

Summary
We show that entecavir, an FDA-approved drug used to treat chronic hepatitis B, potently inhibits HIV replication in vitro and leads to a 1 log decline in HIV RNA in vivo. Detailed analysis of two HIV-HBV co-infected patients receiving up to seven months of entecavir monotherapy demonstrated accumulation of HIV variants with the lamivudine-resistance mutation, M184V, in one of them. In vitro experiments demonstrated that M184V confers resistance to entecavir. Thus, entecavir has clinically relevant anti-HIV activity and can select for variants resistant to anti-HIV drugs. Until more is known about HIV resistance patterns and their selection by entecavir, caution is needed when using entecavir in HIV-HBV co-infected individuals not receiving fully suppressive antiretroviral regimens.
Introduction
Entecavir, an analogue of 2’-deoxyguanosine, is converted intracellularly to an active 5’-triphosphate form which inhibits hepatitis B virus (HBV) polymerase (1-3). Entecavir is approved by the FDA for the treatment of chronic hepatitis B. A previous study (1) indicated that it has no clinically relevant activity against human immunodeficiency virus - type 1 (HIV), and this conclusion is included in the package insert. This selective activity against HBV is unique amongst the nucleoside/nucleotide analogues available for hepatitis B treatment (4-7). Thus, several treatment guidelines, including the United States Public Health Service Guidelines, recommend entecavir for hepatitis B treatment in HIV-infected individuals who do not meet criteria for HIV treatment (8-11). Here, we report that entecavir has clinically relevant activity against HIV and can select HIV variants resistant to the widely used antiretroviral drugs lamivudine and emtricitabine.
Case reports
Patient #1
Patient #1 is a 31 year old Caucasian male co-infected with HIV and HBV who tested seropositive for HIV in 1990. In 2000, he was prescribed zidovudine, lamivudine, and nevirapine, which he took intermittently for <1 year. In February 2002, his CD4 cell count was 596 cells/mm3 with a plasma HIV RNA of 14,602 copies/ml (Figure 1). His CD4 cell counts and HIV RNA levels remained stable for the next four years without therapy. In March, 2006, he commenced entecavir therapy for chronic hepatitis B. At this time, his HIV RNA level was 34,088 copies/ml and his CD4 cell count was 574 cells/mm3. In the first two months on entecavir, HBV DNA declined from 9.60 to 3.95 log IU/ml. Simultaneously, the HIV RNA declined by approximately 1 log to 2193 copies/ml, and his CD4 cell count increased to 634 cells/mm3 with a parallel rise in the percentage of CD4 cells from 17.8% to 18.9%. His HIV RNA has remained below baseline with the most recent value after 10 months of entecavir at 5435 copies/ml and a CD4 cell count of 603 cells/mm3.
Figure 1.
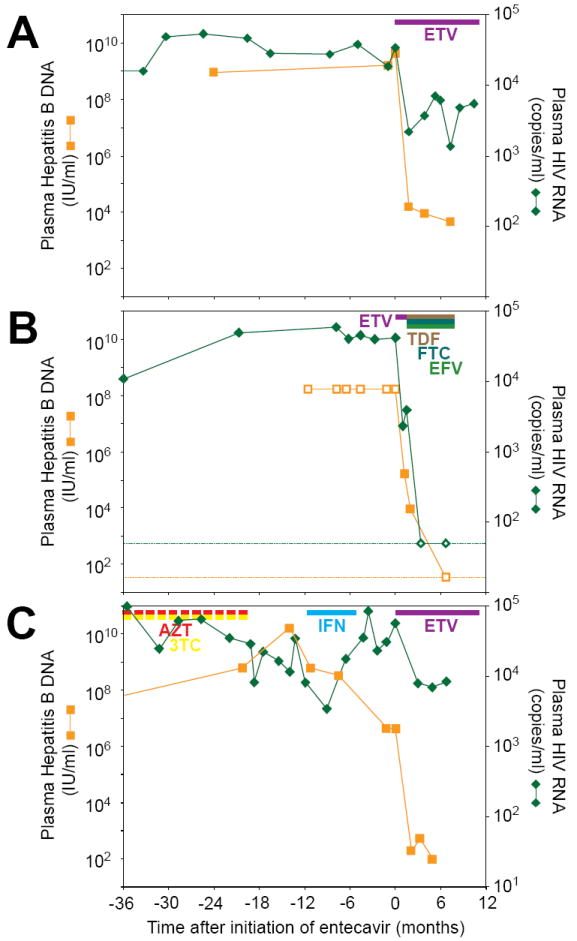
Entecavir therapy causes an acute drop in plasma levels of HBV and HIV-1. Viral loads for patients #1 (A), #2 (B), and #3 (C) are shown as a function of time after starting entecavir. Colored bars indicate treatment with entecavir (ETV), tenofovir (TDF), emtricitabine (FTC), efavirenz (EFV), zidovudine (AZT), lamivudine (3TC), and pegylated-interferon alpha-2a (IFN). The AZT and 3TC bars are interrupted to indicate the intermittent nature of this treatment. Dotted lines indicate the limits of detection of the relevant viral loads assays. Open symbols indicate assay values above or below the range of the relevant assays. Time zero values for patient #2 were estimated from the geometric mean of the 3 prior values. The Hepatitis B DNA value 1.2 months before entecavir therapy was used as the time zero value for patient #3.
