

Abstract
TraI, a bifunctional enzyme containing relaxase and helicase activities, initiates and drives the conjugative transfer of the Escherichia coli F plasmid. Here, we examined the structure and function of the TraI helicase. We show that TraI binds to single-stranded DNA (ssDNA) with a site size of ∼25 nucleotides, which is significantly longer than the site size of other known superfamily I helicases. Low cooperativity was observed with the binding of TraI to ssDNA, and a double-stranded DNA-binding site was identified within the N-terminal region of TraI 1–858, outside the core helicase motifs of TraI. We have revealed that the affinity of TraI for DNA is negatively correlated with the ionic strength of the solution. The binding of AMPPNP or ADP results in a 3-fold increase in the affinity of TraI for ssDNA. Moreover, TraI prefers to bind ssDNA oligomers containing a single type of base. Finally, we elucidated the solution structure of TraI using small angle x-ray scattering. TraI exhibits an ellipsoidal shape in solution with four domains aligning along one axis. Taken together, these data result in the assembly of a model for the multidomain helicase activity of TraI.
Keywords: DNA Helicase, Nucleic Acid Enzymology, Protein DNA-Interaction, Protein Structure, X-ray Scattering, F Plasmid TraI, Bacterial Conjugation
Introduction
Conjugative plasmid transfer is a central mechanism for the horizontal exchange of genetic material between bacterial cells, as well as for the spread of antibiotic resistance genes and virulence factors (1–3). Conjugative plasmid transfer requires both a DNA relaxase and a DNA helicase. The relaxase initiates DNA transfer by cleaving the transferred strand at a specific site within the oriT, forming a 5′-phosphotyrosine intermediate. Following nicking, the helicase uses the energy from ATP hydrolysis to unwind the plasmid and drive the transfer of DNA into the recipient cell. The relaxase completes plasmid transfer by breaking the covalent phosphotyrosine linkage and releasing the transferred DNA for replication in the recipient (2, 4, 5).
The relaxase and helicase activities necessary for F plasmid transfer are located on a 190-kDa multidomain protein, TraI (6, 7). TraI requires the assistance of two additional F plasmid-encoded proteins, TraY and TraM, as well as integration host factor for conjugative transfer. These four proteins bind to the 500-bp oriT site and form a protein complex called the relaxosome (2). TraI contains three major functional domains, including the N-terminal relaxase domain (residues 1–309), a central helicase domain (residues 310 to 1476), and a C-terminal domain (CTD,4 residues 1477–1756) that is responsible for interactions with TraY and TraM (Fig. 1) (8–11). The region between residues 310 and 950 is not fully characterized, although it has been proposed to share a helicase-like fold (see below). The covalent linkage between these functional domains is required for efficient conjugation (12).
FIGURE 1.

Schematic representations of the domain organization of TraI and the TraI constructs used in this study. The N-terminal relaxase domain contains residues 1–309; helicase domain contains residues 310–1476, and the CTD contains residues 1477–1504. The asterisk indicates the location of the catalytic residue Lys-998. RecD-like domain I (residues 303–844) and RecD-like domain II (residues 830–1473) identified by sequence analysis (17) are highlighted by dark lines. TraI constructs are shown in open boxes, whose first and last residues are indicated.
TraI is a superfamily I helicase with ssDNA-dependent NTPase and helicase activities (13, 14). The translocation of TraI along DNA has 5′ to 3′ polarity (15, 16). TraI requires a 5′ single-stranded overhang for the initiation of DNA unwinding (15). In addition, TraI functions as a highly processive helicase as a monomer (14). Unlike other helicases, the processivity of TraI helicase requires neither oligomerization nor the presence of a processivity factor. The underlying mechanism of the processivity of TraI has remained unclear (14).
Although the interaction of TraI with DNA is essential to its helicase activity, little is known about the DNA-binding properties of the TraI helicase. Matson and co-workers (8) showed that deletion of the region 309–349 disrupts helicase-associated ssDNA binding and the helicase activity. Recent work from Dostal and Schildbach (17) suggested that the helicase domain of TraI consists of two RecD-like subdomains (Fig. 1). The C-terminal RecD-like domain (residues 830–1473) contains helicase motifs and is the motor domain, whereas the N-terminal RecD-like domain (residues 303–844) lacks critical helicase motifs and likely specializes in ssDNA binding (17). Numerous fundamental aspects of the interaction of the TraI helicase with DNA, such as the length of DNA bound by TraI, the intrinsic binding affinity, binding cooperativity, dependence of affinity on ionic strength, nucleotide binding, and base specificity, are still unclear. This knowledge is important for the quantitative understanding of the interaction between TraI helicase and DNA, as well as the molecular mechanism and regulation of TraI helicase activity.
Here, we used fluorescence anisotropy-based equilibrium binding assays to investigate TraI helicase-associated DNA-binding properties. Our studies have elucidated the intrinsic binding affinity, binding cooperativity, and the lengths of ssDNA and dsDNA bound by TraI. Also, we identified the location on TraI of the dsDNA binding site. Furthermore, we investigated the effect of ionic strength, nucleotide binding, and base composition on the affinity of TraI for DNA. Finally, we examined the spatial organization of TraI domains in solution. Together, these results advance our understanding of the interaction between TraI helicase and DNA, providing insights into the molecular mechanism by which TraI performs its helicase function.
EXPERIMENTAL PROCEDURES
DNA Oligomers
DNA oligomers used in this study were purchased from Integrated DNA Technology (IDT, Coralville, IA). Labeled oligomers have the fluorescent probe 6-FAMTM covalently attached to their 5′ end and were purified using HPLC by IDT. Unlabeled oligomers were desalted without further purification. The sequences of DNA oligomers are listed in Table 1. All DNA oligomers were dissolved in oligomer annealing buffer containing 50 mm Tris-HCl, pH 8.0, 50 mm NaCl, and 5 mm MgCl2. To prepare duplex DNA, two complementary DNA oligomers were mixed in a 1:1 molar ratio, heated to 95 °C, and slowly cooled down to 20 °C with a speed of 1 °C/min in a thermocycler.
TABLE 1.
Sequences of DNA oligomers used in this study
6FAM, or 6-carboxyfluorescein, is a fluorescent probe that is covalently attached to the 5′ end of DNA oligomers. The subscript indicates the length of the DNA oligomer. For a double-stranded DNA (dsDNA) oligomer, the sequence of the complementary strand is not shown.
| FL-T17 | 6FAM-5′-TTTTTTTTTTTTTTTTT-3′ |
| 25-mer | 5′-CACTGACCGTCTGACTGCGATCCGA-3′ |
| 39-mer | 5′-TCGGATCGCAGTCAGATGGTAAGAGAGACGCATAGATGC-3′ |
| 45-mer | 5′GCGAACTGTCGAGTCGGCATCCGGATCTAGGGTAACCGGTACTGC-3′ |
| 51-mer | 5′TCGGATCGCAGTCAGATGGTAAGAGAGACGCATAGATGCTGAGTGAGAGAT-3′ |
| 63-mer | 5′TCGGATCGCAGTCAGATGGTAAGAGAGACGCATAGATGCTGAGTGAGAGATGCTCAGGTACAG-3′ |
| FL-dsDNA15 | 6FAM-5′-TCG GAT CGC AGT CAG-3′ |
| dsDNA9 | 5′-TCG GAT CGC-3′ |
| dsDNA11 | 5′-TCG GAT CGC AG-3′ |
| dsDNA15 | 5′-TCG GAT CGC AGT CAG-3′ |
| dsDNA19 | 5′-TCG GAT CGC AGT CAG ACG G-3′ |
| dsDNA21 | 5′-TCG GAT CGC AGT CAG ACG GTC-3′ |
| dsDNA23 | 5′-TCG GAT CGC AGT CAG ACG GTC AG-3′ |
| dsDNA27 | 5′-TCG GAT CGC AGT CAG ACG GTC AGT GAC-3′ |
DNA Binding Buffers
Standard DNA binding buffer contained 25 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5 mm magnesium acetate, and 0.1% BSA. Buffer A contained 25 mm Tris-HCl, pH 7.5, 50 mm NaCl, 5 mm magnesium acetate, and 0.1% BSA. Buffer B contained 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5 mm magnesium acetate, and 0.1% BSA. Buffer C contained 25 mm Tris-HCl, pH 7.5, and 0.1% BSA.
