

Abstract
Because syndecan-4 (SD-4) on effector and memory T cells inhibits T-cell activation by binding dendritic cell–associated heparan sulfate proteoglycan-integrin ligand (DC-HIL) on antigen presenting cells and because malignant cells of the cutaneous T-cell lymphoma (CTCL) subset, Sézary syndrome (SS), exhibit memory T-cell phenotype, we posited SS cells to express SD-4. Indeed, malignant T cells from patients with SS and from CTCL cell lines constitutively expressed SD-4 at high levels, in contrast to T cells from healthy volunteers and patients with other inflammatory skin diseases and to non-CTCL cell lines that did not. SS cells also bound to DC-HIL at a level higher than normal T cells activated in vitro, resulting in their inhibited proliferation to anti–CD3 antibody. SD-4 on SS cells also trapped transforming growth factor-β1 to their cell surface, enhancing their ability to inhibit activation of syngeneic and allogeneic normal T cells. All of these inhibitory properties were dependent on overexpression of distinct heparan sulfate (HS) moieties by SD-4 on SS cells. Finally, we showed toxin-conjugated DC-HIL to abrogate the ability of SS cells to proliferate in vitro. These findings indicate that SD-4 bearing distinct HS moieties plays a pathogenic role in SS and may be targeted for treatment.
Introduction
Cutaneous T-cell lymphomas (CTCLs) comprise a spectrum of malignant clonal proliferation of T lymphocytes with a predilection for skin involvement, and which have an increasing incidence.1 CTCL presents commonly as mycosis fungoides (MF) and rarely as the leukemic variant, Sézary syndrome (SS); malignant cells in these disorders display effector/memory and central/memory T-cell phenotype, respectively.2 CTCL cells are typically positive for CD4, CD45RO, cutaneous lymphocyte-associated antigen (CLA), and CC chemokine receptors (CCRs), but negative for CD7 and CD26.3 Because existing treatments for CTCL rarely achieve complete remissions, especially for patients with advanced MF and SS,4–6 it is important to better understand pathogenesis, identify more specific diagnostic markers, and develop better treatments.
T-cell activation is dependent on signals delivered by antigen presenting cells (APCs) to the antigen (Ag)–specific T-cell receptor (TCR) and to accessory receptors on T cells. Accessory receptors can be costimulatory or coinhibitory. A costimulatory signal is transmitted by CD80 or CD86 on APCs to the CD28 receptor on T cells. By contrast, a variety of molecules transmit coinhibitory signals including programmed cell death-1 (PD-1) and its ligands PD-L1 and PD-L27,8; B- and T-lymphocyte attenuator (BTLA) and herpes virus entry mediator;9,10 and Tim-3 ligand/Galectin-9 and Tim-3.11,12 Different types of tumors have been shown to express PD-L1,13 and malignant cells of acute myeloid leukemia and CTCL were reported to overexpress cytolytic T lymphocyte–associated antigen 4 (CTLA-4),14,15 thereby suggesting that tumors can use coinhibitory molecules to suppress host antitumor immunity.
The dendritic cell–associated heparan sulfate proteoglycan-integrin ligand (DC-HIL) is a type I transmembrane receptor (95-120 kDa) expressed constitutively at high levels by many different APCs and at lower levels by particular nonlymphoid cells.16 DC-HIL is also known as glycoprotein nmb,17 osteoactivin,18 and hematopoietic growth factor-inducible neurokinin-1 type.19 We have shown DC-HIL to bind heparan sulfate (HS) chains on syndecan-4 (SD-4) expressed on activated (but not resting) T cells, and this binding attenuates strongly TCR-induced activation.20–22 Activated T cells appear to express unique HS on this SD-4 because DC-HIL does not bind to B cells that also express SD-4 constitutively at high levels.
Having shown SD-4 to be expressed primarily by effector and memory T cells,21 we suggested that CTCL cells also express it. Indeed, we found SD-4 to be overexpressed by CTCL cell lines and by malignant T cells in patients with SS, whereas T cells from healthy controls and patients with atopic dermatitis or psoriasis did not. Moreover, SD-4 on CTCL cell lines and SS cells featured distinct HS moieties at levels much greater than levels expressed by normal T cells activated in vitro. These HS moieties were responsible for the ability of SD-4 to inhibit activation of T cells via the following 2 independent mechanisms: binding to DC-HIL and trapping the immunosuppressive cytokine transforming growth factor-β (TGF-β) on the cell surface. Finally, we took advantage of the exclusivity of DC-HIL binding to SD-4 and showed toxin-conjugated DC-HIL to be lethal for SS cells in a SD-4–dependent manner. Our studies suggest a pathogenic role for SD-4 in SS and raise the therapeutic potential of targeting SD-4 in this disease.
Methods
Cell lines
The human CTCL cell lines MJ (G11), HuT-78, and HH, obtained from American Type Culture Collection, were derived from peripheral blood of patients with MF, SS, and aggressive non-MF/SS CTCL, respectively.23–25 MyLa and SeAx CTCL lines were provided by Keld Kaltoft (University of Aarhus, Aarhus, Denmark).26 Other T-cell leukemia lines (Jurkat and Molt-4) were also obtained from American Type Culture Collection. Cells were maintained in RPMI 1640 medium (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal calf serum (HyClone Laboratories) with or without human recombinant interleukin-2 (IL-2, 50 U/mL; PeproTech).
Blood and T-cell culture
Our study was approved by the University of Texas Southwestern Medical Center Institutional Review Board or the University of Texas M. D. Anderson Cancer Center Institutional Review Board, and was conducted according to principles of the Declaration of Helsinki. Participants gave written informed consent in accordance with the Declaration of Helsinki. Blood samples were taken from 4 healthy donors and 8 patients with SS in different clinical stages of their disease (Table 1). Peripheral blood mononuclear cells (PBMCs) were isolated using cell preparation tube with sodium citrate (BD Vacutainer CPT; BD Biosciences). To activate normal T cells, CD4+ T cells were isolated from the PBMCs of healthy donors using a CD4+ T-cell isolation kit (Miltenyi Biotec), and then cultured with phorbol 12-myristate 13-acetate (5 ng/mL; Sigma-Aldrich) plus ionomycin (250 ng/mL; Sigma-Aldrich) for 3 days.
Table 1.