Patient #2
Patient #2 is a 24 year old male diagnosed with HIV infection in February 2003. He developed acute HBV infection in March 2005, but he did not meet criteria for antiretroviral therapy with HIV RNA levels ranging from 40,000 to 60,000 copies/ml and CD4 cell counts generally >500 cells/mm3. In February, 2006, he started entecavir for treatment of chronic hepatitis B. After one month, the HIV RNA declined ~1 log from 40,273 to 2,347 copies/ml (Figure 1), and his CD4 cell count rose from 490 to 568 cells/mm3. While on entecavir, HBV DNA declined from approximately 9 to 5 log IU/ml. Given the concern for activity of entecavir against HIV and for the possible generation of drug-resistant HIV, he was prescribed tenofovir, emtricitabine and efavirenz after 45 days of entecavir.
Patient #3
Patient #3 is a 46 year old HIV-HBV co-infected male who became HIV seropositive in the 1980s. In 1993, he received zidovudine monotherapy for an undetermined period of time followed by Combivir (zidovudine plus lamivudine) intermittently between 1997-2004. He was not fully compliant to this therapy. Prior to Combivir in 1996, his HIV RNA was 30,075 copies/ml and his CD4 cell count was 375 cells/mm3. From September 2005-February 2006, he received pegylated-interferon alpha-2a to treat his chronic hepatitis B and had a poor response. In August 2006, he began entecavir monotherapy for chronic hepatitis B. At the time entecavir was started, he had an HIV RNA of 55,451 copies/ml and a CD4 cell count of 399 cells/mm3. After 2 months of entecavir, his HIV RNA declined approximately 1 log to 7797 copies/ml with a concomitant rise in his CD4 cell count to 480 cells/mm3. His HIV RNA has remained below baseline after 7 months of entecavir monotherapy. During this period, his HBV DNA declined from 6.63 to 1.99 log IU/ml.
Methods
This study was approved by the Johns Hopkins University Institutional Review Board and was designed by Moira McMahon, Robert Siliciano, and Chloe Thio. All of the authors participated in gathering and analyzing data and writing and reviewing the paper.
Patient Samples
Peripheral blood was obtained from Patient #1 and Patient #3 after they provided written informed consent. HIV RNA was extracted from plasma and a segment of the pol gene was amplified by reverse transcriptase (RT)-PCR, as described previously (12). The products were cloned and multiple independent clones from each time point were sequenced. A maximum likelihood phylogenetic tree was constructed using PAUP, as previously described (13).
Phenotypic Analysis
Dose response curves for inhibition of HIV infection were generated in vitro using a previously described phenotypic assay (12). Briefly, recombinant CXCR4-tropic HIV pseudoviruses were generated by co-transfecting HEK 293T cells with a plasmid containing the HIV genome with a portion of envelope gene replaced by eGFP (pNL4-3-ΔE-GFP) and with a plasmid encoding the HIV envelope (pCXCR4). Culture supernatants were collected, spun at 335×g, and filtered to clear cell debris. Virus was pelleted at 100,000×g for 2 hrs at 4°C. Viral supernatants were standardized by p24 ELISA (Beckman Coulter, Fullerton, CA).
