

Abstract
Novel peptidomimetic pharmacophores to PAR2 were designed based on the known activating peptide SLIGRL-NH2. A set of 15 analogues was evaluated with a model cell line (16HBE14o-) that highly expresses PAR2. Cells exposed to the PAR2 activating peptide with N-terminal 2-furoyl modification (2-furoyl-LIGRLO-NH2) initiated increases in intracellular calcium concentration ([Ca2+]i EC50 = 0.84 μM) and in vitro physiological responses as measured by the xCELLigence real time cell analyzer (RTCA EC50 = 138 nM). We discovered two selective PAR2 agonists with comparable potency: compound 1 (2-aminothiazol-4-yl; Ca2+ EC50 = 1.77 μM, RTCA EC50 = 142 nM) and compound 2 (6-aminonicotinyl; Ca2+ EC50 = 2.60 μM, RTCA EC50 = 311 nM). Unlike the previously described agonist, these novel agonists are devoid of the metabolically unstable 2-furoyl modification and thus provide potential advantages for PAR2 peptide design for in vitro and in vivo studies. The novel compounds described herein also serve as a starting point for structure–activity relationship (SAR) design and are, for the first time, evaluated via a unique high throughput in vitro physiological assay. Together these will lead to discovery of more potent agonists and antagonists of PAR2.
INTRODUCTION
Protease-activated receptors (PARs) 1–4 are a family of four G-protein-coupled receptors (GPCRs) that are self-activated by a tethered sequence exposed by proteolysis of the extracellular domain.1 The four members of the GPCR family of PARs are expressed on a wide variety of cell types. PARs are activated by proteolysis in response to endogenous and exogenous proteases and can contribute to both cellular homeostasis and pathology.1,2 In the case of PAR2, the exposed peptides such as SLIGVK-NH2 (human) or SLIGRL-NH2 (murine) remain tethered on the receptor and activate an orthosteric binding site located on second loop of the receptor.1 The different N-termini of the PARs present substrates for a variety of proteases that create selective activation mechanisms for signal transduction. This enzyme-linked self-activation is limited to PARs among GPCRs.3 There are a variety of enzymes that can expose the tethered ligand on PAR, but a key difference between PAR2 and the other PARs is the lack of activation by thrombin.4 As an obvious consequence of its activation mechanism, PAR2 is associated with pathologies with a strong protease release. The involvement of PAR2 in inflammatory diseases such as arthritis, lung inflammation (asthma), inflammatory bowel disease, sepsis, and pain disorders makes PAR2 an attractive target for drug intervention.
Significant tools used to study PARs are small peptides or peptidomimetics that mimic the ligand binding properties of the tethered ligand exposed by proteolysis of the N-terminus from the natural receptor (reviewed in ref 1b): SFLLRN-NH2 activates PAR1; human-derived SLIGKV-NH2 and murine-derived SLIGRL-NH2 activate PAR2; GYPQVN-NH2 activates PAR4. However, these short peptides activate cognate receptors with lower potency compared with native tethered ligands, which require lower binding energy as a consequence of the covalent link to the receptor. Also, untethered activating peptides have the potential to react across PARs. For example, the PAR1 activating peptide SFLLRN-NH2 can also activate PAR2.5 In lieu of this caveat, activating peptides/peptidomimetics provide useful tools for establishment of SAR and rational drug design because they limit off-target effects that are often a complication of natural protease activation and allow for a more potent and specific activation of individual PARs. Indeed, the most potent PAR2 agonist reported to date was developed by modifying a known activating peptide sequence.6 The N-terminal serine of SLIGRL-NH2 was substituted with 2-furoyl, and the resulting analogue 2-furoyl-LIGRLO-NH2 is 10–25 times more efficient at increasing [Ca2+]i in human and rat PAR2 expressing cells.6,7 The development of 2-furoyl-LIGRLO-NH2 significantly moved research toward a better understanding of PAR2 agonist activity and established a platform for rational peptidomimetic design. Such peptides, peptidomimetics, and other small molecules8 have been demonstrated to have increased specificity and affinity for PAR2 and have been used to both activate PAR2 and, in high concentrations, desensitize cells to subsequent PAR2 responses in model cells.6,7,9 Besides therapeutic intervention, agonists of PAR2 can serve as tools for SAR and for defining the cellular and molecular signaling mechanisms that may contribute to pathology.
