

Abstract
Receptor-induced targeting of exchange factors to specific cellular membranes is the predominant mechanism for initiating and compartmentalizing signal transduction by Ras GTPases. The exchange factor RasGRP1 has a C1 domain that binds the lipid diacylglycerol and thus can potentially mediate membrane localization in response to receptors that are coupled to diacylglycerol-generating phospholipase Cs. However, the C1 domain is insufficient for targeting RasGRP1 to the plasma membrane. We found that a basic/hydrophobic cluster of amino acids within the plasma membrane-targeting domain of RasGRP1 is instead responsible for plasma membrane targeting. This basic/hydrophobic cluster binds directly to phospholipid vesicles containing phosphoinositides via electrostatic interactions with polyanionic phosphoinositide headgroups and insertion of a tryptophan into the lipid bilayer. B cell antigen receptor ligation and other stimuli induce plasma membrane targeting of RasGRP1 by activating the phosphoinositide 3-kinase signaling pathway, which generates phosphoinositides within the plasma membrane. Direct detection of phosphoinositides by the basic/hydrophobic cluster of RasGRP1 provides a novel mechanism for coupling and co-compartmentalizing phosphoinositide 3-kinase and Ras signaling and, in coordination with diacylglycerol detection by the C1 domain, gives RasGRP1 the potential to serve as an integrator of converging signals from the phosphoinositide 3-kinase and phospholipase C pathways.
Keywords: Inositol Phospholipid, Membrane Proteins, Phosphatidylinositol Signaling, Phosphatidylinositol 3-Kinase, Plasma Membrane, Protein Targeting, Ras, Signal Transduction
Introduction
Ras GTPases are membrane-bound signal-transducing proteins that control a wide range of cellular responses to external stimuli. Their attachment to specific cellular membranes allows Ras GTPases to confine, concentrate, and organize networks of signal detectors and transmitters (1). Guanine nucleotide exchange factors directly activate Ras GTPases by promoting their conversion to the active GTP-bound state (2). Additionally, exchange factors regulate Ras activation by detecting and integrating inputs from signal transducers coupled to cell-surface receptors. Conversion of receptor-initiated signals into Ras activation is often achieved by spatial regulation of an exchange factor (2, 3). In the absence of signal detection, the exchange factor is sequestered away from its Ras GTPase substrates, whereas the activating signal induces membrane binding of the exchange factor and thus co-localizes it with Ras GTPase substrates. RasGRP1 is a Ras-activating exchange factor that is expressed in many cell types and can be activated by diverse receptors (2, 4). In lymphocytes, initiation of Ras signaling by RasGRP1 controls progression through developmental and immunoselection checkpoints, whereas misregulation of RasGRP1 can trigger autoimmunity or oncogenic transformation (5–10). Depending on the quality and intensity of signaling from receptors, RasGRP1 can be specifically targeted to the plasma membrane (11–15) or to endomembranes such as Golgi or the endoplasmic reticulum (11, 13, 16–19). The site of RasGRP1 localization can have important consequences for how cells respond to the resulting activation of Ras GTPases. Effective immunoselection of thymocytes requires RasGRP1 and is dictated by the strength of signaling from the T cell antigen receptor; strong signaling induces plasma membrane targeting of RasGRP1 and deletion of the self-reactive thymocyte, although moderate signaling from the antigen receptor induces targeting of RasGRP1 to endomembranes and leads to survival and differentiation of the thymocyte (20). Localization of RasGRP1 at the plasma membrane versus endomembranes can also result in its co-localization with or evasion of negative regulators of Ras GTPases, such as the CAPRI GTPase-activating protein (16), and thus can determine whether its activation of Ras is effective or attenuated.
RasGRP1 is a complex protein with REM, GEF, EF-hand, C1, SuPT, and PT domains (Fig. 1A), and remarkably, each of these can affect the cellular membranes to which RasGRP1 localizes (11, 17, 19, 21–23). The C1 domain was the first membrane-localizing domain identified in RasGRP1, making it the focus of the initial models of membrane-selective targeting of RasGRP1. The C1 domain binds to the lipid second messenger diacylglycerol (DAG)3 (24, 25) and thus has the potential to mediate translocation of RasGRP1 to membranes when DAG-generating phospholipase Cs (PLC) or phosphatidic acid phosphatases are activated (11, 13, 21, 26). By this mechanism, membrane-selective targeting of RasGRP1 could be attained by generation or removal of DAG at specific membranes. As examples, selective removal of DAG from the plasma membrane by localized DAG kinases could restrict RasGRP1 to endomembranes (14, 27), and the generation of DAG specifically at the plasma membrane via receptor-induced PLCs or phosphatidic acid phosphatases could drive translocation of RasGRP1 to the plasma membrane, as seen in T cells co-stimulated via antigen receptor and LFA-1 (13). However, the expectation that membrane-selective targeting of RasGRP1 is dictated by the C1-DAG interaction was confounded by a series of findings indicating that antigen receptor-induced targeting of RasGRP1 to the plasma membrane can occur by a C1-independent mechanism. Expressed on its own, the C1 domain typically accumulates at endomembranes even under stimulatory conditions where RasGRP1 is highly concentrated at the plasma membrane (11, 21, 24). When the C1 domain is deleted, or PLC signaling is eliminated, antigen receptor-induced plasma membrane targeting of RasGRP1 still occurs, although with reduced efficiency (11). Thus, another domain must be primarily responsible for selectively targeting RasGRP1 to the plasma membrane, with the C1 domain playing a quantitatively important but subsidiary role in this process.
FIGURE 1.

BCR-induced plasma membrane targeting of RasGRP1 is mediated by its BHC. A, domain structure of RasGRP1, the sequence of the PT domain and the boundaries of the PT-N and PT-C segments, and the BHC and leucine zipper (LZ). Numbering corresponds to the sequence of full-length murine RasGRP1. The sequence changes for mutants are shown below the PT sequence. B and D, DT40 cells expressing GFP-tagged RasGRP1 (RG1) or the indicated derivatives were untreated (nil) or treated with α-IgM for 10 min and then imaged, as described under “Experimental Procedures.” The single cells shown in this and subsequent figures are representative of the majority of cells in the population, with the exception of those expressing PT-C that were chosen to represent the subset of cells with detectable plasma membrane localization of PT-C (19% of the cell population for nil, and 20% for α-IgM-treated). Quantifications of signal at plasma membrane versus cytoplasm are in Table 1. Cells shown have diameters of 12 ± 1 μm unless otherwise indicated by the presence of a scale bar. Scale bars, 5 μm. C, sequence comparison of the PT-N segment of vertebrate RasGRP1s. Conserved amino acids are boxed (R = K and D = E for conservation). Basic amino acids are blue, and large hydrophobic amino acids are green.
The catalytic GEF domain and the REM domain that regulates the GEF domain are both required for membrane localization, probably through their direct interactions with membrane-anchored Ras GTPases (23, 28). Each of these domains affects both endomembrane and plasma membrane targeting of RasGRP1, reflecting the ubiquitous distribution of Ras GTPases at these membranes. In contrast, the EF-hands and the SuPT domain only affect plasma membrane targeting of RasGRP1 (11, 23). However, neither of these two domains directly mediate plasma membrane targeting. Instead, they act through the PT domain, which was found to be both sufficient and essential for antigen receptor-induced plasma membrane targeting of RasGRP1 in several B and T cell lines (11). Although the C1, EF-hand, SuPT, REM, and GEF domains all provide quantitative inputs, it is the PT domain that is directly responsible for specifying the plasma membrane as the site of RasGRP1 targeting.
The objective of this study was to identify the mechanism by which the PT domain mediates receptor-induced plasma membrane targeting of RasGRP1. We mapped the plasma membrane targeting function of the PT domain to a small segment enriched in basic and hydrophobic residues, the BHC. Similar motifs have been found in other proteins that bind directly to membranes enriched in anionic phospholipids, particularly phosphoinositides (29–34). We showed that plasma membrane targeting of RasGRP1 correlates with generation at the plasma membrane of phosphoinositide 3-kinase (PI3K) products, phosphatidylinositol 3,4-bisphosphate (PI(3,4)P2), or phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3). PT domain-mediated plasma membrane targeting of RasGRP1 was induced by a variety of activators of PI3K and in each case was impeded by inhibition of PI3K. Purified PT domain bound directly to phospholipid vesicles containing phosphoinositides. Mutation of basic or hydrophobic residues in the BHC of the PT domain blocked this phosphoinositide-dependent vesicle binding and also prevented PI3K-dependent plasma membrane targeting of RasGRP1. Thus, direct detection of phosphoinositides by the BHC within the PT domain provides the mechanism by which PI3K regulates RasGRP1 localization. This reveals a novel organizational link between two major signal transduction pathways, with input from the PI3K pathway specifying if and where Ras signaling occurs.