To obtain primary CD4+ T lymphoblasts for infection, peripheral blood mononuclear cells (PBMC) from healthy donors were activated with PHA for 48-72 hours. CD4+ lymphoblasts were isolated using Miltenyi Biotech beads (Auburn, CA) and pretreated for 16-19 hrs with increasing amounts of the indicated drugs prior to infection with standardized amounts of virus. The extent of infection was determined by quantifying the number of GFP+ cells after 72 hours using FACS analysis. These experiments were repeated in triplicate with three different healthy blood donors. The M184V mutant plasmid was generated by site directed mutagenesis (Stratagene, La Jolla, CA) of pNL4-3-ΔE-GFP, and virus was made and standardized as above. The 1.5kb patient-derived pol sequence was amplified using RT-PCR and inserted into pNL4-3-ΔE-GFP as previously described (12).
Analysis of the effect of entecavir on reverse transcription
CXCR4-tropic recombinant HIV pseudovirus was treated with DNase and used to infect CD4+ lymphoblasts from healthy donors in the presence or absence of 50μM entecavir. Total DNA was isolated at 8 and 24 hours after infection and products of reverse transcription were quantified by real time PCR using three separate sets of primers (available upon request).
Results
Entecavir is a potent inhibitor of HIV replication in vitro
To determine whether the observed in vivo declines in HIV RNA levels were due to direct effects of entecavir on HIV replication, we used a precise HIV infectivity assay (12). This assay is related to commercial phenotypic assays (14) but has single cell sensitivity and utilizes primary CD4+ T cells rather than transformed cell lines. HIV pseudoviruses carrying pol gene sequences from a reference HIV isolate or from patient-derived isolates were used to infect CD4+ lymphoblasts in the presence of increasing concentrations of antiviral drugs. Entecavir potently inhibited infection by both the reference strain (IC50 between 0.1 and 1 nM) and the Patient #1-derived wild type pseudoviruses (IC50 ~1 nM) (Figure 2A). Although the patient #1-derived pseudovirus had a slightly reduced susceptibility to entecavir compared to that of the reference strain, the ability of entecavir to inhibit infection at very low concentrations was still apparent. Three additional patient-derived isolates including that from Patient #3 were also tested in this system, and infection by all three was inhibited by entecavir with an IC50 consistently in the low nanomolar range (Figure 2B). The in vitro inhibition is consistent with the one log decline in HIV viral load observed in the patients. Interestingly, the IC50 of entecavir for HIV is 100-1000 fold lower than that of zidovudine in this system (~0.2 μM). Importantly, the IC50 of entecavir in vitro is below the plasma concentrations achieved in vivo at doses given for hepatitis B (Cmax = 28 nM) (15).
Figure 2.

Entecavir is a potent inhibitor of HIV infection in vitro. (A) Effect of entecavir and zidovudine on infection of primary CD4+ T cells in vitro with pseudoviruses carrying pol sequences from the wildtype HIV reference strain NL4-3 (solid lines) or from a wild type clone from patient #1 (dotted lines). Results are expressed as % of maximal infection = (number of infected cells observed in the presence of the indicated concentration of drug/the maximal number of infected cells observed in the absence of a drug effect) × 100. The dotted black line represents a 50% decrease in the number of GFP+ cells. Where they would be obscured by a symbol, error bars are shown at the edge of the symbol. Infection by pseudoviruses carrying Patient #1-derived pol sequence was inhibited by entecavir with an IC50 of ~0.001 μM. Inhibition of infection by zidovudine is also shown for purposes of comparison to entecavir. (B) Effect of entecavir on infection of primary CD4+ T cells in vitro with pseudoviruses carrying HIV pol sequences isolated from other patients. Patient #3 is described herein. Patients #139 and #154 have been previously described (13). The dose-response curves were similar for all isolates. The isolate from patient #154 had no drug resistance mutations. The isolate from patient #3 had the non-nucleoside RT inhibitor resistance mutation K103N in RT. The isolate from patient #139 had the V108I and T215D mutations in RT. (C) Dose response curves for wildtype (wt, open symbols) and M184V point mutant (M184V, closed symbols) forms of the reference NL4-3 pseudovirus in the presence of increasing concentrations of entecavir, zidovudine, or lamivudine. The data represent three experiments. (D) Dose response curves for pseudoviruses carrying pol sequences from wild type (wt, open symbols) and M184V mutant (M184V, closed symbols) isolates from patient #1. The experiment was repeated three times with very similar results, and representative data from one experiment are shown. The curves in (A) for wildtype viruses in the presence of entecavir and zidovudine are identical to those in (B) and (C).
