our mechanistic studies suggest that the activation of p38-MAPK, working by way of Ser727 phosphorylation of Stat1, mediated signal transduction and transcriptional up-regulation of Duox2. In our model system, Ser727 phosphorylation occurred very quickly, within 30 min of IFN-γ treatment, and this activation persisted for 24 h. Although several kinases have been implicated in the phosphorylation of this site, our experiments suggest that p38-MAPK is a likely candidate responsible for Ser727 phosphorylation of Stat1 in BxPC-3 cells. The p38-MAPK-specific inhibitor SB203580 inhibited not only the sustained activation of Ser727 of Stat1 but also completely suppressed IFN-γ-induced Duox2 expression. Knocking down p38-MAPK with p38-MAPK-specific siRNA not only suppressed endogenous p38-MAPK expression but also decreased total Stat1 expression and inhibited IFN-γ-enhanced Duox protein expression. In related experiments, we observed that p42/44-MAPK was phosphorylated along with p38-MAPK (data not shown) following IFN-γ exposure. However, pre-treatment of BxPC-3 cells with p42/44-MAPK siRNA, whereas completely inhibiting total p42/44-MAPK expression, did not suppresses Ser727 phosphorylation of Stat1, or the transcriptional up-regulation of Duox2 following IFN-γ treatment. These studies indicate that p42/44-MAPK is not responsible for the transcriptional regulation of Duox2 expression induced by IFN-γ in BxPC-3 cells. We have also found that IFN-γ activates Stat1 by phosphorylating Tyr701 and Ser727 in the PANC-1 and MIA PaCa-2 human pancreatic cancer cell lines, but without a concomitant induction of Duox2 (data not shown). Thus, as suggested by our cycloheximide experiment, unidentified protein factors may be necessary for the up-regulation of Duox2 transcripts; and thus, Stat1 activation may be necessary but not sufficient for IFN-γ-induced Duox2 expression.
The importance of Stat1 in the activation of Duox2 by IFN-γ was further supported by chromatin immunoprecipitation assays. Although no canonical Stat1 binding sites were identified within 3.0 kb from the transcription start site of the human Duox2 promoter, several potential Stat1 binding sites with similarity to the consensus Stat1 binding site do exist in this region. It has been suggested that Stat1 homodimers can bind to some elements of Stat1 target genes with only half GAS sites (TTCNNNG/TAA) (38). Indeed, we found time-dependent binding of Stat1 to the endogenous human Duox2 promoter following IFN-γ stimulation. Thus, ChIP confirms that the Duox2 promoter is a target of Stat1, regardless of the binding site location within 3.0 kb from the transcription start site.
In summary, our experiments demonstrate that the inflammatory cytokine IFN-γ enhances the transcription of Duox2 and its cognate maturation factor DuoxA2 in BxPC-3 and AsPC-1 human pancreatic carcinoma cell lines, in a Stat1- and p38-MAPK-dependent fashion. Duox2/DuoxA2 up-regulation leads to a significant increase in both intracellular ROS and extracellular H2O2 formation. In light of recent studies that demonstrate a novel role for Duox2-mediated H2O2 gradients in leukocyte recruitment (21), as well as the role of these inflammatory cells, in addition to ROS themselves, in the creation of a pro-angiogenic local environment (51–53), the regulation of Duox2 by IFN-γ could play an important role in pancreatic cancer pathophysiology. Our ongoing studies are focused on understanding the biological consequences of enhanced Duox2 expression in pancreatic and other gastrointestinal malignancies, both as a result of IFN-γ exposure and in the context of enhanced Duox2/DuoxA2 expression in the absence of cytokine activation.
This work was supported, in whole or in part, by a NCI, National Institutes of Health, grant.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
H. Grasberger, University of Chicago, personal communication.
Y. Wu, S. Antony, A. Juhasz, J. Lu, G. Jiang, K. Roy, S. Hewitt, and J. H. Doroshow, unpublished observations.
- ROS
- reactive oxygen species
- Nox
- reduced nicotinamide adenine dinucleotide phosphate oxidase
- Duox
- dual oxidase
- IRF
- interferon regulatory factor
- BAPTA
- 1,2-bis-(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- H2-DCF-DA
- 2′,7′-dichlorodihydrofluorescin diacetate
- WCE
- whole cell extract
- JNK
- c-Jun N-terminal kinase.
