

Abstract
A detailed study of nuclear import mediated by the HIV-1 Tat peptide (47YGRKKRRQRRR57, TatRRR) is reported. Fluorescence-based measurements, calibration of protein concentrations, and binding assays are exploited to address the physicochemical mechanisms of Tat peptide recognition by the classical importin α (Impα) and importin β (Impβ) receptors both in vitro and in intact cells. We show that TatRRR is an unconventional nuclear localization sequence that binds directly to both Impα and Impβ carriers in the absence of competitors (in vitro), whereas this property is silenced in the actual cellular environment. In the latter case, Impα/β-dependent nuclear import can be successfully restored by replacing the “RRR” stretch with “GGG”. We apply a recently developed method to determine quantitatively TatGGG affinity for each receptor. Based on these results, we can rationalize previous controversial reports on the Tat peptide and provide coherent guidelines for the design of novel intracellular targeting sequences.
Keywords: Biophysics, Fluorescence Resonance Energy Transfer (FRET), Intracellular Trafficking, Nuclear Transport, Peptide Interactions, FLIM FRAP, Tat Peptide, Binding Assay, In Vivo, Nuclear Import
Introduction
Cellular compartments are the defining feature of eukaryotic cells. The nucleus is surrounded by a double membrane called the nuclear envelope, which separates the genetic material and transcriptional activity from the translational and metabolic processes of the cytoplasm. Communication between nucleus and cytoplasm is mediated by nuclear pore complexes (1, 2), large macromolecular assemblies that punctuate the nuclear envelope. Nuclear pore complexes form a selective barrier that inhibits translocation of large cargo molecules (>40 kDa) (3), unless they possess specific targeting signals called nuclear localization sequences (NLS).4 The best characterized NLS consist of either one (monopartite) or two (bipartite) stretches of basic amino acids (4, 5). Monopartite NLS are exemplified by the SV40 large T antigen NLS (126PKKKRRV132), and bipartite NLS are exemplified by the nucleoplasmin NLS (155KRPAATKKAGQAKKKK170). These sequences are specifically recognized by a heterodimer of proteins named importin α (Impα) and importin β (Impβ) (6). Impα binds the NLS specifically (7), whereas Impβ both enhances the affinity of Impα for the NLS (8) and mediates the transfer of the cargo-Impα complex across the nuclear pore complex (9). The cargo is then released in the nucleus upon RanGTP binding to Impβ (10). Because of the surfeit of known classical NLS-containing proteins, it is assumed that this pathway is the most prevalent in the cell; yet, to date no studies have empirically established the proportion of cargoes imported via this mechanism. Furthermore, there is evidence that NLS sequences with unconventional nuclear import properties may exist, most of which are derived from viral proteins. Among these, we have been interested for a long time in the HIV-1 Tat protein. Tat is an unusual transcriptional transactivator that attaches to cell surface heparan sulfate proteoglycans (11), enters cells by endolysosomal pathways (12), reaches the nucleus (13), and dramatically enhances the processivity of transcription directed by the viral long terminal repeat promoter element (14, 15). The nuclear localization properties of Tat protein are commonly ascribed to the stretch 47YGRKKRRQRRR57 (16, 17) (also named TatRRR hereafter). Notably, the same sequence was also shown to be responsible for the cell-penetrating properties of the full-length protein (for a review, see Ref. 18) and for its RNA-binding specificity (19–21). As mentioned above, several reports ascribe to Tat NLS (and homologous viral sequences) novel nuclear import properties, albeit with contrasting results. In particular, Efthymiadis et al. (16) reported that the Tat NLS is able to mediate nuclear import in vitro in the absence of both Impα and Impβ, through binding to nuclear components. In turn, Truant and Cullen (17) observed that Tat NLS directly interacts with Impβ but not Impα in vitro and showed that Impβ is both necessary and sufficient for the nuclear import of Tat into isolated nuclei. Contrary to these in vitro experiments, we recently demonstrated that passive diffusion is the dominant mechanism of Tat peptide-mediated nuclear transport in live cells (22). This apparent paradox is accounted for by the overwhelming binding affinity of the C-terminal RRR stretch toward negatively charged biomolecules (e.g. RNAs) that hinders Tat-peptide interactions with the transport machinery. Indeed, the NLS properties of Tat can be recovered in engineered mutants where the RRR stretch is substituted by other motifs (e.g. GGG; sequence: YGRKKRRQGGG, also named TatGGG hereafter) (23). Overall, however, the molecular details of the nuclear import process mediated by wild-type and mutant Tat NLS remain elusive. We recently established a novel and straightforward method that combines fluorescence lifetime imaging microscopy (FLIM) and fluorescence recovery after photobleaching (FRAP) with in vivo calibration of protein concentrations, to gain access to both the thermodynamic (binding specificity and affinity) and kinetic (import rate) details of the nuclear transport process in intact cells (24). The broad applicability of the method was demonstrated for the interaction between NLS of SV40 and the transport receptor Impα (24). Here, we extend this quantitative approach to the study of wild-type and mutant Tat-NLS interactions with the classical import carriers Impα, Impβ, and their dual complex. We show that TatRRR is not able to establish interactions with either Impα or Impβ in the intact cellular environment, in keeping with our previous results (22, 23). Conversely, we demonstrate that the TatGGG mutant binds directly to both Impα and Impβ. Note that the conventional NLS from SV40 can establish direct interactions solely with Impα (activated by Impβ). Finally, by a complementary in vitro binding assay, we find that in the absence of competitors (i.e. intracellular cytosolic and nuclear factors) TatRRR does bind to Impα and Impβ. Overall, these results indicate that TatRRR is characterized by a nonclassical NLS that is silenced in the cellular environment but can be observed easily in vitro (in the absence of competitors) or restored in vivo in engineered mutants (TatGGG). We believe that these findings rationalize the picture of previous controversial results on Tat peptide nuclear import properties and can provide the basic knowledge for the rational design of localization sequences better tailored to the nucleus.