Although 2-furoyl-LIGRLO-NH2 has been useful in many PAR2 cellular and molecular studies, the furane ring is not considered as a safe structural element for drug design.10 Furane-containing drugs (including the structurally similar 2-acetyl furane and at least eight other drugs like the diuretic drug furosemide) can be metabolically activated by cytochrome P450 to form electrophilic intermediates (γ-ketocarboxylic acids) by oxidative opening of the furane heterocycle. These reactive metabolites have been shown to bind covalently to liver proteins and cause hepatotoxicity in vivo. Because furane-based structures present a potential risk for hepatotoxicity, substitutes for the furane in 2-furoyl-LIGRLO-NH2 may provide a more optimal drug design paradigm.
Despite the potential for drug development from known peptide/peptidomimetic PAR2 agonists as starting points for drug discovery (e.g., SLIGVK-NH2, SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2), there are limited reports on SAR5,8b,11 and a limited selection of small molecule compounds that can be used as tools to understand the contribution of PAR2 to cellular and tissue function. We hypothesized that the 2-furoyl residue could be substituted by other metabolically stable heterocycles at the N-terminus of the peptide. Consistent with this hypothesis, Barry et al.8b has presented a small compound library based on a X-LIGRLI-NH2 hexapeptide motif. Results using a Ca2+ mobilization cellular assay to evaluate the X-LIGRLI-NH2 library showed that (i) no heterocyclic substitution was better than 2-furoyl-LIGRLI-NH2, (ii) aliphatic residues lead to less Ca2+ mobilization, (iii) aromatic residues were required, but electron donating effects of the heteroatom can potentiate the activity (2-furoyl-LIGRLI-NH2, EC50 = 0.16 μM, compared to 4-phenyl-LIGRLI-NH2, EC50 = 0.8 μM, and (iv) steric effects can diminish activity (2-furoyl-LIGRLI-NH2, EC50 = 0.16 μM, compared to 2-benzofuranyl-LIGRLI-NH2, EC50 = 0.25 μM; phenyl-LIGRLI-NH2, EC50 = 0.8 μM, compared to 4-biphenyl-LIGRLI-NH2, EC50 = 15.8 μM). These results supported our hypothesis that a metabolically stable modification of 2-furoyl-LIGRLO-NH2 can be found. We focused our initial design on truncation and conformational constraint of the peptide chain in an effort to move toward small molecule drug design for PAR2. Here, we present potential PAR2 ligands based on the shortened pentapeptide sequence X-LIGRL-NH2.
RESULTS
Synthesis
Compound peptidomimetics were synthesized using standard Fmoc chemistry on Rink amide resin.12 Compounds were cleaved off the resin with TFA-scavenger cocktails and purified by PR-HPLC and/or size-exclusion chromatography. All compounds were 95%+ pure from analytical HPLC and expected MS results.
Agonist Design
Our goal was to design potent and selective agonists of PAR2 that contain a metabolically stable substitution of the furane ring at the N-terminus. Furthermore, we strived to modify the peptide portion of the agonist into a peptidomimetic structure to increase stability for systemic applications. In contrast to a previous reported library used to help guide our design,8b we used the LIGRL-NH2 pentapeptide instead of the more active but longer hexapeptides X-LIGRL-NH2.