REFERENCES
- 1. Masamune A., Watanabe T., Kikuta K., Satoh K., Shimosegawa T. (2008) Am. J. Physiol. Gastrointest. Liver Physiol. 294, G99–G108 [DOI] [PubMed] [Google Scholar]
- 2. Yu J. H., Lim J. W., Kim H., Kim K. H. (2005) Int. J. Biochem. Cell Biol. 37, 1458–1469 [DOI] [PubMed] [Google Scholar]
- 3. Farrow B., Evers B. M. (2002) Surg. Oncol. 10, 153–169 [DOI] [PubMed] [Google Scholar]
- 4. Greer J. B., Whitcomb D. C. (2009) Curr. Opin. Pharmacol. 9, 411–418 [DOI] [PubMed] [Google Scholar]
- 5. Lee J. K., Edderkaoui M., Truong P., Ohno I., Jang K. T., Berti A., Pandol S. J., Gukovskaya A. S. (2007) Gastroenterology 133, 1637–1648 [DOI] [PubMed] [Google Scholar]
- 6. Ostman A., Hellberg C., Böhmer F. D. (2006) Nat. Rev. Cancer 6, 307–320 [DOI] [PubMed] [Google Scholar]
- 7. Krause K. H. (2004) Jpn. J. Infect. Dis. 57, S28–S29 [PubMed] [Google Scholar]
- 8. Arnold R. S., Shi J., Murad E., Whalen A. M., Sun C. Q., Polavarapu R., Parthasarathy S., Petros J. A., Lambeth J. D. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 5550–5555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Juhasz A., Ge Y., Markel S., Chiu A., Matsumoto L., van Balgooy J., Roy K., Doroshow J. H. (2009) Free Radic. Res. 43, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambeth J. D., Kawahara T., Diebold B. (2007) Free Radic. Biol. Med. 43, 319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caillou B., Dupuy C., Lacroix L., Nocera M., Talbot M., Ohayon R., Dème D., Bidart J. M., Schlumberger M., Virion A. (2001) J. Clin. Endocrinol. Metab. 86, 3351–3358 [DOI] [PubMed] [Google Scholar]
- 12. El Hassani R. A., Benfares N., Caillou B., Talbot M., Sabourin J. C., Belotte V., Morand S., Gnidehou S., Agnandji D., Ohayon R., Kaniewski J., Noel-Hudson M. S., Bidart J. M., Schlumberger M., Virion A., Dupuy C. (2005) Am. J. Physiol. Gastrointest. Liver Physiol. 288, G933–942 [DOI] [PubMed] [Google Scholar]
- 13. Harper R. W., Xu C., McManus M., Heidersbach A., Eiserich J. P. (2006) FEBS Lett. 580, 5150–5154 [DOI] [PubMed] [Google Scholar]
- 14. Fischer H. (2009) Antioxid. Redox Signal. 11, 2453–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bedard K., Krause K. H. (2007) Physiol. Rev. 87, 245–313 [DOI] [PubMed] [Google Scholar]
- 16. Grasberger H., Refetoff S. (2006) J. Biol. Chem. 281, 18269–18272 [DOI] [PubMed] [Google Scholar]
- 17. Geiszt M., Witta J., Baffi J., Lekstrom K., Leto T. L. (2003) FASEB J. 17, 1502–1504 [DOI] [PubMed] [Google Scholar]
- 18. Harper R. W., Xu C., Eiserich J. P., Chen Y., Kao C. Y., Thai P., Setiadi H., Wu R. (2005) FEBS Lett. 579, 4911–4917 [DOI] [PubMed] [Google Scholar]
- 19. Leto T. L., Geiszt M. (2006) Antioxid. Redox Signal. 8, 1549–1561 [DOI] [PubMed] [Google Scholar]
- 20. Ha E. M., Lee K. A., Seo Y. Y., Kim S. H., Lim J. H., Oh B. H., Kim J., Lee W. J. (2009) Nat. Immunol. 10, 949–957 [DOI] [PubMed] [Google Scholar]
- 21. Niethammer P., Grabher C., Look A. T., Mitchison T. J. (2009) Nature 459, 996–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saha B., Jyothi Prasanna S., Chandrasekar B., Nandi D. (2010) Cytokine 50, 1–14 [DOI] [PubMed] [Google Scholar]
- 23. Kumatori A., Yang D., Suzuki S., Nakamura M. (2002) J. Biol. Chem. 277, 9103–9111 [DOI] [PubMed] [Google Scholar]
- 24. Gough D. J., Levy D. E., Johnstone R. W., Clarke C. J. (2008) Cytokine Growth Factor Rev. 19, 383–394 [DOI] [PubMed] [Google Scholar]
- 25. Andrianifahanana M., Singh A. P., Nemos C., Ponnusamy M. P., Moniaux N., Mehta P. P., Varshney G. C., Batra S. K. (2007) Oncogene 26, 7251–7261 [DOI] [PubMed] [Google Scholar]
- 26. Wen Z., Zhong Z., Darnell J. E., Jr. (1995) Cell 82, 241–250 [DOI] [PubMed] [Google Scholar]
- 27. Kovarik P., Stoiber D., Eyers P. A., Menghini R., Neininger A., Gaestel M., Cohen P., Decker T. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 13956–13961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xia C., Meng Q., Liu L. Z., Rojanasakul Y., Wang X. R., Jiang B. H. (2007) Cancer Res. 67, 10823–10830 [DOI] [PubMed] [Google Scholar]
- 29. Alexandratou E., Yova D., Loukas S. (2005) Free Radic. Biol. Med. 39, 1119–1127 [DOI] [PubMed] [Google Scholar]