EXPERIMENTAL PROCEDURES
Cells and Reagents
The origin, culture, and retroviral transduction of DT40, NIH 3T3, and WEHI-231 cells were described previously (11), except that DT40 cells were cultured in media supplemented with 2% chicken serum (Invitrogen).
RasGRP1 and PH Domain Constructs
The N-terminally GFP-tagged form of full-length murine RasGRP1 (RG1) was described previously (11), with the green fluorescent protein (GFP) coding sequences from pEGFP-C1 (Clontech) fused to amino acid 2 of RasGRP1 (NCBI reference sequence NP 035376). The cDNAs encoding mutants and other derivatives of RasGRP1 were constructed by PCR-directed mutagenesis. The sequences of the encoded proteins are shown in supplemental Methods. The N-terminally GFP-tagged TAPP1 PH domain and the C-terminally GFP-tagged Btk PH domain were provided by Aaron Marshall (35).
The cDNAs were inserted into CTV retroviral vectors (36), which were packaged by transient transfection of BOSC 23 cells and stably transduced into cells by infection (37). Transduced cells were selected by drug resistance conferred by the vector and then selected for GFP expression by flow cytometry.
Fluorescence Microscopy
DT40 B cells were plated on glass coverslips coated with poly-l-lysine, rinsed with PBS, and serum-starved for 4 h in RPMI 1640 medium. Stimulation with 5 μg/ml anti-chicken IgM (Bethyl Laboratories) or 1 mm H2O2 in Hanks' buffer was for 10 min at 20 °C. WEHI-231 cells were plated on coverslips coated with poly-d-lysine, rinsed once with PBS, and serum-starved for 6–8 h in RPMI 1640 medium. Stimulation with 10 μg/ml anti-mouse IgM (Jackson ImmunoResearch) was for 10 min at 20 °C. NIH 3T3 cells were seeded on glass coverslips and allowed to attach for 3–6 h. Cells were then serum-starved in DMEM for 24 h. Stimulation with 50 ng/ml human recombinant PDGF-BB (Invitrogen) was for 5 min at 20 °C. Starvation times were determined as the minimal time required to remove or decrease the serum-dependent plasma membrane localization of RG1 and/or the TAPP1 PH domain. For experiments using inhibitors, cells were pretreated with vehicle, control compound, or inhibitor for 15 min (30 min for WEHI-231) at 37 °C, and subsequent stimulation was as described above, in the presence of vehicle, control compound, or inhibitor. Concentrations of drugs used were as follows: 50 μm LY303511 and LY294002 (Calbiochem); 100 nm wortmannin (Calbiochem); 3 μm PI3Kα inhibitor 2 (Cayman); 100 nm rapamycin (Invitrogen); and 100 μm m-3M3FBS (Sigma). For neomycin experiments, DT40 cells were cultured in the presence of 2 mm neomycin (Sigma) for 2 days, then serum-starved in the presence of 2 mm neomycin for 4 h, and then treated with 2 mm neomycin plus LY303511, LY294002, wortmannin, or DMSO (vehicle for wortmannin) for 15 min at 37 °C.
Prior to microscopy, cells were fixed for 1 h with 4% formaldehyde in PBS at 20 °C. DT40 and WEHI-231 B cells (100× objective) and NIH 3T3 cells (63× objective) were imaged using a DeltaVision imaging station, and images were captured and deconvolved using the default settings of the SoftWorks deconvolution software (Applied Precision). Image brightness and contrast were adjusted by Photoshop to make all features in each cell optimally visible. The individual cells displayed in the figures were chosen to be representative of the majority of the population of cells, unless otherwise noted in the figure legends.
The percentages of cells with detectable signal at the plasma membrane were determined by counting fields of cells from at least two independent experiments. Averages of these values are shown in Tables 1 and 2 and in the supplemental tables. Ratios of GFP fluorescence intensities at the plasma membrane versus cytoplasm were determined as follows. For each of 7–13 cells representative of those with detectable concentration of GFP signal at the plasma membrane (pm+ cells), fluorescence intensities were measured for 10 2 × 2 pixel squares within the plasma membrane, within the cytoplasm, and from outside the cell (background). Mean background intensities were subtracted from mean plasma membrane or mean cytoplasm intensities, and the background-subtracted means were used to calculate plasma membrane/cytoplasm ratios for the pm+ cells. The plasma membrane/cytoplasm ratio for the entire population of cells was then calculated using the formula ((plasma membrane/cytoplasm of pm+ cells − 1) × pm+ cells/total cells) + 1.
TABLE 1.
BHC within the PT domain mediates BCR-induced plasma membrane targeting of RasGRP1
| % of cells with detectable signal at pma |
Signal intensity, pm/cytoplasmb |
||
|---|---|---|---|
| Nil | +αIgM | +αIgM | |
| RG1 | 0 | 100 | 7.7 ± 2.7c |
| RG1ΔPT | 0 | 0 | 1.0 ± 0.1 |
| RG1ΔLZ | 0 | 95 | 3.3 ± 0.9 |
| PT-C | 19 | 20 | 1.1 ± 0.1 |
| BHC | 0 | 90 | 2.4 ± 0.5 |
| RG1-Wμ | 0 | 35 | 1.4 ± 0.3 |
| RG1-FμWμ | 0 | 10 | 1.1 ± 0.1 |
| RG1-RKR/Kμ | 0 | 30 | 1.2 ± 0.2 |
a 200+ DT40 cells were scored for detectable concentration of GFP signal at the plasma membrane.
b Signal intensities at plasma membrane and cytoplasm were determined as described under “Experimental Procedures.”
c Values are the mean ± S.D.
TABLE 2.
PI3K mediates BCR-induced targeting of RasGRP1 to the plasma membrane
| % of cells with detectable signal at plasma membranea αIgM |
Signal intensity, plasma membrane/cytoplasmb + αIgM |
|||
|---|---|---|---|---|
| LY303511 | LY294002 | LY303511 | LY294002 | |
| RG1 | 100 | 50 | 6.7 ± 2.5c | 1.9 ± 0.6 |
| TAPP1 PH | 90 | 28 | 1.7 ± 0.2 | 1.1 ± 0.0 |
a 200+ DT40 cells were scored for detectable concentration of GFP signal at the plasma membrane.
b Signal intensities at plasma membrane and cytoplasm were determined as described under “Experimental Procedures.”
c Values are the mean ± S.D.
Analysis of ERK2 Phosphorylation
DT40 cells were grown to 2–2.5 × 106 cells/ml and then serum-starved in RPMI 1640 medium for 4 h at a concentration of 2–2.5 × 106 cells/ml. The cells were transferred to activation buffer (11) for 15 min at 37 °C and then treated with or without anti-IgM for 10 min at 20 °C to mimic microscopy conditions. When used, PI3K inhibitors or their controls were present during pre-stimulation and stimulation. Cells were then lysed with ice-cold lysis buffer (11). Western blot analysis of RasGRP1 activation was performed as described previously (11) with the exception that 12 μg of lysate protein were loaded. Anti-ERK1/2 anti-phospho-specific ERK1/2 antibodies were from Cell Signaling Technology; anti-GFP was from Roche Applied Science; anti-γ-tubulin antibody was from Sigma. Horseradish peroxidase-conjugated secondary antibodies were from Jackson ImmunoResearch. Phospho-ERK2 band intensities were quantified using Quantity One Imaging software (Bio-Rad). The effect of PI3K inhibitors on RasGRP1 (RG1)-mediated ERK2 phosphorylation relative to controls was calculated as RG1αIgM + inhibitor − GFPαIgM +inhibitor/RG1αIgM+ control − GFPαIgM+control, using data from P-ERK2 Western blots as illustrated in Fig. 2C.
FIGURE 2.

BCR-induced plasma membrane targeting and activation of RasGRP1 is dependent on PI3K. A, DT40 cells expressing GFP-tagged TAPP1 PH domain or RG1 were either untreated (nil) or pretreated for 15 min with LY303511 (LY30, used as a control) or the PI3K inhibitors LY294002 (LY29) or wortmannin (Wm) and then stimulated with α-IgM for 10 min in the presence of LY303511, LY294002, or wortmannin. Cells shown have diameters of 12 ± 1 μm unless otherwise indicated by the presence of a scale bar. Scale bars, 5 μm. B, effect of PI3K inhibitors on BCR-induced RasGRP1 activation in DT40 cells. ERK2 phosphorylation was used to indirectly measure RasGRP1 activity in the presence of the PI3K inhibitors LY294002 or wortmannin or their respective controls LY303511 or DMSO. Data are mean ± S.E. from three (LY294002) or five (wortmannin) independent experiments. The calculation is described under “Experimental Procedures.” C, one of the Western blots from which the data in B was derived. DT40 cells expressing GFP alone (as a control) or GFP-tagged RG1 were untreated (nil) or pretreated for 15 min with LY303511, LY294002, wortmannin, or DMSO and then stimulated with α-IgM for 10 min in the presence of LY303511, LY294002, wortmannin, or DMSO as indicated. Whole cell lysates were then analyzed for phosphorylated ERK2 levels by Western blot. The numbers below the P-ERK2 blot are relative quantities of each band. Total ERK2, RG1, and γ-tubulin (as a loading control) were determined by probing strips from the same blot (RG1 and γ-tubulin) or a second blot (ERK) with appropriate antibodies as described under “Experimental Procedures.”