Ca2+ Mobilization Assay
A set of 15 compounds based on the X-LIGRL-NH2 sequence were first screened in model epithelial cells (16HBE14o-) by monitoring changes in [Ca2+]i in response to an initial compound dose of 100 μM (based on activity of native SLIGRL-NH2 peptide activation). [Ca2+]i was determined for 80–100 cells using the Ca2+-sensitive fluorescent dye fura-2 and digital imaging microscropy.13 To quantify the response, cells were determined to be “activated” if their [Ca2+]i reached 200 nM or more (from a resting value of 50–75 nM) in a 3 min time period. The percentage of activated cells for each experiment was determined. Compounds 11–16 lacked activity in the Ca2+ mobilization assay (0% activated cells) at 100 μM dose and were eliminated from further screening. Compounds 5–10 activated cells at doses of 100 μM but failed to activate cells at doses of 2.5 μM (for details see Supporting Information Table S1). Compounds 1–4 displayed activated Ca2+ responses in a significant percentage of cells (>5%) at doses of ≤2.5 μM. EC50 values were calculated for these four agonists. As a point of reference, SLIGRL-NH2 has an approximate EC50 of >40 μM in this assay (data not shown).
Because there are a variety of mechanisms that can lead to [Ca2+]i mobilization in epithelial cells, we examined the specificity of the novel compounds for PAR2. We redeployed the Ca2+ mobilization assay in either low expressing PAR2 HeLa cells and HeLa cells transfected with human PAR2 (HeLa-PAR2) or low expressing kNRK and cells transfected with human PAR2 (kNRK PAR2). We found that 2-furoyl-LIGRLO-NH2 and the novel compounds 1 and 2 were able to quickly stimulate increases in [Ca2+]i in HeLa-PAR2 cells (not shown) or kNRK-PAR2 cells (Figure 1). In the absence of PAR2 overexpression, agonist-induced [Ca2+]i changes were minimized and representative of background PAR2 expression in these cell types (Figure 1). From these data we conclude that compounds 1 and 2 represent novel, potent, and selective full agonists at PAR2.
Figure 1.

Ca2+ signaling induced by novel agonists in the presence or absence of PAR2: typical experiments showing the average [Ca2+]i (±SEM) plotted over time for all cells (~90) in a single experiment following agonist exposure. In each experiment 5 μM agonist was added (denoted by line) to PAR2 expressing kNRK cells (top) or plasmid expressing control kNRK cells (bottom). In the PAR2-transfected cells, agonists induce rapid and robust changes in [Ca2+]i. However, in the absence of PAR2 overexpression, [Ca2+]i response is minimal. Similar traces are observed with PAR2-transfected/nontransfected HeLa cells (not shown).
In Vitro Physiological Response Assay
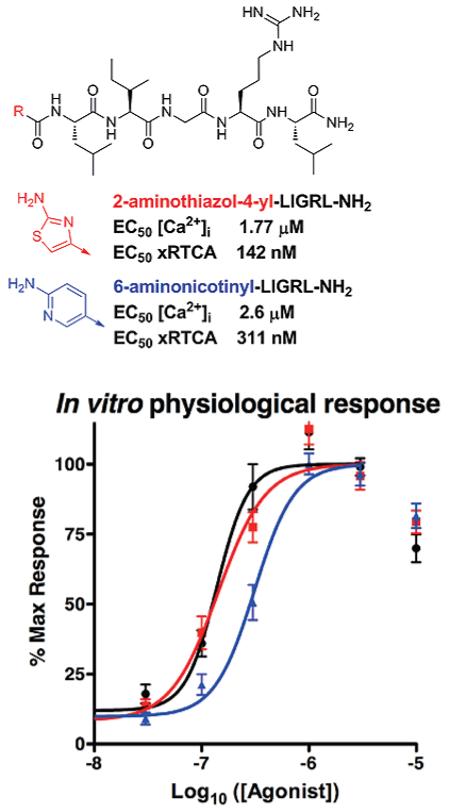
Novel agonists 1 and 2 with Ca2+ mobilization assay EC50 values approaching that determined for 2-furoyl-LIGRLO-NH2 were evaluated along with 2-furoyl-LIGRLO-NH2 in an in vitro physiological response assay using the xCELLigence real time cell analyzer (RTCA, Table 1).14 The RTCA records changes in impedance (reported as a cell index) over a prolonged time course in a noninvasive system. These readings represent a physiological output that reflects the combined cellular effects of multiple signaling pathways such as those activated by PAR2.1,4 Each agonist was applied to the cells over an appropriate dose range and cell index was monitored every 15 s for 2 h. We found that agonist activity was similar among the three compounds, with 2-furoyl-LIGRLO-NH2 and compound 1 displaying nearly identical RTCA EC50 values and compound 2 displaying less potent activity (Figure 2).