Demographic and phenotypic characteristics in patients with SS
| Patient no. | Age, y/sex | Diagnosis | TCR clone (%)* | Tumor burden expression, % |
|||
|---|---|---|---|---|---|---|---|
| CD4+CD26− | CD3+CD4+, %/μL | CD3+CD8+ | CD4+SD4+ | ||||
| 1 | 73/M | SS/MF IVA | Vβ 18.0 (98) | 83.4 | 93.2/1968 | 1.8 | 92.4 |
| 2 | 80/M | SS/MF IVB | Vβ 13.6 (98) | 74.8 | 89.8/5496 | 2.4 | 82.5 |
| 3A | 76/M | SS/MF III | Vβ 7.2 (89.0) | 79.5 | 84.2/1061 | 2.8 | 94.7 |
| 3B | 75/M | SS | Vβ 7.2 (88) | 79.6 | 84.4/1464 | 1.8 | 71.4 |
| 4 | 71/M | SS IVA | Vβ 17 (94) | 17.9 68.1 (CD26+) | 80.9/1573 | 10.4 | 60.9 |
| 5 | 75/F | SS IVB | αβ (91.5) | 80.4 | 84.9/1467 | 8.7 | 81.1 |
| 6 | 67/F | SS IVB | Vβ 2.0 (98) | 90.9 | 93.0/8229 | 4.0 | 71.0 |
| 7 | 56/M | SS IVB | ND | 33.8 | ND | 13.3 | 32.0 |
| 8 | 62/M | SS IVB | ND | 24.9 | ND | 10.3 | 23.0 |
Patients (Pt.) with SS ranged in age from 56 to 80 years with median age of 70 years, and forming 2 groups: Pt. 1 through Pt. 6 who had high tumor burden (CD4+CD26− T cells was ≥ 74.8%) and Pt. 7 and Pt. 8 who had low tumor burden (≤ 33.8%). Tumor burden of Pt. 4 was much less (CD4+CD26− 17.9%) but with 68.1% of CD4+CD26+ T cells.
IVA indicates stage IV A SS; IVB, stage IV B SS; and ND, not determined.
All patients were shown to have a TCR clone except for Pt. 5, Pt. 7, and Pt. 8 in whom the procedure was not performed. Pt. 3A and Pt. 3B are the same patient, whose samples were taken at different times during progression of disease.
Reverse transcription-polymerase chain reaction analysis
Total RNA (1 μg) isolated from T cells was converted to the cDNA and an aliquot (5%) was used for polymerase chain reaction (PCR) amplification using following the primers: for CD160, 5′-GTTCACCATAAGCCAAGTCACACC-3′ and 5′-TTGCCCCAGCTTATATTTCCACAG-3′; for CTLA-4, 5′-GACCTGGCCCTGCACTCTCCT-3′ and 5′-AAAAACAACCCCGAACTAACTGCT-3′; for BTLA, 5′-ATGCCCTGTGAAATACTGTGCTAA-3′ and 5′-TGCCTGGTGCTTGCTTCTGT-3′; or for PD-1, 5′-GGGCCCGGCGCAATGACA-3′ and 5′-GCGGGCGGGGGATGAGGT-3′. Primers for SD-4 and glyceraldehyde-3-phosphate dehydrogenase were as described previously.20 After 30 cycles of amplification, PCR products were separated electrophoretically on 1% agarose gel.
Antibody and flow cytometry
Antibody (Ab) raised against SD-4 (5G9), CD4 (RPA-T4), CD3 (UCHT1), CD69 (FN50), CD26 (M-A261), and PD-1 (MIH4) were purchased from eBioscience; 3 different monoclonal antibodies (mAbs) against HS (F58-10E4, HepSS-1, and F69-3G10) were from Seikagaku Corporation; anti–TGF-β1 and anti–TGF-β type II receptor Ab were from R&D Systems; and anti–TCR-Vβ subtype Ab was from Beckman Coulter.
For immunophenotyping, T cells or PBMCs (1 × 105) were incubated with primary Ab (1-5 μg/mL) or control immunoglobulin G (IgG) and labeled fluorescently with 5 μg/mL phycoerythrin (PE)–conjugated or fluorescein isothiocyanate-conjugated secondary Ab (Jackson ImmunoResearch Laboratories). After extensive washing, fluorescence intensity of stained cells was analyzed by FACSCalibur (BD Biosciences).
DC-HIL binding
CTCL lines, normal activated CD4+ T cells, or PBMCs from patients with SS (1 × 105 cells) were treated with human IgG (5 μg/mL) before incubation with 10 μg/mL DC-HIL–Fc (the extracellular domain fused with the Fc portion of mouse IgG) produced in transfected COS-1 cells.20 After washing, treated cells were stained fluorescently with PE-conjugated anti–mouse IgG F(ab′)2 (5 μg/mL) and then examined by flow cytometry. To examine involvement of HS, HuT-78 T cells (5 × 105) were pretreated with heparinase I (0.1 U/mL) and heparinase III (0.2 U/mL; Sigma-Aldrich) at 37°C for 2 hours before incubation with DC-HIL–Fc. We also performed blocking experiments in which MJ cells or PBMCs freshly isolated from patients with SS (5 × 105 cells) were pretreated with anti–HS Ab, or control immunoglobulin M (IgM) plus IgG (each 10 or 40 μg/mL) for 30 minutes on ice, followed by DC-HIL binding assay.
Assay for inhibitory function of DC-HIL
Cell line HH, HuT-78 cells, or PBMCs from patients with SS were cultured in microculture wells (1 × 105 cells/well) precoated with anti–CD3 Ab (increasing doses) plus DC-HIL–Fc or control immunoglobulin (Ig, each 10 μg/mL) for 2 days. To measure activation of the HH line, treated cells were stained with anti–CD69 mAb and assayed by flow cytometry for frequency of CD69+ cells in culture. For the HuT-78 cell line or PBMCs, culture supernatant was recovered and examined for IL-2 production using an enzyme-linked immunosorbent assay kit (eBioscience).
TGF-β binding and immunosuppression
CTCL cell lines or freshly isolated PBMCs (1 × 105 cells) were incubated with or without 1 μg/mL TGF-β1 (PeproTech) on ice for 30 minutes. After extensive washing, cells were pretreated with 5 μg/mL mouse IgG on ice for 30 minutes (blocking) and then stained with biotinylated anti–TGF-β1 Ab (2 μg/mL) and PE-streptavidin. Surface-bound TGF-β1 was measured by flow cytometry. Binding was assessed by mean fluorescence intensity (MFI). To assess duration of binding, cells treated with TGF-β1 were incubated for time periods indicated before incubation with anti–TGF-β1 Ab. For inhibition experiments, CTCL cells were pretreated with 20 μg/mL of control IgG, anti–SD-4, anti–HS, or anti–TGF-β type II receptor Ab on ice for 30 minutes before incubation with TGF-β1.
To examine the effect of surface-bound TGF-β1 on T-cell activation, MJ cells or freshly isolated PBMCs (varying numbers) were treated with or without TGF-β1 (1 μg/mL), extensively washed, treated with 25 μg/mL mitomycin C (Sigma-Aldrich) at 37° for 20 minutes. After extensive washing, cells were cocultured with CD4+ T cells (2 × 105 cells/well) freshly isolated from healthy donors in the presence of various doses of anti–CD3 Ab for 3 days. Cells were pulsed with 3H-thymidine (1 μCi/well) for the last 20 hours of culture.