EXPERIMENTAL PROCEDURES
Plasmids and Cell Culture
Plasmids expressing the mCherry-tagged NLSSV40, TatRRR and TatGGG sequences were obtained by subcloning starting from their EGFP-tagged counterparts described previously (22). The cDNA encoding for mCherry obtained from the laboratory of Roger Y. Tsien (25) was amplified by PCR introducing the HindIII and EcoRI restriction sites at the 5′ and 3′ extremities, respectively. These sites were used to replace EGFP with mCherry. TatMUT-AARRR-mCherry and TatMUT-AAGGG-mCherry mutants were obtained by site-directed mutagenesis using a QuikChange Lightning site-directed mutagenesis kit (Stratagene). In both constructs, the first moiety of Tat sequence MYGRKKRRQ was substituted with MYGRAARRQ. To introduce the two mutations, the following primer (Invitrogen) was used: 5′-ATGTATGGCAGGGCGGCGCGGAGACAG-3′. The antisense primer has the respective reverse complementary sequence. The plasmid encoding for the EGFP-tagged importin α (mouse full-length mNPI2) was kindly provided by Yoshihiro Yoneda (Department of Frontier Biosciences, Osaka University) (26). The plasmid encoding for the EGFP-tagged human importin β1 was kindly provided by Marilena Ciciarello (Institute of Molecular Biology and Pathology, National Research Council, Rome, Italy) (27). CHO-K1 were purchased from American Type Culture Collection (CCL-61 ATCC) and were grown in Ham's F12K medium supplemented with 10% of fetal bovine serum at 37 °C and in 5% CO2. Transfections were carried out by using Lipofectamine Reagent (Invitrogen) according to the manufacturer's instructions. For live imaging, ∼105 cells were plated 24 h before experiments onto 35-mm glass bottom dishes (WillCo-dish GWSt-3522). For energy depletion measurements, cells were incubated in medium supplemented with sodium azide and 2-deoxy-d-glucose, as described elsewhere (22).
Cloning, Extraction, and Purification of Recombinant Proteins
Importin β was amplified by using PCR and ligated to pGEX-6P1 vector into BamHI and NotI sites. Importin α was subcloned into pGEX-6P1 vector into EcoRI and SalI sites. Expression of importin α and importin β recombinant proteins were induced in the Escherichia coli BL21 DE3 strain (Invitrogen) growing in the log-phase upon treatment with 1 mm isopropy-B-d-galactoside for 14 h at 20 °C. Bacteria expressing recombinant proteins were recovered by centrifugation, resuspended in TE lysis buffer (50 mm Tris-HCl pH 8.3, 1 mm EDTA, 2 mm DTT, 500 mm NaCl, 1 mm PMSF, and protease inhibitors and lysed on ice by sonication. Lysates were clarified by centrifugation. The resulting supernatant was incubated with glutathione-agarose beads at 4 °C with gentle rotation. cDNA encoding for TatRRR-EGFP, TatGGG-EGFP, NLSSV40-EGFP, and EGFP (22) were cloned by PCR into pASK-IBA33plus His Tag vectors (IBA vectors). Protein expression is induced upon addition of 200 μg anhydrotetracycline per 1 liter of E. coli shaking culture (A550 nm of 0.5). Purification of His6 tag proteins was performed according to standard protocols by using gravity flow nickel-nitrilotriacetic acid Superflow columns (IBA BiotagTechnology).
In Vitro Protein-Protein Binding Assays
First, His6-tagged fusion proteins were incubated with glutahione-agarose beads for 1 h at 4° C to avoid aspecific binding of fusion proteins to the matrix. GST-tagged importin α and GST-tagged importin β were incubated with a stoichiometric amount of His6-tagged fusion proteins (EGFP-HIS tag, NLSSV40-EGFP-HIS tag, TatRRR-EGFP-HIS tag, and TatGGG-EGFP-HIS tag) in IP buffer (50 mm Hepes, pH 7.4, 150 mm NaCl, 1.5 mm MgCl2, 1 mm EGTA, 20 mm NaF, 10% glycerol, 1% Nonidet P-40, and protease inhibitors) at 4 °C for 2 h. Then beads were washed four times with IP buffer and incubated with 100 μl of 4× SDS gel loading buffer at 95 °C for 5 min. Proteins were analyzed by SDS-PAGE and Western blotting. Filter was incubated with anti-GFP monoclonal antibody (JL-8, Clontech, Mountain View, Ca). Purified proteins were also analyzed by Comassie Blue staining.