Table 1.
Ca2+ Mobilization and in Vitro Physiological (RTCA) Assays
| [Ca2+]i measurements |
||||||
|---|---|---|---|---|---|---|
| R-CO-Leu-Ile-Gly-Arg-Leu-NH2a | % response 100 μM |
% response 10 μM |
% response 2.5 μM |
EC50 (μM) | xRTCAb EC50 (nM) |
|
| 2-furoyl-Leu-Ile-Gly-Arg-Leu-Orn(Aloc)-NH2 | 100 ± 0 | 97.8 ± 1.0 | 93.6 ± 3.3 | 0.84 ± 0.08 | 138 ± 13 | |
| 1 | 2-aminothiazol-4-yl | 100 ± 0 | 96.2 ± 1.4 | 67.5 ± 7.5 | 1.77 ± 0.24 | 142 ± 18 |
| 2 | 6-aminonicotinyl | 100 ± 0 | 93 ± 2.0 | 55 ± 11.6 | 2.60 ± 0.32 | 311 ± 26 |
| 3 | 6-aminopyridin-2-yl | 99.3 ± 0.7 | 94.4 ± 2.6 | 27.1 ± 6.9 | 3.50 ± 0.36 | ND |
| 4 | 4-aminophenyl | 93.0 ± 0.3 | 86.9 ± 4.1 | 16.5 ± 5.3 | 6.59 ± 1.7 | ND |
The structures do not contain C-terminal Orn as 2-furoyl-Leu-Ile-Gly-Arg-Leu-Orn(Aloc)-NH2.
EC50 = concentration of compound that was able to generate 50% maximal intracellular activity. Values are expressed ± SEM (N ≥ 2 for 100 μM; N ≥ 4 for all others in Ca2+ assay; data are expressed % response ± SEM; N ≥ 8 in the RTCA assay). ND = not determined.
Figure 2.

Physiological signaling induced by novel agonists. Dose response curves were calculated based on area under the curve for 2 h of agonist exposure and calculated from ½ log dose steps from 10 μM down to 10 nM for each agonist.
DISCUSSION
To date, there has not been a comprehensive SAR study on N-terminal modifications of short pentapeptide sequences (e.g., X-LIGRL-NH2) built as agonists to PAR2. Therefore, we have designed and synthesized a set of 15 analogues to investigate these N-terminal modifications and their ability to activate PAR2 (Scheme 1). The analogues were first evaluated using a Ca2+ mobilization screen. This initial screen indicated a subset of N-terminal modifications that approached the potency of the furan analogue 2-furoyl-LIGRLO-NH2 with modifications that have structural similarity to be used in further SAR.
Scheme 1.

Solid-Phase Synthesis of Compound 1 and Structures of PAR-2 Agonistsa
a (a) (i) Fmoc/tBu synthetic strategy; (ii) 10% piperidine in DMF, 2 + 20 minutes; (iii) HBTU coupling of 2-aminothiazole-4-carboxylic acid;
(b) (i) 91% TFA, 3% thioanisole, 3% 1,2-ethanedithiol, 3% water, 4 h; (ii) ether extraction; (iii) HPLC and SEC purifications.
