

Abstract
Sphingolipids constitute a class of lipids defined by their eighteen carbon amino-alcohol backbones which are synthesized in the ER from nonsphingolipid precursors. Modification of this basic structure is what gives rise to the vast family of sphingolipids that play significant roles in membrane biology and provide many bioactive metabolites that regulate cell function. Despite the diversity of structure and function of sphingolipids, their creation and destruction are governed by common synthetic and catabolic pathways. In this regard, sphingolipid metabolism can be imagined as an array of interconnected networks that diverge from a single common entry point and converge into a single common breakdown pathway. In their simplest forms, sphingosine, phytosphingosine and dihydrosphingosine serve as the backbones upon which further complexity is achieved. For example, phosphorylation of the C1 hydroxyl group yields the final breakdown products and/or the important signaling molecules sphingosine-1-phosphate, phytosphingosine-1-phosphate and dihydrosphingosine-1-phosphate, respectively. On the other hand, acylation of sphingosine, phytosphingosine, or dihydrosphingosine with one of several possible acyl CoA molecules through the action of distinct ceramide synthases produces the molecules defined as ceramide, phytoceramide, or dihydroceramide. Ceramide, due to the differing acyl CoAs that can be used to produce it, is technically a class of molecules rather than a single molecule and therefore may have different biological functions depending on the acyl chain it is composed of. At the apex of complexity is the group of lipids known as glycosphingolipids (GSL) which contain dozens of different sphingolipid species differing by both the order and type of sugar residues attached to their headgroups. Since these molecules are produced from ceramide precursors, they too may have differences in their acyl chain composition, revealing an additional layer of variation. The glycosphingolipids are divided broadly into two categories: glucosphingolipids and galactosphingolipids. The glucosphingolipids depend initially on the enzyme glucosylceramide synthase (GCS) which attaches glucose as the first residue to the C1 hydroxyl position. Galactosphingolipids, on the other hand, are generated from galactosylceramide synthase (GalCerS), an evolutionarily dissimilar enzyme from GCS. Glycosphingolipids are further divided based upon further modification by various glycosyltransferases which increases the potential variation in lipid species by several fold. Far more abundant are the sphingomyelin species which are produced in parallel with glycosphingolipids, however they are defined by a phosphocholine headgroup rather than the addition of sugar residues. Although sphingomyelin species all share a common headgroup, they too are produced from a variety of ceramide species and therefore can have differing acyl chains attached to their C-2 amino groups. Whether or not the differing acyl chain lengths in SMs dictate unique functions or important biophysical distinctions has not yet been established. Understanding the function of all the existing glycosphingolipids and sphingomyelin species will be a major undertaking in the future since the tools to study and measure these species are only beginning to be developed. The simple sphingolipids serve both as the precursors and the breakdown products of the more complex ones. Importantly, in recent decades, these simple sphingolipids have gained attention for having significant signaling and regulatory roles within cells. In addition, many tools have emerged to measure the levels of simple sphingolipids and therefore have become the focus of even more intense study in recent years. With this thought in mind, this chapter will pay tribute to the complex sphingolipids, but focus on the regulation of simple sphingolipid metabolism.
Introduction to Sphingolipid Metabolism
Sphingolipids constitute a class of lipids defined by their eighteen carbon amino-alcohol backbones which are synthesized in the ER from non-sphingolipid precursors. Modification of this basic structure is what gives rise to the vast family of sphingolipids that play significant roles in membrane biology and provide many bioactive metabolites that regulate cell function. Despite the diversity of structure and function of sphingolipids, their creation and destruction are governed by common synthetic and catabolic pathways. In this regard, sphingolipid metabolism can be imagined as an array of interconnected networks that diverge from a single common entry point and converge into a single common breakdown pathway.
In their simplest forms, sphingosine, phytosphingosine, and dihydrosphingosine serve as the backbones upon which further complexity is achieved. For example, phosphorylation of the C1 hydroxyl group yields the final breakdown products and/or the important signaling molecules sphingosine-1-phosphate, phytosphingosine-1-phosphate, and dihydrosphingosine-1-phosphate, respectively. On the other hand, acylation of sphingosine, phytosphingosine, or dihydrosphingosine with one of several possible acyl CoA molecules through the action of distinct ceramide synthases produces the molecules defined as ceramide, phytoceramide, or dihydroceramide. Ceramide, due to the differing acyl CoAs that can be used to produce it, is technically a class of molecules rather than a single molecule and therefore may have different biological functions depending on the acyl chain it is composed of.
At the apex of complexity is the group of lipids known as glycosphingolipids (GSL) which contain dozens of different sphingolipid species differing by both the order and type of sugar residues attached to their headgroups. Since these molecules are produced from ceramide precursors, they too may have differences in their acyl chain composition, revealing an additional layer of variation. The glycosphingolipids are divided broadly into two categories: glucosphingolipids and galactosphingolipids. The glucosphingolipids depend initially on the enzyme glucosylceramide synthase (GCS) which attaches glucose as the first residue to the C1 hydroxyl position. Galactosphingolipids, on the other hand, are generated from galactosylceramide synthase (GalCerS), an evolutionarily dissimilar enzyme from GCS. Glycosphingolipids are further divided based upon further modification by various glycosyltransferases which increases the potential variation in lipid species by several fold. Far more abundant are the sphingomyelin species which are produced in parallel with glycosphingolipids, however they are defined by a phosphocholine headgroup rather than the addition of sugar residues. Although sphingomyelin species all share a common headgroup, they too are produced from a variety of ceramide species, and therefore can have differing acyl chains attached to their C-2 amino groups. Whether or not the differing acyl chain lengths in SMs dictate unique functions or important biophysical distinctions has not yet been established. Understanding the function of all the existing glycosphingolipids and sphingomyelin species will be a major undertaking in the future since the tools to study and measure these species are only beginning to be developed (see figure 1 for an illustrated depiction of the various sphingolipid structures).
Figure 1. Simple and Complex Sphingolipid Structures.
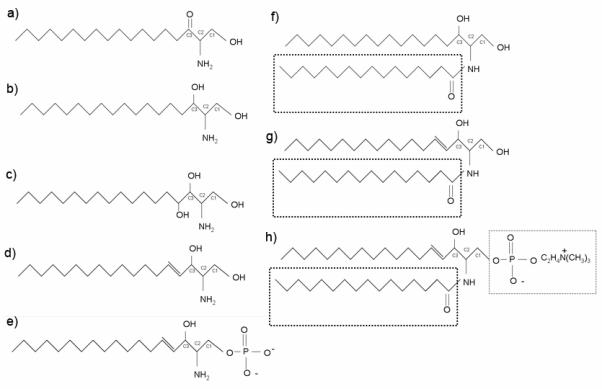
Structures shown: (A) 3-Ketodihydrosphingosine, (B) Dihydrosphingosine, (C) Phytosphingosine, (D) Sphingosine, (E) Sphingosine-1-Phosphate, (F) Dihydroceramide: Boxed region shows variable acyl chain, (G) Ceramide, (H) Complex Sphingolipids: Sphingomyelin shown with phosphocholine R group. Substitute R for glucose=Glucosylceramide, Substitute R for galactose=Galactosylceramide.
The simple sphingolipids serve both as the precursors and the breakdown products of the more complex ones. Importantly, in recent decades, these simple sphingolipids have gained attention for having significant signaling and regulatory roles within cells. In addition, many tools have emerged to measure the levels of simple sphingolipids and therefore have become the focus of even more intense study in recent years. With this thought in mind, this chapter will pay tribute to the complex sphingolipids, but focus on the regulation of simple sphingolipid metabolism.
Sphingolipid Properties in Membranes
An important feature of lipid biology is that many of these molecules are restricted to biological membranes and therefore lipids are governed by a set of rules based on their biophysical properties. For example, compartmentalization of a lipid can mean access to both sides of a membrane, access to only a single leaflet of a bilayer and therefore only a single compartment, or a molecule could be sufficiently amphipathic that it could diffuse from a membrane and freely traverse the cytosol, the lumen of an organelle, or enter the extracellular space. This becomes important to keep in mind when understanding how compartmentalized enzymes only have effects on a specific pool of lipid metabolites. It is also important to understand the enzymes that regulate the lipids since cytosolic proteins are generally restricted to a single compartment and transmembrane domain containing enzymes generally have their catalytic sites facing only one of the compartments in which a bilayer divides.