For all HIV isolates tested, the dose response curve for entecavir was atypical in that although the drug partially inhibited HIV infection at very low concentrations, the anti-HIV activity of entecavir reaches a plateau at drug concentrations in the low nanomolar range and concentrations above 10 nM do not give increased inhibition (Figure 2A and 2B). In contrast, zidovudine shows higher levels of inhibition of HIV replication with increasing concentrations (Figure 2A). The fact that the inhibitory activity of entecavir plateaus may explain why anti-HIV activity was not detected using conventional assays.
Because entecavir is a nucleoside analogue, we hypothesized that the observed anti-HIV activity was due to inhibition of HIV reverse transcriptase. To test this hypothesis, we used real time PCR to quantify reverse transcripts in cells infected in the presence of entecavir. Entecavir decreased the levels of early, intermediate, and late reverse transcripts (Supplemental Figure 1) suggesting that it may inhibit HIV replication at or before the reverse transcription step.
The M184V mutant virus is selected by and is resistant to entecavir
To determine if drug-resistant HIV variants were selected by entecavir, we cloned and sequenced HIV RNA from the plasma of Patient #1 and Patient #3 immediately before the start of entecavir and at various time points after initiation of entecavir therapy. At the time entecavir was started in Patient #1, none of 19 independent clones had mutations in HIV pol known to confer resistance to anti-HIV drugs. At 2, 4 and 6 months after initiation of entecavir, 12%, 61% and 96% of the clones, respectively, harbored the M184V mutation (Figure 3B). This mutation confers high level resistance to the anti-HIV drugs lamivudine and emtricitabine (16-18). No other known resistance mutations were observed. The clinical genotype obtained at 6 months after starting entecavir confirmed the presence of the M184V mutation. By phylogenetic analysis (Figure 3C), the M184V mutants detected while on entecavir therapy showed greater divergence from the most recent common ancestor than did most of the wild type sequences present before entecavir therapy. These results are consistent with the selection of M184V viruses by entecavir.
Figure 3.

Selection for the M184V mutation in HIV-1 RT in a patient on entecavir. (A) Time course of sampling for patient #1. Plasma was obtained for genotypic analysis on the day that entecavir was started (time point 0) and after 2, 4, and 6 months of entecavir monotherapy (time points 1, 2, and 3, respectively). The plasma HIV-1 RNA levels during this period are indicated in the graph. See Figure 1 for full details on the clinical course of patient #1. (B) Fraction of independent plasma virus isolates carrying the M184V mutation at time points 0, 1, 2, and 3. The number of independent clones analyzed was 19, 41, 18, and 27 for time points 0, 1, 2 and 3, respectively. * denotes no resistant mutations detected. (C) Maximum likelihood phylogenetic tree of wild type (open symbols) and M184V mutants (closed symbols) from time points 0, 1, 2, and 3. All isolates from patient #1 cluster together and are clearly distinct (bootstrap value =99) from reference clade B isolates obtained from other patients (HXB2, SF2, RF, and NL4-3)
In the case of Patient #3, who had more extensive prior lamivudine therapy than Patient #1, no clones with the M184V mutation were detected at the time entecavir was started. Interestingly, after months of entecavir therapy, only 1 out of 175 clones had the M184V. The only other known resistance mutation detected in this patient was the non-nucleoside RT inhibitor resistance mutation K103N, which was present in all the clones pre- and post-entecavir. Taken together, these results indicated that entecavir can select the M184V mutation in some but not all patients.
Since entecavir can select the M184V mutation in vivo, we hypothesized that HIV variants with the M184V mutation would be less susceptible to entecavir. The reference strain HIV carrying the M184V mutation was resistant to entecavir (Figure 2C). In control experiments (Figure 2C), we showed that the same M184V mutant virus had markedly decreased susceptibility to lamivudine and the expected hypersusceptiblity to zidovudine (16, 17, 19, 20). A post-entecavir isolate from patient #1 containing the M184V mutation behaved similarly (Figure 2D).