Anti–CTCL activity
DC-HIL–Fc or mouse IgG (as control) was biotinylated using EZ-Link NHS-Biotin (Pierce) following the manufacturer's recommendations (typically, 1 to 2 biotin molecules/protein). Biotinylated protein was then conjugated with streptavidin-saporin (Advanced Targeting System) by mixing together at a protein to saporin ratio of 1:1. Growing HH cells, HuT-78 cells, or freshly isolated PBMCs were harvested, washed, seeded to microculture wells (5 × 104 cells/well), and cultured with 10% fetal calf serum medium containing 100 U/mL IL-2 (eBioscience) and saporin conjugates at indicated concentrations. Proliferation was measured by 3H-thymidine.
Statistical analysis
Results from healthy donors and patients with SS were compared using the Student t test. All experiments were performed at least 2 times with representative results illustrated.
Results
MF/SS cell lines express constitutively high levels of SD-4
Having found effector and central memory T cells to express SD-4 in immunized mice,21 we posited that CTCL cells also express SD-4. We examined SD-4 mRNA expression from total RNA of 3 CTCL lines (HH, MJ, and HuT-78) and 2 non-CTCL leukemia lines (Jurkat and Molt-4, Figure 1A). SD-4 mRNA was expressed constitutively at highest levels by HuT-78 and at a lower level by MJ, but not by HH nor by non-CTCL lines. Note that HH cells were derived from a patient with non-MF/SS CTCL. Flow cytometric analyses (Figure 1B) revealed MJ and HuT-78 cells to express SD-4 protein on the surface at levels similar to that seen for CD4+ T cells from healthy donors activated in vitro. Other CTCL lines (MyLa and SeAx) also expressed high levels of SD-4 (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Finally, we examined CTCL cells for mRNA expression of other coinhibitory receptors (CD160, CTLA-4, BTLA, and PD-1), and found none. Because this absence of PD-1 expression differs from a previous report showing PD-1 to be markedly up-regulated on the CD4+ T cells of patients with SS,27 we assessed the expression by freshly isolated PBMCs from patients with SS using flow cytometry. PD-1 was not expressed by CD26−CD4+ malignant cells (data not shown). Our results suggest that SD-4 (but not other coinhibitory receptors) is a marker for malignant T cells in patients with MF and SS.
Figure 1.

Expression of SD-4 by CTCL lines and PBMCs from SS patients. (A) Total RNA isolated from 3 CTCL or 2 non-CTCL T-cell leukemia (TCL) lines was examined by reverse transcription-PCR for mRNA expression of SD-4, coinhibitors (CD160, CTLA-4, BTLA, and PD-1) or glyceraldehyde-3-phosphate dehydrogenase. (B) The 5 cell lines and normal in vitro–activated CD4+ T cells (CD4-T) were also assayed by flow cytometry for surface expression of SD-4. Positive (open histogram) and control (shaded histogram) staining are shown. (C-D) PBMCs freshly isolated from 7 patients with SS in different stages of lymphomalignancy (Table 1) or 4 normal donors (ND) were examined by flow cytometry for surface expression of SD-4 on CD4+ T cells. Dot plots shown are representative for healthy donors and patients with SS (Pt. 6). (C). Frequency of SD-4+CD4+ cells in total PBMCs of each patient and normal donor is shown as a percentage and plotted in a scatter chart as mean (± SD). P was evaluated by the Student t test. (D-E) PBMCs from 3 patients with SS (Pt. 1 to Pt. 3B) were also examined for SD-4 expression on clonal malignant T cells (TCR-Vβ subtype-positive). (F) PBMCs from Pt. 7 and Pt. 8 (with low tumor burden) contained CD4low and CD4high subpopulations, each of which was examined for SD-4 expression. CD4low and CD4high cells were negative and positive, respectively, for CD26 expression (data not shown). (G) PBMCs freshly isolated from patients with atopic dermatitis, psoriasis, or MF (each 3 patients) were also assayed for SD-4 expression on CD4+ T cells. Data shown represent each disease group.
SD-4 is overexpressed by malignant T cells in patients with SS
PBMCs freshly isolated from 6 patients with SS (Table 1) and from 4 healthy donors were assayed by flow cytometry for expression of SD-4 on CD4+ T cells (Figure 1C). There was little-to-no expression of SD-4 on CD4+ cells from healthy donors with an average frequency 0.4% (± 0.17%) in total PBMCs, whereas expression was found highly on CD4+ cells from all 6 patients with SS at 79% (± 12%, P = 1.7 × 10−11 by the Student t test; Figure 1D). We then examined whether SD-4 expression is confined to malignant T cells in patients with SS who have high tumor burdens (> 90% Vβ+ cells, Figure 1E). PBMCs from 3 patients were stained fluorescently with anti–SD-4 Ab and Ab directed at TCR-Vβ subtype specific for each patient. Almost all SD-4+ cells were also positive for the relevant TCR-Vβ in each of 3 patients. Moreover, all SD-4+/CD4+ cells in patients with SS were negative for CD26, which is another marker of the malignant cells (supplemental Figure 2). Patient no. 7 (Pt. 7) and Pt. 8 who had low tumor burdens differed from Pt. 1 through Pt. 6 who had high tumor burdens, with the former bearing CD26−CD4low malignant cells and CD26+/CD4high normal cells among their PBMCs, and the latter exhibiting only CD26−CD4low malignant cells (Figure 1F and data not shown for CD26 expression). The phenotypic dichotomy of PBMCs in Pt. 7 and Pt. 8 provided an opportunity to evaluate the SS malignant cell–associated expression of SD-4 within the same patient. We found SD-4 to be expressed by CD4low (but not CD4high) cells. Finally, we noted all patients who had low tumor burdens not only to bear the aforementioned 2 malignant cell populations in varying proportions but also to show the just cited restriction of SD-4 expression to the CD4low population.
We next questioned whether such high SD-4 expression can be seen in circulating CD4+ T cells from patients with atopic dermatitis, psoriasis, or MF in which malignant T cells are restricted to skin (Figure 1G). PBMCs were freshly isolated from 3 patients for each disease and assayed for SD-4 expression on CD4+ T cells. Although the frequency of CD4+ T cells varied among diseases, PBMCs from all patients tested were devoid of SD-4 expression. Thus, constitutive SD-4 expression at high levels by circulating T cells is characteristic for SS cells.
Distinct HS moieties overexpressed by CTCL lines and SS cells mediate DC-HIL binding
Having shown SD-4 to be the ligand for DC-HIL, we examined the ability of SD-4+ CTCL cells to bind soluble DC-HIL receptor (DC-HIL–Fc) consisting of the extracellular domain fused to the Fc portion of IgG (Figure 2A). As predicted by expression levels of SD-4, DC-HIL bound MJ at the highest levels and HuT-78 at a lower level. DC-HIL did not bind HH or other non-CTCL leukemia lines (Jurkat and Molt-4). MyLa and SeAx CTCL lines also showed DC-HIL binding (supplemental Figure 1). We also noted that although in vitro–activated CD4+ T cells from normal controls express SD-4 at a high level similar to expression levels of MJ and HuT-78 cells, their ability to bind DC-HIL was much lower.