Protein Constructs, Expression, and Purification
Standard ligation independent cloning techniques, as described by Stols et al. (18), were employed in the construction of expression plasmids encoding the segments of TraI used in this study. The amplified DNA fragments were treated and cloned into empty pMCSG7-Lic-MBP expression vector (18). The His6 maltose-binding protein tag was cleaved off after purification using tobacco etch virus protease. All expression plasmids used in this study were sequence-verified.
The expression plasmids were transformed into Escherichia coli BL21 (DE3) Gold (Stratagene). Bacteria were grown in LB medium supplemented with 50 μg/ml ampicillin at 37 °C with shaking. After the A600 reached 0.6, isopropyl β-d-thiogalactoside was added to a final concentration of 0.2 mm, and bacteria were grown for another 12 h at 16 °C with shaking. Bacteria were harvested and resuspended in loading buffer (50 mm sodium phosphate, pH 7.6, 500 mm NaCl, 25 mm imidazole) supplemented with 0.5 mm EDTA, 0.1% Triton X-100, 1 mm PMSF, 1 tablet of a protease inhibitor mixture (Roche Applied Science), and 1 mg/ml lysozyme. After 1 h of incubation on ice, the resuspended cells were sonicated on ice for 2 min, and the lysate was centrifuged at 45,000 × g for 90 min at 4 °C. The supernatant was passed through a 0.2-μm filter (Millipore) and then loaded onto a 5-ml high performance HisTrapTM column (GE Healthcare), equilibrated with loading buffer. The column was washed with 100 ml of loading buffer before bound protein was eluted with elution buffer (50 mm sodium phosphate, pH 7.6, 500 mm NaCl, 500 mm imidazole). The eluted protein was pooled and loaded onto a HiPrepTM 26/10 desalting column (GE Healthcare) equilibrated with desalting buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm DTT, and 0.5 mm EDTA). Fractions containing protein were collected. Tobacco etch virus protease was added to the pooled protein fractions with a ratio of 1:100 (w/w) tobacco etch virus to TraI. After 16 h of incubation at 4 °C, the mixture was reloaded onto a 5-ml HisTrapTM column equilibrated with loading buffer. The flow-through fractions were collected and concentrated in a Centricon YM30 (Amicon) concentrator. Finally, concentrated protein was loaded on a HiLoadTM 16/60 Superdex 200 column (GE Healthcare) equilibrated with sizing buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5% glycerol). Protein-containing fractions were concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C. Purified protein was >95% pure by SDS-PAGE.
Small Angle X-ray Scattering (SAXS) and Data Analysis
SAXS data were collected for protein solutions and their matched buffers using the standard SAXS instrument at the beamline 18-ID (Bio-CAT, Advanced Photon Source, Argonne, IL). The experiments were carried out at 16 °C using an x-ray photon energy of 12 keV. The Mar165 CCD detector with an active area of ∼160 × 80 mm2 was used for data collection. The sample-to-detector distances were set to 2.3 m to make scattering vectors, q, range from 0.007 to 0.38 Å−1. The scattering vector here is defined as q = 4πsinθ/λ, where 2θ is the scattering angle. Samples were flowed back and forth during SAXS measurements with a Hamilton programmable syringe pump to minimize radiation damage. Twenty frames of short exposure (2 s) were taken on each sample and averaged to improve the signal/noise ratio. Data were reduced using Bio-CAT SAXS data reduction macros installed in the program Igor (WaveMetrics, Portland, OR). Guinier plots, made using the program Igor, were used for detection of aggregation in protein samples. Guinier approximation I(q) = I(0) exp(−q2Rg2/3) with the limits qRg <1.3 was used to determine the radius of gyration, Rg, and the scattering intensity extrapolated to zero angle I(0). The molecular weight of TraI was determined from I(0) on a relative scale using cytochrome c as a reference. The pair distribution function, P(r), and the maximum dimension of the protein, Dmax, were computed by the program GNOM (19).
Ab Initio Shape Restoration and Structural Modeling
The low resolution shape of TraI was restored from the experimental SAXS profile using the program DAMMIN (20). The data within the q range of 0.0104 to 0.2129 Å−1 were used for the analysis. Forty individual models were generated by DAMMIN (21). The 20 models with the best χ2 were aligned using SUBCOMB and averaged using DAMMAVER (21). The averaged model was then filtered based on occupancy and volume using DAMFILT to generate the final model of TraI. The volumetric representation of the SAXS model was prepared by SITUS (22).
Crystal structures of the relaxase domain (residues 1–307) and the C-terminal domain (residues 1476–1628) were obtained from Protein Data Bank (PDB) using the accession numbers 1P4D and 3FLD, respectively. Homology models of TraI 309–842 and TraI 846–1473 were constructed by PHYRE (23) using the crystal structure of RecD from E. coli RecBCD complex (PDB accession number 1W36) as a template. The atomic models were placed into the ab initio shape by visual judgment using the program Chimera (24). The scattering intensities of the structural model were calculated using CRYSOL (25). The goodness of the structural model was assessed by CRYSOL based on the discrepancy (χ2) between the calculated scattering intensities and the experimental scattering intensities. The orientation and position of individual domains in the structural model were manually adjusted to minimize the discrepancy.
Direct DNA Binding Assays
Fluorescence anisotropy (FA)-based DNA binding assays were employed to study DNA binding by TraI as described by Shildbach and co-workers (26) with some modifications. To perform the direct DNA binding assay, a 6FAM-labeled DNA oligomer was mixed with increasing concentrations of protein in DNA binding buffer in 384-well plates (Corning Glass). The fluorescence anisotropy of each well was measured by a Pherastar plate reader (BMG Labtech, Offenburg, Germany) using the excitation and emission wavelengths of 485 and 520 nm, respectively. The fluorescence anisotropy (FA) was defined as shown in Equation 1,
where Ivv and Ivh represent the fluorescence intensity signal parallel and perpendicular to the excitation polarization. All experiments were done in triplicate. To calculate the macroscopic DNA binding constant (KN), normalized data were plotted as average FA versus total protein concentration and fit to Equation 2,
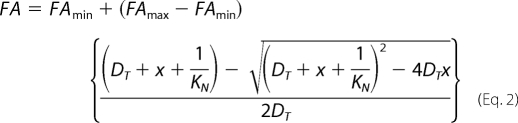 |
using nonlinear regression in SigmaPlot 11.0 (Systat Software, Inc.), where FA is the observed FA signal; FAmin is the FA signal in the absence of protein; FAmax is the FA signal in the presence of saturating concentration of protein; DT is the total concentration of 6FAM-labeled DNA; x is the total protein concentration; and KN is the macroscopic binding constant. The error bars represent the standard deviation of three replicates.
Macromolecule Competition Titration (MCT) Method
The binding of TraI to unlabeled DNA oligomers was investigated using the MCT method as described by Jezewska and Bujalowski (27). In brief, a 6FAM-labeled reference DNA oligomer (total concentration, DTR) was mixed with increasing concentrations of protein in DNA binding buffer in 384-well plates in the presence of a competing unlabeled DNA oligomer (total concentration, DTS). The fluorescence anisotropy of each well was measured using a Pherastar plate reader with the excitation and emission wavelengths of 485 and 520 nm, respectively. The total protein concentration in the presence of a competing DNA oligomer, PT1, was defined as Equation 3,
where (Σϴi)R, (Σϴi)S, and PF represent the binding density of the protein on the fluorescent DNA oligomer, the binding density on the unlabeled competing DNA oligomer, and the free protein concentration, respectively. In the absence of a competing DNA oligomer, Equation 3 can be simplified as shown in Equation 4,
When the same FA signal is observed in the absence and in the presence of a competing DNA oligomer, the binding density of the protein on the unlabeled competing DNA oligomer, defined as Equation 5, can be derived by solving the set of Equations 3 and 4.