Expression and Purification of Proteins for Vesicle Binding Assays
GST-tagged PT and PT mutant recombinant proteins were expressed in E. coli BL21 Gold (DE3) (Stratagene) from the pGEX6P-1 vector (GE Healthcare) and purified using glutathione-conjugated agarose (Sigma) beads. Bound protein was cleaved from the GST tag with PreScission protease (GE Healthcare) according to the supplier's instructions, aliquoted, and stored at −80 °C in the cleavage buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol, pH 7.0). Proteins were centrifuged at 40,000 × g at 4 °C to remove aggregates immediately before use in the binding assay. Protein concentration was measured using the BCA assay kit (Pierce) after centrifugation.
Preparation of Phospholipid Vesicles
All lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Egg PC and 1-palmitoyl, 2-oleoylphosphatidyl-l-serine were stored in CHCl3. Tetra-ammonium salts of 1,2-dioleoyl-sn-glycero-3-(phosphoinositol-3,4- or 4,5-bisphosphate or 3,4,5-trisphosphate) were reconstituted and stored in CHCl3/MeOH/H2O (20:9:1). All lipid stocks were stored at −20 °C under nitrogen gas, and their concentrations were determined by quantifying phosphorus (38). The integrity of phosphoinositides was assessed by thin layer chromatography in a solvent system composed of chloroform, methanol, 4 m NH4OH (9:7:2). Small unilamellar vesicles were prepared by rotary evaporation of the solvent from lipid mixtures, hydration of dried lipids in buffer (10 mm Tris, 100 mm NaCl, pH 7.0), followed by probe sonication on ice under a stream of nitrogen for 10–12 min. Titanium debris was removed by centrifugation at 10,000 × g for 3 min. Vesicles were then used immediately for binding assays. The integrity of PI(3,4,5)P3 in the vesicles, before and after the sonication step, was confirmed by electrospray mass spectrometry in negative ion mode where the mass ratios of PC/PIP3 varied by only 7%.
Binding of the PT Domain to Sucrose-loaded Vesicles
Methods were adapted from those described previously (24, 39), except that sonicated rather than extruded sucrose-loaded vesicles (SLVs) were used to match the conditions used for the tryptophan fluorescence analysis of protein-lipid interactions. Lipids were mixed with 0.5 μCi of [3H]dipalmitoyl-PC, and films prepared as described (24). The films were hydrated in 20 mm Tris, pH 7.2, 170 mm sucrose to a concentration of 2 mm and were sonicated under argon at 4 °C for 6–10 min. Vesicles were diluted 5-fold in 20 mm Tris, pH 7.2, 0.12 m NaCl and were collected by centrifugation for 3 h at 125,000 × g at 20 °C. SLVs, which are very loosely pelleted, were resuspended in the bottom 0.2 volume of a spun lipid sample. The recovery of lipid (75–80%) was determined by liquid scintillation counting of aliquots before and after each step, and the final concentration was determined based on this recovery. Purified PT domain was pre-spun at 4 °C for 45 min at 125,000 × g. The supernatant, containing >90% of the original protein concentration, was immediately diluted to 2.5 μm into SLV buffer containing 0–0.6 mm SLV, 20 mm Tris, pH 7.2, 0.1 m NaCl, 20 mm sucrose, and 8 mm DTT in a total volume of 60 μl. The samples were centrifuged for 45 min at 20 °C and 125,000 × g. The upper 40 μl was separated from the lower 20 μl, and aliquots were transferred for assessment of lipid recovery by scintillation counting (>95% in lower fraction) and protein content by gel electrophoresis on 16% Tricine gels, Coomassie staining, and densitometry of scanned gels using ImageQuant 5.2. The fraction of PT domain bound to SLVs was calculated after corrections for the fraction of PT that sedimented in the absence of lipid (0.29 ± 0.07; n = 7) and for the contamination of the pellet fraction with supernatant.
Fluorescence Detection of Tryptophan Insertion into Phospholipid Vesicles
Recombinant proteins (3 μm final concentration) and vesicles were mixed and incubated at 20 °C for 5 min and then tryptophan fluorescence spectra were acquired on an SLM 4800C spectrophotometer (SLM Instruments, Urbana, IL) with an excitation wavelength of 290 nm to minimize contributions from tyrosine. Excitation and emission slits were 8 nm. The spectra were obtained at 1-nm intervals, smoothened, and corrected for buffer and/or lipid vesicle fluorescence by spectral subtraction. The peak fluorescence (λmax) was determined to 0.25 nm by interpolation.
The partition coefficient for binding of the PT domain to 75% PC, 20% PS, and 5% PI(3,4,5)P3 vesicles equals the reciprocal of the concentration of accessible lipid giving half-maximal binding (39). Accessible lipid concentration = 0.6 × total lipid concentration. Maximum binding was obtained from the end plateau value.
RESULTS
Basic/Hydrophobic Cluster in the PT Domain Mediates Plasma Membrane Targeting of RasGRP1
As shown previously (11, 12), RasGRP1 is cytoplasmic in unstimulated DT40 cells but translocates to the plasma membrane when the BCR is ligated with anti-IgM antibody (Fig. 1B). Deletion of the PT domain from RasGRP1 completely prevents BCR-induced plasma membrane targeting (Fig. 1B and Table 1). How does the PT domain specifically recognize the plasma membrane as its site of localization, and what BCR-initiated signal drives this process? As a first step toward addressing these questions, we expressed GFP fusions of RasGRP1 and various derivatives in DT40 B cells and used them to identify the components of the PT domain that are responsible for BCR-induced plasma membrane targeting.
The C-terminal portion of the PT domain contains a predicted leucine zipper (Fig. 1A) (11), which has the potential to mediate plasma membrane targeting either by heterodimerizing with a compatible leucine zipper on a plasma membrane-localized protein (40) or by direct interaction with the membrane (41). However, when the leucine zipper was deleted from RasGRP1, BCR-induced plasma membrane targeting still occurred, although with moderately reduced effectiveness (Fig. 1B and Table 1). When the C-terminal portion of the PT domain containing the leucine zipper was expressed on its own, it did not translocate to the plasma membrane in response to BCR ligation (PT-C in Fig. 1B and Table 1). In contrast, the N-terminal portion of the PT domain by itself was capable of BCR-induced plasma membrane targeting (PT-N in Fig. 1B), and deletion of this segment from RasGRP1 completely eliminated BCR-induced translocation to the plasma membrane, despite retention of the leucine zipper (Fig. 1B).
The N-terminal portion of the PT domain does not have significant sequence similarity to other proteins but is enriched in basic and hydrophobic residues (Fig. 1C). Unstructured basic/hydrophobic clusters can mediate membrane binding by presenting positive charges that electrostatically interact with anionic phospholipid headgroups and aromatic or long aliphatic side chains that insert into the lipid bilayer (29–34, 42). To test whether this mechanism was operative, we surveyed candidate basic and hydrophobic residues in the N-terminal portion of the PT domain for their contributions to plasma membrane targeting. The Leu732–Ile733 pair of aliphatic residues was not required for plasma membrane targeting of the PT domain nor was the LHLRLR segment of alternating aliphatic/basic residues (supplemental Fig. 1). The N terminus of the PT domain contains two aromatic residues, a tryptophan (Trp726), which is conserved among vertebrate RasGRP1s (Fig. 1C), and a nearby phenylalanine (Phe723, a tyrosine in birds or amphibians). Induction of plasma membrane targeting by BCR ligation was very inefficient when Trp726 was mutated to alanine (RG1-Wμ in Fig. 1A), such that RasGRP1 was undetectable at the plasma membrane in most cells and in the other cells only a minor portion of the RasGRP1 relocalized to the plasma membrane (Fig. 1D and Table 1). Mutation of Phe723 had no noticeable effect on its own (RG1-Fμ in Fig. 1, A and D) but further reduced the residual plasma membrane targeting that occurred when the Trp726 was mutated (Table 1). Thus, Trp726 in the N-terminal portion of the PT domain is a critical determinant of plasma membrane targeting, although Phe723 makes a minor contribution.