- 30. Vaquero E. C., Edderkaoui M., Pandol S. J., Gukovsky I., Gukovskaya A. S. (2004) J. Biol. Chem. 279, 34643–34654 [DOI] [PubMed] [Google Scholar]
- 31. Dikalov S., Griendling K. K., Harrison D. G. (2007) Hypertension 49, 717–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brar S. S., Corbin Z., Kennedy T. P., Hemendinger R., Thornton L., Bommarius B., Arnold R. S., Whorton A. R., Sturrock A. B., Huecksteadt T. P., Quinn M. T., Krenitsky K., Ardie K. G., Lambeth J. D., Hoidal J. R. (2003) Am. J. Physiol. Cell Physiol. 285, C353–C369 [DOI] [PubMed] [Google Scholar]
- 33. Hancock J. T., Jones O. T. (1987) Biochem. J. 242, 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Valledor A. F., Sánchez-Tilló E., Arpa L., Park J. M., Caelles C., Lloberas J., Celada A. (2008) J. Immunol. 180, 4523–4529 [DOI] [PubMed] [Google Scholar]
- 35. Ramsauer K., Sadzak I., Porras A., Pilz A., Nebreda A. R., Decker T., Kovarik P. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 12859–12864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nair J. S., DaFonseca C. J., Tjernberg A., Sun W., Darnell J. E., Jr., Chait B. T., Zhang J. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5971–5976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quandt K., Frech K., Karas H., Wingender E., Werner T. (1995) Nucleic Acids Res. 23, 4878–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chatterjee-Kishore M., Wright K. L., Ting J. P., Stark G. R. (2000) EMBO J. 19, 4111–4122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edderkaoui M., Hong P., Vaquero E. C., Lee J. K., Fischer L., Friess H., Buchler M. W., Lerch M. M., Pandol S. J., Gukovskaya A. S. (2005) Am. J. Physiol. Gastrointest. Liver Physiol. 289, G1137–G1147 [DOI] [PubMed] [Google Scholar]
- 40. Pandol S., Edderkaoui M., Gukovsky I., Lugea A., Gukovskaya A. (2009) Clin. Gastroenterol. Hepatol. 7, S44–S47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mochizuki T., Furuta S., Mitsushita J., Shang W. H., Ito M., Yokoo Y., Yamaura M., Ishizone S., Nakayama J., Konagai A., Hirose K., Kiyosawa K., Kamata T. (2006) Oncogene 25, 3699–3707 [DOI] [PubMed] [Google Scholar]
- 42. Kita H., Hikichi Y., Hikami K., Tsuneyama K., Cui Z. G., Osawa H., Ohnishi H., Mutoh H., Hoshino H., Bowlus C. L., Yamamoto H., Sugano K. (2006) J. Gastroenterol. 41, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 43. Chiera F., Meccia E., Degan P., Aquilina G., Pietraforte D., Minetti M., Lambeth D., Bignami M. (2008) Free Radic. Biol. Med. 44, 332–342 [DOI] [PubMed] [Google Scholar]
- 44. Linderholm A. L., Onitsuka J., Xu C., Chiu M., Lee W. M., Harper R. W. (2010) Am. J. Physiol. Lung Cell Mol. Physiol. 299, L215–L221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ushio-Fukai M., Nakamura Y. (2008) Cancer Lett. 266, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luxen S., Belinsky S. A., Knaus U. G. (2008) Cancer Res. 68, 1037–1045 [DOI] [PubMed] [Google Scholar]
- 47. Gattas M. V., Forteza R., Fragoso M. A., Fregien N., Salas P., Salathe M., Conner G. E. (2009) Free Radic. Biol. Med. 47, 1450–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morand S., Ueyama T., Tsujibe S., Saito N., Korzeniowska A., Leto T. L. (2009) FASEB J. 23, 1205–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hill T., 3rd, Xu C., Harper R. W. (2010) Biochem. Biophys. Res. Commun. 395, 270–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kishimoto T. (2010) Int. Immunol. 22, 347–352 [DOI] [PubMed] [Google Scholar]
- 51. Esposito I., Menicagli M., Funel N., Bergmann F., Boggi U., Mosca F., Bevilacqua G., Campani D. (2004) J. Clin. Pathol. 57, 630–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matsuo Y., Sawai H., Funahashi H., Takahashi H., Sakamoto M., Yamamoto M., Okada Y., Hayakawa T., Manabe T. (2004) Pancreas 28, 344–352 [DOI] [PubMed] [Google Scholar]
- 53. Arbiser J. L., Petros J., Klafter R., Govindajaran B., McLaughlin E. R., Brown L. F., Cohen C., Moses M., Kilroy S., Arnold R. S., Lambeth J. D. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]