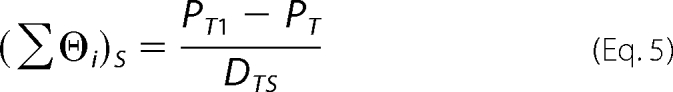 |
When (Σϴi)S is known, PF can be obtained by using Equation 6,
The binding stoichiometry is equal to the value of (Σϴi)S when FA reaches its maximum, as determined from the plot of FA versus (Σϴi)S.
Determination of the Binding Parameters
The model-independent and thermodynamically rigorous binding isotherm can then be constructed by plotting (Σϴi)S against PF. The binding isotherms were analyzed using the Epstein combinatorial approach for binding of a protein to a linear nucleic acid (28). The relation between (Σϴi) and PF can be expressed as Equations 7 and 8,
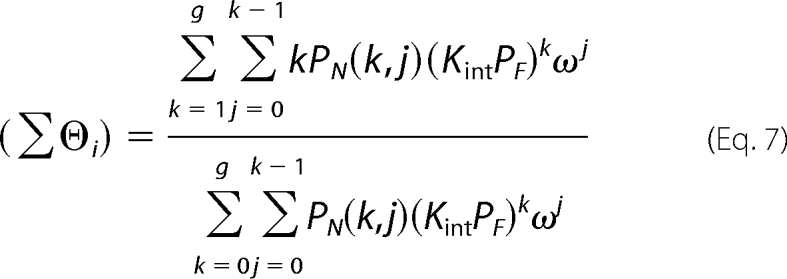 |
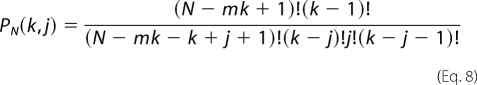 |
Where k is the number of protein bound per nucleic acid; g is the maximum number of k; N is the total length of the nucleic acid; m is the size of nucleic acid-binding site; Kint is the intrinsic binding constant; ω is the cooperativity parameter, and j is the number of cooperative contacts formed between protein molecules bound to the nucleic acid. The combinatorial factor PN (k, j) defines the number of distinct ways that k protein molecules, each of which form j cooperative contacts, can bind to a nucleic acid.
When the nucleic acid can maximally bind one protein molecule, Equations 7 and 8 can be simplified as Equations 9 or 10,
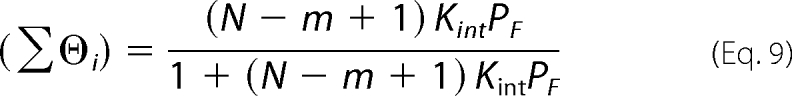 |
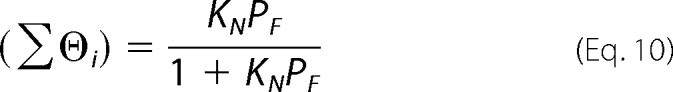 |
Competition Binding Assay
A 6FAM-labeled DNA oligomer was used as the reference DNA. Protein was mixed with the reference DNA and titrated with increasing concentrations of an unlabeled DNA oligomer (the competitor DNA). The decrease in fluorescence anisotropy values with the titration of the competitor DNA was monitored. The IC50, or the concentration of competitor DNA required to displace 50% of the complex formed by protein and reference DNA, was determined by plotting the anisotropy value as a function of competitor DNA concentration and fitting the curve using Equation 11.
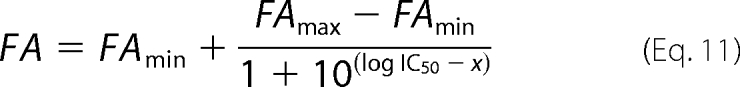 |
RESULTS
The interaction between protein and DNA can be studied conveniently by quantitative titrations of a fluorescently labeled DNA with the protein of interest. However, the binding parameters obtained in this manner are often inaccurate due to the interference from the fluorescent probe. To understand the intrinsic DNA-binding property of TraI, we have studied the interaction of TraI with unlabeled DNA oligomers using the MCT method as described by Jezewska and Bujalowski (27) (see also “Experimental Procedures”). This method has been successfully applied to the analysis of the interactions of the PriA and RepA helicases with DNA (29, 30). In brief, a fluorescently labeled DNA oligomer, or the reference DNA, is titrated with TraI in the absence or presence of an unlabeled DNA oligomer, or the competing DNA, whose binding parameters are to be determined. Because the association of the unlabeled ssDNA oligomer with TraI does not give significant anisotropy signal, the presence of the unlabeled DNA oligomer will shift the titration curve to higher TraI concentration as compared with the curve generated in the absence of competitor. Based on the shift of the titration curve, this method allows the determination of the binding stoichiometry of TraI with an unlabeled DNA oligomer. The MCT method also enables the construction of a model-independent binding isotherm, which can be used for the determination of thermodynamically rigorous binding parameters for the unlabeled DNA oligomer. The protein constructs and DNA oligomers used for this study are listed in Fig. 1 and Table 1 respectively.
Binding of TraI Helicase to ssDNA: Determination of the Site Size and DNA Binding Parameters
The interaction of TraI with a series of unlabeled ssDNA oligomers of different lengths was studied using the MCT method (27). Titration of 50 nm FL-T17 (fluorescently labeled T17) with TraI in the absence or presence of 200 or 350 nm ssDNA oligomers of lengths ranging from 13 to 63 nucleotides were conducted in standard DNA binding buffer (Fig. 2, A and B). The unlabeled 13-, 15-, and 17-mer ssDNA did not cause significant shift of the titration curves to higher TraI concentration, suggesting that TraI cannot bind tightly to unlabeled 13-, 15-, and 17-mer under our assay conditions.
FIGURE 2.

Determination of the stoichiometry of ssDNA binding by TraI. Titration of 50 nm FL-T17 with TraI was performed in the presence or absence of 200 nm Tn (n ranges from 13 to 31) (A) and 350 nm 39-, 45-, 51-, and 63-mer ssDNA (B) in standard DNA binding buffer at 25 °C. The solid lines are used to separate the data and do not have any theoretical basis. C, dependence of observed anisotropy on the total averaged binding density of TraI on the 39-mer. This plot was constructed based on the titration curve in B using the MCT analysis (27). The short solid line indicates the maximum value of the observed anisotropy. The stoichiometry was estimated to one TraI molecule per 39-mer from this plot.
The binding stoichiometry of TraI with the 39-mer was determined by examining the dependence of anisotropy values on the binding density from the titration curves (Fig. 2C). The binding stoichiometry of TraI with the 39-mer was estimated to be one, because binding density equaled one at the maximum anisotropy value of 291. Analogous analyses of the binding stoichiometry were performed for DNA oligomers varying in length from 17 nucleotides to 63 nucleotides (data not shown). The dependence of maximum stoichiometry on the length of ssDNA oligomers indicated that 51- and 63-mers can accommodate two TraI molecules, whereas ssDNA oligomers shorter than 45 nucleotides can only accommodate one TraI molecule.
To determine the binding parameters of TraI for the ssDNA oligomers, we constructed a model-independent binding isotherm for each ssDNA oligomer. Because ssDNA 13-, 15-, and 17-mer did not create a significant shift in the titration curve, their binding isotherms could not be accurately determined. The binding isotherms for the 19-mer and longer oligomers are shown in Fig. 3, A and B. The macroscopic binding constants (KN) for 31-mer and below were determined by analyzing the binding isotherms with Equation 10 because these DNA oligomers can only accept one TraI molecule; the results are summarized in Table 2. The dependence of KN on the length of ssDNA oligomers (N) can be clearly divided into two phases. KN increased relatively slowly when N < 25 but faster when N >25. A roughly linear relationship between KN and N was observed when N ranges from 25 to 31 nucleotides (Fig. 3C). An explanation for this linear relationship is that TraI experiences multiple potential binding sites on an ssDNA longer than 25 nucleotides, indicating that the ssDNA-binding site size of TraI is about 25 nucleotides. The observed dramatic increase in KN was caused by a statistical factor arising from the existence of multiple potential binding sites, instead of the formation of more contacts between TraI and ssDNA oligomers (28, 31, 32). This statistical factor can be defined in terms of the intrinsic binding constant (Kint) and the total site size (m) (29, 30) as shown in Equation 12.