Figure 2.

DC-HIL binds SD-4+ CTCL cells through distinct HS moieties. (A) All 3 CTCL lines, 2 non-CTCL lines, or normal CD4+ T cells (in vitro–activated) were incubated with control Ig (shaded histogram) or DC-HIL-Fc (open histograms) and stained with PE-conjugated anti–mouse IgG. Binding of DC-HIL was assayed by flow cytometry. (B) HuT-78 cells were left untreated (None) or treated with heparinase before incubation with DC-HIL-Fc. Binding of DC-HIL was assayed as before. (C) The 3 CTCL lines or normal activated CD4+ T cells were stained with 3 different anti–HS (F58-10E4, HepSS-1, and F69-3G10) mAb (open histogram) or control Ig, as a mixture of IgM plus IgG (shaded histogram). Expression of each epitope was assessed by flow cytometry. (D) MJ cells were pretreated separately with the 3 anti–HS mAb or control IgG (10 or 40 μg/mL) before DC-HIL binding assay. Data shown is MFI of DC-HIL-bound cells.
Having shown previously that DC-HIL binding to SD-4 on normal T cells can be abrogated by pretreatment of DC-HIL–Fc with heparin or pretreatment of T cells with heparinase (which cleaves HS from the cell),22 we questioned whether the same is true for CTCL cells. Thus, we pretreated DC-HIL with heparin or HuT-78 cells with heparinase, before binding assay. Either of these pretreatments abrogated binding of DC-HIL to HuT-78 cells (Figure 2B, data for heparinase pretreatment). Because HS is structurally heterogeneous among different cell and tissue types,28 we analyzed HS in CTCL cells versus activated normal T cells using 3 different mAbs, each recognizing a distinct moiety of HS (Figure 2C). We found the F58-10E4 and HepSS-1 (but not F69-3G10) epitopes to be expressed at highest levels by MJ cells, at intermediate levels by HuT-78 cells, and not at all by HH, again correlating with levels of SD-4 expression. Surprisingly, all 3 epitopes were either not expressed or only expressed at a very low level by activated normal CD4+ T cells. We next tested the ability of anti–HS mAb to inhibit DC-HIL binding (Figure 2D). MJ cells were pretreated with or without each mAb before the binding assay. Again, DC-HIL bound untreated MJ cells at high levels, but this binding was blocked markedly (by 70%) by pretreatment with F58-10E4 or HepSS-1 mAb (10 or 40 μg/mL), but not by F69-3G10 mAb for which epitope is absent on CTCL cells. Similar results were seen for HuT-78 cells (data not shown).
We confirmed these data using PBMCs freshly isolated from Pt. 2 and Pt. 3B (Figure 3). DC-HIL bound these PBMCs at levels higher than MJ cells (Figure 3A), and these cells expressed F58-10E4 and HepSS-1 epitopes (but not F69-3G10 epitope) constitutively at high levels, albeit lower than for MJ cells (Figure 3B). DC-HIL binding was blocked by pretreatment with 2 positive mAbs (but not the mAb whose expression is absent) almost completely (Figure 3C). All data are consistent with those data from experiments using CTCL lines. These results indicate that SD-4 on CTCL cells binds to DC-HIL at an affinity much greater than on normal activated T cells because of high expression levels of distinct HS moieties.
Figure 3.
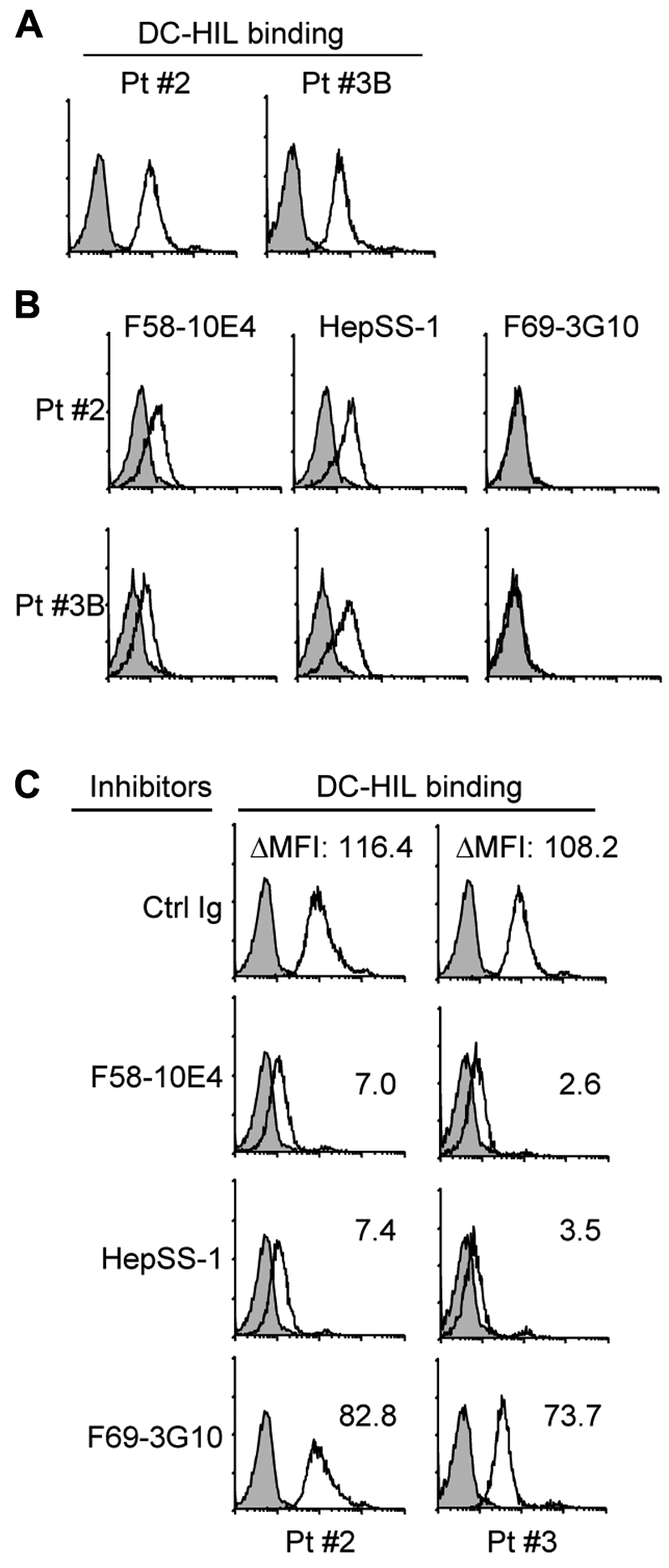
PBMCs of patients with SS use distinct HS moieties to bind DC-HIL at high levels. PBMCs freshly isolated from patients with SS (Pt. 2 with Vβ+ cells at 93%, in which SD-4+ cells were 95%, and Pt. 3B with Vβ+ cells at 98.3%, in which SD-4+ cells were 96%) were assayed by flow cytometry for DC-HIL binding (A) and expression of HS epitopes (B) as in Figure 2. (C) DC-HIL binding by these cells was blocked by pretreatment with 40 μg/mL anti–HS mAb or control Ig (IgM plus IgG). DC-HIL binding is assessed by MFI left after subtracting MFI of control staining from MFI of positive staining (ΔMFI).