Fluorescence Microscopy and Concentration Analysis
Cell fluorescence was measured using a Leica TCS SP2 inverted confocal microscope (Leica Microsystems AG, Wetzlar, Germany) interfaced with an argon laser for excitation at 488 nm, and with a HeNe laser for excitation at 561 nm. Glass-bottomed Petri dishes containing transfected cells were mounted in a temperature-controlled chamber at 37 °C (Leica Microsystems) and viewed with a 40 × 1.25 numerical aperture oil-immersion objective (Leica Microsystems). Images were collected at low excitation power and monitoring emission by means of the acousto-optical beam splitter detection system of the confocal microscope. The following collection ranges were adopted: 500–550 nm (EGFP) and 580–650 nm (mCherry). The global concentrations of intracellular EGFP- and mCherry-linked proteins were determined by using the synthetic adduct fluorescein-glycine, as described thoroughly in a previous publication (24).
FRAP Experiments
Each FRAP experiment started with a four-time line-averaged image (prebleach) of the cell followed by a single-point bleach (nonscanning) near the center of the nucleus with laser pulse at full power to photobleach most of the nuclear fluorescence. Fluorescence recovery was measured by starting a time-lapse acquisition within few milliseconds after bleaching, with the imaging settings described above. Hence, under the assumption of fluorescence proportionality to concentration, the collected FRAP curves in both compartments were fitted to a monoexponential equation (Equation 1),
where F0 and F∞ label the fluorescence intensity collected at time 0 and asymptotically after bleaching, respectively. Fluorescence values were normalized by the signal of the entire cell at the same time to correct for bleaching caused by imaging and by prebleach fluorescence to verify the presence of an immobile fraction of fluorescent molecules within the nucleus. As described in Ref. 24, the excess flux of cargo toward the nucleus solely due to active transport (ΦC→N), the concentration of cargo molecules bound to the importin carrier ([NLS:Imp]) in the cytoplasm, and the nuclear envelope permeability (PX) are linked by Equation 2,
where vC→N (μm3/s) is the maximum rate for active transport toward the nucleus (i.e. the rate achievable when all of the cargo molecules are bound to import carriers). Thus, the calculated ΦC→N was plotted against the cytoplasmic cargo concentration (CNLS/Tat) for each cell. Finally, if we assume a single binding equilibrium between the NLS/Tat cargo and the import carrier, [NLS:Imp] can be expressed as a function of CNLS/Tat, the global cytoplasmic concentration of import carriers (CImp), and the binding dissociation constant (KD*), according to Equation 3.
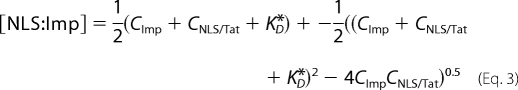 |
To recover the biochemically relevant parameter KD*, we fitted the ΦC→N versus CNLS/Tat curve with Equations 2 and 3, setting CImp = 1 μm (see Ref. 24 for more details).
FLIM Measurements
FLIM measurements were performed illuminating the sample with a 468-nm pulsed laser diode at 40 MHz repetition rate. Fluorescence emission was detected by means of fast photon-counting heads (H7422P-40, Hamamatsu) and time-correlated single photon counting electronics (SPC-830, Becker & Hickl, Berlin, Germany) at 500–540 nm (band pass filter 510AF23, Omega Optical, Brattleboro, VT). Measurements were performed in living cells with the confocal system described previously with a 40× oil immersion objective (Leica Microsystems). Additional measurements were carried out using a Leica TCS SP5 inverted confocal microscope (Leica Microsystems AG, Wetzlar, Germany) coupled to a PicoQuant single-molecule detection module. Laser power was adjusted to yield photon-counting rates of ∼105 cps. Fluorescence decay was analyzed by the SPC Image (Becker & Hickl, Berlin Germany) software package. Time-correlated single photon counting detection was used to generate a lifetime map by fitting the fluorescence decay curve in each pixel of the image. We used EGFP as the “donor” fluorophore (high brightness and photostability and monoexponential lifetime) fused to transport carriers (Impα and Impβ) and mCherry as the “acceptor” (fast maturation, large absorption, and high photostability) fused to the candidate localization sequences (NLS, TatGGG, and TatRRR). Fluorescence decay curves of biological samples containing only isolated donor molecules (i.e. donor alone or in the presence of a noninteracting acceptor) were fitted within a monoexponential decay model. The result of the fitting procedure is thus a single fluorescence lifetime (τF), characteristic of that donor form. When a mix of unbound and bound donor molecules was present (i.e. donor in presence of an interacting acceptor), lifetime data were fitted to a bi-exponential decay law (Equation 4),
where XB and XF (XB + XF = 1) are the amplitude coefficients corresponding to the individual lifetime components of bound (τB) and free donor molecules (τF). We set τF to the calculated value for isolated donor (see above) and analyzed the distribution of the average lifetime τm, according to the relation τm = (XB × τB + XF × τF)/(XB + XF). A decrease in the τm value highlights the appearance of a fraction of donor molecules bound to acceptor molecules. To quantitatively address the equilibrium constant (KD*) of Impα-TatGGG and Impβ-TatGGG interactions, we used a combination of fluorophore concentration analysis and FLIM measurements, as described previously (24). Briefly, we calculated the characteristic lifetime of the complex (τB) by using the ATP depletion assay (see supplemental Fig. 2); then, by setting τB and τF to the calculated values, we extracted XB and XF molar fractions from Equation 4. Finally, XB and XF were combined with EGFP/mCherry absolute concentrations to derive the actual KD* value of the studied interactions by Equation 5.