We found that aliphatic modifications such as diglycolyl (15), 1-piperidinethyl (14), and 2-cyanomethyl (10) were ineffective (structures are depicted in Scheme 1). Heteroaromatic substituents also did not guarantee PAR2 activity as documented for indole derivatives (11 and 13) or 2-hydroxypyridinyl (12). However, a subset of heteroaromatic substituents (5–9) elicited [Ca2+]i activation at the highest doses tested (100 μM) but failed at doses shown to be effective for highest activity compounds (e.g., 10 or 2.5 μM). In this manner, they were as effective as the native peptide sequence (SLIGRL-NH2). The most potent agonists created (1–4) share a common structural feature, nitrogen based heterocycle (thiazole or pyridine) with an amino group orientated in the ortho position to the nitrogen (Table 1). 2-aminothiazol-4-yl (1) induced Ca2+ mobilization EC50 (1.77 μM) that was higher but not significantly different from that induced by 2-furoyl-LIGRLO-NH (Ca2+ EC50 = 0.84 μM). In contrast, 6-aminonicotinyl (Ca2+ EC50 = 2.6 μM) (2) and 6-aminopyridin-2-yl (3) (Ca2+ EC50 = 3.5 μM) derivatives induced Ca2+ mobilization EC50 values that were significantly higher than that observed for 2-furoyl-LIGRLO-NH2. In 4-aminophenyl (4), the nitrogen atom was removed from the aromatic ring of the nicotinyl residue and the Ca2+ mobilization EC50 dropped to 6.5 μM. Other isomers of aminonicotinic acid analogues such as 6-aminopyridin-4-yl (6) and 6-aminopyridin-5-yl (7) or 3-aminotriazine (8) were ineffective at 2.5 μM. From this short peptapeptide SAR we conclude that both the amino group and the aromatic nitrogen contributed to the PAR2 Ca2+ signaling agonistic activity.
Because PAR2 activation can lead to downstream signaling pathways in addition to Ca2+ signaling, we evaluated the most potent PAR2 agonists on 16HBE14o- cells using RTCA, an in vitro physiological assay. This high-throughput assay detects changes in cell membrane interaction with a tissue culture surface by continually measuring impedance across the tissue culture surface.14 We selected the two high potency novel agonists (1 and 2) and compared them to the most potent PAR2 agonist described to date, 2-furoyl-LIGRLO-NH2. Both novel agonists (1 and 2) stimulated an increase in cell index in a dose-dependent manner and reached their peak responses within 30 min (not shown). Each compound induced the greatest increase in cell index at a dose of 3 μM, which is approximately 1/10 the concentration required for a maximum response to the native ligand SLIGRL-NH2 (not shown). Physiological signaling outputs induced by novel agonists (1 and 2) and 2-furoyl-LIGRLO-NH2 are plotted as dose response curves in Figure 2. The calculated physiological EC50 values are summarized in Table 1. The physiological EC50 of the novel agonist 1 was 142 nM, nearly identical to that of 2-furoyl-LIGRLO-NH2 (138 nM). A significantly higher physiological EC50 (311 nM) was determined for compound 2. Both agonists (1 and 2) achieved equal Emax values equivalent to that of the 2-furoyl-LIGRLO-NH2,6 indicating that these novel ligands are full agonists at PAR2. This RTCA analysis demonstrates potent activation of model epithelial cells by known and novel PAR2 agonists and presents a high-throughput in vitro physiological assay that allows better comparison of full activity of PAR2 agonists than traditional single pathway analyses (e.g., Ca2+ mobilization).
EXPERIMENTAL SECTION
Synthesis
Peptidomimetics were prepared as previously published by solid-phase synthesis as summarized in Scheme 1 on Rink amide TentaGel resin (0.23 mmol/g) using Fmoc/tBu synthetic strategy and standard DIC and HBTU or HCTU activations.12c,15 The synthesis was performed in fritted syringes using a Domino manual synthesizer obtained from Torviq (Niles, MI). N-Terminal heterocyclic acids were obtained from Sigma-Aldrich or TCI and coupled using HBTU activation. For compound 4, the Fmoc-protected version (Fmoc-4-aminobenzoic acid) was used and otherwise was acid coupled as purchased. All compounds were fully deprotected and cleaved from the resin by treatment with 91% TFA (3% water, 3% EDT, and 3% TA). After ether extraction of scavengers, compounds were purified by HLPC and/or size-exclusion chromatography (Sephadex G-25, 0.1 M acetic acid) to >95% purity. All compounds were analyzed for purity by analytical HPLC and MS by ESI or MALDI-TOF.