Bilayer penetration of aromatic side chains is often coupled with electrostatic interactions between neighboring basic side chains and phospholipid headgroups (34). There is a conserved cluster of basic residues in the N-terminal portion of the PT domain as follows: three arginines or lysines at positions 719–721 and a lysine at 725, adjacent to the tryptophan (Fig. 1, A and C). Mutation of these four basic residues to neutral hydrophilic residues (RG1-RKR/Kμ in Fig. 1A) reduced plasma membrane targeting of RasGRP1, equivalently to mutation of Trp726 (Fig. 1D and Table 1). The tryptophan and the basic amino acids were also required for plasma membrane targeting of the isolated PT domain (supplemental Fig. 1). A 17-amino acid segment containing the arginines, lysines, tryptophan, and phenylalanine, which we refer to as the BHC (Fig. 1A), was sufficient for BCR-induced plasma membrane targeting (Fig. 1B and Table 1). Thus, the BHC is the core component of the PT domain that detects BCR-initiated signaling and directly mediates plasma membrane targeting. The essential roles of the tryptophan and basic residues indicate that membrane localization of the BHC could be occurring by the combination of electrostatic and hydrophobic interactions, which characterizes other membrane-binding basic/hydrophobic clusters (29–34).
Phosphoinositide 3-Kinase Positively Regulates BCR-induced Plasma Membrane Targeting of RasGRP1
What is the BCR-induced signal to which the BHC in the PT domain is responding? Phosphoinositides are particularly effective at mediating membrane binding of proteins with basic/hydrophobic clusters, because they present a high negative charge density on the membrane surface that concentrates around the positively charged adsorbed peptide (43). Following BCR ligation, PI3K is activated at the plasma membrane, and the predominant phosphoinositide that is generated in response is PI(3,4)P2 (35, 44, 45), and so we examined the possible roles of PI3K and PI(3,4)P2 in BCR-induced plasma membrane targeting of RasGRP1. Using the C-terminal PH domain of TAPP1 as a fluorescent probe specific for PI(3,4)P2 (46, 47), we found that treatment of BCR-stimulated DT40 cells with the PI3K inhibitor LY294002 reduced PI(3,4)P2 generation at the plasma membrane and also reduced plasma membrane targeting of RasGRP1 (Fig. 2A and Table 2). LY303511, an inactive analog of LY294002, did not affect either PI(3,4)P2 generation or RasGRP1 targeting to the plasma membrane in response to BCR ligation (Fig. 2A and Table 2). Wortmannin and PI3Kα inhibitor 2, two other inhibitors of PI3K, also reduced both PI(3,4)P2 generation and plasma membrane targeting of RasGRP1 (Fig. 2A and supplemental Fig. 2). PI3K inhibitors can also inhibit the TORC signaling complexes, but rapamycin, an inhibitor of one of the TORC complexes, TORC1, had no effect on plasma membrane targeting of RasGRP1 (supplemental Fig. 2). Thus, PI3K appears to be responsible for the relocalization of RasGRP1 from the cytoplasm to the plasma membrane that occurs following BCR ligation.
Membrane localization positively regulates the activity of RasGRP1, by bringing it into contact with its membrane-bound Ras substrates. Elimination of plasma membrane targeting by deletion of the PT domain reduces BCR-induced RasGRP1 activation, as detected by phosphorylation of ERK2, a downstream component of the Ras signaling pathway (11). Preventing plasma membrane targeting by inhibiting PI3K also reduced activation of RasGRP1 by BCR ligation (Fig. 2, B and C). Thus, PI3K-induced plasma membrane targeting of RasGRP1 via its PT domain is required for optimal activation of signaling through the Ras pathway.
PI3K Is Required for BHC-mediated Plasma Membrane Targeting of RasGRP1 in Response to Hydrogen Peroxide and PDGF
Hydrogen peroxide activates PI3K in DT40 cells (48), resulting in the generation of PI(3,4)P2 at the plasma membrane (Fig. 3A). This provides an alternative system for testing the roles of PI3K and phosphoinositides in plasma membrane targeting of RasGRP1. As predicted, hydrogen peroxide induced translocation of RasGRP1 to the plasma membrane (Fig. 3A), and this was impeded when PI3K was inhibited by either LY294002 or wortmannin (Fig. 3A and supplemental Table 1).
FIGURE 3.

PI3K is required for plasma membrane targeting of RasGRP1 in response to hydrogen peroxide or PDGF. A, DT40 cells expressing GFP-tagged TAPP1 PH domain or RG1 were either untreated (nil) or pretreated for 15 min with LY303511 (LY30), LY294002 (LY29), or wortmannin (Wm) and then stimulated with H2O2 in the presence of LY303511, LY294002, or wortmannin for 10 min. Scale bar, 5 μm. B, NIH 3T3 cells expressing GFP-tagged PH domains of TAPP1, Btk, or RG1 were untreated (nil) or pretreated for 15 min with LY303511, LY294002, or wortmannin prior to stimulation with PDGF-BB in the presence of LY303511, LY294002, or wortmannin for 5 min. Scale bar, 10 μm.
In the NIH 3T3 fibroblast line, PDGF-induced PI3K activation results in the generation of both PI(3,4)P2 and PI(3,4,5)P3 at the plasma membrane (Fig. 3B), the former being detected by the PH domain of TAPP1 and the latter being detected by the PH domain of Btk (47, 49). PDGF stimulation of NIH 3T3 cells also induces partial translocation of RasGRP1 to the plasma membrane (Fig. 3B). The PI3K inhibitors LY294002 and wortmannin blocked PDGF-induced generation of PI(3,4)P2 and PI(3,4,5)P3 at the plasma membrane of the NIH 3T3 cells, and it also blocked plasma membrane targeting of RasGRP1 in response to PDGF (Fig. 3B and supplemental Table 2). This provides a third example of induced plasma membrane targeting of RasGRP1 being dependent on induced activation of PI3K.
Constitutive Activation of PI3K Decouples Plasma Membrane Targeting of RasGRP1 from BCR Ligation
PI3K is active in WEHI-231 B cells in the absence of BCR ligation, and the phosphoinositide 3-phosphatase PTEN is not expressed in these cells, resulting in constitutive activation of the PI3K pathway (50, 51). A substantial portion of RasGRP1 is at the plasma membrane in WEHI-231 cells even in the absence of BCR ligation (Fig. 4A) (11). This BCR-independent plasma membrane targeting of RasGRP1, which occurs independently of the C1 domain (11), required both the tryptophan and the basic cluster within the BHC (Fig. 4A) and was reduced by inhibition of PI3K (Fig. 4B and supplemental Table 3). This demonstrates that the BHC and PI3K can drive plasma membrane targeting of RasGRP1 in the absence of other BCR-induced signals, and it illustrates how plasma membrane targeting of RasGRP1 can be deregulated by constitutive activation of PI3K.
FIGURE 4.

Constitutive plasma membrane localization of RasGRP1 in WEHI-231 B cells is mediated by PI3K and the BHC. A, unstimulated WEHI 231 cells expressing GFP-tagged RG1 or the indicated mutants. Scale bar, 5 μm. B, unstimulated WEHI 231 cells expressing GFP-tagged RG1 were treated with LY294002 (LY29) to inhibit PI3K or with LY303511 (LY30) as a control for 30 min. Scale bar, 5 μm.
Both PI3K-generated Phosphoinositides and PI(4,5)P2 Contribute to Constitutive Plasma Membrane Localization of the PT Domain
In contrast to RasGRP1, the isolated PT domain is partially at the plasma membrane in unstimulated DT40 B cells, although there is additional targeting of the PT domain to the plasma membrane following BCR ligation (supplemental Fig. 1). This constitutive plasma membrane localization of the PT domain is reduced but not eliminated by PI3K inhibition (Fig. 5 and supplemental Table 4), indicating that PI3K-generated phosphoinositides contribute to but are not solely responsible for its targeting to the plasma membrane. The phosphoinositide PI(4,5)P2 is constitutively generated at the plasma membrane independently of PI3K, and thus could be responsible for the plasma membrane targeting of the PT domain that occurs even when PI3K is inhibited. We tested this by reducing PI(4,5)P2 levels at the plasma membrane using m-3M3FBS, an activator of the PI(4,5)P2-hydrolyzing enzyme phospholipase C (52). Treatment of DT40 cells with m-3M3FBS lowered PI(4,5)P2 at the plasma membrane, as detected by the PH domain of PLCδ (47, 53), and reduced the intensity of the PT domain localization at the plasma membrane (Fig. 5). Co-treatment with m-3M3FBS and the PI3K inhibitor LY294002 (Fig. 5 and supplemental Table 4) or wortmannin (supplemental Table 4) completely removed the PT domain from the plasma membrane. Co-treatment of DT40 cells with PI3K inhibitor plus neomycin, which binds to PI(4,5)P2 (54), also eliminated constitutive localization of the PT domain at the plasma membrane (supplemental Table 4). These experiments indicate that plasma membrane targeting of the PT domain can be driven by a combination of PI3K-generated phosphoinositides and PI(4,5)P2, with this combination of phosphoinositides being sufficient for partial plasma membrane targeting of the PT domain even in the absence of receptor ligation. The insufficiency of PI(4,5)P2 to localize full-length RasGRP1 to the plasma membrane in the absence of BCR ligation (Fig. 1B) is presumably due to the presence of the SuPT and GEF domains, both of which serve as attenuators of PT domain-mediated plasma membrane targeting (11, 23).