In the case of sphingolipids, sphingosine and dihydrosphingosine are sufficiently amphipathic to diffuse between membranes and to flip between membrane leaflets; however, they also are likely to accumulate in acidic pH organelles due to ionization of their free amino group. All of the known enzymes to act upon sphingosine have their catalytic sites facing the cytosolic compartment suggesting that only cytosolic sphingosine is in a modifiable form. Ceramide, on the other hand is restricted to membranes, but has a relatively rapid flip rate. Ceramide is therefore likely to be restricted to the organelle in which it was created, but may have access to enzymes or binding proteins on either side of the bilayer in which it was produced. This is important because these enzymes are distributed in discrete compartments and therefore ceramide will most likely be modified by whichever enzyme is within the same compartment in which the ceramide was generated or transported to. Sphingomyelins and glycosphingolipids are the most spatially restricted sphingolipids of all since their bulky headgroups make flipping between membrane leaflets extremely unlikely without the aid of specific flippases. One such flippase is thought to be present in the Golgi apparatus to aid glucosylceramide in gaining access to lumenal glycosyltransferases. In the absence of a flippase, sphingomyelin and glycosphingolipids are restricted to whichever leaflet they are generated in, and since they are generated in the Golgi lumen, they are mainly present in the lumenal Golgi leaflet or on the outer leaflet of the plasma membrane after vesicular transport to that location. Finally, the ultimate catabolic products of all sphingolipids are sphingosine-1-phosphate and dihydrosphingosine-1-phosphate which are soluble in a hydrophilic environment, but are unable to traverse membranes without the aid of lipid transporters. Therefore, S1P and DHS1P are also restricted to the hydrophilic compartments in which they are generated, but can be exported to the extracellular space with the aid of specific transporters or be dephosphorylated into a more hydrophobic compound.
Sphingolipid structures differ significantly from kingdom to kingdom and sphingolipid diversity within the animal kingdom itself has been recognized. Despite this diversity being extremely fascinating for its implications in evolutionary biology, this chapter will only discuss mammalian enzymes involved in sphingolipid metabolism. Although many of the critical enzymes in mammalian systems could not have been identified without the aid of yeast and other model organisms, for the sake of brevity, unless otherwise stated, the enzymes discussed in this chapter are to be assumed to be the mammalian form (fig 2). By discussing the synthesis and the catabolism of mammalian sphingolipids we hope to bring to light an understanding of sphingolipids, how sphingolipids are created and destroyed, and to define more clearly the process by which sphingolipids become distributed to their respective membranes within mammalian cells.
Figure 2.

The Sphingolipid Metabolic Network
I) De novo Synthesis in the ER
De novo sphingolipid synthesis begins at the cytosolic leaflet of the ER where a set of four enzyme groups coordinately generate ceramides of different acyl chain lengths from non-sphingolipid precursors. Through the coordinated action of Serine palmitoyltransferase, 3-Ketodihydrosphingosine Reductase, and (dihydro)Ceramide Synthases, the ER is able to convert cytosolic serine and palmitoyl CoA molecules into a single membrane bound lipid, dihydroceramide. After its generation, dihydroceramide is acted on by a desaturase which introduces a double bond. This coordinated anabolic pathway generates the precursors to complex sphingolipids that can serve such diverse functions as providing electrical insulation to axons, act as an important hydrophobic barrier within the epidermis essential for decreasing water loss, and regulate red blood cell surface charge to prevent agglutination, to name just a few important functions.
In this phase of synthesis, complex sphingolipids begin to be diversified through differential addition of fatty acyl chains at the C2-amino group of the dihydrosphingosine/sphingosine backbone through the action of ceramide synthases. Variations in ceramide acyl chain length as well as the use of alpha hydroxylated fatty acids could potentially alter membrane bilayer dynamics, or have differential signaling properties by recruiting different binding partners. The effects of different acyl chain lengths on ceramide or complex sphingolipid biology are not yet understood.
i) Serine Palmitoyltransferase and 3-Ketodihydrosphingosine Reductase
The initial reaction in sphingolipid synthesis requires the enzyme serine palmitoyltransferase (SPT). This reaction occurs through cytosolic serine and palmitoyl CoA condensation to produce 3-ketodihydrosphingosine [1]. SPT is encoded by the genes SPTLC1, SPTLC2, and the recently identified SPTLC3 [2] Each SPT subunit contains several putative transmembrane domains and displays type I topology with its N-terminus directed into the ER lumen, C-terminus facing the cytosol, and its catalytic site facing the cytosol [3]. SPT1 and SPT2 form a heterodimer in the ER membrane which is likely the active form of the enzyme [4]. Although SPT3 shares 40% homology to SPT2, and likely can substitute for the SPT2 subunit in the SPT complex, it has not yet been shown to dimerize with SPT1 and still needs further investigation [2]. In yeast, a third subunit of SPT, TSC3 plays a major role in regulation of SPT activity by forming a heterotrimer with the SPT1 and SPT2 homologues, however, no mammalian homologue to TSC3 has yet been identified [5]. SPT is a member of the α-oxoamine synthase family, a group of enzymes that catalyze the condensation of amino acids with carboxylic acid CoA thioesters [6]. Like other members of this family, SPT requires the cofactor pyridoxal 5′-phosphate (PLP) for catalysis [7]. SPT2 is the only subunit which binds PLP, however both subunits are required for catalytic activity [4]. Hanada et. al have proposed that SPT2 requires SPT1 for stabilization and that up regulation of SPT1 expression subsequently leads to an increase in SPT2 expression through protein stabilization. Therefore, a secondary function of SPT1 other than its contribution to catalytic activity may be simply to stabilize SPT2 in the ER [6]. Whether or not regulation of SPT1 is the primary means by which SPT activity is regulated is yet to be determined.
Some insight into the functional significance of SPT comes from human patients with Hereditary Sensory and Autonomic Neuropathy Type I (HSNI), a disease which, in some families, has been mapped to a mutation in the SPTLC1 gene on chromosome 9 [8]. HSNI is an autosomal dominant disease that is characterized by progressive degeneration of motor neurons and dorsal ganglia with symptoms initiating after the first or second decade [9], [10]. Measurements of total SPT activity in patient lymphoblasts showed less than 50% SPT activity within these cells [11]. It is significant that patients still have a significant amount of SPT activity since in mice, complete lack of either SPT1 or SPT2 was shown to be embryonic lethal [4], [12]. The mutations responsible for this disease have been identified as point mutations in Cys133 or Val144 in the SPT1 protein which act in a dominant negative fashion on SPT activity [8], [11]. These mutations have been predicted to be localized, based on tertiary modeling of other known α-oxoamine synthases, near the catalytic interface of SPT1 and SPT2. HSNI mutant forms of SPT1 are able to form heterodimers with SPT2, but lack catalytic activity [13]. Interestingly, mutations in the SPTLC2 gene were not identified in any of the tested families affected by HSNI [14]. The recently identified SPTLC3 gene product has homology to SPTLC2 suggesting that SPT3 may be able to functionally substitute for SPT2 if expressed [2]. It is possible that a functional SPT3 subunit could mask a partial defect in the SPT2 subunit if they are functionally redundant. Although speculative, this could be one explanation for why SPTLC2 mutants are not associated with HSNI. Future studies into the interplay between the three SPT subunits will provide further insights into how SPT function is determined by its components.