This empirical linear relationship usually appears only when the length of the DNA oligomer is longer than the total site size. In contrast, the slow increase of KN with N, when N <25, likely reflects the formation of increasing contacts between TraI and DNA. The intrinsic binding constants (Kint) for the ssDNA that bind one TraI molecule were determined by analyzing the binding isotherms in Fig. 3A using Equation 9 under the condition m = 25. The results were summarized in Table 2. The Kint did not change significantly with the length of the ssDNA oligomers, ranging from 1.8 ± 0.5 to 2.6 ± 0.3 × 107 m−1. This result supports the estimation of the total site size to be 25 nucleotides.
FIGURE 3.

Determination of binding parameters for TraI binding to ssDNA. A, model-independent binding isotherms of TraI to unlabeled ssDNA Tn (n ranges from 19 to 31). The binding density and the corresponding concentration of free TraI were calculated from the titration curve in Fig. 2A using the MCT analysis (27). The solid lines are computer fits of the data using Equation 9 under the condition m = 25 or Equation 10. The macroscopic binding constants (KN) and intrinsic binding constants (Kint) derived from curve-fitting are summarized in Table 2. B, model-independent binding isotherm of TraI to 63-mer ssDNA. The solid line is computer fit of the data using the Epstein combinatorial approach (Equations 7 and 8) under the condition m = 25 and g = 2. The best fit gives Kint = 6.7(±0.2) ×105 m−1 and ω = 33.4 ± 2.4. C, dependence of KN on the length of the ssDNA oligomers. The solid line is the linear fit of the data points for T25, T27, T29, and T31.
TABLE 2.
Thermodynamic parameters of TraI binding to ssDNA oligomers of length ranging from 19 to 31 nucleotides in standard DNA binding buffer at 25 °C
The following abbreviations are used: n, stoichiometry; KN, macroscopic binding constant; Kint, intrinsic binding constant; NA, not applicable.
| T19 | T21 | T23 | T25 | T27 | T29 | T31 | |
|---|---|---|---|---|---|---|---|
| n | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| KN ×107 (m−1) | 0.9 ± 0.1 | 1.6 ± 0.1 | 2.5 ± 0.1 | 2.6 ± 0.3 | 6.2 ± 0.3 | 10 ± 1 | 12 ± 1 |
| Kint × 107 (m−1) | NA | NA | NA | 2.6 ± 0.3 | 2.1 ± 0.1 | 2.1 ± 0.1 | 1.8 ± 0.5 |
Because the total site size is ∼25 nucleotides, complete engagement of the ssDNA-binding site of two TraI molecules should require ssDNA of at least 50 nucleotides. To determine the cooperativity parameter (ω) for TraI binding to ssDNA, we analyzed the interaction of TraI with the ssDNA 63-mer. The corresponding binding isotherm, shown in Fig. 3B, was analyzed using the Epstein equation under the condition m = 25 and g = 2. The Kint and ω were determined to be 6.7 (±0.2) ×105 m−1 and 33.4 (±) 2.4, respectively. Our data clearly indicate that TraI binds to ssDNA oligomers with low cooperativity.
Binding of TraI to dsDNA, Determination of the Site Size and Binding Parameters
We next examined the interaction of TraI with unlabeled dsDNA using the MCT method (27). Titration of 100 nm FL-dsDNA15 with TraI in the absence or presence of 500 nm unlabeled dsDNA oligomers of length ranging from 9 to 27 bp in buffer A was performed (Fig. 4A). Oligomers shorter than 9 bp were not studied here because they have low melting temperatures and are unstable at the temperature of the experiment (25 °C). The binding stoichiometry of TraI with dsDNA15 was estimated from the plot of fluorescence anisotropy versus binding density as described above (Fig. 4B). Analogous analyses were performed to determine the binding stoichiometry of TraI with other dsDNA oligomers (data now shown). Our results show that the 19-, 23-, and 27-mer dsDNA can accept two TraI molecules, whereas 11- and 15-mer dsDNA can only bind one TraI molecule (Table 3). The fact that a 19-mer, but not a 15-mer, dsDNA can accommodate two TraI molecules indicates that each TraI molecule encompasses at least 8–9 bp when forming a stable complex with a dsDNA.
FIGURE 4.

Determination of stoichiometry and binding parameters for TraI binding to dsDNA. A, titration of 100 nm FL-dsDNA15 with TraI was performed in the absence or presence of 500 nm dsDNA of different lengths (ranging from 9 to 27 residues) in buffer A at 25 °C. The solid lines are used to separate the data and do not have any theoretical basis. B, dependence of observed fluorescence anisotropy on the total averaged binding density of TraI on the 15-mer dsDNA. The short solid line indicates the maximum value of the observed anisotropy. C, model-independent binding isotherms of TraI to unlabeled dsDNA of different lengths, ranging from 9 to 27 bp. The binding density and the concentration of free TraI were calculated from the titration curves in A using the MCT analysis (27). The solid lines are computer fits of the data using the Epstein combinatorial approach (Equations 7 and 8). The parameters were m = 9 and g = 1 for dsDNA9, dsDNA11, and dsDNA15 and m = 9, g = 2 for dsDNA19, dsDNA23, and dsDNA27. The intrinsic binding constants (Kint) and cooperativity parameters (ω) derived from computer fits are summarized in Table 3.
TABLE 3.
Thermodynamic parameters of TraI binding to dsDNA oligomers of length ranging from 9 to 27 bp in buffer A at 25 °C
The following abbreviations are used: n, maximum stoichiometry; Kint, intrinsic binding constant; ω, cooperativity parameter; NA, not applicable.
| dsDNA9 | dsDNA11 | DsDNA15 | dsDNA19 | DsDNA23 | dsDNA27 | |
|---|---|---|---|---|---|---|
| n | 1 | 1 | 1 | 2 | 2 | 2 |
| Kint ×105 (m−1) | 9.9 ± 7.9 | 4.7 ± 0.2 | 5.1 ± 0.2 | 5.2 ± 0.5 | 6.2 ± 0.3 | 5.4 ± 0.4 |
| ω | NA | NA | NA | 18.1 ± 3.8 | 38.6 ± 7.7 | 14.8 ± 2.2 |
Next, the intrinsic binding constant (Kint) was determined by fitting the binding isotherms (Fig. 4C) with the Epstein equation under the conditions that the site size m = 9 and the maximum binding density g = 1 (for 9-, 11-, and 15-mer) or g = 2 (for 19-, 23-, and 27-mer). These results are summarized in Table 3. The large error in Kint for the 9-mer resulted from the low binding density of TraI on the 9-mer under our assay conditions (Fig. 4C). Our data show that TraI has a similar Kint for dsDNA of lengths ranging from 11 to 27 bp, supporting the hypothesis that TraI contacts ∼9 bp when it forms a complex with dsDNA. TraI does not bind dsDNA oligomers in a highly cooperative fashion, as evidenced by ω values ranging from 14.8 ± 2.2 to 38.6 ± 7.7 for dsDNA ranging in length from 19 to 27 bp.
Identification of the dsDNA-binding Site
To identify the dsDNA-binding site, we designed a series of TraI constructs based on the proposed domain organization of TraI (Fig. 1) and examined their interaction with FL-dsDNA15 using the direct DNA binding assay (Fig. 5A) (26). Macroscopic binding constants, KN, were determined by fitting the titration curves using a one-site binding model (Equation 2) and are summarized in Table 4. The affinity of TraI 1–1476 and TraI 1–858 for FL-dsDNA15 were comparable with that of full-length TraI, suggesting that the dsDNA-binding site is located N-terminal to residue 858. TraI 1–330, a construct that contains the relaxase domain, cannot bind FL-dsDNA15 under our assay condition (Fig. 5A). However, the deletion of residues 1–301 dramatically reduced the affinity of TraI for FL-dsDNA15. The binding affinity of TraI 302–1756 for FL-dsDNA15 is about 20-fold lower than that of full-length TraI. These results indicate that although the dsDNA-binding site is primarily located between residues 302 and 858, the relaxase domain facilitates the binding of dsDNA.