Ca2+ Mobilization Assay
Model epithelial cells (16HBE14o-, HeLa, kNRK) were grown on matrix coated coverslips for 5–7 days with appropriate growth and selection medium. Cells were loaded with the Ca2+ sensitive dye fura-2 and evaluated for intracellular calcium concentration ([Ca2+]i) over 3 min using digital imaging microscopy.13,16 For calculation of Ca2+ mobilization EC50, response was quantified by the percent of cells responding in the field of view with an increase of [Ca2+]i of ≥200 nM. Compounds were initially tested at 100 μM and down to 250 nM, as appropriate (Table 1, Supporting Information Table S2). To demonstrate specificity, the average [Ca2+]i among all cells (±SEM) in the field of view was calculated over time with [Ca2+]i measured every 0.6 s (Figure 1).
In Vitro Physiological Response Assay
16HBE14o- cells were plated onto 96-well E-plates (Roche) and grown in fully supplemented growth medium to 95% confluence overnight at 37 °C, 5% CO2, and monitored for proliferation using the xCELLigence real time cell analyzer (RTCA; Roche Diagnostics).14a Prior to the experiment, well cultures were washed and replaced with 100 μL of modified Hank’s balanced saline solution (HBSS) prewarmed to 37 °C and allowed to come to room temperature (30–45 min). Each well was supplemented with 100 μL of HBSS containing appropriate agonists to measure seven concentrations for three agonists in quadruplicate. Additional wells (eight) were used for HBSS controls (0 mM agonists). Concentrations were from 10 μM to 10 nM in ½ log steps. Relative impedance in each well was monitored every 15 s over 2 h. For evaluation purposes, relative impedance at any given time is expressed as a “cell index.” Cell index is defined as (Zi – Zo)/(15 Ω), where Zi is impedance at a given time point during the experiment and Zo is impedance before the addition of the agonist. Average cell index for each agonist/dose (n = 4) was graphed over time. Physiological EC50 values were calculated from the area under the curve values.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the Department of Chemistry, University of Arizona, and more specifically Arpad Somogy for mass spectrometry results. The authors also thank Renata Patek and Zhenyu Zhang for technical assistance, Daniel X. Sherwood for his program that allowed for quick quantification of Ca2+ data, and Cara L. Sherwood for her help in the laboratory in getting this work going. This work is part of a multiprincipal investigator collaboration between J.V., T.J.P., and S.B. This work was supported in part by the following grants: NIEHS Superfund Research Grant ES 04940 (S.B.), SRC Project 425.024 (S.B.), NIH Grant R01NS065926 (T.J.P.), Technology and Research Initiative Fund from Arizona State Proposition 301 (J.V.), and NIH Training Grant T32-HL007249 (A.N.F.). S.M.S. is a UA UBRP scholar (Grant HHMI 52005889).
ABBREVIATIONS USED
- Aloc
allyloxycarbonyl
- Boc
tert-butyloxycarbonyl
- BB
bromophenol blue
- CH3CN
acetonitrile
- DCM
dichloromethane
- DI
deionized
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- DIC
diisopropylcarbodiimide
- DMEM
Dulbecco’s modified Eagle medium
- Fmoc
fluorenylmethoxycarbonyl
- FTICR
Fourier Transform ion cyclotron resonance
- ESI-MS
electrospray ionization mass spectrometry
- EDT
1,2-ethanedithiol
- Et2O
diethyl ether
- HBSS
Hank’s balanced saline solution buffered with 25 mM Hepes
- HCTU
O-[1H-(6-chlorobenzotriazol-1-yl)(dimethylamino)ethylene]uranium hexafluorophos phate N-oxide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethane-sulfonic acid
- HOBt
N-hydroxybenzotriazole
- HOCt
6-chloro-1-hydroxybenzotriazole
- GPCR
G-protein-coupled receptor
- MALDI-TOF
matrix assisted laser desorption ionization time of flight
- PAR2
protease-activated receptor type 2