The second step in the synthesis of all sphingolipids is performed by the enzyme 3-Ketodihydrosphingosine Reductase (KDHR). 3-Ketodihydrosphingosine (KDHSph), the direct product of SPT is reduced at its ketone group to a hydroxyl group by KDHR in a NADPH dependent manner. Only recently were the human and murine genes cloned for this enzyme based on a homology screen for the yeast gene TSC10. TSC10 deficient yeast were identified based on their build up of KDHSph and their inability to grow on media deficient in KDHSph [15]. In humans the KDHR gene was identified through a homology screen as FVT-1, a gene which was originally identified and named for its juxtaposition to the Ig-κ gene in a human follicular lymphoma [16]. Whether or not the FVT-1 translocation was merely coincidental or that it conferred any advantage to the follicular lymphoma is unclear. KDHR is predicted to have three transmembrane domains and display type I topology. Like SPT, KDHR has its catalytic site on the cytosolic leaflet of the ER where it is likely to encounter newly generated KDHSpH [16]. KDHSph is a minor lipid within cells due to the rapid conversion of KDHSph into dihydrosphingosine by the action of KDHR. Although poorly studied, KDHR is a critical step in the synthesis of sphingolipids. Its importance to mammalian physiology has been highlighted by a breed of cattle, recently identified with a missense mutation in FVT-1, that become afflicted with bovine spinal muscular atrophy and die shortly after birth [17].
ii) Dihydroceramide Synthases/Ceramide Synthases and Dihydroceramide Desaturase
Dihydrosphingosine (DHSph) is further acylated by the action of six distinct (Dihydro) ceramide synthases. In mammals, six distinct (dihydro)ceramide synthases abbreviated as CerS1-6 have been identified and are encoded by six distinct genes [18], [19]. No other step in sphingolipid metabolism has as many genes devoted to it as dihydroceramide synthesis, suggesting that the different CerS have distinct functions. There is a significant amount of evidence that each CerS has a distinct, but overlapping acyl CoA preference, and that each CerS can produce different dihydroceramide/ceramide species profiles. For example, CerS1 has been shown to prefer stearoyl CoA as a substrate and mainly produces C18-ceramide species [20]. On the other hand, CerS2 utilizes C20-C26 acyl CoA species and is one of the major CerS responsible for very long chain ceramide species [21]. Cers5 and CerS6 both prefer palmitoyl CoA as substrates and generate predominantly C16-ceramide species [22], [23]. Finally, CerS3, which is predominantly expressed in the testis and weakly in the epidermis, prefers middle and long chain acyl CoAs and is thought to be a contributor to large structural sphingolipid molecules that maintain the water barrier in the epidermis [24], [25]. It is still an outstanding question whether or not the bioactive properties associated with ceramide are sensitive to differences in acyl chain length and hence different CerS can influence the cellular fate of cells by modulating bioactive molecules. Another possibility is that the six different CerS exist because differences in the biophysical properties of various ceramide species provide advantages for specific tissue functions (e.g. epidermal barrier maintenance or myelination). It has already been established that the CerS have a significant variation in their tissue expression and that this correlates with differences in their sphingolipid acyl chain compositions [21].
All CerS studied to date have been localized to the ER with their catalytic sites facing the cytosol. In this manner, CerS are in a position to acylate newly generated DHSph molecules at their C2-amino groups in the presence of available fatty acyl CoAs. Very little is known about the regulation of CerS activity in cells although there are clear differences in CerS expression between different tissues. It is unclear if CerS are predominantly regulated at the transcriptional level or if significant post-translational regulation also occurs. Recently, a S1P binding site was identified on CerS2, which in vitro inhibited CerS2 activity. This suggests that sphingolipid breakdown could negatively regulate a specific subset of CerS activity within cells, in this case very long chain ceramide synthesis. Although de novo synthesis of dihydroceramide has repeatedly been shown to occur in response to various stress stimuli, the mechanisms by which this occurs remain opaque. Those that have studied it have suggested that the regulation is post-translational (e.g. not inhibited by cycloheximide) [26].
Although the family name of sphingolipids was named after the molecule sphingosine, this molecule is not actually generated during de novo synthesis. Only through the desaturation of dihydroceramide is the molecule sphingosine eventually generated.
Dihydroceramide Δ4-desaturase (DES) is the member of the desaturase family responsible for converting the dihydrosphingosine backbone within ceramide into a sphingosine backbone [27]. DES utilizes molecular oxygen to first introduce a hydroxyl group into the C4 position of the dihydrosphingosine backbone, and following a dehydration reaction, with the aid of NADPH, produces a double bond in the C4-C5 position of dihydroceramide [28], [29], [30]. Dihydroceramide with a double bond introduced at this position is referred to as ceramide. Dihydroceramide Δ4-desaturase (DES1), encoded by the DES1 gene contains multiple transmembrane domains and was recently shown to require myristoylation on its N-terminus for full activity [31], [32]. Like the previous three enzymes in the sphingolipid biosynthetic pathway, DES1 is embedded in the ER membrane where it has access to newly synthesized dihydroceramide species [27]. It is interesting to note that an intermediate reaction product in the conversion of dihydroceramide to ceramide is 4-hydroxyceramide which is also known as phytoceramide. Phytoceramide is the predominant ceramide species in plants and yeast. Although the enzyme DES1 only converts dihydroceramide species into fully desaturated ceramide, a second family member, dihydroceramide C4 hydroxylase/ Δ4-desaturase (DES2), is capable of creating either phytoceramide or ceramide from dihydroceramide precursors [33]. Therefore, it is not surprising that DES2 is highly expressed in the intestines, kidneys, and skin where phytoceramides are present in high abundance [34], [33]. The differences in biophysical properties between dihydroceramide, ceramide, and phytoceramide are not entirely clear, however, one may speculate that the addition of a hydroxyl group into the sphingosine backbone may increase lipid packing in the membrane by increasing the amount of hydrogen bonding at the interfacial region of the membrane. It has been shown repeatedly that ceramide has distinct signaling properties from dihydroceramide and phytoceramide, suggesting that, if nothing else, cells have evolved to recognize ceramide as a more significant determinant to initiate a cellular response to in most cells [35]. The recent report of the phenotype of the Des1 −/− mice suggests that the inability to form ceramide leads to serious consequences for mammalian physiology. Des1 −/− mice have highly elevated dihydroceramide, low levels of ceramide, multi-organ dysfunction, and failure to thrive [36].
II) Ceramide Transport from the ER to the Golgi
Ceramide is a membrane bound molecule that has very low solubility in an aqueous environment and therefore, a cell must find a way to transport it from one membrane to another. The cell employs two major mechanisms to mobilize ceramide; either through vesicular transport or through the protein ceramide transfer protein (CERT). CERT is a cytosolic protein that transfers ceramide from the ER, where it is generated, to the Golgi apparatus where it can be modified into sphingomyelins and possibly glycosphingolipids. The CERT protein is composed of at least four functional domains that determine its function. The N-terminus of CERT contains a PH domain which is able to recognize PI4P on acceptor Golgi membranes and therefore allows for directed transport to the Golgi. A FFAT domain in the middle of the protein serves an analogous function to the PH domain but for donor membrane recognition. The FFAT domain is thought to allow its binding to ER resident VAP proteins, and therefore CERT can only accept ceramides from the ER, something that may have implications for cellular signaling [37]. The C-terminus of CERT contains a START domain which provides a hydrophobic pocket responsible for the direct binding of ceramide and allows for its delivery to the Golgi through an aqueous environment. In vitro studies with CERT have shown that phosphorylation of CERT at multiple serine residues, by an unidentified kinase, result in an autoinhibitory binding event that occurs between both the START domain and the PH domain [38]. The in vivo significance of CERT phosphorylation is unclear and remains to be seen. On the other hand, a globular domain between the PH and START domain has been shown to be responsible for homotrimer formation during UV stress in keratinocytes, however, this was shown to be phosphorylation independent. It is unclear if oligomerization, or potentially phosphorylation, is a general mechanism by which cellular stresses can inactivate CERT [39].
CERT was originally identified as the responsible mutant in a CHO cell line, LY-A, that was resistant to hemolysis by the sphingomyelin-dependent fungal toxin lysenin [40], [41]. CERT displays a preference for ceramide species with acyl chains less than C22. Although CERT still transfers C22 and C24:1 ceramide, it does so with 40% the efficiency of shorter chain species [38], [42]. In addition, CERT showed minimal to no transfer of C24 ceramide. CERT is also able to recognize dihydroceramide and phytoceramide although less effectively than ceramide [38]. Ceramide which is transported to the Golgi by CERT is preferentially incorporated into SM over glycosphingolipids [41]. Since CERT has preference for specific chain lengths, this may have implications for which forms of ceramide are preferentially utilized for SM synthesis and which ceramide species are preferred for glycosphingolipid utilization. If this is true, then one could speculate that relative SM and glycosphingolipid synthesis could be regulated by shifting CerS expression from predominantly long chain specific CerS to very long chain specific CerS and vice versa.