FIGURE 5.

Identification of the location of the dsDNA-binding site. A, titration of 50 nm FL-dsDNA15 with different TraI deletion mutants was performed using the direct DNA binding assay in standard DNA binding buffer at 25 °C. The solid lines are computer fits of the binding curves using a one-site binding model (Equation 2). The macroscopic binding constants, KN, derived from the computer fits are summarized in Table 4. B, competition of dsDNA and ssDNA for the binding of TraI. Titration of a mixture of 100 nm FL-dsDNA15 and 100 nm TraI with increasing concentration of 25-mer ssDNA, T25, was performed in buffer A at 25 °C. The solid line is the computer fit of the curve using nonlinear least square regression.
TABLE 4.
Macroscopic binding constants of different TraI constructs for FL-dsDNA15 in standard DNA binding buffer at 25 °C
KN is the macroscopic binding constant.
| TraI constructs | KN × 106 |
|---|---|
| m−1 | |
| 1–1756 | 1.6 ± 0.1 |
| 1–1476 | 0.78 ± 0.7 |
| 1–858 | 1.1 ± 0.1 |
| 1–330 | <0.02 |
| 302–1756 | 0.08 ± 0.01 |
A helicase-associated ssDNA-binding site is located at the N-terminal region of TraI 302–820 (17). Therefore, it is possible that the dsDNA-binding site identified here overlaps with the ssDNA-binding site. To test this possibility, we performed a competition binding assay using FL-dsDNA15 as the reference DNA and the ssDNA oligomer T25 as the competitor DNA. Under our experimental conditions, a complex of TraI with FL-dsDNA15, but not with T25, can generate a fluorescence anisotropy signal. If T25 effectively competes with FL-dsDNA15 for the dsDNA-binding site of TraI, the total anisotropy value will drop with an increase in the concentration of T25. Indeed, the total anisotropy value decreased with the increase of T25 concentration with an apparent IC50 of 1.5 μm (Fig. 5B). This suggests that the binding of T25 and FL-dsDNA15 by TraI occurs along an overlapping if not identical binding surface.
Modulation of DNA Binding Affinity by Nucleotides
We evaluated the effect of nucleotides on the affinity of TraI for unlabeled ssDNA oligomers using the MCT method (27). Titration of 50 nm FL-T17 with TraI in the absence or presence of T25 and in the absence or presence of ADP or the nonhydrolyzable ATP analog AMPPNP was performed in buffer B (Fig. 6, A–C), and a model-independent binding isotherm was constructed (Fig. 6D). The Kint for T25 in the absence of any nucleotides was determined to be 2.8 (±0.4) × 107 m−1 by analyzing the binding isotherm using Equation 9 under the condition m = 25. Because TraI can slowly hydrolyze ATP in the presence of T25 under our assay conditions, the nonhydrolyzable ATP analog (AMPPNP) was used instead of ATP. The presence of 1 mm ADP or AMPPNP increased the Kint of TraI for T25 to 1.2 (±0.3) × 108 and 1.4 (±0.4) × 108 m−1, respectively. These results demonstrate that the binding of ADP and AMPPNP slightly enhances the affinity of TraI for ssDNA.
FIGURE 6.

ADP and AMPPNP increased TraI affinity for ssDNA. Titration of 50 nm FL-T17 with TraI was performed in the absence or presence of 350 nm T25 in buffer B (A), supplemented with 1 mm ADP (B) or 1 mm AMPPNP (C) at 25 °C. The solid lines are used to separate the data and do not have theoretical basis. D, model-independent binding isotherms of TraI to T25 in the absence or presence of 1 mm ADP or AMPPNP. The binding density and the concentration of free TraI were calculated from the titration curves in A–C using the MCT analysis (27). The solid lines are computer fits of the data using the Epstein combinatorial approach (Equation 9) under the condition m = 25. The intrinsic binding constants (Kint) for T25 in the absence of any nucleotide or in the presence of 1 mm ADP or AMPPNP are 2.8 (±0.4) ×107, 1.2 (±0.4) ×108, and 1.4 (±0.3) ×108 m−1, respectively.
Effect of Ionic Strength on DNA Binding
To further define the DNA-binding properties of TraI, we examined the effect of ionic strength on the affinity of TraI for ssDNA and dsDNA using the direct DNA binding assay (26). Titration of 50 nm FL-T25 was performed in buffer C supplemented with increasing concentrations of NaCl: 50, 75, 100, and 125 mm. To avoid the interference of Mg2+, buffer C did not contain magnesium acetate and was supplemented with 1 mm EDTA. Analogous experiments were performed for FL-dsDNA15. The affinity of TraI for ssDNA and dsDNA decreased with the increase of NaCl concentration (Fig. 7A). The Kint for FL-T25 is 1.1 (±0.3) × 108 m−1 when [NaCl] = 50 mm. A 3.4-fold decrease in Kint was observed when [NaCl] was increased to 125 mm NaCl. More dramatic change in Kint was observed with FL-dsDNA15, with Kint decreasing about 36-fold when [NaCl] increased from 50 to 125 mm (Fig. 7A). Within experimental error, a linear relationship between ln(Kint) and ln[NaCl] was detected for both FL-T25 and FL-dsDNA15, with slopes of −1.2 ± 0.2 and −3.7 ± 0.2, respectively, suggesting that the binding of TraI to FL-T25 and FL-dsDNA15 was accompanied with the net release of about one and four ions, respectively (33). These results suggest that electrostatic interactions are crucial for the stability of TraI-DNA complex. Moreover, electrostatic interactions play a more important role in the interaction of TraI with dsDNA.
FIGURE 7.

Role of ionic strength and base specificity in TraI DNA binding. A, dependence of binding affinity on NaCl concentration. 50 nm FL-T25 or FL-dsDNA15 was titrated with TraI in buffer C supplemented with different concentrations of NaCl as follows: 50, 75, 100, and 125 mm. The titration curves are not shown here. The intrinsic binding constants, Kint, at different NaCl concentrations were determined by fitting the titration curves using the Epstein combinatorial approach (Equation 9). The data presented here are averages from three independent experiments. The solid lines are linear least square fit of the data. The slopes of the computer fits for FL-T25 and FL-dsDNA15 are −1.2 ± 0.2 and −3.7 ± 0.2 respectively. B, binding of TraI to ssDNA with different base compositions. Titration of 50 nm FL-T17 with TraI was performed in the presence or absence of 250 nm 25-mer, T25, and C25, in standard DNA binding buffer at 25 °C. The solid lines are used to separate the data and do not have any theoretical basis. C, dependence of binding density on the concentration of free TraI. The binding density and the concentration of free TraI were calculated from the binding curve in B using the MCT analysis (27). The solid lines are computer fits of the data using the Equation 9. The Kint for 25-mer, T25, and C25, are 8.7 (±0.3) ×106, (2.5 ± 0.2) ×107, and 5.8 (±0.1) ×107 m−1, respectively.