RESULTS
Subcellular Localization of Fusion Proteins
First, the fluorescent chimeras used in this study were individually transfected in CHO-K1 cells and analyzed by confocal microscopy (Fig. 1). EGFP-tagged Impα and Impβ were detected in both the nucleus and the cytoplasm, with a local enrichment on the nuclear envelope (Fig. 1, A and B). This localization is consistent with their ability to shuttle across the nuclear envelope (28) and bind protein components of the nuclear pore complex (29). As expected, NLSSV40-mCherry protein was predominantly localized in the nucleus (Fig. 1C), owing to the contribution of carrier-mediated active transport, whereas passively diffusing TatRRR-mCherry is uniformly distributed across nuclear envelope, with a slight enrichment in nucleolar fluorescence (Fig. 1D). Mutation of the last three arginines into glycines conferred to the Tat peptide the ability to perform active transport; accordingly, TatGGG-mCherry was localized predominantly in the nucleus (Fig. 1E). The behavior of mCherry-tagged localization signals is in keeping with previous results obtained on their EGFP-tagged counterparts (22, 23).
FIGURE 1.

Subcellular localization of fusion proteins. Confocal images of transfected EGFP-Impα (A), Impβ-EGFP (B), NLSSV40-mCherry (C), TatRRR-mCherry (D), and TatGGG-mCherry (E) are shown. Scale bar, 10 μm.
Analysis of Impα/β Direct Binding to Tat Peptides in Intact Cells
As a first test, we performed FLIM analysis of cells expressing the donor molecule alone (EGFP-Impα or Impβ-EGFP, as shown in Fig. 2, A and B). As expected, the obtained lifetime decays are well fitted to a monoexponential function (see “Experimental Procedures”) yielding the characteristic decay constant of the unquenched donor (τF = 2.56 ± 0.03 ns for EGFP-Impα, 2.57 ± 0.03 ns for Impβ-EGFP, mean ± S.D. for n = 12 cells). Subsequently, we measured EGFP lifetime in cells co-expressing EGFP-Impα and NLSSV40-mCherry (Fig. 2C). We quantitatively addressed this interaction in a previous publication (24); in this context, it is displayed as a control for the classical import pathway through the adaptor carrier Impα. As expected, two exponential components (Equation 4) are necessary for a satisfactory fit of the EGFP lifetime decays (supplemental Fig. S1), revealing the appearance of a fraction of NLSSV40-bound Impα undergoing FRET (Fig. 2C). The average lifetime values measured show FRET occurrence in all analyzed cells (τm = 2.37 ± 0.13 ns, n = 45). An analogous set of measurements on cells co-expressing Impβ-EGFP and NLSSV40-mCherry (Fig. 2D), yielded no detectable FRET signal in n = 24 analyzed cells (accordingly, EGFP lifetime was adequately fitted by a monoexponential function, τF = 2.55 ± 0.13 ns, see also example in supplemental Fig. S1). This result confirms that this classical monopartite NLS does not bind directly to Impβ; in this case, in fact, the transfected Impβ-EGFP and NLSSV40-mCherry can form a ternary complex with the endogenous Impα. According to the molecular model, Impα binds directly to NLSSV40-mCherry, whereas importin β binding domain (IBB)-EGFP turn binds to the IBB of Impα. Note that FRET efficiency varies nonlinearly with the distance between fluorophores; thus, we can argue that the adaptor protein Impα leads to a distance between tagged NLSSV40 and Impβ in the ternary complex that strongly reduces FRET efficiency. The same FLIM-based approach was used to test the capability of Tat-based sequences to bind importins. Consistently with all the results we obtained so far, we found no detectable interaction of TatRRR-sequence with either Impα or Impβ in intact cells (examples are reported in Fig. 2, E and F), as EGFP decays were adequately fitted by a monoexponential function yielding lifetime value close to that of the donor alone (τF = 2.55 ± 0.03 ns, n = 12 for Impα and τF = 2.55 ± 0.02 ns, n = 12 for Impβ). On the other hand, we observed direct interaction of TatGGG mutant with both Impα and Impβ (Fig. 2, G and H); FLIM measurements yielded shorter average lifetimes compared with the donor-only sample (τm = 2.37 ± 0.11 ns, n = 21 for Impα and τm = 2.32 ± 0.13 ns, n = 26 for Impβ), revealing the presence of a fraction of TatGGG-bound import carriers.