Discussion
This study demonstrates that entecavir is a potent but partial inhibitor of HIV replication in vitro and in vivo and that in some patients it can select for viruses bearing the M184V mutation, which confers high level resistance to entecavir and to the widely used anti-HIV drugs lamivudine and emtricitabine. These conclusions are supported by the striking temporal association of entecavir therapy with 1 log reductions in HIV RNA as well as in vitro studies showing inhibition of HIV infectivity at clinically relevant concentrations. In addition, one patient clearly demonstrated selection of M184V variants in vivo while on entecavir monotherapy and in vitro data demonstrate reduced activity of entecavir on the M184V variants.
These data have important implications for treatment of hepatitis B in HIV-infected patients. Current guidelines recommend entecavir as the first line treatment in HIV-HBV co-infected persons who do not require anti-HIV therapy (8, 9). Since this recommendation is predicated on the assumption that entecavir does not have activity against HIV, our data indicate that this recommendation should be reconsidered, especially since entecavir can select for the M184V mutation in some patients.
Our data contradict a previous report that entecavir does not have anti-HIV activity at clinically relevant concentrations (1). That report used an assay described in 1989 by Weislow et al., which relies on cytopathic effects resulting from infection (21). We believe the quantitative infectivity assay we used has several advantages over previous assays that allow it to detect subtle anti-HIV effects such as that shown by entecavir. First, the assay used here allows a more direct measure of drug inhibition of early steps in HIV replication from virus attachment through virus gene expression than older assays that used cell death as a surrogate measure of infection. Second, since the assay relies on a single round of infection, it allows rapid and precise quantification of individual infection events without the complications introduced by multiple rounds of infection in an extended culture. A third advantage is that drug inhibition of HIV replication is measured in primary CD4+ T lymphoblasts, which are the in vivo target cells of HIV, rather than in transformed cell lines, which may metabolize entecavir differently.
Further work is needed to understand why the M184V mutation clearly emerged in one of the two patients who was studied in detail. It is unlikely that this patient was taking lamivudine since it was not being prescribed to him at the time or in the recent past and since complete emergence of this mutation from lamivudine occurs within weeks (22). Rather, the appearance of the M184V mutation likely reflects the fact that it abolishes the anti-HIV activity of entecavir (Figures 2C and 2D). Interestingly, selection of the M184V mutation in HIV RT could be anticipated since structural models show that the M184V corresponds to the HBV pol mutation, rtM204V (23), which decreases HBV susceptibility to entecavir (24). In both cases, the targeted methionine is in the YMDD motif at the active site.
The failure of the M184V mutation in HIV RT to become dominant in Patient #3 may reflect the fact that this mutation has a well known negative effect on viral fitness (25). In some patients, this reduction in fitness may outweigh the modest benefit conferred by entecavir resistance since entecavir only partially inhibits HIV replication. Thus in some patients, selection for M184V may not occur or may occur very slowly.
It is also unknown whether the M184V variants that appeared in Patient #1 following entecavir treatment were initially generated by exposure to a lamivudine-containing HAART regimen several years earlier. Phylogenetic analysis suggests that the M184V variants evolved recently, most likely as a result of entecavir treatment. The appearance of M184V could also reflect emergence of an archived variant from the latent reservoir (26), but in either case there is clear evidence that entecavir selected for this variant in patient #1.
Several other questions are also raised by this study. One question is whether control of HBV replication lead to diminished lymphocyte activation or alterations in cytokine release that affected HIV replication. Although this is theoretically possible, treatment of hepatitis B with adefovir dipivoxil in HIV-infected individuals does not support this hypothesis since the patients had stable HIV RNA levels despite declines in HBV DNA (27). Although our data demonstrate that entecavir affects HIV replication, we cannot rule out that such secondary effects may have contributed to the decline in HIV RNA. Other questions include why the anti-HIV activity of entecavir plateaus at low nanomolar concentrations, whether modifications of the compound could overcome the plateau effect, whether entecavir could play a clinically meaningful role as an antiretroviral agent for HIV, and whether entecavir selects for other HIV drug-resistant mutants. Lastly, our data raise the question of the generality of the observations. We are aware of nine HIV-HBV co-infected patients who received entecavir monotherapy (three of whom are included in this report), and for all nine of them, the same simultaneous decline in HIV RNA was apparent.