FIGURE 5.
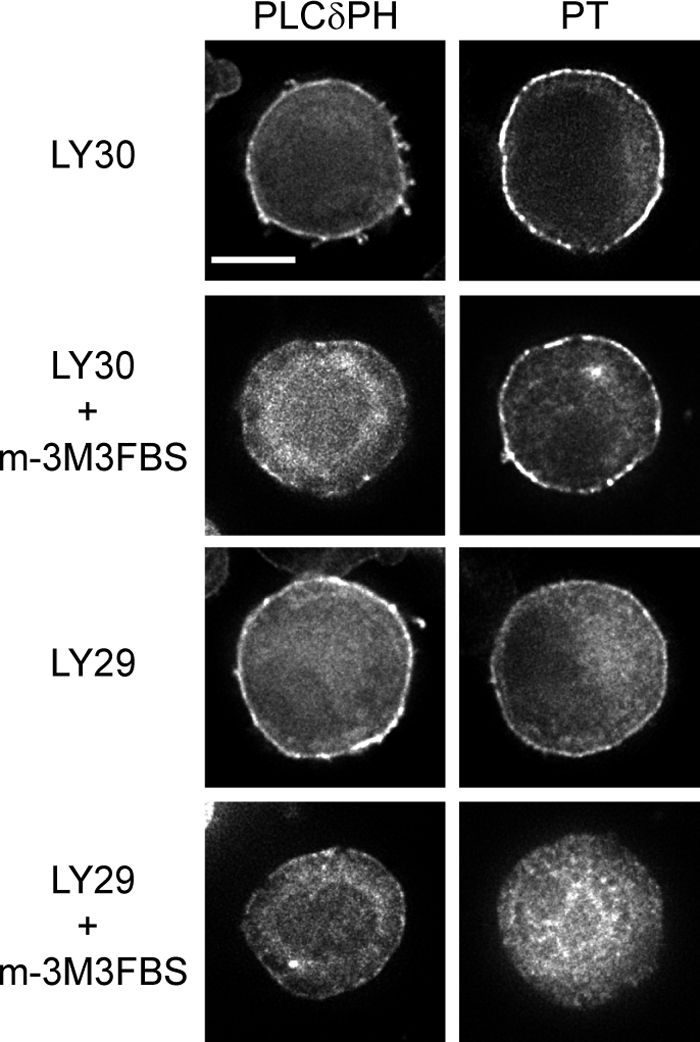
Constitutive plasma membrane targeting of the PT domain is mediated by PI(4,5)P2 as well as PI3K-generated phosphoinositides. Unstimulated DT40 cells expressing GFP-tagged PLCδ PH domain or PT domain were treated for 15 min with LY303511 (LY30), LY294002 (LY29), and/or m-3M3FBS. Scale bar, 5 μm.
Phosphoinositides Are Direct Ligands for the PT Domain
The mutational analysis of the PT domain suggests that the mechanism of plasma membrane targeting could involve direct interaction of the BHC with membranes enriched in anionic phospholipids, and the requirement for PI3K in BHC-mediated plasma membrane targeting implies that PI3K-generated phosphoinositides would be the critical anionic phospholipids if this mechanism was operative.
We tested the ability of the PT domain to bind to membranes by examining its co-sedimentation with unilamellar vesicles containing the neutral phospholipid PC supplemented with 20% of the anionic phospholipid PS to approximate the negative charge density of the inner leaflet of the plasma membrane in unstimulated cells (55). Even at the highest lipid concentration tested, only a minor fraction of the PT domain bound to the PC/PS vesicles (Fig. 6). However, addition of 5% PI(3,4,5)P3 to the vesicles resulted in complete binding of the PT domain at high lipid concentration (Fig. 6). By varying the lipid concentration, the partition coefficient of the PT domain for PC/PS vesicles containing 5% PI(3,4,5)P3 was calculated to be 2.5 × 104 m−1, an affinity that could provide physiologically significant membrane binding in cells (56, 57).
FIGURE 6.
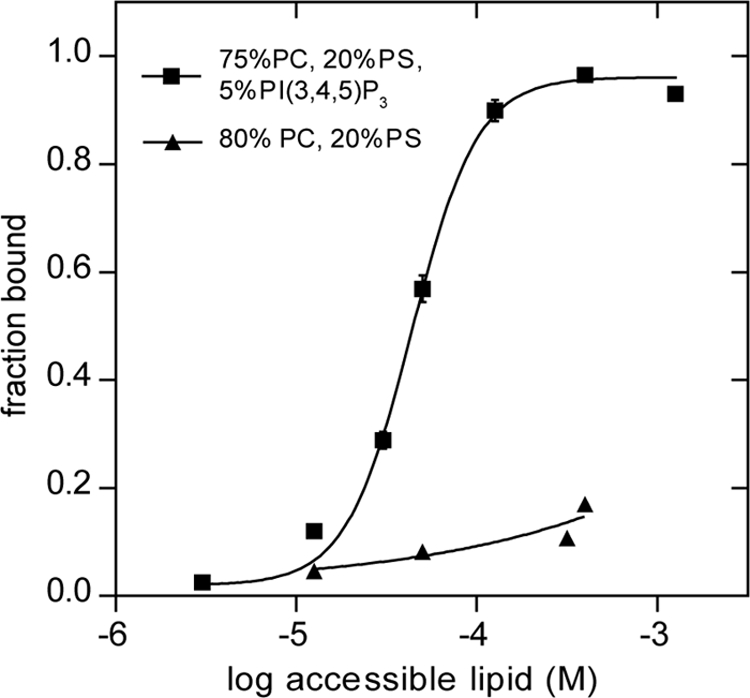
PT domain co-sediments with phospholipid vesicles containing PI(3,4,5)P3. Purified PT domain (2.5 μm) was incubated with sucrose-loaded vesicles composed of the indicated lipids. Bound and free PT proteins were separated by sedimentation, and the fraction bound was determined as described under “Experimental Procedures.” The data, compiled from two independent experiments with separately prepared SLVs, were fitted to a nonlinear variable slope regression line using GraphPad Prism. The R2 value for the fits were 0.99 for binding to vesicles containing PI(3,4,5)P3, and 0.80 for binding to the vesicles lacking PI(3,4,5)P3. Bars indicate ranges of two independent experiments.
In most basic/hydrophobic clusters that mediate membrane binding, insertion of hydrophobic, particularly aromatic, side chains into the lipid bilayer makes a major contribution to membrane binding (29–34, 42). The tryptophan in the BHC of RasGRP1, which is essential for effective plasma membrane targeting, could directly facilitate membrane binding by inserting into the lipid bilayer (58). Transfer of the indole group of tryptophan from the aqueous phase into the reduced polarity environment of a lipid bilayer results in a blue shift in its fluorescence emission spectrum, accompanied by an increase in emission intensity (59), and this spectral shift can be used to detect binding of tryptophan-containing proteins to model membranes (60–64). When excited at 290 nm in the absence of vesicles, the PT domain had an emission peak at 348 nm, because of its single tryptophan Trp726 (supplemental Fig. 3A, comparing PT versus PT-Wμ). Addition of PC/PS vesicles did not cause a significant change in the tryptophan emission spectrum of the PT domain, but inclusion of 3 or 5% PI(3,4,5)P3 in the PC/PS vesicles induced an emission blue shift of about 5 nm, as well as an increase in emission intensity (Fig. 7A and supplemental Fig. 3A). These fluorescence shifts indicate that binding of the PT domain to phosphoinositide-containing vesicles involves insertion of Trp726. Binding of the PT domain to PC, PS, 5% PI(3,4,5)P3 vesicles had the same partition coefficient (2.5 × 104 m−1) when measured by tryptophan blue shift versus vesicle sedimentation (Fig. 6 versus 7B). This demonstrates that the tryptophan blue shift provides an effective assay for detecting binding of the PT domain to vesicles, with the advantage that this method, unlike vesicle sedimentation, detects binding in real time without perturbing the binding equilibrium and therefore is preferable for assessing a protein-vesicle interaction (62, 65). We used the tryptophan blue shift assay to further characterize how the PT domain interacts with phospholipid vesicles.