FIGURE 2.

FLIM analysis of Impα/β direct binding to nuclear localization signals. A and B, intensity image (gray), lifetime image (color), and lifetime distribution histogram (graphs) of EGFP-Impα and Impβ-EGFP, respectively. C and D, same FLIM analysis shown in A and B applied to NLSSV40-mCherry co-transfected with either EGFP-Impα or Impβ-EGFP. E and F, FLIM analysis of TatRRR-mCherry co-transfected with EGFP-Impα and Impβ-EGFP, respectively. G and H, TatGGG-mCherry co-transfected with either EGFP-Impα or Impβ-EGFP, respectively. Scale bar in all images, 10 μm. Monoexponential fit of decay curves is applied in A, B, D, E, and F (τF is displayed). A biexponential fit is applied elsewhere (τm is displayed).
Calculation of TatGGG-Impα and TatGGG-Impβ Effective Dissociation Constant (KD*) from FLIM Data
As shown recently for the case of NLSSV40-Impα interaction, affinity values can be extracted from FLIM data (24). Briefly, the KD* value can be calculated if the characteristic lifetime of the complex (τB) is known (τF is easily derived by measuring the unquenched donor, as shown above). The τB values for TatGGG-Impα and TatGGG-Impβ complexes were derived by FLIM measurements in energy-depleted cells (supplemental Fig. S2) and combined to the corresponding τF values to calculate the molar fraction of bound (XB) and unbound (XF) import carriers (Equation 4, “Experimental Procedures”). These XB and XF values can be used together with intracellular protein concentrations to derive the effective KD* through Equation 5. For what concerns TatGGG binding to Impα, we found two characteristic ranges of affinity depending on Impα cytoplasmic concentration (Table 1 and Fig. 3A). At an EGFP-Impα concentration close to the endogenous value (≤1 μm (30)), we obtained KD* = 26 ± 5 μm, whereas at high EGFP-Impα expression levels (>10 μm), we found KD* = 175 ± 35 μm. As recently discussed for NLSSV40-Impα (24), the two ranges of TatGGG affinity for Impα can be interpreted as the result of endogenous Impβ intervention in altering the binding affinity. This hypothesis was further strengthened by an in vitro binding assay using purified importins and the recombinant fusion protein NLSSV40-EGFP as a model substrate (supplemental Fig. S3). Remarkably, in the case of TatGGG binding to Impβ, we found only one characteristic equilibrium constant (KD* = 320 ± 75 μm, Table 1). This KD* value shows no significant correlation with the cytoplasmic carrier concentration (Fig. 3B) and confirms the absence of additional partners modulating Impβ affinity toward NLS-endowed molecules.
TABLE 1.
Affinity constants derived by FLIM
For what concerns TatGGG binding to Impα, were found for TatGGG-Impα binding depending on the Impα cytoplasmic concentration (≤1 μm or >10 μm). In the case of TatGGG binding to Impβ, only one affinity constant was extracted from FLIM data, independently of the carrier cytoplasmic concentration.
| Protein | KD* |
|---|---|
| μm | |
| EGFP-Impα | |
| ≤1 μm | 26 ± 5 |
| >10 μm | 175 ± 35 |
| Impβ-EGFP | |
| ≥1 μm | 320 ± 75 |
FIGURE 3.

Affinity values for TatGGG interaction with import carriers. A, the KD* value for TatGGG interaction with Impα is plotted against the calculated Impα cytoplasmic concentration. Two average values of affinity are clearly distinguishable; KD* ∼ 26 μm for low Impα levels (∼1 μm), where the endogenous Impβ may play a role in modulating Impα autoinhibition (schematic); KD* ∼ 175 μm for Impα levels >10 μm, where the contribution of endogenous Impβ can be considered negligible. B, in the case of the TatGGG interaction with Impβ, we find a broad distribution of affinity values (plot) but with no clear dependence on Impβ expression levels. This can be explained by the lack of any possible modulation for the direct interaction to Impβ carrier (schematic).