In summary, entecavir, at doses used to treat chronic hepatitis B, is a potent partial inhibitor of HIV replication and can select for the M184V resistance mutation in HIV RT. Since the full extent of HIV RT mutations selected by entecavir monotherapy is not known, caution should be used in treating chronic hepatitis B with entecavir in HIV-infected patients not on fully suppressive antiretroviral regimens. Furthermore, these data underscore the importance of careful study of agents with potential for anti-HIV activity prior to licensure.
Supplementary Material
Entecavir inhibits HIV infection at or before the reverse transcription step. Primary CD4+ T cells were infected in vitro in the presence (ETV) or absence (Ctrl) of 50μM entecavir. At the indicated times post infection, DNA was isolated, and early, intermediate, or late products of the reverse transcription reaction were detected by real time PCR using appropriate primer pairs (28). As a negative control, DNA was isolated from cells after infection at 4°C.
Acknowledgments
This work was funded by NIH grants R01AI060449, A143222, and A151178.
We would like to thank David Thomas for critical reading of this manuscript, Stuart Ray and Meghdad Rahdar for helpful discussions and advice, and the patients for their participation in this study.
Footnotes
Disclosure None of the authors have any disclosures relevant to this manuscript.
The views expressed in this article are those of the authors and do not reflect the official policy or position of the Department of the Navy, Department of Defense, or the US Government.
References
- 1.Innaimo SF, Seifer M, Bisacchi GS, Standring DN, Zahler R, Colonno RJ. Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob Agents Chemother. 1997;41(7):1444–8. doi: 10.1128/aac.41.7.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marion PL, Salazar FH, Winters MA, Colonno RJ. Potent efficacy of entecavir (BMS-200475) in a duck model of hepatitis B virus replication. Antimicrob Agents Chemother. 2002;46(1):82–8. doi: 10.1128/AAC.46.1.82-88.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamanaka G, Wilson T, Innaimo S, et al. Metabolic studies on BMS-200475, a new antiviral compound active against hepatitis B virus. Antimicrob Agents Chemother. 1999;43(1):190–3. doi: 10.1128/aac.43.1.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doong SL, Tsai CH, Schinazi RF, Liotta DC, Cheng YC. Inhibition of the replication of hepatitis B virus in vitro by 2’,3’-dideoxy-3’-thiacytidine and related analogues. Proc Natl Acad Sci U S A. 1991;88(19):8495–9. doi: 10.1073/pnas.88.19.8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heijtink RA, De Wilde GA, Kruining J, et al. Inhibitory effect of 9-(2-phosphonylmethoxyethyl)-adenine (PMEA) on human and duck hepatitis B virus infection. Antiviral Res. 1993;21(2):141–53. doi: 10.1016/0166-3542(93)90050-s. [DOI] [PubMed] [Google Scholar]
- 6.Ying C, De Clercq E, Neyts J. Lamivudine, adefovir and tenofovir exhibit long-lasting anti-hepatitis B virus activity in cell culture. J Viral Hepat. 2000;7(1):79–83. doi: 10.1046/j.1365-2893.2000.00192.x. [DOI] [PubMed] [Google Scholar]
- 7.Karayiannis P. Hepatitis B virus: old, new and future approaches to antiviral treatment. J Antimicrob Chemother. 2003;51(4):761–85. doi: 10.1093/jac/dkg163. [DOI] [PubMed] [Google Scholar]
- 8.Benson CA, Kaplan JE, Masur H, et al. Treating opportunistic infections among HIV-exposed and infected children: recommendations from CDC, the National Institutes of Health, and the Infectious Diseases Society of America. MMWR Recomm Rep. 2004;53(RR-15):1–112. [PubMed] [Google Scholar]
- 9.Soriano V, Miro JM, Garcia-Samaniego J, et al. Consensus conference on chronic viral hepatitis and HIV infection: updated Spanish recommendations. J Viral Hepat. 2004;11(1):2–17. doi: 10.1046/j.1365-2893.2003.00491.x. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett JG, Lane HC. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Bethesda, MD: US Department of Health and Human Services; 2006. [December 16, 2006]. at http://AIDSinfo.nih.gov. [Google Scholar]