An alternative pathway exists for the transport of ceramide species to the Golgi which is coatomer protein dependent and is based on vesicular transport [43]. Less is known about how this pathway is regulated, however, this is thought to be the major pathway responsible for delivering ceramide to the cis-Golgi for glycosphingolipid synthesis. Clearly, our knowledge of how ceramide species can be transported from the ER to the Golgi for regulated glycosphingolipid synthesis is incomplete.
III) Synthesis of Complex Sphingolipids
Complex sphingolipids are divided into three major groups based on the primary residue attached to their C1-hydroxy headgroup. This classification also captures the three biosynthetic pathways, spatially separated within the ER and the Golgi complex, that generate an immense diversity of glycosphingolipids and sphingomyelins. The three major enzymes that regulate complex sphingolipid biosynthesis are ceramide galactosyltransferase, glucosylceramide synthase, and sphingomyelin synthase.
i) Ceramide galactosyltransferase and Galactosphingolipids
Ceramide galactosyltransferase (CGT) utilizes UDP-galactose and ceramide to create galactosylceramide. CGT is an ER transmembrane protein that has its catalytic site facing the lumen of the ER. It is structurally related to UDP-glucuronosyltransferases, an enzyme critical to type II biotransformation of xenobiotics and porphyrin metabolism [44]. CGT has a limited tissue distribution with expression being detected primarily in schwann cells, oligodendrocytes, kidneys, testis, and intestines. In the central nervous system, the product galactosylceramide (and its subsequent metabolite, sulfatide) is highly enriched in myelin. CGT knockout mice display a tremor phenotype, severe motor weakness due to loss of nerve conduction, male infertility, and premature death [45], [46]. Interestingly, the neuronal phenotype in mice lacking CGT can be rescued by expression of an oligodendrocyte specific CGT gene suggesting that galactosylceramide is extremely important for oligodendrocyte function [47]. Galactosylceramide is a precursor for sulfatides and many of the myelination defects may be due to a lack of sulfatide production. Evidence for this comes from mice deficient in the enzyme galactosylceramide sulfotransferase, the enzyme responsible for sulfatide production from galactosylceramide, which have major defects in myelination, although their pathology is less severe than an outright CGT knockout mouse. [48].
ii) Glucosylceramide Synthase and Derivatives of Glucosylceramide
Glucosylceramide is synthesized in the cis-Golgi from ceramide and UDP-glucose by the enzyme glucosylceramide synthase (GCS) [49]. GCS is a transmembrane protein present on the cis-Golgi, and it has its catalytic site facing the cytosol where newly produced glucosylceramide can be recognized by the lipid transport protein FAPP2 [50], [51]. Some reports suggest that FAPP2 transports glucosylceramide back to the ER where it is translocated from the inner leaflet to the outer leaflet, however, this point remains to be resolved.
Unlike galactosylceramide, glucosylceramide (GC) is an absolutely essential sphingolipid for the development of mammals [52]. Mice lacking GCS do not survive to term. The loss of GCS results in embryonic lethality at embryonic day 6.5-7.5 when gastrulation is occurring [52]. This specific defect can be rescued by the addition of exogenous GC to the embryos. Glucosylceramide is the precursor for the majority of all glycosphingolipids that can be produced by a mammal and these glycosphingolipids are likely to play an essential role in cell-cell recognition during embryonic and post-natal development [52].
Tissue specific knockouts of GCS within the nervous system and the skin have been created. Absence of GCS in the epidermis leads to defects in lamellar body formation which are major contributors to the hydrophobic barrier of the skin. These lamellar body defects lead to rapid water loss due to excessive evaporation and eventual lethality several days after birth [53]. Absence of GCS specifically in neuronal tissue, on the other hand, leads to premature death 11-24 days after birth, suggesting that glycosphingolipids are necessary for neuronal function and proper brain maturation. Unusually, no histological defects could be identified in the brains of the neuron specific GCS knockout mice through light microscopy examination or electron microscopic evaluation of synapses despite their obvious phenotypic differences. However, in vitro studies of primary hippocampal neurons from GCS deficient mice showed defects in neurite outgrowth in culture [54], [55].
iii) Sphingomyelin Synthesis
The most abundant complex sphingolipids in mammalian cells are the sphingomyelin species. Evidence for the essential role that sphingomyelin has in eukaryotic cell viability is displayed by the inability of mammalian or yeast cells to survive in culture when they are unable to produce sphingomyelin either through CERT mutation or defects in de novo sphingolipid synthesis [56]. It is interesting to note that this absolute requirement for a sphingolipid is not true for glucosylceramide or galactosylceramide which, although critical for mammalian development and tissue specific functions are not required for the viability of cells in culture. The precise single function that sphingomyelin fulfills which is absolutely necessary for cell survival is not clear due to sphingomyelin’s many known functions in membrane biology.
Sphingomyelin is produced by the action of sphingomyelin synthases. There are at least two members of the sphingomyelin synthase family in most mammalian species [57] and possibly a third family member known as SMSr [58]. The sphingomyelin synthases (SMS) are evolutionarily similar to the lipid phosphate phosphatase family which have six transmembrane domains and have their catalytic domains facing the luminal or exoplasmic leaflet of the membrane [58]. Like their cousins, the SMS family constituents also have six transmembrane domains and are oriented with their catalytic sites facing the Golgi lumen or extracellular space. Sphingomyelin synthases 1 and 2 are both present in the trans-Golgi, however, SMS2 is also localized to the plasma membrane. Therefore, SMS2 may also have a unique function in maintaining plasma membrane sphingomyelin content directly at the plasma membrane. Generation of sphingomyelin occurs through the transfer of a phosphocholine headgroup from phosphatidylcholine to ceramide yielding the products diacylglycerol (DAG) and sphingomyelin (SM). Since both ceramide and DAG have been identified as bioactive lipids with opposing effects on cellular proliferation and survival, SMS has also been proposed to play an essential role in regulating cellular fate. . A recent study has implicated SMS in generating DAG in the Golgi with effects on PKC [59] Because SMS activity directly regulates the level of sphingomyelin, ceramide, DAG, and PC simultaneously, direct effects of SMS products on biological processes have been difficult to elucidate to date.
IV) Ceramide Kinase and Ceramide-1-Phosphate
Although ceramide is primarily converted into more complex sphingolipids in the Golgi, ceramide can also be phosphorylated to produce ceramide-1-phosphate (C1P). C1P is produced in the trans-Golgi, and potentially the plasma membrane, by ceramide kinase (CERK). CERK, a member of the DAG kinase family, was originally identified based on its homology to sphingosine kinase. Unlike the sphingosine kinases, CERK only utilizes ceramide as a substrate and has no activity for sphingosine or DAG (Sugiura 2002). CERK activity is enhanced in the presence of calcium or magnesium and contains a putative calmodulin-like domain. In addition, ceramide kinase has specificity for sphingosine contai[60]ning ceramides since it has very low activity against dihydroceramide and phytoceramide species. [61]. Among the ceramide species it recognizes, CERK prefers ceramide species with acyl chain lengths greater than 12 carbons long, however, no preference was observed for the degree of saturation [61]. The measurement of C1P levels in A549 lung adenocarcinoma cells revealed an enrichment of C1P species containing acyl chain lengths of C16, C18, and C20 relative to their respective ceramide species. [62]. The enrichment for particular ceramide species for C1P was suggested to be due to specific delivery of ceramides to the trans-Golgi by CERT, a lipid transport protein which is biased for ceramide species with acyl chain lengths less than 22 carbons [63]. Knockdown of CERT using RNA interference lead to a decrease in C1P levels in A549 cells. A separate study using a pharmacological approach for CERT inhibition, found that inhibiting CERT had no effect on C1P production, but still inhibited sphingomyelin production [62], [64]. It is unclear if these differences were due to intrinsic differences in the cell types studied (human lung versus mouse macrophage) or due to non-specific effects of either approach. Future studies are needed to resolve this discrepancy.
CERK displays significant homology to other DAG kinases, however it also contains a N-terminal myristoylation site and a pleckstrin homology (PH) domain [65]. The PH domain targets CERK to PIP2 containing membranes, but is also necessary for enzymatic activity. Several studies have shown that CERK is localized to the trans-Golgi in a PH dependent manner. C1P generated in the Golgi can act as a docking site for cytosolic PLA2 and enhances arachidonic acid release. In addition, CERK translocates to the plasma membrane in response to osmotic swelling, an insult that enhances PIP2 on the plasma membrane. Translocation of CERK to the plasma membrane was shown to also be dependent on its PH domain.