FRAP Analysis of TatGGG Nuclear Import Kinetics and Binding Specificity
To this point, we showed that TatGGG is a functional NLS capable of direct binding to both Impα and Impβ carriers in live cells, albeit with different relative affinities. Here, we validated our conclusions by investigating the kinetics of TatGGG nuclear import by FRAP and relating it to the thermodynamics of binding to Importins. Quantitative FRAP analysis of TatGGG-GFP nucleocytoplasmic shuttling was performed as described in “Experimental Procedures” (example in Fig. 4A). By means of our mathematical model, we could derive the excess flux of cargo toward the nucleus solely due to active transport (ΦC→N, mol/s) and plot it in Fig. 4B against the cytoplasmic cargo concentration (Ccargo) for each cell. We used the variability of protein expression levels to examine the relationship between cargo concentration and import fluxes. As demonstrated previously (24) and showed here by the green dots in Fig. 4B, NLSSV40-GFP import fluxes follow a simple linear relationship with respect to the available cytoplasmic cargo concentrations up to 10–15 μm of NLSSV40-GFP, where they begin to level off, reaching a plateau for high cargo concentrations. Fitting the ΦC→N plot to Equations 2 and 3 (“Experimental Procedures”) yields an estimate of the maximum rate for active transport toward the nucleus (vC>N ∼ 300 μm3/s) and, in the case of NLSSV40, the binding dissociation constant to Impα (KD*∼16 μm). Remarkably, replacement of NLSSV40 with TatGGG leads to a similar “saturation-like” behavior but to much higher maximum rates of cargo delivery to the nucleus (vC>N > 1000 μm3/s; compare plateau levels of red and green dots in Fig. 4B). This evidence points at the presence of a different molecular mechanism for TatGGG-driven import into the nucleus, relying on two importins rather than just one. Note that the difference between TatGGG and NLSSV40 becomes particularly evident for cargo concentrations above ∼50 μm. On the basis of the affinities for import carriers calculated by FLIM, we know that this behavior can be linked to TatGGG direct binding to Impβ; above 50 μm cargo concentration, in fact, we can assume that TatGGG binding to Impα (KD* ∼ 26 μm, by FLIM) almost reached saturation, whereas its binding to Impβ (KD*∼320 μm, by FLIM) starts to play a role in the nuclear import process. We confirmed this hypothesis by fitting TatGGG import fluxes above 50 μm cargo concentration; we obtained KD* = 285 ± 45 μm (supplemental Fig. S4), in keeping with FLIM. In Fig. 4C, we show that the purported additional interaction of TatGGG with Impβ is effectively leading to functional transport; TatGGG-GFP nuclear accumulation above 50 μm cargo concentrations is still sustained (Keq close to 2), whereas NLSSV40-GFP distribution is almost uniform in the cells. (Keq drops to ∼1 above 50 μm cargo concentration.) Furthermore, in Fig. 4D, we show that sequential addition of arginine residues to TatGGG decreases the relative affinity for the import carriers involved (see the change in slope for the ΦC→N curve) but not the maximum rate of transport allowed (plateau values of vC>N), thus suggesting a similar import mechanism for all tested sequences. This behavior supports the hypothesis that the first eight residues of the Tat peptide (YGRKKRRQ) operate as an NLS, whereas the remaining three arginine residues (RRR) hinder active transport. We can further clarify this point by showing that mutations introduced within the first eight residues are able to completely abolish the importin-binding capability of Tat peptides (supplemental Fig. S5).
FIGURE 4.

FRAP analysis of TatGGG-driven nuclear import kinetics. A, example FRAP measurement conducted on TatGGG-EGFP. Representative images are depicted. Scale bar, 10 μm. B, excess active fluxes (ΦC→N, mol/s) are calculated cell-by-cell and plotted against the corresponding cytoplasmic cargo concentration, obtaining the whole population data plot for TatGGG-EGFP (red dots) (compared here with NLSSV40-EGFP, green dots). C, plot of calculated Keq against cargo cytoplasmic concentration. D, ΦC→N plot for TatGGG, TatRGG, and TatRRG mutants, showing the decrease in overall affinity (slope of the curve) and the conservation of the maximal import rate allowed (plateau).
In Vitro Analysis of Impα/β Direct Binding to Tat Peptides
The relationship between the import properties of TatGGG to those of its precursor TatRRR is crucial and deserves further investigation. Because TatRRR is unable to perform active transport in intact cells, we address its interaction with import carriers in vitro in the absence of competitors. To this end, we performed a binding assay using recombinant purified proteins. His6-tagged TatRRR-EGFP, TatGGG-EGFP, NLSSV40-EGFP, and EGFP as well as GST-tagged Impα and Impβ were expressed bacterially and purified as described under “Experimental Procedures” (Fig. 5A). The proteins were mixed, and GST fusion proteins were pulled down by means of glutathione-Sepharose beads. The bound proteins were detected by Western blotting by means of an EGFP antibody. Remarkably, both Impα and Impβ interact with TatRRR-EGFP in our assay (Fig. 5B, lanes 2 and 3). The TatGGG mutant shows approximately the same ability of TatRRR and NLSSV40-EGFP to interact with Impα (lane 5, compare with lanes 3 and 8), but partly loses its ability to directly target Impβ (lane 6); quantification of the obtained signals, in fact, reveals an almost 6-fold decrease of binding capability in the latter case. Despite the different experimental conditions, this behavior closely resembles what observed in the actual cellular environment, where TatGGG direct binding to Impα (KD* = 175 μm, in the range where Impβ contribution is not relevant) is less efficient than to Impβ (KD* = 320 μm). This set of results also definitely clarifies that the observed unconventional binding properties are maintained by the wild-type Tat peptide, but only in the absence of cytosolic and nuclear factors.
FIGURE 5.

In vitro binding assay. A, purified His-tagged proteins composed by TatRRR, TatGGG, and NLSSV40 sequences fused to EGFP and purified recombinant Impα and Impβ fused to glutathione S-transferase. B, Western blot (WB) filter showing the direct interaction of TatRRR and TatGGG with Impα and Impβ. The NLS of SV40 was used as a control for the interaction with Impα and not Impβ, whereas the His-tagged EGFP protein was used as a control for the absence of interaction with import carriers.