SD-4 on CTCL cells mediates the inhibitory function of DC-HIL
We next addressed whether SD-4 on CTCL cells inhibits T-cell activation after binding to DC-HIL (Figure 4). HuT-78 or HH (SD-4− control) cells were cultured with immobilized anti–CD3 Ab (increasing doses) plus DC-HIL–Fc or control Ig (a constant dose). Activation was measured by IL-2 production for HuT-78 cells or by a percentage of the expression of CD69 for HH cells (the latter does not produce IL-2 on activation). HuT-78 cells produced IL-2 after anti–CD3 stimulation in a dose-dependent manner, and this production was inhibited by immobilized DC-HIL–Fc up to 70% (Figure 4A). Expression of CD69 on HH cells was up-regulated by anti–CD3 Ab in a dose-dependent manner, although it was not inhibited by DC-HIL–Fc (Figure 4B). MJ cells were not used in these assays because they do not express CD3. We were unable to examine effects on proliferation because HuT-78 cells are incapable of proliferating in response to anti–CD3 Ab.
Figure 4.

SD-4 on CTCL cells or PBMCs of patients with SS inhibits their anti–CD3 response following binding to DC-HIL. HuT-78 (A), HH cells (B), or PBMCs (C-D) from 2 patients with SS (Pt. 2 and Pt. 3A with Vβ+ cells at 98%, in which SD-4+ cells were 94%) were cultured with immobilized anti–CD3 Ab (increasing doses) plus 5 μg/mL DC-HIL-Fc (●) or control Ig (○). Activation was measured by IL-2 production for the HuT-78 cells (A) and PBMCs (C), by the frequency of CD69+ cells as a percentage for the HH line (B), or by 3H-thymidine (TdR) incorporation (D) as mean (± SD) for n = 3 experiments.
We then addressed whether these results were also true for freshly isolated SS cells. PBMCs freshly isolated from patients with SS were cultured with immobilized anti–CD3 Ab plus control or DC-HIL–Fc, and activation was measured by IL-2 production (Figure 4C) and proliferation (3H-thymidine incorporation, Figure 4D). SS cells proliferated to anti–CD3 and produced IL-2, but at a 70% lower level than that of normal CD4+ T cells. These responses were inhibited markedly by DC-HIL. Thus, SD-4 on CTCL cell lines and SS cells mediates the inhibitory function of DC-HIL to anti–CD3 stimulation.
Epitopes help SD-4 trap TGF-β1 on the cell surface
SD-4 traps TGF-β1 on the cell surface using F58-10E4/HepSS-1 HS epitopes. Because negative regulation of the proliferation of CTCL by SD-4 appears to be independent of its malignancy, we surmised that SD-4 must exert another function independent of DC-HIL. Because SD-4 on macrophages was reported to bind TGF-β1,29 we questioned this possibility for CTCL cells. Having shown each of 3 CTCL lines to constitutively express TGF-β1 mRNA (supplemental Figure 3A), we examined whether CTCL cells bear endogenous TGF-β1 on the surface (Figure 5A). None of the 3 CTCL lines showed significant levels of surface-bound TGF-β1 constitutively. However, after addition of exogenous TGF-β1, both SD-4+ MJ and HuT-78 cells displayed significant surface binding, with SD-4− HH cells showing some binding but at a much lower level (Figure 5A). Such binding was not blocked by pretreatment with anti–SD-4 Ab, whereas F58-10E4 or HepSS-1 anti–HS mAb blocked binding almost completely (Figure 5B). Specificity for HS was supported by inability of control Ig or F69-3G10 (this epitope is absent) to block TGF-β1 binding. We do not think TGF-β receptors were involved in binding because (1) mRNA for TGF-β type I receptor is not expressed by the CTCL lines (supplemental Figure 3A); (2) TGF-β type II receptor protein is not present on the surface of these cells (although it is expressed intracellularly at low levels in HH and HuT-78 cells, supplemental Figure 3B-C); and (3) anti–TGF-β type II receptor Ab does not block binding (Figure 5B).
Figure 5.

HS-mediated binding of TGF-β1 to SD-4+ CTCL cells and PBMCs of patients with SS. (A) CTCL cells were untreated (None) or treated with recombinant TGF-β1 (TGF-β1 added) and stained with anti–TGF-β1 Ab (open histogram) or control IgG (shaded histograms), followed by flow cytometry for surface-bound TGF-β1. The bound amount is expressed as ΔMFI. (B) MJ or HuT-78 cells were pretreated with control Ig, anti–SD-4 Ab, anti–TGF-β type II receptor or anti–HS Ab before addition of TGF-β1. Binding of TGF-β1 was examined and expressed as ΔMFI. (C) MJ cells were loaded with TGF-β1 and incubated for the indicated time periods. Cell surface–bound TGF-β1 was assayed by flow cytometry and expressed as a percentage relative to MJ cells just before incubation. Similarly, PBMCs from 3 different patients were assayed for cell surface binding of TGF-β1 (D), blocking of the binding by Ab pretreatment (E), or retention of cell-bound TGF-β1 (F).
We also examined retention of surface-bound TGF-β1 on MJ cells (Figure 5C). After culturing TGF-β1–loaded CTCL cells for various time periods, surface-bound TGF-β1 was measured by flow cytometry. MJ cells retained 60% of initial surface-bound TGF-β for up to 3 days in culture. TGF-β1 binding, blocking by anti–HS mAb, and their retention were also examined using PBMCs freshly isolated from Pt. 2 and Pt. 3A with SS (Figure 5D-F). The 2 PBMC samples showed binding of TGF-β1 at levels 10-fold greater than MJ and HuT-78 cells (Figure 5D) and this binding was also blocked by pretreatment with anti–HS mAb completely (Figure 5E). Finally, 70% of PBMCs from Pt. 3A with SS held TGF-β1 on the cell surface for at least 3 days (Figure 5F).