The generation of a Cerk −/− mouse has provided some clues to CERK function in vivo. Cerk −/− mice are fully viable and show no gross phenotypic changes suggesting that CERK is not essential for development. Lipid analysis of serum from Cerk −/− revealed greatly elevated ceramides, but decreased dihydroceramides suggesting that CERK contributes significantly to ceramide metabolism in the serum [60]. Upon closer examination, Cerk −/− mice were found to have a normal level of C1P in their brains, despite a lack of CERK activity, suggesting that C1P can be produced through a mechanism independent of CERK [66]. Behavioral testing of the mice did, however, show abnormal emotional behavior based on an increase in ambulation and defecation frequencies in an open field test [66]. More recently, the Cerk −/− mice were found to have significant neutropenia under basal conditions. When these mice were challenged with S. pneumoniae they succumbed to lethal pneumonia earlier than wildtype mice and had a higher bacterial burden in their lungs [67].
A CERK homologue formerly named retinitis pigmentosa 26, RP26, but recently renamed CERKL, or CERK-like, has been identified. Since this gene has been implicated in a human form of retinitis pigmentosa it was initially suspected that C1P might play an essential role in retinal biology. Initial expression studies of CERKL expressed in cells failed to detect any CERK activity drawing into question if CERKL was an actual ceramide kinase [68], [69]. After generation of a CERKL knockout mouse, it was determined that Cerkl −/− mice had no alterations in retinal C1P, ceramide, or CERK activity [60]. CERK −/− mice, on the other hand, had 80% less C1P levels and elevated ceramides suggesting that CERK is the major enzyme responsible for C1P in the retina [60]. Taken together, it is unlikely that CERKL is a true ceramide kinase, but may have a separate function which is essential for retinal cells. Also, it is interesting to note that this study found that the retina of Cerk −/− mice had greatly reduced C1P levels, whereas previous studies have shown that whole brain C1P in these mice was unchanged [60], [66]. Future studies will help address the contribution of CERK to C1P levels and potentially identify other enzymes involved in C1P production.
C1P levels are regulated both by its synthesis through CERK, but also by its dephosphorylation back into ceramide. It is unclear how ceramide-1-phosphate is dephosphorylated, however, several groups have reported C1P phosphatase activity in plasma membrane fractions in both the liver and brain [70], [71], [72]. C1P has been shown to traffic through the secretory pathway to reach the plasma membrane where it could potentially be dephosphorylated by C1P phosphatases [64]. Also, C1P appears to be a substrate for non-specific lipid phosphatases of the LPP family.
V) Catabolizing Complex Sphingolipids and Sphingomyelins into Ceramide
There is a significant tradeoff between the ability of an organism to produce a novel advantageous lipid and the potential that the organism is incapable of catabolizing the same lipid, resulting in accumulation of a lipid product in cells or tissues. Lipids pose an additional problem in that they can not be excreted as readily as other more hydrophilic molecules and therefore tend to accumulate within cells when they can not be destroyed. As such, it is not surprising that for every enzyme capable of generating a specific sphingolipid, there exists an ‘opposing’ enzyme capable of breaking down the generated product. Indeed, the mutation of specific catabolic enzymes is the general principle behind lipid storage diseases, and defects in sphingolipid catabolizing enzymes are responsible for a significant number of these diseases. Due to the limited scope of this chapter, the reader is referred to several recent reviews on lipid storage diseases for more expansive information on the variety of lipid storage diseases due to glycosphingolipid enzyme mutations [73], [74]. Suffice it to say that many lysosomal hydrolases are necessary for the coordinated breakdown of complex glycosphingolipids and the absence of any single one of these results in the accumulation of its respective substrate.
Sphingomyelin is the most abundant complex sphingolipid in human cells. Therefore, coordinated breakdown of sphingomyelin is an essential part of membrane homeostasis. Breakdown of sphingomyelin occurs through the hydrolysis of the phosphocholine headgroups by the sphingomyelinase family. The direct result of sphingomyelin hydrolysis is the production of ceramide and free phosphocholine. The mammalian sphingomyelinases fall into three major categories based upon their pH optimum: acid sphingomyelinase, alkaline sphingomyelinase, and the neutral sphingomyelinases. Although all three forms of sphingomyelinases catalyze a similar reaction, these three groups of enzymes are evolutionarily unrelated and have different subcellular distributions. Alkaline sphingomyelinase, which is exclusively expressed in the intestine and liver, plays a role in the digestion of dietary sphingomyelin and will not be discussed further in this section [75]. Acid sphingomyelinase and neutral sphingomyelinase are ubiquitously expressed and serve as the major regulators of SM catabolism in most tissues and will be discussed in more detail.
Acid sphingomyelinase (ASMase) was the first sphingomyelinase to be characterized in mammalian cells. ASMase is predominantly a lysosomal protein which metabolizes sphingomyelin present on endosomal membranes. Lysosomal ASMase becomes N-glycosylated on at least six residues within the ER which stabilizes the enzyme structure and provides protection from proteolysis within the lysosomes. ASMase also becomes mannose-6 phosphorylated within the Golgi which directs it into the lysosomal compartment where it is most active [76]. The ability of ASMase to reach the lysosomal compartment is essential for its ability to catabolize sphingomyelin.
ASMase is also secreted into the extracellular space (where it is often referred to as secretory SMase) where it has access to sphingomyelin-containing lipoproteins which are abundant in the plasma [77]. In addition, secretory ASMase can metabolize outer leaflet SM on the plasma membrane. Secretory ASMase, unlike the lysosomal form, requires zinc for sphingomyelinase activity. It is unclear how the secretion of ASMase is regulated, but it appears to be through the constitutive secretory pathway. Moreover, the function of secretory ASMase is still unclear, but is thought to play a role in reducing plasma SM content and may play a role in cellular stress responses by generating plasma membrane localized ceramide with a specific signaling role.
Insights into the significance of the acid sphingomyelinase protein, encoded by the gene SMPD1, come from a human lysosomal storage disorder Niemann Pick Disease Types A (NPD A) and B (NPD B) [78]. Complete absence of a functional ASMase gene product results in NPD A which is characterized by a progressive neurodegenerative disease with psychomotor retardation, retinal cherry red spots, hepatosplenomegaly, lung disease, and premature death. Patients afflicted with NPD A usually do not live past the age of three. A mouse model of NPD A, Smpd1 −/−, exhibits growth defects similar to the human disease and also dies prematurely around 4 months of age [79]. NPD B, on the other hand, is a less severe form of the disease that lacks neuronopathic symptoms, however, hepatosplenomegaly and lung disease still occur. Fortunately, individuals afflicted with NPD B are able to survive into adulthood. The NPD B phenotype has been mimicked in mice by fusing a Smpd1 transgene to the Lamp1 gene [80]. In these mice there is complete absence of secretory SMase, but low level lysosomal ASMase activity. Future studies will dissect which symptoms of NPD are likely due to a lack of secretory ASMase function and which symptoms are associated with lysosomal ASMase function, thereby elucidating further some of the specific functions of each form of the enzyme.
Within the past decade, three different mammalian neutral sphingomyelinase (NSMase) genes have been identified, SMPD2, SMPD3, and SMPD4, although, the first NSMase gene discovered, SMPD2, is not likely to function as a SMase, but rather as a lyso-PAF phospholipase C [81]. To further muddy the point, mice lacking the Smpd2 gene have a decreased tissue SMase activity, but have no alteration in tissue SM levels [82].
The best characterized NSMase to date is NSmase 2. NSMase 2 contains two highly hydrophobic domains, but it is still unclear if these regions are bona fide transmembrane domains or peripherally associated with membranes [83]. Unusually, N-SMase2 is thought to have its catalytic site facing the cytosolic leaflet of either the Golgi or plasma membrane. This orientation for a SMase is unusual since SM is thought to be excluded from the cytosolic leaflet [83]. Interestingly, NSMase2 localizes to the Golgi under subconfluent conditions, however upon reaching confluence, NSMase2 translocates to the plasma membrane [84]. It was later shown that plasma membrane association is highly dependent upon its palmitoylation on multiple cysteine residues [83].