DISCUSSION
The thorough understanding of any signal-dependent nuclear import mechanism requires a quantitative analysis of both the thermodynamic and kinetic aspects of the phenomenon. To this end, we recently presented a method that combines FLIM and FRAP measurements with protein concentration calibration and showed its application to the analysis of the well known NLSSV40-Impα interaction (24). Here, we apply the same approach to the study of Tat peptide-mediated nuclear transport. The motivation for this study is our demonstration that the mechanism driving TatRRR nuclear permeation in live cells is passive diffusion (22), a result contrasting with previous in vitro studies that suggest that active processes are involved (16, 17). We recently linked this discrepancy to the observation that the first eight residues of Tat peptide (YGRKKRRQ) can indeed operate as an NLS in engineered mutants (e.g. TatGGG), but the remaining three arginine residues (RRR) hinder active transport by promoting binding to intracellular moieties, including RNAs (23). Accordingly, we show here that mutation of the purported NLS stretch of Tat leads to inhibition of active import (i.e. inhibition of importin-binding capabilities) (supplemental Fig. S5). However, the identity of the nuclear import carriers potentially involved in Tat peptide transport is still a matter of debate. In this article, we combine FLIM microscopy and protein concentration calibration to directly monitor Tat peptide-importin interactions and measure the corresponding effective dissociation constant (KD*) in the actual cellular environment. In keeping with all our previous results in live cells, the wild-type TatRRR sequence shows no detectable interaction with importins. On the contrary, we find that the mutated TatGGG sequence is a direct target of both Impα and Impβ. It is worth noting that the KD* of TatGGG-Impα binding is dependent on Impα expression level, analogously to what we observed for the NLS of SV40 (24). This effect is a consequence of the fact that endogenous Impβ can modulate this affinity through direct binding to the autoinhibitory IBB domain of Impα (8, 24). On the contrary, the KD* value of TatGGG-Impβ binding is not dependent on the Impβ expression level, as expected for a nonmediated interaction. Thanks to FRAP experiments, we obtained independent proof of TatGGG transport mechanism. By measuring TatGGG import rate as a function of cargo concentration, we, in fact, recovered a saturation behavior markedly different from that of the classical NLS of SV40. In particular, the much higher TatGGG import rates suggest the presence of a different molecular mechanism of transport that we argued relies on two importins rather than just one. Accordingly, fitting FRAP data to our model of nucleocytoplasmic shuttling revealed an additional (low affinity) interaction of TatGGG with the import machinery (i.e. with Impβ, as suggested by FLIM). This interaction proved to be functional, as showed by the sustained nuclear accumulation of TatGGG at high cargo concentrations (>50 μm), compared with the NLSSV40 case. Furthermore, the FRAP assay was used to test the effect of addition of arginines to TatGGG; in addition to showing the expected decrease of affinity for the import machinery, our results suggest that all the Tat mutants tested share the same import mechanism. We thus speculate that TatGGG shares TatRRR properties and that these are merely progressively unveiled by arginine substitution by restoring its capability to bind importins. Finally, we emphasize that the FRAP assay validates FLIM results in the absence of Impα/β overexpression. This in turn discounts the possibility that many other cellular importins bind Tat peptides (perhaps with high affinity) and thereby mediate transport under conditions when Impα/β are not overexpressed, as this would produce a detectable effect on the slope of ΦC→N versus Ccargo. Our hypothesis of a dual functionality of Tat peptide sequence implies that the importin-binding capability observed for TatGGG in living cells be fully recovered for TatRRR in vitro. In the latter case, the absence of cellular components would make the RRR stretch irrelevant and let the “YGRKKRRQ” domain operate as an NLS. This prediction was tested and confirmed based on an in vitro binding assay. We observed that the wild-type Tat peptide can function as an NLS with unconventional properties because it is a direct target of both Impα and Impβ. Interestingly, we found that TatRRR binds Impα and Impβ with comparable affinity, whereas TatGGG shows a clear preference for Impα. Although the latter result is consistent with the data reported in living cells (dissociation constants calculated by FLIM), the former reveals that the YGRKKRRQ and RRR domains act cooperatively in determining importin-binding specificity and affinity (as we already demonstrated for the complementary binding to intracellular moieties (23)). The observation that Impα directly contributes to Tat peptide transport to the nucleus is new but somewhat expected, as it was recently showed that the “KKRR” domain is widely conserved as an optimal target of Impα (31). We believe that these findings complement previous reports on the Tat peptide properties and lead to a coherent picture on the molecular details of its nuclear import process. More importantly, they provide useful knowledge for the rational design and the accurate in vivo testing of a new generation of localization sequences.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- NLS
- nuclear localization sequence
- FLIM
- fluorescence lifetime imaging microscopy
- FRAP
- fluorescence recovery after photobleaching
- EGFP
- enhanced GFP
- Impα
- importin α.