Base Specificity of DNA Binding
Base specificity in interactions of TraI with unlabeled ssDNA oligomers was addressed using the MCT method (27). Titration of 50 nm FL-T17 with TraI in the absence or presence of 250 nm unlabeled 25-mer ssDNA (see Table 1 for the sequence) was performed (Fig. 7B). This 25-mer ssDNA contains a mixture of four types of bases and does not form any secondary structure or duplex under our assay conditions. The presence of the 25-mer ssDNA shifted the titration to a higher protein concentration, suggesting competition between the 25-mer ssDNA and FL-T17 (Fig. 7B). A model-independent binding isotherm for the 25-mer ssDNA and subsequent analysis using Equation 9 under the condition m = 25 gave an intrinsic binding constant, Kint = 8.7 (±0.3) ×106 m−1 (Fig. 7C). Analogous titrations of FL-T17 in the presence of T25 or C25 indicate a pronounced shift in titration curves (Fig. 7B). These data indicate that C25 and T25 compete more efficiently with FL-T17 for TraI than does the 25-mer ssDNA. Binding of TraI to T25 and C25 were characterized by a Kint = 2.5 (±0.2) ×107 and 5.8 (±0.1) ×107 m−1, respectively. These data indicate that TraI prefers to bind ssDNA oligomers containing a single type of base, with a further preference for pyrimidine oligomers. G25 was not examined here because it tends to form a cruciform structure and is not easily synthesized. The presence of A25 dramatically interfered with the fluorescence anisotropy signal, making the titration curves in the absence or presence of A25 incomparable. As a result, the binding constant for A25 was not determined by the MCT method.
Spatial Organization of the TraI Domains
Although the crystal structures of several domains of TraI are available, there is no known structure of a full-length TraI. To elucidate the spatial organization of the TraI domains, we examined full-length TraI by SAXS, which allows for the construction of a low resolution structural envelope for a macromolecule. Many important structural parameters, such as the radius of gyration (Rg) and the maximum dimension (Dmax), can also be obtained by analyzing the SAXS profile. The SAXS experiments were performed on TraI solutions over a concentration range of 0.5–2 mg/ml. The SAXS profile and the linearity of the Guinier region indicated that TraI was well behaved and free of aggregation (Fig. 8A). The Rg for TraI as obtained by the Guinier approximation was 58.5 ± 0.9 Å (Fig. 8A, inset). The Dmax of TraI was 220 Å as determined from the P(r) function, calculated using the program GNOM (Fig. 8B). The P(r) function reflects the probable distribution of inter-atomic distances within the scattering particles. The asymmetric feature of the P(r) function suggested that TraI exists in an elongated shape in solution.
FIGURE 8.

Solution structure of TraI. A, SAXS profiles of TraI. The theoretical scattering profile of the rigid -body model for TraI (D) is shown by the red dashed line. The inset is the Guinier plot of the experimental scattering profile. The solid lines are least square linear fits of the data under the condition Rg × q <1.3. B, P(r) plot of TraI. The maximum dimension (Dmax) is derived to be 220 Å. C, DAMMIN model of the TraI shown in volumetric representation. D, DAMMIN model of TraI superimposed with the atomic structures of the four structural domains of TraI (blue, the crystal structure of the relaxase domain residues 1–307, PDB entry 1P4D; yellow, the homology model of the RecD-like domain I, residues 310–844, from this work; pink, a homology model of the RecD-like domain II, residues 845–1476, from this work; green, the crystal structure of TraI 1476–1628, PDB entry 3FLD).
To better define the geometric shape of TraI, we performed ab initio shape restoration of TraI using the program DAMMIN (20). Forty independent runs were performed, and initial parameters were intentionally varied between runs. Independent runs generated very similar structural envelopes, signifying consistency between different runs and reliability of the generated structural envelopes. The 40 structural envelopes were ranked based on their respective χ2. The top 20 envelopes (χ2 ≈0.34) were averaged to generate the final SAXS envelope that is ∼220 × 82 × 65 Å (Fig. 8C). The envelope features a highly extended structure with two protuberances at each end of the longest dimension. This structural envelope was used as a guide for the determination of the spatial organization of TraI domains in the following analysis.
In combination with atomic models, the structural envelope generated from SAXS data can be used to determine the domain organization of a multidomain protein. Although crystal structures are available for the relaxase domain (residues 1–236 and 267–307) and part of the C-terminal domain (1476–1628), atomic structures are missing for the rest of the protein. We used the program PHYRE (23) to generate homology models for these two regions. The homology models of TraI 309–842 and TraI 846–1473 were successfully generated by PHYRE using the crystal structure of E. coli RecD (PDB entry, 1W36) as a template. Due to the lack of homology, 56 and 74 residues are missing from the final models of TraI 309–842 and TraI 846–1473, respectively. Also, PHYRE failed to generate a model for TraI 1629–1756 due to the lack of a homologous template. As a result, the region encompassing residues 1629–1756 was not incorporated into the following analysis.
A rigid body model of TraI was generated by manual placement of TraI domains into the SAXS envelope. The flat and extended shape of the TraI envelope, and the constraints of inter-domain connectivity, allowed relatively little ambiguity in determining the position of the four structural domains of TraI. The crystal structure of the relaxase domain was manually placed into the protuberance at one end of the envelope initially. Then homology models of the two RecD-like domains were placed into the main body of the envelope, which was the only portion of the envelope that could accommodate the combined size of these two domains. Numerous positions and orientations of these two RecD-like domains were examined to minimize the discrepancy (χ2) between the calculated scattering intensities and the experimental scattering intensities. The connectivity of contiguous domains was used as a constraint for placement. Finally, the crystal structure of the C-terminal domain was modeled into the protuberance at the other end of the envelope. Part of the envelope at the C-terminal end is empty due to the missing of residues 1629–1756. The rigid body model of TraI generated here has acceptable geometry and gave a satisfactory χ2 of 5.0 (Fig. 8A), especially considering that 292 of 1756 residues are missing from the atomic models. In our model, the four domains of TraI aligned along a single axis, with the relaxase domain and the C-terminal domain at the opposite ends, and the two RecD-like domains juxtaposed in the middle (Fig. 8D).
DISCUSSION
TraI Has a Long ssDNA-binding Site
Most helicases require the presence of a single-stranded tail for the unwinding of duplex DNA. Typical superfamily I helicases, such as UvrD and RecD, can efficiently unwind a duplex DNA with a single-stranded tail of 10–12 nucleotides (34, 35). However, TraI requires at least a 27-nucleotide tail for efficient unwinding and cannot unwind duplex DNA if the tail is shorter than 20 nucleotides (36). Quantitative analysis in this work suggests that the unusually long single-stranded tail required by TraI for DNA unwinding arises from its unique ssDNA-binding properties. TraI only weakly binds ssDNA oligomers shorter than 19 nucleotides, potentially explaining why TraI cannot unwind duplex DNA when its single-stranded tail is shorter than 20 nucleotides. Considering that the ssDNA-binding site of TraI can encompass up to 25 nucleotides, and that, for optimal helicase activity, the minimal single-stranded overhang requirement is 27 nucleotides, it appears that full engagement of the ssDNA-binding site by DNA may trigger efficient DNA unwinding by TraI.
TraI Binds to ssDNA with Low Cooperativity
TraI displayed low cooperativity (ω = 33.4 ± 2.4) when binding to a ssDNA oligomer that can accept two TraI molecules, indicating that no significant cooperative interactions existed between the two bound TraI molecules. High cooperativity (ω values between 102 and 105) is often associated with multimeric DNA-binding proteins and proteins whose main function is to cover DNA (37, 38). Previous studies show that TraI functions as a processive helicase in vitro (14). The low DNA binding cooperativity observed here supports the monomeric nature of TraI. Low cooperativity during DNA binding is also associated with many other helicases and DNA-binding proteins that function as a monomer, including helicase RecQ (39) and ssDNA-binding protein ICP8 (40). It is interesting to note that the low cooperativity during DNA binding might also reflect a physiological adaptation of TraI to function as a single protein in the context of the relaxosome. Many DNA helicases, which function as a part of a large machinery, such as PriA and DnaB, have been reported to display low cooperativity during DNA binding (41–43).
dsDNA-binding Site Might Play a Role in the Assembly of the Relaxosome
It is of interest to find that TraI can bind dsDNA in the absence of any other protein in vitro. The intrinsic binding constant for dsDNA was determined to be about 5 × 105 m−1 (Table 3), which is significantly lower than that for ssDNA (Table 2). The low affinity of TraI for dsDNA might explain why TraI-dsDNA binding was not detected in the study by Matson and co-workers (44), in which the highest TraI concentration examined was 100 nm. Our deletion studies show that the dsDNA-binding site is located at the N-terminal region (residues 1–858) of TraI and likely overlaps with the helicase-associated ssDNA-binding site. Given that the affinity of TraI for dsDNA is at least one order of magnitude lower than its affinity for ssDNA, dsDNA binding will likely be outcompeted by ssDNA binding when ssDNA, long enough to form stable complex with TraI, is available. Therefore, we consider that this dsDNA-binding site will not be engaged during the unwinding of duplex DNA when a long stretch of ssDNA is available. However, this dsDNA-binding site might play a role in the assembly of the relaxosome when ssDNA is not available. Biochemical studies indicate that TraI alone can bind to a supercoiled dsDNA that contains the oriT site in vitro (45). The interaction, however, is sensitive to NaCl concentration and completely inhibited when the NaCl concentration is above 75 mm (44). The binding of TraI to supercoiled dsDNA might be mediated by the dsDNA-binding site identified here. Our observation that the affinity of TraI to dsDNA is very sensitive to NaCl concentration explains the sensitivity of relaxosome formation to NaCl concentration.