NSMase2 overexpression, in the absence of a specific stimulus causes degradation of sphingomyelin into ceramide with a preference for C24:0 and C24:1 species [84]. Generation of C24 and C24:1 ceramide during confluence dependent growth arrest was dependent on NSMase 2 suggesting that generation of specific ceramide species could have specific effects on growth arrest [84]. These results also suggest that NSMase2 either has specificity for very long chain sphingomyelin species or that very long chain sphingomyelins are enriched in the inner leaflet of the plasma membrane. Since most sphingomyelin species are thought to be localized to the outer leaflet of the plasma membrane or the luminal side of the Golgi, it is a mystery how NSMase2 with an inner leaflet catalytic site could access its substrate. The condundrum of how NSMase2 reaches its substrate undoubtedly puts into question some existing paradigms about sphingomyelin localization in membrane leaflets.
The generation of a Smpd3 −/− mice has provided some insights into the physiological role of NSMase 2 [85]. Smpd3 −/− mice display severe growth retardation, organ hypoplasia, delayed puberty, and skeletal defects. Upon further examination, a combined pituitary hormone deficiency was discovered in these mice and a significant decrease in serum IGF-1, TSH, and GnRH was found. Due to the complex interactions between the pituitary hormones and target tissues it is difficult to dissect which symptoms are due to hormone deficiency and which defects are due to primary organ defects. Some insight into this was provided by the generation of a chondrocyte specific SMPD3 expression mouse. When the chondrocyte specific SMPD3 mouse was crossed with the Smpd3 −/− mouse, there was an absence of skeletal defects, suggesting that chondrocyte specific NSMase2 activity is essential for skeletal development [86]. Despite the dramatic pathology observed with the Smpd3 −/− mouse, it is still difficult to understand how the NSMase2 knockout phenotype correlates with an intracellular role of NSMase 2 in sphingomyelin generation. Further characterizing the cellular dysfunction in the Smpd3 −/− mouse in parallel with further characterizing N-SMase 2 in vitro will shed light on a murky area of sphingolipid biology and provide new insights into cell biology as a whole.
Finally, a third NSMase, NSMase3 has recently been identified which is encoded by the SMPD4 gene [87]. Interestingly, NSMase3 is localized to the ER and possibly the Golgi. It was found to contain at least one transmembrane domain and potentially more along with an ER retention signal [88]. NSMase3 is predominantly expressed in skeletal and cardiac muscle with minor expression in many other tissues. It will be interesting to see how an ER localized SMase can affect sphingolipid metabolism since SM is not thought to be present in the ER compartment.
VI) The Catabolism of Ceramides and the Final Common Breakdown Pathway
Just as a few sphingolipid precursors are generated to produce hundreds of different sphingolipids, all sphingolipids are eventually catabolized to ceramide, sphingosine, and finally, sphingosine-1-phosphate. The deacylation of ceramide species is achieved through the family of enzymes known as ceramidases. These ceramidases have organelle specific expression and may have specificity for different forms of ceramide to bias a cell towards the generation of complex sphingolipids with specific sphingoid bases. Organelle specific expression also allows for the possibility to serve as negative regulators of organelle specific ceramide signaling. After ceramide is deacylated into sphingosine, the conversion of sphingosine to sphingosine-1-phosphate is achieved through one of two sphingosine kinases localized in the cytosol or peripherally associated with specific membrane compartments. In the final step of sphingolipid breakdown, sphingosine-1-phosphate is degraded by the enzyme sphingosine-1-phosphate lyase in the ER to produce hexadecenal and phosphoethanolamine.
i) Acid, Neutral, and Alkaline Ceramidases
The ceramidases, like many other sphingolipid enzymes, have been classified biochemically according to their pH optima. Acid ceramidase (AC), as its name suggests, is a lysosomal enzyme which deacylates ceramide species produced from the degradation of plasma membrane sphingolipids. AC, encoded by the ASAH1 gene, is a member of the N-terminal nucleophile (Ntn) hydrolase superfamily. Members of the Ntn family are characterized by their ability to undergo autoproteolytic cleavage through cysteine dependent proteolysis. Interestingly, this processing was shown to be accelerated at pH 4.5 when compared to neutral pH. This pH dependent maturation is likely a mechanism that has evolved to prevent premature activation of AC prior to it reaching the lysosomal compartment. AC activity was shown to be higher against C12 and C14 ceramide species compared with C6 or C18 ceramide species [89]. Although it has been reported that AC has the greatest activity against medium and long- chain ceramide species, it is difficult to imagine how very long-chain ceramides would be processed in a compartment where AC is the only known ceramidase [89]. Therefore, it is likely that very long-chain ceramide containing sphingolipids are either excluded from the endolysosomal compartment or that AC is able to degrade these very long chain ceramides albeit at a slower rate or with the aid of an accessory protein.
The significance of AC in mammalian systems is reinforced by its role in the human lipid storage disease Farber lipogranulomatosis. Farber disease is an autosomal recessive disorder due to a dysfunctional AC gene product. Farber’s disease is characterized by early onset arthritis, swollen lymph nodes, psychomotor difficulties, and vocal cord pathology. Complete absence of AC expression in a mouse model results in embryonic lethality at a very early stage in development [90]. Mice heterozygous for AC develop a progressive lipid storage disease due to ceramide accumulation in the liver, skin, lungs, and bones. The striking phenotype of the AC heterozygous mice raises questions about its implications for human disease. One could imagine that a heterozygous defect in AC within a human population may lead to a progressive lipid storage disorder that may only express itself with advanced age. Answers to questions like these will most likely be revealed when human genome sequencing becomes more widespread.
Neutral ceramidase, the most active ceramidase at neutral pH is encoded by the ASAH2 gene. Neutral ceramidase (NC) is synthesized through the secretory pathway as a type II integral membrane protein. NC can be cleaved at its N-terminus to produce a soluble protein that peripherally associates with the outer leaflet of the plasma membrane. NC contains mucin box domains which are highly O-glycosylated and are necessary for plasma membrane association [91]. NC associated with the plasma membrane is an important regulator of sphingosine and sphingosine-1-phosphate (S1P) production and for S1P release [92]. In addition, NC is highly expressed in the intestinal epithelium and contributes to the digestion of dietary sphingolipids [93], [94]. Asah2 −/− mice showed an inability to metabolize dietary ceramides. Although, sphingosine and ceramide levels were normal in the brain, liver, and kidney of Asah2 −/− mice, the intestines had a significantly increased C16:0 ceramide content but a reduced sphingosine content. Due to an absence of visible pathology in the Asah2 −/− mice, it is unclear what role NC plays in non-intestinal tissues, if any [93]. Future studies with the Asah2 −/− mice may reveal subtle or previously uncharacterized, but important functions for this gene under specific stresses.
The alkaline ceramidases (ACERs) contain three separate family members: alkaline ceramidases 1, 2, and 3 are encoded by the ASAH3 [95], ASAH3L [96], and PHCA [97] genes respectively, which share significant homology to one another. These ACERs differ in subcellular localizations and substrate specificity although they have alkaline pH optima for their in vitro activity and are activated by calcium ion in vitro [98]. ACER1 is mainly expressed in the epidermis, whereas ACER2 and ACER3 are expressed in various tissues. ACER1 has multiple putative transmembrane domains and is localized to the ER [99], [95]. Interestingly, ACER1 has marked substrate specificity for C24 and C24:1 ceramides, but has no activity against dihydroceramides or phytoceramides [99], [95]. This highly restricted substrate specificity may prevent phytoceramide species from being degraded because the skin is one of the limited tissues that is enriched in these types of ceramides.
ACER2, also referred to as Golgi alkaline ceramidase, is highly expressed in the placenta, but its low expression can be detected in most tissues. Like its homologue ACER1, ACER2 has several putative transmembrane domains, however, ACER2 is localized to the Golgi complex. ACER2 has a less restricted substrate specificity since it also metabolizes other long-chain ceramides C16, C18, and C20 ceramide species, and long-chain dihydroceramides and phytoceramides with an unsaturated acyl chain, in addition to C24 and C24:1 species (Cungui Mao personal communication). ACER2 requires calcium ions but not other cations for its activity.