REFERENCES
- 1. Weis K. (2003) Cell 112, 441–451 [DOI] [PubMed] [Google Scholar]
- 2. Fahrenkrog B., Aebi U. (2003) Nat. Rev. Mol. Cell Biol. 4, 757–766 [DOI] [PubMed] [Google Scholar]
- 3. Ribbeck K., Görlich D. (2002) EMBO J. 21, 2664–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalderon D., Roberts B. L., Richardson W. D., Smith A. E. (1984) Cell 39, 499–509 [DOI] [PubMed] [Google Scholar]
- 5. Robbins J., Dilworth S. M., Laskey R. A., Dingwall C. (1991) Cell 64, 615–623 [DOI] [PubMed] [Google Scholar]
- 6. Lange A., Mills R. E., Lange C. J., Stewart M., Devine S. E., Corbett A. H. (2007) J. Biol. Chem. 282, 5101–5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Conti E., Uy M., Leighton L., Blobel G., Kuriyan J. (1998) Cell 94, 193–204 [DOI] [PubMed] [Google Scholar]
- 8. Fanara P., Hodel M. R., Corbett A. H., Hodel A. E. (2000) J. Biol. Chem. 275, 21218–21223 [DOI] [PubMed] [Google Scholar]
- 9. Bayliss R., Littlewood T., Stewart M. (2000) Cell 102, 99–108 [DOI] [PubMed] [Google Scholar]
- 10. Görlich D., Panté N., Kutay U., Aebi U., Bischoff F. R. (1996) EMBO J. 15, 5584–5594 [PMC free article] [PubMed] [Google Scholar]
- 11. Tyagi M., Rusnati M., Presta M., Giacca M. (2001) J. Biol. Chem. 276, 3254–3261 [DOI] [PubMed] [Google Scholar]
- 12. Serresi M., Bizzarri R., Cardarelli F., Beltram F. (2009) Anal Bioanal Chem 393, 1123–1133 [DOI] [PubMed] [Google Scholar]
- 13. Stauber R. H., Pavlakis G. N. (1998) Virology 252, 126–136 [DOI] [PubMed] [Google Scholar]
- 14. Berkhout B., Silverman R. H., Jeang K. T. (1989) Cell 59, 273–282 [DOI] [PubMed] [Google Scholar]
- 15. Brady J., Kashanchi F. (2005) Retrovirology 2, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Efthymiadis A., Briggs L. J., Jans D. A. (1998) J. Biol. Chem. 273, 1623–1628 [DOI] [PubMed] [Google Scholar]
- 17. Truant R., Cullen B. R. (1999) Mol. Cell. Biol. 19, 1210–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brooks H., Lebleu B., Vivès E. (2005) Adv. Drug Deliv. Rev. 57, 559–577 [DOI] [PubMed] [Google Scholar]
- 19. Calnan B. J., Biancalana S., Hudson D., Frankel A. D. (1991) Genes Dev. 5, 201–210 [DOI] [PubMed] [Google Scholar]
- 20. Delling U., Roy S., Sumner-Smith M., Barnett R., Reid L., Rosen C. A., Sonenberg N. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 6234–6238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weeks K. M., Crothers D. M. (1991) Cell 66, 577–588 [DOI] [PubMed] [Google Scholar]
- 22. Cardarelli F., Serresi M., Bizzarri R., Giacca M., Beltram F. (2007) Mol Ther 15, 1313–1322 [DOI] [PubMed] [Google Scholar]
- 23. Cardarelli F., Serresi M., Bizzarri R., Beltram F. (2008) Traffic 9, 528–539 [DOI] [PubMed] [Google Scholar]
- 24. Cardarelli F., Bizzarri R., Serresi M., Albertazzi L., Beltram F. (2009) J. Biol. Chem. 284, 36638–36646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N., Palmer A. E., Tsien R. Y. (2004) Nat. Biotechnol. 22, 1567–1572 [DOI] [PubMed] [Google Scholar]
- 26. Miyamoto Y., Hieda M., Harreman M. T., Fukumoto M., Saiwaki T., Hodel A. E., Corbett A. H., Yoneda Y. (2002) EMBO J. 21, 5833–5842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ciciarello M., Mangiacasale R., Thibier C., Guarguaglini G., Marchetti E., Di Fiore B., Lavia P. (2004) J. Cell Sci. 117, 6511–6522 [DOI] [PubMed] [Google Scholar]
- 28. Görlich D., Mattaj I. W. (1996) Science 271, 1513–1518 [DOI] [PubMed] [Google Scholar]
- 29. Görlich D., Kostka S., Kraft R., Dingwall C., Laskey R. A., Hartmann E., Prehn S. (1995) Curr. Biol. 5, 383–392 [DOI] [PubMed] [Google Scholar]
- 30. Percipalle P., Butler P. J., Finch J. T., Jans D. A., Rhodes D. (1999) J. Mol. Biol. 292, 263–273 [DOI] [PubMed] [Google Scholar]
- 31. Yang S. N., Takeda A. A., Fontes M. R., Harris J. M., Jans D. A., Kobe B. (2010) J. Biol. Chem. 285, 19935–19946 [DOI] [PMC free article] [PubMed] [Google Scholar]