- Pbf
2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- SPPS
solid-phase peptide synthesis
- RP-HPLC
reverse-phase high performance liquid chromatography
- RCTA
xCELLigence real-time cell analyzer
- TA
thioanisole
- THF
tetrahydrofuran
- TIS
triisopropylsilane
- TFA
trifluoroacetic acid
Footnotes
ASSOCIATED CONTENT
Supporting Information. Solid phase synthesis, purification and characterization procedures, spectral and HPLC data, Ca2+ mobilization, and in vitro physiological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol. Rev. 2004;84(2):579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]; (b) Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br. J. Pharmacol. 2008;153(Suppl. 1):S263–S282. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Rattenholl A, Steinhoff M. Proteinase-activated receptor-2 in the skin: receptor expression, activation and function during health and disease. Drug News Perspect. 2008;21(7):369–381. doi: 10.1358/dnp.2008.21.7.1255294. [DOI] [PubMed] [Google Scholar]; (b) Soreide K. Proteinase-activated receptor 2 (PAR-2) in gastrointestinal and pancreatic pathophysiology, inflammation and neoplasia. Scand. J. Gastroenterol. 2008;43(8):902–909. doi: 10.1080/00365520801942141. [DOI] [PubMed] [Google Scholar]; (c) Bueno L. Protease activated receptor 2: a new target for IBS treatment. Eur. Rev. Med. Pharmacol. Sci. 2008;12(Suppl. 1):95–102. [PubMed] [Google Scholar]; (d) Mackie EJ, Loh LH, Sivagurunathan S, Uaesoontrachoon K, Yoo HJ, Wong D, Georgy SR, Pagel CN. Protease-activated receptors in the musculoskeletal system. Int. J. Biochem. Cell Biol. 2008;40(6–7):1169–1184. doi: 10.1016/j.biocel.2007.12.003. [DOI] [PubMed] [Google Scholar]; (e) Russo A, Soh UJ, Trejo J. Proteases display biased agonism at protease-activated receptors: location matters! Mol. Intervensions. 2009;9(2):87–96. doi: 10.1124/mi.9.2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vergnolle N. Protease-activated receptors as drug targets in inflammation and pain. Pharmacol. Ther. 2009;123(3):292–309. doi: 10.1016/j.pharmthera.2009.05.004. [DOI] [PubMed] [Google Scholar]
- (4).Soh UJ, Dores MR, Chen B, Trejo J. Signal transduction by protease-activated receptors. Br. J. Pharmacol. 2010;160(2):191–203. doi: 10.1111/j.1476-5381.2010.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Blackhart BD, Emilsson K, Nguyen D, Teng W, Martelli AJ, Nystedt S, Sundelin J, Scarborough RM. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996;271(28):16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- (6).McGuire JJ, Saifeddine M, Triggle CR, Sun K, Hollenberg MD. 2-Furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. J. Pharmacol. Exp. Ther. 2004;309(3):1124–1131. doi: 10.1124/jpet.103.064584. [DOI] [PubMed] [Google Scholar]
- (7).Hollenberg MD, Renaux B, Hyun E, Houle S, Vergnolle N, Saifeddine M, Ramachandran R. Derivatized 2-furoyl-LIGRLO-amide, a versatile and selective probe for proteinase-activated receptor 2: binding and visualization. J. Pharmacol. Exp. Ther. 2008;326(2):453–462. doi: 10.1124/jpet.108.136432. [DOI] [PubMed] [Google Scholar]
- (8).(a) Barry GD, Le GT, Fairlie DP. Agonists and antagonists of protease activated receptors (PARs) Curr. Med. Chem. 2006;13(3):243–265. doi: 10.2174/092986706775476070. [DOI] [PubMed] [Google Scholar]; (b) Barry GD, Suen JY, Low HB, Pfeiffer B, Flanagan B, Halili M, Le GT, Fairlie DP. A refined agonist pharmacophore for protease activated receptor 2. Bioorg. Med. Chem. Lett. 2007;17(20):5552–5557. doi: 10.1016/j.bmcl.2007.08.026. [DOI] [PubMed] [Google Scholar]