We then questioned whether surface-bound TGF-β enhances inhibition of T cell activation by MJ cells (Figure 6A). MJ cells were coated with and without TGF-β, treated with mitomycin C to inhibit DNA synthesis, and finally cocultured with CD4+ T cells (from allogeneic healthy donors) in the presence of immobilized anti–CD3 Ab. Activation was measured by proliferation and IL-2 production (Figure 6A). Untreated MJ cells inhibited proliferation of CD4+ T cells by 60% at the highest dose (compared with control), and TGF-β1 coating enhanced greatly the inhibitory function at each dose tested. Similar results were observed for IL-2 production. Again, we addressed whether this enhancement is also true for PBMCs from patients with SS (Figure 6B). Experiments using these cells showed similar results. Moreover, this effect was not seen for TGF-β–treated SD-4−/CD4+ T cells isolated from PBMCs of healthy donors (supplemental Figure 4). Enhanced inhibition by TGF-β coating was also seen in the syngeneic response (supplemental Figure 5). Altogether, our findings suggest that SD-4 on CTCL cells acts as a cell-surface reservoir for TGF-β1, and this property may potentiate inhibition of T-cell activation.
Figure 6.

Coating with TGF-β1 augments inhibition of T-cell activation by CTCL cells or PBMCs from patients with SS. Varying cell numbers of MJ cells (A) or PBMCs (B) from Pt. 3A were treated with or without TGF-β1 and cocultured with CD4+ T cells isolated from healthy donors in the presence of anti–CD3 Ab. T-cell activation was assessed by 3H-thymidine incorporation or IL-2 production.
Toxin-bearing DC-HIL suppresses proliferation of CTCL cells
Having shown DC-HIL to bind CTCL cells, we questioned whether toxin-bearing DC-HIL can block proliferation of CTCL cells using 3H-thymidine incorporation assay (Figure 7). DC-HIL–Fc (or control Ig) was conjugated to the toxin saporin, a potent type I ribosome-inactivating protein.30 Saporin-conjugated DC-HIL inhibited IL-2–driven proliferation of HuT-78 cells in a dose-dependent manner, with highest inhibition of 60% at a concentration of 80nM (Figure 7A). Specificity for DC-HIL was supported by failure of saporin-conjugated control protein to inhibit proliferation of SD-4+ HuT-78 cells and by no detectable deleterious effect of DC-HIL-saporin on SD-4− HH cells (Figure 7B). We also examined anti–CTCL activity of DC-HIL-saporin to primary SS cells (Figure 7C). More impressively than for HuT-78 cells, DC-HIL-saporin inhibited IL-2–driven proliferation of SS cells by 80% inhibition at 80nM. Again, control saporin had almost no effect on proliferation. These results support the utility of exploiting the DC-HIL/SD-4 pathway to develop new treatments for patients with SS using SD-4+ malignant T cells.
Figure 7.

Saporin-conjugated DC-HIL inhibits proliferation of SD-4+ CTCL cells and PBMCs of patients with SS. HuT-78 (A), HH cells (B), or PBMCs from a patient (Pt. 3A) with SS (C) were cultured for 2 days with varying nanomolar concentrations of saporin-conjugated DC-HIL or control saporin in the presence of IL-2. Proliferation of treated cells was measured by 3H-thymidine uptake. The effect of saporin conjugates on proliferation is expressed as a percentage relative to counts per minute taken by untreated cells (at 0nM saporin conjugate).
Discussion
CTCL cells are commonly positive for CD4, CD45RO, CLA, and CCR4, and negative for CD7 and CD26. Skin-homing memory CD4+CD45RO+CLA+CCR4+ T cells are found frequently in patients with cutaneous inflammation.31 Even PBMCs from healthy donors contain CCR4+ or CLA+ T cells, albeit comprising a minor population.32,33 Because CD7 and CD26 are expressed by most mature CD4+ T cells in healthy donors, their negativity is a useful screening tool for CTCL,34 although positive markers are needed to identify the cells. We now show high SD-4 expression to be characteristic of clonal malignant T cells in SS cells (but not of T cells from patients with atopic dermatitis, psoriasis, and MF or from healthy donors). Normal T cells express SD-4 only after in vitro activation, and in skin-resident T cells of patients with inflammatory skin diseases (our unpublished data). These findings indicate that SD-4 can be a valuable diagnostic marker for circulating SS cells.
The exclusivity of DC-HIL binding to SD-4 on T cells is an opportunity for exploiting toxin-bearing DC-HIL as a Trojan horse for treating SS. A precedent for this approach is denileukin diftitox (Ontak), an IL-2 recombinant protein fused to diphtheria toxin that is approved by the United States Food and Drug Administration to treat CTCL.4 Overexpression by CTCL of the high affinity IL-2 receptor (CD25high) allows denileukin to bind to CTCL cells, deliver the toxin into the tumor cells, and kill the tumor.5 A recent phase 3 study showed denileukin to have a 30% positive (10% complete and 20% partial) response rate in patients treated for CTCL compared with patients receiving placebo.5,35 However, one downside is the high prevalence of adverse side effects especially capillary leak, and constitutional and gastrointestinal symptoms.35 Potentially more serious is the ability of denileukin to kill activated normal T cells and B cells including those in the memory pool important for protective immunity. By contrast, SD-4 is not expressed by circulating normal CD4+ T cells in patients with SS who have low tumor burdens nor in patients with inflammatory skin diseases. Thus, toxin-bearing DC-HIL may hold theoretical advantages over denileukin by sparing normal T cells and B cells, including some activated T cells, from killing.21
SD-4 belongs to the syndecan family of transmembrane receptors heavily laden with HS chains consisting of alternating disaccharide residues (glucuronic acid or iduronic acid with glucosamine).36 The structure of HS moieties varies widely among different cell types.28 In fact, F58-10E4 and HepSS-1 (but not F69-3G10) epitopes are expressed by malignant T cells of SS at extremely high levels compared with normal T cells activated in vitro that express SD-4 at similarly high levels. These epitopes are also expressed by activated mouse T cells but not by mouse B cells that express SD-4 constitutively at high levels (our unpublished data). The F58-10E4 epitope is sensitive to N-desulfation of glycosaminoglycan and its expression is regulated developmentally.37 HepSS-1 epitope includes a HS glycosaminoglycan that correlates negatively with metastatic potential and proliferative capacity of mouse B16 melanoma cells.38 Thus, overexpression of these distinct HS epitopes may reflect abnormal carbohydrate metabolism in SS malignant T cells, akin to that in many malignant cells expressing tumor-associated polysaccharides.39
We showed SD-4 on CTCL cells to inhibit the anti–CD3 response of CTCL lines and SS cells. CTLA-4, another coinhibitory receptor, has been reported to be expressed by CTCL (vs normal T cells) after treatment with the protein kinase C–stimulator phorbol myristate acetate15 or activated by dendritic cells loaded with apoptotic CTCL cells.40 Unfortunately, the function of CTLA-4 on CTCL cells has yet to be characterized. More recently, the SC5 receptor was reported to be expressed by CTCL cells and capable of inhibiting its anti–CD3 response.41 If so, then CTCL cells may possess autocrine regulatory mechanisms that may be separate from their malignant potential.