ACER3 was previously called phytoceramidase because it was found to have higher in vitro activity towards the artificial fluorescent phytoceramide D-ribo-C12-NBD-phytoceramide, than towards NBD-ceramide or NBD-dihydroceramide [97]. Mao et. al recently found that ACER3 only catalyzes the hydrolysis of natural phytoceramide, dihydroceramide, and ceramides carrying an unsaturated fatty acid (≤C20) (Cungui Mao personal communication). ACER3 appears to be the only ceramidase identified in mammals which has a preference for phytoceramide species. Its tissue expression is widespread, but, like ACER2, is highly expressed in the placenta. ACER3 is localized to both the ER and Golgi complex, with a C-terminal ER retention sequence [97]. Interestingly, its activity is inhibited by sphingosine, but not by dihydrosphingosine or phytosphingosine. The significance of this mammalian ceramidase remains to be seen, but its unusual specificity for phytoceramide raises questions about the unique functions of phytoceramide containing sphingolipids.
Although investigation has begun to classify and characterize the different biochemical properties of the ceramidases, much still remains to be done to define the specific functions of each alkaline ceramidase. The generation of the three alkaline ceramidase knockout mice will undoubtedly shed further light on this subject.
ii) Sphingosine-1-Phosphate and Sphingosine Kinases 1 and 2
The two sphingosine kinases (SK), SK1 and SK2 are members of the DAG kinase family [100]. Both enzymes utilize ATP to phosphorylate the C-1 hydroxy group of free sphingosine, dihydrosphingosine, or, in the case of SK2, also phytosphingosine. Both sphingosine kinases are cytosolic enzymes that peripherally associate with membranes. Regulation of sphingosine kinase localization within the cell is thought to be the primary mode by which these enzymes acutely affect sphingolipid metabolism since only modest changes in their activity can be detected after stimulation by various agonists [101], [102]. In addition, SK enzymes are regulated transcriptionally by a variety of stimuli [103], [104], [105]. Although SK1 and SK2 both catalyze the same reaction, they have slight differences in their substrate specificities and have distinct, but overlapping, subcellular localizations which determine their effects on specific sphingolipid compartments within cells.
Sphingosine kinase 1 (SK1) is a cytosolic enzyme, but it can associate with the plasma membrane, move into the nucleus, and even be secreted from cells [106], [101], [107], [108], [109]). The first major insight into the regulation of SK1 activity first came when it was shown that SK1 translocates from the cytoplasm to the plasma membrane in response to phorbol ester treatment [101]. Shortly after this, translocation to the plasma membrane was shown to be phosphorylation dependent and that ERK2 was the kinase responsible for this phosphorylation [102]. Later work showed that phosphorylation of SK1 lead to its enhanced affinity to anionic phospholipids such as PS, PI, and PA which are in high abundance on the inner leaflet of the plasma membrane [110]. The metabolic significance of SK1 translocation to the plasma membrane was shown to be enhancement of S1P production and extracellular release of S1P [101], [111]. This agonist induced translocation of SK1 through phosphorylation by ERK2, has become the general paradigm for S1P signaling and receptor activation in response to various agonists. It is worth noting that many different growth actors converge on SK1 as an intracellular target for downstream signaling. These include, but are not limited to, PDGF, VEGF, NGF, EGF, Insulin, and IGF-1. Activation of SK1 was shown to be necessary for the proliferative effects of many different growth factors [112], [113], [114], [115], [116], [117]. Whether or not all of these growth factors act through the same intracellular signaling pathway to activate SK1 remains to be tested. The reader is referred to several good reviews on SK1 for more detailed information on its signaling role in proliferation and survival. [118], [119], [35]).
Although phosphorylation dependent translocation of SK1 to the plasma membrane has been the best defined mechanism by which SK1 activity is regulated, SK1 also traffics through the nucleus. SK1 was shown to have two putative nuclear export sequences (NES) which are necessary for SK1 to leave the nucleus [108]. It is unclear how SK1 introduction into the nucleus is regulated, but deletion of the two nuclear export sequences present on SK1, lead to nuclear accumulation of SK1. The effect nuclear trafficking of SK1 has on sphingolipid metabolism is unclear, but attachment of a nuclear localization sequence on SK1 inhibited cellular proliferation through an unknown mechanism [120].
In addition, SK1 has been shown to be secreted from endothelial cells, however, the physiological significance of this is not clear. An interesting discovery will be the route by which an exclusively cytosolic protein gets secreted from endothelial cells. Analysis of SK in endothelial cells showed that secretion was constitutive and not regulated by agonist stimulation [107]. It has been suggested that extracellular SK can generate S1P in the extracellular space following coordinated hydrolysis of plasma SM by secretory SMase and NCDase [121]. Some evidence for SK secretion occurring in vivo comes from Venkataraman et. al. A significant amount of soluble SK activity was found in the serum of wildtype, but not SphK1 −/− mice suggesting that SK1 is indeed released into the blood of mice [122]. Once again, the physiological significance of SK1 release remains to be determined.
In addition to its effects on promoting proliferation, SK1 has plays an important role in survival and resistance to chemotherapy [123]. Overexpression of SK1 correlates with a resistance to chemotherapeutic agents [124]. In addition, overexpressing SK1 in sensitive cells can induce resistance to chemotherapy [106]. Importantly, it has been shown that SK1 degradation is a downstream event in the DNA damage response [125]. Apoptosis inducing agents, such as TNF-α or various DNA damaging agents lead to degradation of SK1 through a Cathepsin B dependent mechanism [126]. In vitro characterization of SK1 degradation has shown that cathepsin B degrades SK1 through a stepwise cleavage, first at Histidine 122 followed by cleavage at Arginine 199 [127]. How DNA damaging agents or death inducing ligands, such as TNF-α, induce degradation of cytosolic SK1 through lysosomal localized cathepsin B is not yet clear. Future studies will better define how SK1 contributes to survival and how its degradation is involved in apoptosis.
Sphingosine kinase 2, the lesser studied isoform of the two, is predominantly localized in the nucleus or perinuclear region of a cell. SK2 has broader substrate specificity than SK1 and is able to phosphorylate phytosphingosine as well as sphingosine and dihydrosphingosine. Early reports on SK2 function suggest that it enhances apoptotic responses and can induce apoptosis through overexpression [128]. This is in contrast to SK1, which enhances survival of cells when overexpressed and allows for resistance to apoptosis inducing agents [123]. Due to its perinuclear localization, SK2 is thought to produce phosphate bases distal from the plasma membrane, where ABC transporters are less likely to export them into the extracellular space [129]. Another explanation for their disparate functions comes from a recent study suggesting that SK2 uniquely couples to sphingosine phosphate phosphatase 1 (SPP1) which generates free sphingosine for ceramide synthesis [130]. Interestingly, predominantly C16, C18, and C20 ceramide species were produced through this pathway, suggesting that the recycling pathway through SK2 and SPP1 may couple to specific ceramide synthases. Importantly, SK1 was found not to have the same function in enhancing sphingosine recycling into ceramide. Therefore, SK2 may have a distinct function from SK1 by having an enhanced ability to recycle sphingoid bases for ceramide synthesis. The structural basis for this difference has not yet been characterized, but will help elucidate how two enzymes with the same catalytic activity can have dramatic differences on cell biology [128].
Insight into the physiological role of S1P was hoped to be gained through generation of sphingosine kinase 1 knockout mice, however, in the absence of stresses they displayed no obvious phenotypic abnormalities [131]. In contrast, SphK1 −/− mice develop less intestinal adenomas when crossed with the Apcmin/+ mice, a model of intestinal adenocarcinoma [132]. SphK1 −/− mice also develop less adenocarcinomas when treated with DSS/AOM [133]. Moreover, SphK1 −/− mice are less susceptible to DSS induced colitis and have a marked decrease in intestinal inflammation in that model [134].