- 11.Rathbun RC, Lockhart SM, Stephens JR. Current HIV treatment guidelines--an overview. Curr Pharm Des. 2006;12(9):1045–63. doi: 10.2174/138161206776055840. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Zhou Y, Alcock C, et al. Novel Single-Cell-Level Phenotypic Assay for Residual Drug Susceptibility and Reduced Replication Capacity of Drug-Resistant Human Immunodeficiency Virus Type 1. J Virol. 2004;78(4):1718–29. doi: 10.1128/JVI.78.4.1718-1729.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailey JR, Sedaghat AR, Kieffer T, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol. 2006;80(13):6441–57. doi: 10.1128/JVI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petropoulos CJ, Parkin NT, Limoli KL, et al. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2000;44(4):920–8. doi: 10.1128/aac.44.4.920-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sims KA, Woodland AM. Entecavir: a new nucleoside analog for the treatment of chronic hepatitis B infection. Pharmacotherapy. 2006;26(12):1745–57. doi: 10.1592/phco.26.12.1745. [DOI] [PubMed] [Google Scholar]
- 16.Schinazi RF, Lloyd RM, Jr, Nguyen MH, et al. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob Agents Chemother. 1993;37(4):875–81. doi: 10.1128/aac.37.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tisdale M, Kemp SD, Parry NR, Larder BA. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3’-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc Natl Acad Sci U S A. 1993;90(12):5653–6. doi: 10.1073/pnas.90.12.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao Q, Gu Z, Parniak MA, et al. The same mutation that encodes low-level human immunodeficiency virus type 1 resistance to 2’,3’-dideoxyinosine and 2’,3’-dideoxycytidine confers high-level resistance to the (-) enantiomer of 2’,3’-dideoxy-3’-thiacytidine. Antimicrob Agents Chemother. 1993;37(6):1390–2. doi: 10.1128/aac.37.6.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallant JE, Gerondelis PZ, Wainberg MA, et al. Nucleoside and nucleotide analogue reverse transcriptase inhibitors: a clinical review of antiretroviral resistance. Antivir Ther. 2003;8(6):489–506. [PubMed] [Google Scholar]
- 20.Boyer PL, Sarafianos SG. Arnold E and Hughes SH. The M184V Mutation Reduces the Selective Excision of Zidovudine 5’-Monophosphate (AZTMP) by the Reverse Transcriptase of Human Immunodeficiency Virus Type 1. J Virol. 2002;76(7):3248–56. doi: 10.1128/JVI.76.7.3248-3256.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weislow OS, Kiser R, Fine DL, Bader J, Shoemaker RH, Boyd MR. New soluble-formazan assay for HIV-1 cytopathic effects: application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J Natl Cancer Inst. 1989;81(8):577–86. doi: 10.1093/jnci/81.8.577. [DOI] [PubMed] [Google Scholar]
- 22.Schuurman R, Nijhuis M, van Leeuwen R, et al. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with lamivudine (3TC) JID. 1995;171(6):1411–9. doi: 10.1093/infdis/171.6.1411. [DOI] [PubMed] [Google Scholar]
- 23.Bartholomeusz A, Tehan BG, Chalmers DK. Comparisons of the HBV and HIV polymerase, and antiviral resistance mutations. Antivir Ther. 2004;9(2):149–60. [PubMed] [Google Scholar]
- 24.Levine S, Hernandez D, Yamanaka G, et al. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob Agents Chemother. 2002;46(8):2525–32. doi: 10.1128/AAC.46.8.2525-2532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Back NKT, Nijhuis M, Keulen W, et al. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase enzyme. EMBO J. 1996;15:4040–4049. [PMC free article] [PubMed] [Google Scholar]
- 26.Ruff CT, Ray SC, Kwon P, et al. Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J Virol. 2002;76(18):9481–92. doi: 10.1128/JVI.76.18.9481-9492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheldon JA, Corral A, Rodes B, et al. Risk of selecting K65R in antiretroviral-naive HIV-infected individuals with chronic hepatitis B treated with adefovir. AIDS. 2005;19(17):2036–38. doi: 10.1097/01.aids.0000189563.79976.05. [DOI] [PubMed] [Google Scholar]
- 28.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 1990;61(2):213–22. doi: 10.1016/0092-8674(90)90802-l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Entecavir inhibits HIV infection at or before the reverse transcription step. Primary CD4+ T cells were infected in vitro in the presence (ETV) or absence (Ctrl) of 50μM entecavir. At the indicated times post infection, DNA was isolated, and early, intermediate, or late products of the reverse transcription reaction were detected by real time PCR using appropriate primer pairs (28). As a negative control, DNA was isolated from cells after infection at 4°C.