Spatial Organization of TraI Domains
TraI is a bifunctional protein that contains four major structural domains. Understanding the spatial organization of these domains will provide insight into the relative contribution of each domain to TraI function. The SAXS-generated model of TraI reveals that the protein assumes an extended shape in solution with the four structural domains aligning along one axis. The relaxase domain is located at one end of the model and creates a limited number of contacts with the remainder of the protein (Fig. 8D). This domain configuration provides the relaxase domain significant flexibility that is likely essential for the function of TraI. The two RecD-like helicase domains are modeled into the middle of the SAXS envelope and together form multiple intimate contacts, suggesting they may function as a unit. The CTD is at the opposite end of the model relative to the relaxase domain, consistent with the independent activities of these two domains. The extended conformation of TraI allows for the formation of contacts between each sequential domain of TraI, as well as between TraI and other proteins. This network of contacts may play a role in the regulation of the relaxase and helicase activities of TraI.
A model of the TraI helicase in complex with a DNA substrate containing a dsDNA-ssDNA junction was assembled based on the SAXS envelope (Fig. 9). In this model, the location of the dsDNA-ssDNA junction was determined based upon the location of the “pin” domain, or subdomain 1B, of RecD-like domain II, which has been proposed to contact duplex DNA in structural studies of RecD proteins (46). The 5′ ssDNA overhang binds to the long ssDNA binding groove formed by the two RecD-like domains. Two possible end structures of the 5′ ssDNA overhang are presented here. Based on the common belief that one TraI molecule carries out both nicking at oriT and ensuing duplex DNA unwinding, the 5′ end of the ssDNA is modeled as covalently attached to the relaxase domain. However, recent data from Schildbach and co-workers (47) suggest nicking at oriT and ensuing duplex DNA unwinding could be performed by two different TraI molecules; thus, we have also modeled the ssDNA 5′ end positioned away from the depicted TraI, as it may be associated with another TraI (Fig. 9). In our model, the 3′ ssDNA overhang does not contact the protein because we have found that the presence of a 3′ ssDNA overhang does not enhance the affinity of TraI for DNA (Fig. 9).5
FIGURE 9.

Model of TraI association with a dsDNA-ssDNA junction. The relaxase domain is shown in blue, the RecD-like domain I in yellow, the RecD-like domain II in pink, and the C-terminal domain (CTD) in green. The two strands of dsDNA are colored in orange and dark green. The four subdomains (N-terminal domain (NTD), 1A, 2A, and 2B) of RecD-like domain I and the five subdomains (N-terminal domain, 1A, 2A, 1B, and 2B) of RecD-like domain II are shown and labeled. The known NTP-binding site is highlighted in red. In this model, the 5′ ssDNA overhang binds to the ssDNA binding groove mainly formed by RecD-like domain I and RecD-like domain II. Two possible end structures of the 5′ ssDNA overhang, unattached or covalently attached to the relaxase domain, are presented as dashed lines (47). The 3′ ssDNA tail is not bound by TraI. The dsDNA-ssDNA junction contacts subdomain 1B, or the “pin” domain, of RecD-like domain II.
Contribution of RecD-like Domain I to the Processivity of TraI Helicase
TraI functions as a highly processive monomeric helicase (14). The helicase domain of TraI contains two RecD-like domains, one with intact helicase motifs (domain II) and one in which the helicase motifs are not present (domain I) (Fig. 1) (17). Our SAXS model suggests that the two RecD-like domains are closely associated with one another and may form a continuous ssDNA binding groove. Similar domain organization has been observed in the structure of the multimeric E. coli helicase RecBCD (48). Alone, RecB is a monomeric 3′–5′ helicase with limited processivity. Similar to the RecD-like domain I of TraI, RecC has a helicase-like fold but lacks both helicase activity and key helicase motifs. Together, RecBC, a heterodimer formed by RecB and RecC, has significantly higher processivity than RecD alone (48, 49). The crystal structure of the complex clearly shows that the DNA-binding sites of RecB and RecC are brought together in the heterodimer, forming a long and continuous ssDNA binding groove (48). The extended binding groove likely facilitates the association of RecB with ssDNA during translocation. We propose that in TraI, RecD-like domain I functions as a processivity domain to assist the motor domain, RecD-like domain II, in a manner analogous to the RecBC complex.
AMPPNP and ADP Moderately Enhance the Affinity of TraI for ssDNA
Helicases utilize the energy from NTP hydrolysis to unwind nucleic acids. During each cycle of the NTPase reaction, helicases process through a number of distinct nucleotide binding states, including unbound, NTP-bound, and NDP-bound. In current models, the transitions between nucleotide-binding states during the NTPase reaction result in conformational changes in the nucleic acid-binding site of the helicase, thereby driving the translocation of the helicase along the nucleic acid (50). To understand how the conformational states of TraI are controlled by its nucleotide-binding states, we examined the effects of nucleotide concentration on the affinity of TraI for ssDNA. Our data illustrate that the presence of AMPPNP or ADP results in an approximate 3-fold increase in ssDNA binding affinity by TraI. This indicates that TraI exists in two distinct conformational states during each cycle of the NTPase reaction. When the nucleotide-binding site is empty, TraI stays in a conformational state that has relatively low affinity for ssDNA (the loose state). The binding of ATP switches TraI into a conformational state that binds ssDNA with higher affinity (the tight state). At the end of the cycle, TraI returns to the loose state following the release of ADP. The presence of different conformational states during each NTPase cycle has been observed in most established helicases, although the detailed mechanism can vary. For example, the helicase DnaB exhibits 210-fold greater affinity for ssDNA upon NTP binding (51), whereas nucleotide binding reduced the affinity of PriA for ssDNA about 5-fold (43). Structural studies of SF1 helicases have shown that nucleotide binding induces the close up of the cleft between 1A and 2A domains (52–54). Given the high similarity between the helicase domain of TraI and other SF1 helicases, it is conceivable that TraI helicase may experience a similar structural change upon nucleotide binding.
In summary, we have integrated biochemical and structural data to provide the first comprehensive model of the interaction of the TraI helicase with DNA. This model provides insight into the mechanism of the TraI helicase and in particular suggests how the TraI helicase can achieve such an exceptional processivity through the cooperation of the two RecD-like domains.
Acknowledgments
We thank Dr. Liang Guo at beamline 18-ID (Bio-CAT), Advanced Photon Source, Argonne, IL, for assistance in data collection, analysis, and manuscript preparation. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health Grant P41 RR001081).
This work was supported, in whole or in part, by National Institutes of Health Grant AI78924.
Y. Cheng and M. R. Redinbo, unpublished data.
- CTD
- C-terminal domain
- ssDNA
- single-stranded DNA
- dsDNA
- double-stranded DNA
- SAXS
- small angle x-ray scattering
- PDB
- Protein Data Bank
- MCT
- macromolecule competition titration
- AMPPNP
- adenosine 5′-(β,γ-imino)triphosphate.