FIGURE 7.

PT domain binds to phosphoinositide-containing vesicles via hydrophobic and electrostatic interactions. A, purified PT domain (3 μm) was incubated with PC vesicles containing 20 mol % PS and the indicated mol % of PI(3,4,5)P3, PI(3,4)P2, or PI(4,5)P2. The phosphoinositide content was varied at the expense of the PC. The molar ratio of lipid to protein was 100 in this and subsequent panels. Spectra were collected, and λmax values were obtained and subtracted from that of the PT domain without vesicles to determine tryptophan blue shift values. All data in this and subsequent panels are means ± S.E. of two independent experiments, each with two replicates, with the exception of the 3% phosphoinositides in A that are from three experiments, each with two replicates. B, purified PT domain or BHC was incubated with increasing concentrations of vesicles containing 75% PC, 20% PS, and 5% PI(3,4,5)P3. Spectra were collected, and λmax values were subtracted from that of PT without vesicles to determine blue shift values. The data were fit to a nonlinear variable slopes regression line using GraphPad Prism version 5. The R2 value for the fits were 0.94 (PT) and 0.85 (BHC). Purified proteins of the indicated constructs were incubated with 75% PC, 20% PS, and 5% PI(3,4,5)P3. C and D, wild-type or mutant forms of the PT domain were incubated with vesicles of the indicated composition.
The phenylalanine within the BHC, Phe723, also has the potential to contribute to membrane binding, by insertion of its aromatic side chain into the bilayer. In cells, Phe723 was largely redundant to Trp726, in that mutation of Phe723 had no detectable effect on plasma membrane localization of RasGRP1 unless Trp726 was already mutated (Table 1). However, mutation of Phe723 did reduce PI(3,4,5)P3-induced vesicle binding of the PT domain even when Trp726 was intact (Fig. 7C). This confirms that Phe723 can contribute to the binding mechanism, although the role of Phe723 is predominant.
The basic amino acids adjacent to Phe723 and Trp726 in the BHC were essential for PI3K-dependent targeting of RasGRP1 to the plasma membrane (Fig. 1D). Mutation of the same basic amino acids prevented insertion of Trp726 into PI(3,4,5)P3-containing vesicles (Fig. 7C). Insertion of Trp726 was also prevented when electrostatic forces were masked by elevated NaCl concentration (supplemental Fig. 3B). This confirms that the mechanism of membrane binding involves electrostatic interactions in conjunction with aromatic side chain insertion.
PI(3,4)P2 or PI(4,5)P2 also induced binding of the PT domain to the PC/PS vesicles as detected by the blue shift assay, with similar efficacy to PI(3,4,5)P3 when present in the vesicles at 5%, but with lower relative efficacy when the phosphoinositide concentration was reduced (Fig. 7A). The abilities of phosphoinositide headgroups to facilitate membrane binding by the PT domain could simply be a function of their additive contributions to the net negative charge at the surface of the vesicles. The PT domain can bind to vesicles containing a high molar ratio of PS (supplemental Fig. 3C), indicating that high anionic charge density, rather than specific structural recognition of phosphoinositides, is indeed responsible for PT domain binding. However, when we compared binding of the PT domain to PC vesicles containing PI(3,4)P2 (net charge −4) or PI(3,4,5)P3 (net charge −5.5) (66) versus vesicles carrying the equivalent negative charge provided by PS (net charge −1), the phosphoinositides were more effective at inducing binding of the PT domain to the vesicles (Fig. 7D). This presumably reflects the exceptionally strong electrostatic force between the multiple phosphates on the phosphoinositide headgroups and the clustered basic charges on the BHC (43).
When the leucine zipper within the C-terminal portion of the PT domain was removed, the remaining BHC still bound to PI(3,4,5)P3-containing vesicles (Fig. 7B), demonstrating that the BHC is sufficient for phosphoinositide recognition and membrane binding. But in comparison with the intact PT domain, the BHC had lower affinity for the vesicles, as indicated by the shift toward higher lipid concentration required for an equivalent level of binding (Fig. 7B). This result substantiates what we inferred from the roles of the PT domain components in plasma membrane targeting; it is the BHC that directly detects phosphoinositides in membranes, whereas the leucine zipper within the C-terminal portion of the PT domain plays a secondary role by enhancing the efficiency of BHC-mediated membrane binding.
Considered in combination with the cell experiments showing that plasma membrane targeting of RasGRP1 occurs via the BHC, requires PI3K, involves major contributions from the basic cluster and tryptophan and a subsidiary contribution from the phenylalanine, and is enhanced by the leucine zipper, the experiments with phospholipid vesicles establish a molecular mechanism explaining how the BHC detects phosphoinositides and thereby mediates plasma membrane targeting of RasGRP1 in response to ligation of cell-surface receptors that are coupled to PI3K (Fig. 8).
FIGURE 8.

Proposed mechanism of plasma membrane targeting of RasGRP1. A, in the absence of PI3K activation by receptors (top), there is insufficient negative charge at the inner surface of the plasma membrane (pm) to support stable electrostatic binding of RasGRP1 via its BHC, despite the presence of PI(4,5)P2 (triplet of negative charge in the diagram). When PI3K is activated, the phosphoinositide that is generated (PI(3,4)P2 as shown by the second triplet of negative charge or alternatively PI(3,4,5)P3) provides additional clustered negative charges that drive electrostatic binding of the BHC to the membrane, and this enables insertion of aromatic side chains into the bilayer, which further stabilizes binding. B, potential for coincidence detection and signal amplification by RasGRP1. Binding of the BHC to phosphoinositides generated by PI3K could synergize with binding of the C1 domain to DAG generated by PLCγ to maximize plasma membrane targeting of RasGRP1. In addition to triggering downstream effector pathways such as Raf/MEK/ERK, GTP loading of Ras by plasma membrane-localized RasGRP1 could activate PI3K, thus setting up a positive feedback loop amplifying signaling through both the PI3K and Ras pathways.
DISCUSSION
Like other exchange factors, RasGRP1 is primarily regulated by controlling its access to membrane-bound substrates. Previous investigations have focused on the C1 domain of RasGRP1, which detects signaling from phospholipase Cs and mediates membrane localization by binding the lipid second messenger DAG. We have now shown that plasma membrane localization of RasGRP1 can also be regulated by PI3K, via a BHC within the PT domain of RasGRP1 that directly detects phosphoinositide generation at the plasma membrane by PI3K.
Binding of the BHC of RasGRP1 to membranes involves insertion of a tryptophan into the membrane. The moderate size of the fluorescence blue shift (∼7 nm, Fig. 7B) when binding is saturated indicates that the indole side chain is probably penetrating only as far as the interface between the highly hydrated phosphoinositide headgroups and the hydrophobic acyl chains (67). A phenylalanine within the BHC makes a minor contribution to binding, and this may also involve insertion of the aromatic side chain into the membrane bilayer. Arginines and lysines within the BHC and anionic phospholipid headgroups are additionally required for membrane binding. This synergism of charged and hydrophobic components is typical of the membrane binding mechanisms of other basic/hydrophobic clusters (29, 31–34), with the electrostatic force between the basic amino acid side chains and the anionic headgroups enabling insertion of the aromatic side chains into the bilayer (68). The particular effectiveness of phosphoinositides as ligands for the BHC presumably reflects their high negative charge densities, and perhaps also their abilities to sequester around the cluster of basic charges on the BHC, thus maximizing the electrostatic forces that drive membrane binding (43, 68).
Fig. 8A shows our current model of how the BHC mediates PI3K-induced plasma membrane targeting. In unstimulated cells, the concentrations of accessible anionic phospholipids in the plasma membrane (PI(4,5)P2, PS, PG etc.) are insufficient to support stable binding of the BHC. When signaling from a receptor such as BCR induces PI3K activation, the addition of PI(3,4)P2 and/or PI(3,4,5)P3 to the plasma membrane raises the concentration of anionic phospholipids above the threshold required for stable BHC binding.
The BHC- and PI3K-generated phosphoinositides are central to the mechanism of plasma membrane targeting of RasGRP1, but they are not the only players in this process. Another component of the PT domain, the leucine zipper, enhances the efficiency of BHC-mediated membrane binding and thus lowers the threshold concentration of phosphoinositides that triggers plasma membrane targeting. As a result, the PT domain can weakly bind to the plasma membrane in the absence of receptor ligation by detecting a set of constitutively present phosphoinositides, composed of PI(4,5)P2 plus low levels of PI(3,4)P2 and PI(3,4,5)P3. Basic/hydrophobic clusters in Rit, Rin, and PKCα are similarly targeted to the plasma membrane by PI(4,5)P2 in combination with PI(3,4)P2 and/or PI(3,4,5)P3 (30, 69). Given the large excess of PI(4,5)P2 over PI(3,4,5)P3 plus (PI(3,4)P2 in cells, it is surprising that PI(4,5)P2 does not completely dictate the localization of basic hydrophobic clusters due to their broad specificities for phosphoinositides, but it is possible that the ratio of PI(4,5)P2 to PI(3,4,5)P2 plus PI(3,4)P2 available for BHC binding at the plasma membrane is lower than whole cell quantities would suggest, perhaps due to selective sequestration of PI(4,5)P2 (70, 71).