- (9).(a) Kawabata A, Saifeddine M, Al-Ani B, Leblond L, Hollenberg MD. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J. Pharmacol. Exp. Ther. 1999;288(1):358–370. [PubMed] [Google Scholar]; (b) Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, Hollenberg MD. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol. Pharmacol. 2009;76(4):791–801. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gardell LR, Ma JN, Seitzberg JG, Knapp AE, Schiffer HH, Tabatabaei A, Davis CN, Owens M, Clemons B, Wong KK, Lund B, Nash NR, Gao Y, Lameh J, Schmelzer K, Olsson R, Burstein ES. Identification and characterization of novel small-molecule protease-activated receptor 2 agonists. J. Pharmacol. Exp. Ther. 2008;327(3):799–808. doi: 10.1124/jpet.108.142570. [DOI] [PubMed] [Google Scholar]
- (10).Williams DP, Antoine DJ, Butler PJ, Jones R, Randle L, Payne A, Howard M, Gardner I, Blagg J, Park BK. The metabolism and toxicity of furosemide in the Wistar rat and CD-1 mouse: a chemical and biochemical definition of the toxicophore. J. Pharmacol. Exp. Ther. 2007;322(3):1208–1220. doi: 10.1124/jpet.107.125302. [DOI] [PubMed] [Google Scholar]
- (11).(a) Scarborough RM. Protease-activated receptor-2 antagonists and agonists. Curr. Med. Chem.: Cardiovasc. Hematol. Agents. 2003;1(1):73–82. doi: 10.2174/1568016033356698. [DOI] [PubMed] [Google Scholar]; (b) Maryanoff BE, Santulli RJ, McComsey DF, Hoekstra WJ, Hoey K, Smith CE, Addo M, Darrow AL, Andrade-Gordon P. Protease-activated receptor-2 (PAR-2): structure–function study of receptor activation by diverse peptides related to tethered-ligand epitopes. Arch. Biochem. Biophys. 2001;386(2):195–204. doi: 10.1006/abbi.2000.2207. [DOI] [PubMed] [Google Scholar]
- (12).(a) Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: homooligomers of alpha-MSH. Bioorg. Med. Chem. Lett. 2004;14(1):211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]; (b) Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. Solid-phase synthesis of heterobivalent ligands targeted to melanocortin and cholecystokinin receptors. Int. J. Pept. Res. Ther. 2008;14(4):293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vagner J, Xu L, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Heterobivalent ligands crosslink multiple cell-surface receptors: the human melanocortin-4 and delta-opioid receptors. Angew. Chem., Int. Ed. 2008;47(9):1685–1688. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Isakson BE, Olsen CE, Boitano S. Laminin-332 alters connexin profile, dye coupling and intercellular Ca2+ waves in ciliated tracheal epithelial cells. Respir. Res. 2006;7:105–116. doi: 10.1186/1465-9921-7-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Abassi YA, Xi B, Zhang W, Ye P, Kirstein SL, Gaylord MR, Feinstein SC, Wang X, Xu X. Kinetic cell-based morphological screening: prediction of mechanism of compound action and off-target effects. Chem. Biol. 2009;16(7):712–723. doi: 10.1016/j.chembiol.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xi B, Yu N, Wang X, Xu X, Abassi YA. The application of cell-based label-free technology in drug discovery. Biotechnol. J. 2008;3(4):484–95. doi: 10.1002/biot.200800020. [DOI] [PubMed] [Google Scholar]
- (15).(a) Krchnak V, Vagner J, Lebl M. Noninvasive continuous monitoring of solid-phase peptide synthesis by acid-base indicator. Int. J. Pept. Protein Res. 1988;32(5):415–416. doi: 10.1111/j.1399-3011.1988.tb01276.x. [DOI] [PubMed] [Google Scholar]; (b) Krchnak V, Vagner J. Color-monitored solid-phase multiple peptide synthesis under lowpressure continuous-flow conditions. Pept. Res. 1990;3(4):182–193. [PubMed] [Google Scholar]
- (16).Olsen CE, Liguori AE, Zong Y, Lantz RC, Burgess JL, Boitano S. Arsenic upregulates MMP-9 and inhibits wound repair in human airway epithelial cells. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2008;295(2):L293–302. doi: 10.1152/ajplung.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.