REFERENCES
- 1. Brochet M., Rusniok C., Couvé E., Dramsi S., Poyart C., Trieu-Cuot P., Kunst F., Glaser P. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 15961–15966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de la Cruz F., Frost L. S., Meyer R. J., Zechner E. L. (2010) FEMS Microbiol. Rev. 34, 18–40 [DOI] [PubMed] [Google Scholar]
- 3. Frost L. S., Leplae R., Summers A. O., Toussaint A. (2005) Nat. Rev. Microbiol. 3, 722–732 [DOI] [PubMed] [Google Scholar]
- 4. Gomis-Rüth F. X., Coll M. (2006) Curr. Opin. Struct. Biol. 16, 744–752 [DOI] [PubMed] [Google Scholar]
- 5. Lanka E., Wilkins B. M. (1995) Annu. Rev. Biochem. 64, 141–169 [DOI] [PubMed] [Google Scholar]
- 6. Abdel-Monem M., Taucher-Scholz G., Klinkert M. Q. (1983) Proc. Natl. Acad. Sci. U.S.A. 80, 4659–4663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Traxler B. A., Minkley E. G., Jr. (1988) J. Mol. Biol. 204, 205–209 [DOI] [PubMed] [Google Scholar]
- 8. Byrd D. R., Sampson J. K., Ragonese H. M., Matson S. W. (2002) J. Biol. Chem. 277, 42645–42653 [DOI] [PubMed] [Google Scholar]
- 9. Matson S. W., Ragonese H. (2005) J. Bacteriol. 187, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ragonese H., Haisch D., Villareal E., Choi J. H., Matson S. W. (2007) Mol. Microbiol. 63, 1173–1184 [DOI] [PubMed] [Google Scholar]
- 11. Street L. M., Harley M. J., Stern J. C., Larkin C., Williams S. L., Miller D. L., Dohm J. A., Rodgers M. E., Schildbach J. F. (2003) Biochim. Biophys. Acta 1646, 86–99 [DOI] [PubMed] [Google Scholar]
- 12. Matson S. W., Sampson J. K., Byrd D. R. (2001) J. Biol. Chem. 276, 2372–2379 [DOI] [PubMed] [Google Scholar]
- 13. Abdel-Monem M., Hoffmann-Berling H. (1976) Eur. J. Biochem. 65, 431–440 [DOI] [PubMed] [Google Scholar]
- 14. Sikora B., Eoff R. L., Matson S. W., Raney K. D. (2006) J. Biol. Chem. 281, 36110–36116 [DOI] [PubMed] [Google Scholar]
- 15. Kuhn B., Abdel-Monem M., Krell H., Hoffmann-Berling H. (1979) J. Biol. Chem. 254, 11343–11350 [PubMed] [Google Scholar]
- 16. Lahue E. E., Matson S. W. (1988) J. Biol. Chem. 263, 3208–3215 [PubMed] [Google Scholar]
- 17. Dostál L., Schildbach J. F. (2010) J. Bacteriol. 192, 3620–3628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., Donnelly M. I. (2002) Protein Expr. Purif. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 19. Svergun D. I. (1992) J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 20. Svergun D. I. (1999) Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. VolKov V., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 860–864 [Google Scholar]
- 22. Wriggers W., Chacón P. (2001) J. Appl. Crystallogr. 34, 773–776 [Google Scholar]
- 23. Kelley L. A., Sternberg M. J. (2009) Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 24. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 25. Svergun D. I., Barberato C., Koch M. H. (1995) J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 26. Anderson B. J., Larkin C., Guja K., Schildbach J. F. (2008) Methods Enzymol. 450, 253–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jezewska M. J., Bujalowski W. (1996) Biochemistry 35, 2117–2128 [DOI] [PubMed] [Google Scholar]
- 28. Epstein I. R. (1978) Biophys. Chem. 8, 327–339 [DOI] [PubMed] [Google Scholar]
- 29. Jezewska M. J., Galletto R., Bujalowski W. (2004) J. Mol. Biol. 343, 115–136 [DOI] [PubMed] [Google Scholar]
- 30. Jezewska M. J., Rajendran S., Bujalowski W. (2000) J. Biol. Chem. 275, 27865–27873 [DOI] [PubMed] [Google Scholar]
- 31. Bujalowski W., Lohman T. M., Anderson C. F. (1989) Biopolymers 28, 1637–1643 [DOI] [PubMed] [Google Scholar]
- 32. McGhee J. D., von Hippel P. H. (1974) J. Mol. Biol. 86, 469–489 [DOI] [PubMed] [Google Scholar]
- 33. Datta K., LiCata V. J. (2003) J. Biol. Chem. 278, 5694–5701 [DOI] [PubMed] [Google Scholar]
- 34. Wang J., Julin D. A. (2004) J. Biol. Chem. 279, 52024–52032 [DOI] [PubMed] [Google Scholar]
- 35. Maluf N. K., Fischer C. J., Lohman T. M. (2003) J. Mol. Biol. 325, 913–935 [DOI] [PubMed] [Google Scholar]
- 36. Csitkovits V. C., Zechner E. L. (2003) J. Biol. Chem. 278, 48696–48703 [DOI] [PubMed] [Google Scholar]
- 37. Bujalowski W., Lohman T. M. (1987) J. Mol. Biol. 195, 897–907 [DOI] [PubMed] [Google Scholar]
- 38. Kowalczykowski S. C., Lonberg N., Newport J. W., von Hippel P. H. (1981) J. Mol. Biol. 145, 75–104 [DOI] [PubMed] [Google Scholar]
- 39. Dou S. X., Wang P. Y., Xu H. Q., Xi X. G. (2004) J. Biol. Chem. 279, 6354–6363 [DOI] [PubMed] [Google Scholar]
- 40. Gourves A. S., Tanguy Le Gac N., Villani G., Boehmer P. E., Johnson N. P. (2000) J. Biol. Chem. 275, 10864–10869 [DOI] [PubMed] [Google Scholar]
- 41. Jezewska M. J., Kim U. S., Bujalowski W. (1996) Biochemistry 35, 2129–2145 [DOI] [PubMed] [Google Scholar]
- 42. Bujalowski W., Jezewska M. J. (1995) Biochemistry 34, 8513–8519 [DOI] [PubMed] [Google Scholar]
- 43. Jezewska M. J., Bujalowski W. (2000) Biochemistry 39, 10454–10467 [DOI] [PubMed] [Google Scholar]
- 44. Howard M. T., Nelson W. C., Matson S. W. (1995) J. Biol. Chem. 270, 28381–28386 [PubMed] [Google Scholar]
- 45. Matson S. W., Morton B. S. (1991) J. Biol. Chem. 266, 16232–16237 [PubMed] [Google Scholar]
- 46. Saikrishnan K., Griffiths S. P., Cook N., Court R., Wigley D. B. (2008) EMBO J. 27, 2222–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dostal L., Shao S., Schildbach J. F. (2010) Nucleic Acids Res., [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singleton M. R., Dillingham M. S., Gaudier M., Kowalczykowski S. C., Wigley D. B. (2004) Nature 432, 187–193 [DOI] [PubMed] [Google Scholar]
- 49. Korangy F., Julin D. A. (1993) Biochemistry 32, 4873–4880 [DOI] [PubMed] [Google Scholar]
- 50. Myong S., Ha T. (2010) Curr. Opin. Struct. Biol. 20, 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jezewska M. J., Bujalowski W. (1996) J. Biol. Chem. 271, 4261–4265 [DOI] [PubMed] [Google Scholar]
- 52. Lee J. Y., Yang W. (2006) Cell 127, 1349–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Velankar S. S., Soultanas P., Dillingham M. S., Subramanya H. S., Wigley D. B. (1999) Cell 97, 75–84 [DOI] [PubMed] [Google Scholar]
- 54. Saikrishnan K., Powell B., Cook N. J., Webb M. R., Wigley D. B. (2009) Cell 137, 849–859 [DOI] [PubMed] [Google Scholar]