Although addition of the leucine zipper to the BHC of RasGRP1 hypersensitizes its detection of phosphoinositides and thus causes constitutive plasma membrane targeting, two other domains within RasGRP1 counteract this and thus ensure that plasma membrane targeting only occurs when the phosphoinositide content is elevated by receptor-coupled PI3K. The SuPT domain directly down-modulates the efficiency of plasma membrane targeting of the PT domain, reducing constitutive localization and raising the threshold of receptor signaling required for optimal plasma membrane targeting (11, 23). When not bound to its catalytic product Ras-GTP, the GEF domain of RasGRP1 also attenuates BHC-mediated plasma membrane targeting, further suppressing constitutive targeting while still permitting PI3K-induced targeting (23). Although plasma membrane targeting of RasGRP1 is directly mediated by phosphoinositide detection by the BHC, optimization and stringent regulation of this process requires additional inputs from the leucine zipper and the SuPT and GEF domains.
The efficiency of plasma membrane targeting of RasGRP1 can be amplified by cooperativity between its PT and C1 domains. After RasGRP1 is drawn to the plasma membrane via binding of the BHC to phosphoinositides, its interaction with the membrane is further stabilized by the C1 domain binding to the DAG that is generated at the plasma membrane by receptor-coupled PLCγ2 (11). Through its PT and C1 domains, RasGRP1 can serve as a coincidence detector for PI3K and PLC signaling, with high output of Ras signaling at the plasma membrane occurring only when both PI3K and PLC signaling inputs are received by RasGRP1 (Fig. 8B). If just PI3K signaling is detected through the PT domain, plasma membrane targeting of RasGRP1 and Ras activation at the plasma membrane is suboptimal (11). Conversely, if just PLC signaling is detected through the C1 domain, plasma membrane targeting of RasGRP1 does not occur, and Ras activation is restricted to endomembranes (11).
We have shown that RasGRP1 is a downstream effector of PI3K. GTP-loaded Ras can directly activate PI3K (72), so RasGRP1 could also be an upstream activator of PI3K. Once PI3K signaling has triggered translocation of RasGRP1 to the plasma membrane, the GTP-loaded Ras that is produced there would contribute to the further activation of plasma membrane-localized PI3K. This could set up a locally concentrated positive feedback cycle between PI3K and Ras signaling that would coordinately amplify signal output from these two pathways (Fig. 8B). By serving as the link that completes the positive feedback loop, RasGRP1 could couple together two of the predominant signaling pathways involved in cancer. In cells expressing RasGRP1, a mutation that chronically activates the PI3K pathway would also deregulate Ras signaling, thus promoting oncogenesis via the Ras pathway, whereas the feedback from Ras to PI3K would concurrently exacerbate the other oncogenic effects of PI3K signaling. By spatially restricting this positive feedback between PI3K and Ras, the C1, GEF, SuPT, and EF-hand domains of RasGRP1 that modulate membrane targeting may be as important for avoiding oncogenesis as they are for shaping the normal signaling patterns that flow through RasGRP1.
Acknowledgments
We thank Jeremy Parker (British Columbia Cancer Agency) for providing valuable advice and criticism on the writing of this paper. We also thank Aaron Marshall (University of Manitoba) and Tamas Balla (National Institutes of Health) for PH domain constructs; Pamela Hoodless (British Columbia Cancer Agency) and Lynne Quarmby (Simon Fraser University) for microscopy facilities; Donna Hogge (British Columbia Cancer Agency) for providing the PI3Kα inhibitor 2; Melissa Dennis and Svetla Taneva for technical advice on vesicle binding experiments; Jeremy Parker and Laura Hilton for advice on microscopy; and Bonny Tam for assistance with molecular and cellular procedures.
This work was supported by grants from the Canadian Institutes of Health Research (to R. B. C. and R. J. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Methods, Tables 1–4, and Figs. S1–3.
- DAG
- diacylglycerol
- BHC
- basic/hydrophobic cluster
- BCR
- B cell antigen receptor
- PI(3,4)P2
- phosphatidylinositol 3,4-bisphosphate
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- PC
- phosphatidylcholine
- PS
- phosphatidylserine
- PH
- pleckstrin homology
- SLV
- sucrose-loaded vesicle
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- PLC
- phospholipase C
- PT
- plasma membrane-targeting domain
- GEF
- guanine nucleotide exchange factor
- SuPT
- suppressor of PT.
REFERENCES
- 1. Kholodenko B. N., Hancock J. F., Kolch W. (2010) Nat. Rev. Mol. Cell Biol. 11, 414–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Buday L., Downward J. (2008) Biochim. Biophys. Acta 1786, 178–187 [DOI] [PubMed] [Google Scholar]
- 3. Groves J. T., Kuriyan J. (2010) Nat. Struct. Mol. Biol. 17, 659–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stone J. C. (2006) Biochem. Soc. Trans. 34, 858–861 [DOI] [PubMed] [Google Scholar]
- 5. Dower N. A., Stang S. L., Bottorff D. A., Ebinu J. O., Dickie P., Ostergaard H. L., Stone J. C. (2000) Nat. Immunol. 1, 317–321 [DOI] [PubMed] [Google Scholar]
- 6. Klinger M. B., Guilbault B., Goulding R. E., Kay R. J. (2005) Oncogene 24, 2695–2704 [DOI] [PubMed] [Google Scholar]
- 7. Kool J., Berns A. (2009) Nat. Rev. Cancer 9, 389–399 [DOI] [PubMed] [Google Scholar]
- 8. Layer K., Lin G., Nencioni A., Hu W., Schmucker A., Antov A. N., Li X., Takamatsu S., Chevassut T., Dower N. A., Stang S. L., Beier D., Buhlmann J., Bronson R. T., Elkon K. B., Stone J. C., Van Parijs L., Lim B. (2003) Immunity 19, 243–255 [DOI] [PubMed] [Google Scholar]
- 9. Oki-Idouchi C. E., Lorenzo P. S. (2007) Cancer Res. 67, 276–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Priatel J. J., Chen X., Dhanji S., Abraham N., Teh H. S. (2006) J. Immunol. 177, 1470–1480 [DOI] [PubMed] [Google Scholar]
- 11. Beaulieu N., Zahedi B., Goulding R. E., Tazmini G., Anthony K. V., Omeis S. L., de Jong D. R., Kay R. J. (2007) Mol. Biol. Cell 18, 3156–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caloca M. J., Zugaza J. L., Matallanas D., Crespo P., Bustelo X. R. (2003) EMBO J. 22, 3326–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mor A., Campi G., Du G., Zheng Y., Foster D. A., Dustin M. L., Philips M. R. (2007) Nat. Cell Biol. 9, 713–719 [DOI] [PubMed] [Google Scholar]
- 14. Sanjuán M. A., Pradet-Balade B., Jones D. R., Martínez-A C., Stone J. C., Garcia-Sanz J. A., Mérida I. (2003) J. Immunol. 170, 2877–2883 [DOI] [PubMed] [Google Scholar]
- 15. Zugaza J. L., Caloca M. J., Bustelo X. R. (2004) Oncogene 23, 5823–5833 [DOI] [PubMed] [Google Scholar]
- 16. Bivona T. G., Pérez De Castro I., Ahearn I. M., Grana T. M., Chiu V. K., Lockyer P. J., Cullen P. J., Pellicer A., Cox A. D., Philips M. R. (2003) Nature 424, 694–698 [DOI] [PubMed] [Google Scholar]
- 17. Caloca M. J., Zugaza J. L., Bustelo X. R. (2003) J. Biol. Chem. 278, 33465–33473 [DOI] [PubMed] [Google Scholar]
- 18. Perez de Castro I., Bivona T. G., Philips M. R., Pellicer A. (2004) Mol. Cell. Biol. 24, 3485–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tognon C. E., Kirk H. E., Passmore L. A., Whitehead I. P., Der C. J., Kay R. J. (1998) Mol. Cell. Biol. 18, 6995–7008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daniels M. A., Teixeiro E., Gill J., Hausmann B., Roubaty D., Holmberg K., Werlen G., Holländer G. A., Gascoigne N. R., Palmer E. (2006) Nature 444, 724–729 [DOI] [PubMed] [Google Scholar]