Proetoglycans (including SD-4) are proteins carrying glycosaminoglycans, large carbohydrates with repeating disaccharide units, including HS, and are known to be modulators of growth factor activities (eg, fibroblast growth factor, chemokines, and TGF-β).42 TGF-β1 and TGF-β2 (but not TGF-β3) have been shown to bind to HS, and thereby initiate proliferation of fibroblasts in an anchorage-independent manner.43 This effect is likely because of protection of HS-bound TGF-β from tryptic degradation and more importantly because of blocking of the neutralizing activity of α2-macroglobulin), a serum protein that forms noncovalent complexes in which the effects of TGF-β become latent.42,44 We found that SD-4 on SS cells can bind TGF-β1 via distinct HS moieties (HepSS-1 and F58-10E4 epitopes) and thereby trap it on the cell surface. Cell surface TGF-β enhanced the ability of SS cells to suppress the anti–CD3 response of T cells from healthy donors. These findings led us to speculate that cell surface TGF-β binds to receptors expressed on antitumor effector T cells45 at much higher affinity than soluble TGF-β, thus delivering negative signals persistently to these cells. These effects may be augmented further by the inability of SD-4 to internalize TGF-β1 (longer surface retention), in contrast to types I and II receptors.46 Such a mechanism is also used by regulatory T cells that exert strong immunosuppression via cell-cell interaction involving cell surface TGF-β (not through secreted inhibitory factors).46,47
Overexpression of HS on leukemic cells was reported previously.48 Malignant cells of adult T cell leukemia overexpress F58-10E4 HS moiety that can trap the chemokine macrophage inflammatory protein-1β/CCL4 on the surface. Trapped macrophage inflammatory protein-1β is then presented to the corresponding receptor on the same cell that triggers integrin activation and promotes cell adhesion to endothelial cells. Therefore, although overexpressed HS can mediate extravasation of tumor cells, this function may not be operative in CTCL and SS cells because these cells express little to no CCR1, a receptor for CC chemokine ligand 4.32
In summary, constitutive high expression of SD-4 by circulating malignant cells in SS distinguishes it from other leukemias and certainly from other inflammatory skin diseases. SD-4 on SS cells is not only overexpressed, but also qualitatively different from SD-4 expressed by other cells because it bears distinct HS moieties that act both as the ligand of DC-HIL and as a cell surface reservoir for TGF-β. Our studies provide a rationale for targeting SD-4 to treat SS.
Supplementary Material
Acknowledgments
We thank Irene Dougherty and Megan Randolph for technical and secretarial assistance.
This work was supported by National Institutes of Health grant AI064927-05 and a Pilot and Feasibility Study grant from Galderma.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.-S.C. performed all experiments; L.H.S. organized clinical data for patients with SS and collected peripheral blood from patients; M.D. recruited and characterized the patients with SS who had high tumor burden and contributed advice; A.P. recruited patients with SS who had a low tumor burden; P.D.C. contributed to the intellectual work and editing; and K.A. coordinated overall research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kiyoshi Ariizumi, Department of Dermatology, UT Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-9069; e-mail: Kiyoshi.Ariizumi@UTSouthwestern.edu.
References
- 1.Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143(7):854–859. doi: 10.1001/archderm.143.7.854. [DOI] [PubMed] [Google Scholar]
- 2.Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and MF arise from distinct T cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116(5):767–771. doi: 10.1182/blood-2009-11-251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger CL, Edelson RL. Current concepts of the immunobiology and immunotherapy of cutaneous T cell lymphoma: insights gained through cross-talk between the clinic and the bench. Leuk Lymphoma. 2003;44(10):1697–1703. doi: 10.1080/1042819031000104033. [DOI] [PubMed] [Google Scholar]
- 4.Wong BY, Gregory SA, Dang NH. Denileukin diftitox as novel targeted therapy for lymphoid malignancies. Cancer Invest. 2007;25(6):495–501. doi: 10.1080/07357900701360096. [DOI] [PubMed] [Google Scholar]
- 5.Talpur R, Jones DM, Alencar AJ, et al. CD25 expression is correlated with histological grade and response to denileukin diftitox in cutaneous T-cell lymphoma. J Invest Dermatol. 2006;126(3):575–583. doi: 10.1038/sj.jid.5700122. [DOI] [PubMed] [Google Scholar]
- 6.Apisarnthanarax N, Talpur R, Duvic M. Treatment of cutaneous T cell lymphoma: current status and future directions. Am J Clin Dermatol. 2002;3(3):193–215. doi: 10.2165/00128071-200203030-00006. [DOI] [PubMed] [Google Scholar]
- 7.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe N, Gavrieli M, Sedy JR, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4(7):670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 10.Sedy JR, Gavrieli M, Potter KG, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6(1):90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez-Fueyo A, Tian J, Picarella D, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4(11):1093–1101. doi: 10.1038/ni987. [DOI] [PubMed] [Google Scholar]
- 12.Zhu C, Anderson AC, Schubart A, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(12):1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 13.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 14.Pistillo MP, Tazzari PL, Palmisano GL, et al. CTLA-4 is not restricted to the lymphoid cell lineage and can function as a target molecule for apoptosis induction of leukemic cells. Blood. 2003;101(1):202–209. doi: 10.1182/blood-2002-06-1668. [DOI] [PubMed] [Google Scholar]
- 15.Wong HK, Wilson AJ, Gibson HM, et al. Increased expression of CTLA-4 in malignant T cells from patients with Mycosis fungoides-cutaneous T-cell lymphoma. J Invest Dermatol. 2006;126(1):212–219. doi: 10.1038/sj.jid.5700029. [DOI] [PubMed] [Google Scholar]
- 16.Shikano S, Bonkobara M, Zukas PK, Ariizumi K. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J Biol Chem. 2001;276(11):8125–8134. doi: 10.1074/jbc.M008539200. [DOI] [PubMed] [Google Scholar]
- 17.Weterman MA, Ajubi N, van Dinter I, et al. nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int J Cancer. 1995;60(1):73–81. doi: 10.1002/ijc.2910600111. [DOI] [PubMed] [Google Scholar]