- 21. Carrasco S., Merida I. (2004) Mol. Biol. Cell 15, 2932–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ebinu J. O., Bottorff D. A., Chan E. Y., Stang S. L., Dunn R. J., Stone J. C. (1998) Science 280, 1082–1086 [DOI] [PubMed] [Google Scholar]
- 23. Tazmini G., Beaulieu N., Woo A., Zahedi B., Goulding R. E., Kay R. J. (2009) Biochim. Biophys. Acta 1793, 447–461 [DOI] [PubMed] [Google Scholar]
- 24. Johnson J. E., Goulding R. E., Ding Z., Partovi A., Anthony K. V., Beaulieu N., Tazmini G., Cornell R. B., Kay R. J. (2007) Biochem. J. 406, 223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lorenzo P. S., Beheshti M., Pettit G. R., Stone J. C., Blumberg P. M. (2000) Mol. Pharmacol. 57, 840–846 [PubMed] [Google Scholar]
- 26. Roose J. P., Mollenauer M., Gupta V. A., Stone J., Weiss A. (2005) Mol. Cell. Biol. 25, 4426–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zha Y., Marks R., Ho A. W., Peterson A. C., Janardhan S., Brown I., Praveen K., Stang S., Stone J. C., Gajewski T. F. (2006) Nat. Immunol. 7, 1166–1173 [DOI] [PubMed] [Google Scholar]
- 28. Gureasko J., Galush W. J., Boykevisch S., Sondermann H., Bar-Sagi D., Groves J. T., Kuriyan J. (2008) Nat. Struct. Mol. Biol. 15, 452–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gambhir A., Hangyás-Mihályné G., Zaitseva I., Cafiso D. S., Wang J., Murray D., Pentyala S. N., Smith S. O., McLaughlin S. (2004) Biophys. J. 86, 2188–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heo W. D., Inoue T., Park W. S., Kim M. L., Park B. O., Wandless T. J., Meyer T. (2006) Science 314, 1458–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skwarek L. C., Garroni M. K., Commisso C., Boulianne G. L. (2007) Dev. Cell 13, 783–795 [DOI] [PubMed] [Google Scholar]
- 32. Stahelin R. V., Cho W. (2001) Biochemistry 40, 4672–4678 [DOI] [PubMed] [Google Scholar]
- 33. Takahashi S., Pryciak P. M. (2007) Mol. Biol. Cell 18, 4945–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang W., Crocker E., McLaughlin S., Smith S. O. (2003) J. Biol. Chem. 278, 21459–21466 [DOI] [PubMed] [Google Scholar]
- 35. Marshall A. J., Krahn A. K., Ma K., Duronio V., Hou S. (2002) Mol. Cell. Biol. 22, 5479–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whitehead I., Kirk H., Kay R. (1995) Mol. Cell. Biol. 15, 704–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pear W. S., Nolan G. P., Scott M. L., Baltimore D. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bartlett G. R. (1959) J. Biol. Chem. 234, 466–468 [PubMed] [Google Scholar]
- 39. Murray D., Hermida-Matsumoto L., Buser C. A., Tsang J., Sigal C. T., Ben-Tal N., Honig B., Resh M. D., McLaughlin S. (1998) Biochemistry 37, 2145–2159 [DOI] [PubMed] [Google Scholar]
- 40. Vinson C., Myakishev M., Acharya A., Mir A. A., Moll J. R., Bonovich M. (2002) Mol. Cell. Biol. 22, 6321–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cornell R. B., Taneva S. G. (2006) Curr. Protein Pept. Sci. 7, 539–552 [DOI] [PubMed] [Google Scholar]
- 42. Maass K., Fischer M. A., Seiler M., Temmerman K., Nickel W., Seedorf M. (2009) J. Cell Sci. 122, 625–635 [DOI] [PubMed] [Google Scholar]
- 43. McLaughlin S., Murray D. (2005) Nature 438, 605–611 [DOI] [PubMed] [Google Scholar]
- 44. Cheung S. M., Kornelson J. C., Al-Alwan M., Marshall A. J. (2007) Cell. Signal. 19, 902–912 [DOI] [PubMed] [Google Scholar]
- 45. Gold M. R., Aebersold R. (1994) J. Immunol. 152, 42–50 [PubMed] [Google Scholar]
- 46. Watt S. A., Kimber W. A., Fleming I. N., Leslie N. R., Downes C. P., Lucocq J. M. (2004) Biochem. J. 377, 653–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Várnai P., Balla T. (2008) Methods 46, 167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qin S., Stadtman E. R., Chock P. B. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 7118–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Várnai P., Rother K. I., Balla T. (1999) J. Biol. Chem. 274, 10983–10989 [DOI] [PubMed] [Google Scholar]
- 50. Bilancio A., Okkenhaug K., Camps M., Emery J. L., Ruckle T., Rommel C., Vanhaesebroeck B. (2006) Blood 107, 642–650 [DOI] [PubMed] [Google Scholar]
- 51. Hyun T., Yam A., Pece S., Xie X., Zhang J., Miki T., Gutkind J. S., Li W. (2000) Blood 96, 3560–3568 [PubMed] [Google Scholar]
- 52. Bae Y. S., Lee T. G., Park J. C., Hur J. H., Kim Y., Heo K., Kwak J. Y., Suh P. G., Ryu S. H. (2003) Mol. Pharmacol. 63, 1043–1050 [DOI] [PubMed] [Google Scholar]
- 53. Lemmon M. A., Ferguson K. M., O'Brien R., Sigler P. B., Schlessinger J. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 10472–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arbuzova A., Martushova K., Hangyás-Mihályné G., Morris A. J., Ozaki S., Prestwich G. D., McLaughlin S. (2000) Biochim. Biophys. Acta 1464, 35–48 [DOI] [PubMed] [Google Scholar]
- 55. Leventis P. A., Grinstein S. (2010) Annu. Rev. Biophys. 39, 407–427 [DOI] [PubMed] [Google Scholar]
- 56. McLaughlin S., Aderem A. (1995) Trends Biochem. Sci. 20, 272–276 [DOI] [PubMed] [Google Scholar]
- 57. Nomikos M., Mulgrew-Nesbitt A., Pallavi P., Mihalyne G., Zaitseva I., Swann K., Lai F. A., Murray D., McLaughlin S. (2007) J. Biol. Chem. 282, 16644–16653 [DOI] [PubMed] [Google Scholar]
- 58. Lazaridis T. (2005) Proteins 58, 518–527 [DOI] [PubMed] [Google Scholar]
- 59. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., pp. 205–216, Springer-Verlag Inc., New York [Google Scholar]
- 60. Caesar C. E., Esbjörner E. K., Lincoln P., Nordén B. (2006) Biochemistry 45, 7682–7692 [DOI] [PubMed] [Google Scholar]
- 61. Johnson J. E., Rao N. M., Hui S. W., Cornell R. B. (1998) Biochemistry 37, 9509–9519 [DOI] [PubMed] [Google Scholar]
- 62. Cho W., Bittova L., Stahelin R. V. (2001) Anal. Biochem. 296, 153–161 [DOI] [PubMed] [Google Scholar]
- 63. Masuda M., Takeda S., Sone M., Ohki T., Mori H., Kamioka Y., Mochizuki N. (2006) EMBO J. 25, 2889–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sweede M., Ankem G., Chutvirasakul B., Azurmendi H. F., Chbeir S., Watkins J., Helm R. F., Finkielstein C. V., Capelluto D. G. (2008) Biochemistry 47, 13524–13536 [DOI] [PubMed] [Google Scholar]
- 65. Narayan K., Lemmon M. A. (2006) Methods 39, 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McLaughlin S., Wang J., Gambhir A., Murray D. (2002) Annu. Rev. Biophys. Biomol. Struct. 31, 151–175 [DOI] [PubMed] [Google Scholar]
- 67. Killian J. A., von Heijne G. (2000) Trends Biochem. Sci. 25, 429–434 [DOI] [PubMed] [Google Scholar]
- 68. Mulgrew-Nesbitt A., Diraviyam K., Wang J., Singh S., Murray P., Li Z., Rogers L., Mirkovic N., Murray D. (2006) Biochim. Biophys. Acta 1761, 812–826 [DOI] [PubMed] [Google Scholar]
- 69. Manna D., Bhardwaj N., Vora M. S., Stahelin R. V., Lu H., Cho W. (2008) J. Biol. Chem. 283, 26047–26058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Golebiewska U., Nyako M., Woturski W., Zaitseva I., McLaughlin S. (2008) Mol. Biol. Cell 19, 1663–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Janmey P. A., Lindberg U. (2004) Nat. Rev. Mol. Cell Biol. 5, 658–666 [DOI] [PubMed] [Google Scholar]
- 72. Castellano E., Downward J. (2010) Curr. Top. Microbiol. Immunol. 346, 143–169 [DOI] [PubMed] [Google Scholar]