- 18.Safadi FF, Xu J, Smock SL, et al. Cloning and characterization of osteoactivin, a novel cDNA expressed in osteoblasts. J Cell Biol. 2001;84(1):12–26. doi: 10.1002/jcb.1259. [DOI] [PubMed] [Google Scholar]
- 19.Metz RL, Patel PS, Hameed M, Bryan M, Rameshwar P. Role of human HGFIN/nmb in breast cancer. Breast Cancer Res. 2007;9(5):R58. doi: 10.1186/bcr1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung JS, Sato K, Dougherty I, Cruz PD, Jr, Ariizumi K. DC-HIL is a negative regulator of T lymphocyte activation. Blood. 2007;109(10):4320–4327. doi: 10.1182/blood-2006-11-053769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung JS, Dougherty I, Cruz PD, Jr, Ariizumi K. Syndecan-4 mediates the coinhibitory function of DC-HIL on T cell activation. J Immunol. 2007;179(9):5778–5784. doi: 10.4049/jimmunol.179.9.5778. [DOI] [PubMed] [Google Scholar]
- 22.Chung JS, Bonkobara M, Tomihari M, Cruz PD, Jr, Ariizumi K. The DC-HIL/syndecan-4 pathway inhibits human allogeneic T-cell responses. Eur J Immunol. 2009;39(4):965–974. doi: 10.1002/eji.200838990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gootenberg JE, Ruscetti FW, Mier JW, Gazdar A, Gallo RC. Human cutaneous T cell lymphoma and leukemia cell lines produce and respond to T cell growth factor. J Exp Med. 1981;154(5):1403–1418. doi: 10.1084/jem.154.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starkebaum G, Loughran TP, Jr, Waters CA, Ruscetti FW. Establishment of an IL-2 independent, human T-cell line possessing only the p70 IL-2 receptor. Int J Cancer. 1991;49(2):246–253. doi: 10.1002/ijc.2910490218. [DOI] [PubMed] [Google Scholar]
- 25.Popovic M, Sarin PS, Robert-Gurroff M, et al. Isolation and transmission of human retrovirus (human T-cell leukemia virus). Science. 1983;219(4586):856–859. doi: 10.1126/science.6600519. [DOI] [PubMed] [Google Scholar]
- 26.Kaltoft K, Bisballe S, Dyrberg T, et al. Establishment of two continuous T-cell strains from a single plaque of a patient with mycosis fungoides. In Vitro Cell Dev Biol. 1992;28A(3 Pt 1):161–167. doi: 10.1007/BF02631086. [DOI] [PubMed] [Google Scholar]
- 27.Samimi S, Benoit B, Evans K, et al. Increased programmed death-1 expression on CD4+ T cells in cutaneous T-cell lymphoma: implications for immune suppression. Arch Dermatol. 2010;146(12):1382–1388. doi: 10.1001/archdermatol.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 29.Ishiguro K, Kadomatsu K, Kojima T, et al. Syndecan-4 deficiency leads to high mortality of lipopolysaccharide-injected mice. J Biol Chem. 2001;276(50):47483–47488. doi: 10.1074/jbc.M106268200. [DOI] [PubMed] [Google Scholar]
- 30.Santanche S, Bellelli A, Brunori M. The unusual stability of saporin, a candidate for the synthesis of immunotoxins. Biochem Biophys Res Commun. 1997;234(1):129–132. doi: 10.1006/bbrc.1997.6597. [DOI] [PubMed] [Google Scholar]
- 31.Akdis CA, Akdis M, Simon H-U, Blaser K. Regulation of allergic inflammation by skin-homing T cells in allergic eczema. Int Arch Allergy Immunol. 1999;118(2-4):140–144. doi: 10.1159/000024051. [DOI] [PubMed] [Google Scholar]
- 32.Narducci MG, Scala E, Bresin A, et al. Skin homing of Sézary cells involves SDF-1-CXCR4 signaling and down-regulation of CD26/dipeptidylpeptidase IV. Blood. 2006;107(3):1108–1115. doi: 10.1182/blood-2005-04-1492. [DOI] [PubMed] [Google Scholar]
- 33.Fuhlbrigge RC, Kiefler JD, Armerding D, Kupper TS. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature. 1997;389(6654):978–981. doi: 10.1038/40166. [DOI] [PubMed] [Google Scholar]
- 34.Bernengo MG, Novelli M, Quaglino P, et al. The relevance of the CD4+CD26− subset in the identification of circulating Sézary cells. Br J Dermatol. 2001;144(1):125–135. doi: 10.1046/j.1365-2133.2001.04014.x. [DOI] [PubMed] [Google Scholar]
- 35.Horwitz SM. Novel therapies for cutaneous T-cell lymphomas. Clin Lymphoma Myeloma. 2008;8(suppl 5):S187–S192. doi: 10.3816/CLM.2008.s.015. [DOI] [PubMed] [Google Scholar]
- 36.Baciu PC, Saoncella S, Lee SH, et al. Syndesmos, a protein that interacts with the cytoplasmic domain of syndecan-4, mediates cell spreading and actin cytoskeletal organization. J Cell Sci. 2000;113(Pt 2):315–324. doi: 10.1242/jcs.113.2.315. [DOI] [PubMed] [Google Scholar]
- 37.David G, Bai XM, Van der SB, Cassiman JJ, Van den BH. Developmental changes in heparan sulfate expression: in situ detection with mAbs. J Cell Biol. 1992;119(4):961–975. doi: 10.1083/jcb.119.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kure S, Yoshie O, Aso H. Metastatic potential of murine B16 melanoma correlates with reduced surface heparan sulfate glycosaminoglycan. Jpn J Cancer Res. 1987;78(11):1238–1245. [PubMed] [Google Scholar]
- 39.Ono M, Hakomori S. Glycosylation defining cancer cell motility and invasiveness. Glycoconj J. 2004;20(1):71–78. doi: 10.1023/B:GLYC.0000018019.22070.7d. [DOI] [PubMed] [Google Scholar]
- 40.Berger CL, Tigelaar R, Cohen J, et al. Cutaneous T-cell lymphoma: malignant proliferation of T-regulatory cells. Blood. 2005;105(4):1640–1647. doi: 10.1182/blood-2004-06-2181. [DOI] [PubMed] [Google Scholar]
- 41.Nikolova M, Tawab A, Marie-Cardine A, et al. Increased expression of a novel early activation surface membrane receptor in cutaneous T cell lymphoma cells. J Invest Dermatol. 2001;116(5):731–738. doi: 10.1046/j.0022-202x.2001.doc.x. [DOI] [PubMed] [Google Scholar]
- 42.Ruoslahti E, Yamaguchi Y. Proteoglycans as modulators of growth factor activities. Cell. 1991;64(5):867–869. doi: 10.1016/0092-8674(91)90308-l. [DOI] [PubMed] [Google Scholar]
- 43.Lyon M, Rushton G, Gallagher JT. The interaction of the transforming growth factor-betas with heparin/heparan sulfate is isoform-specific. J Biol Chem. 1997;272(29):18000–18006. doi: 10.1074/jbc.272.29.18000. [DOI] [PubMed] [Google Scholar]
- 44.Feige JJ, Negoescu A, Keramidas M, Souchelnitskiy S, Chambaz EM. Alpha 2-macroglobulin: a binding protein for transforming growth factor-beta and various cytokines. Horm Res. 1996;45(3-5):227–232. doi: 10.1159/000184793. [DOI] [PubMed] [Google Scholar]
- 45.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7(10):1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 46.Miyazono K. TGF-beta receptors and signal transduction. Int J Hematol. 1997;65(2):97–104. doi: 10.1016/s0925-5710(96)00542-7. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+) CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194(5):629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanaka Y, Kimata K, Wake A, et al. Heparan sulfate proteoglycan on leukemic cells is primarily involved in integrin triggering and its mediated adhesion to endothelial cells. J Exp Med. 1996;184(5):1987–1997. doi: 10.1084/jem.184.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.