Sphingosine kinase 2 knockout mice have also been generated and these mice too do not display any obvious abnormalities [135]. Crossing of SphK1 −/− and SphK2 −/− mice revealed that complete absence of SK activity, and therefore S1P, results in embryonic lethality due to improper neural and vascular development [135]. These defects are likely to be due to a lack of S1P and not due to sphingosine accumulation since sphingosine levels were actually below wildtype levels in double knockout embryos. In addition, the vascular defects observed in the double knockout mice resembled previously observed defects in S1P1 receptor knockout mice, suggesting that S1P is absolutely necessary for vascular development [136], [135]. Finally, female SphK1 −/−, SphK2 +/− mice are infertile due to defects in decidualization [137]. Together these studies suggest that SK1 and SK2 have redundant functions during development since neither SK knockout displays any developmental defects alone, but display severe defects when both enzymes are absent. On the other hand, each SK isozyme may have unique functions in mature tissues since SphK1 −/− mice have specific defects in inflammatory responses [134].
iii) Lipid Phosphate Phosphatases, S1P Phosphatases, and the Salvage Pathway
Sphingosine-1-phosphate can be dephosphorylated at the cell surface by a family of broad specificity lipid phosphate phosphatases, LPP1-3. LPPs have six transmembrane domains and have their catalytic sites facing the extracellular space. The LPP family is important for sphingolipid metabolism because they are thought to be the primary mechanism by which extracellular S1P signaling is attenuated ([138], [139]. Overexpression of specific LPPs reduces S1P-dependent signaling events [140], [141]. LPP effects on S1P metabolism is complicated by the fact that LPPs also affect phosphatidic acid (PA) levels which have been shown to regulate SK1 localization in cells [142], [139]. In addition, S1P may require LPP dependent dephosphorylation prior to its uptake by cells, although a separate mechanism for uptake requiring the CFTR protein has also been proposed [143]. For additional information on LPPs, the reader is referred to some recent reviews [144], [139].
In addition to LPPs, cytosolic S1P can be dephosphorylated at the ER by S1P specific phosphatases, SPP1 and SPP2 [145], [146], [147]. SPP1, and perhaps SPP2, plays a role in regulating the reintroduction of sphingoid bases into ceramide species at the ER [148], [130]. Distantly related to LPPs, SPP1 and 2 each contain eight putative transmembrane domains [146], [149], [150]. Interestingly, overexpression of SPP1 results in an increase in ceramide accumulation suggesting that dephosphorylation of S1P is a rate limiting step in the salvage pathway [146], [151], [130]. This increase in ceramide can be exacerbated further by the addition of extracellular S1P. Therefore, regulation of SPP1 levels can change the metabolic fate of S1P to be predominantly recycled into ceramide. This has been shown to have dramatic effects on the biological response of cells to S1P [151]. Recently, SPP1 conversion to ceramide was shown to be enhanced by overexpression of SK2, suggesting that SK2 and SPP1 work coordinately to increase sphingosine salvage [130]. In addition, higher expression of SPP1 reduced extracellular release of S1P, suggesting that SPP1 can negatively regulate extracellular signaling of S1P [150].
A second sphingosine phosphate phosphatase, SPP2, was identified based on homology to SPP1. Both SPP isoforms are expressed ubiquitously with high expression in the kidney. On the other hand, SPP1 is highly expressed in the placenta whereas, SPP2 is highly expressed in the heart [147]. More recently, SPP2 was shown to be upregulated during inflammatory responses, however, it is unclear what effect this has on sphingolipid metabolism [152]. It remains to be seen if SPP2 plays an important role in regulating the sphingolipid recycling pathway like SPP1.
iv) S1P Lyase in the Removal of Sphingoid Bases
Sphingosine-1-phosphate lyase (SPL) is the enzyme responsible for the conversion of phosphorylated sphingoid bases to hexadecenal and phosphoethanolamine, thereby serving as the final step in sphingolipid degradation. SPL is a single pass transmembrane protein, displaying type I topology, and is exclusively localized to the ER. [153]. The catalytic site of SPL faces the cytosolic surface of the ER where it has access to cytosolically produced S1P. Since SPL is exclusively localized to the ER, all sphingoid phosphate bases must reach the ER for their final degradation by SPL. SPL has broad substrate specificity since it can utilize sphingosine-1-phosphate, dihydrosphingosine-1-phosphate, and phytosphingoinse-1-phosphate as substrates and therefore is able to catabolize all sphingoid bases found in mammals [154]. Like SPT, SPL is dependent on pyridoxal 5′-phosphate (PLP) as a cofactor for enzymatic activity, making both the initial and the final steps in the sphingolipid pathway PLP dependent. The relationship between symptoms associated with vitamin B6 deficiency, the dietary precursor to PLP, and alterations in sphingolipid metabolism are unclear, but will be important to assess since inhibition of SPT or S1P lyase could result in demyelination and altered immune function respectively [8], [155].
SPL has a wide tissue distribution with its highest expression in the thymus and intestines and its lowest expression in the brain and skeletal muscle [153]. In addition, no SPL activity can be detected in platelets or red blood cells since these cells lack ER membranes [153]. Immunohistochemical staining for SPL within the intestine revealed high expression on differentiated enterocytes, but low expression in the intestinal cypt cells [156]. In addition, intestinal adenomas from ApcMin/+ mice showed a marked decrease in SPL expression compared with adjacent normal mucosa [132]. Together these data suggest that SPL upregulation is part of the normal differentiation of enterocytes. SPL expression in the thymus was also assessed and was localized almost exclusively to thymic epithelium with low expression in lymphocytes. Thymic expression of SPL seems to be essential for proper lymphocyte trafficking between the blood, primary lymphoid tissue, and secondary lymphoid tissue due its role in establishing S1P gradients between the different trafficking compartments [155].
Several studies have identified S1P lyase as an essential gene for development in Drosophila melanogaster, Caenorhibitis elegans and Dictyostelium discoideum [156], [157], [158]. Recently, the phenotype of the S1P lyase knockout mouse, Sgpl1 −/−, was published [159]. These SPL defective mice display an inability to gain weight and premature death by eight weeks of age. These mice also displayed an array of defects which closely resembled those seen with the PDGF receptor, Pdgfra −/− and Pdgfrb −/−, mutant mice. Some of these defects include vascular hemorrhaging, defective glomeruli formation, and skeletal defects. It is unclear if Sgpl1 −/− mice had defects in any other systems since the study was interested only in PDGF dependent phenotypes [159]. Regardless, this study provides clear evidence that SPL plays an essential role in the development of many tissues in mammals. Future studies will help identify the role of SPL in adult tissues and assess its potential as a therapeutic target for the treatment of disorders ranging from immunological disorders to neoplasias.
Conclusions
Sphingolipids are a diverse group of lipids which serve a variety of functions in both mammalian development and physiology. Only a few of these functions have been highlighted in the chapter, but the scope of sphingolipid biology is vast. New functions for sphingolipids in mammalian physiology continue to be discovered each year and will likely increase as our understanding of sphingolipid biology in whole organisms improves.
It is clear that sphingolipids play an essential role in mammalian systems, therefore, it is essential to understand how sphingolipids are synthesized and degraded to maintain their functional levels at both an organismal and cellular level. Thanks to the hard work of many pioneers in the sphingolipid field, the biochemical characterization of many sphingolipid enzymes was laid out before any genes were identified. Aided by the advancement of recombinant DNA technology and the human genome project, significant progress has been made to clone and characterize the enzymes responsible for sphingolipid metabolism. At least one gene, and in most cases two or more, has been cloned for each of the known enzymatic steps required from the condensation of serine and palmitoyl CoA by SPT to the production of hexadecenal and phosphoethanolamine by S1P lyase. Although it is easy to assume that we have a near complete list of the enzymes involved in sphingolipid metabolism, new isoforms of these enzymes are still being discovered and validated. While having a list of enzymes is comforting, our understanding of their regulation is still lacking. We still have a lot of work to understand how the different sphingolipid enzymes work in concert to determine the sphingolipid composition of the plasma membrane and subcellular organelles. In addition, the breakthrough discoveries of the sphingolipid transfer proteins CERT and FAPP2 have provided a new paradigm for non-vesicular sphingolipid trafficking. These proteins are likely to only be the first members of a class of sphingolipid transfer proteins which regulate the trafficking of sphingolipids to specific compartments within cells.
Although this chapter aimed to provide an abbreviated overview of our current understanding of sphingolipid metabolism in mammalian systems, it also highlights some areas of lipid biology that are poorly understood. It is a healthy scientific practice to regularly reflect on the progress that has been achieved within a field, both, to bring to light significant faults or assumptions we have about it, but also for the sense of awe one gets about our collective accomplishment.
Acknowledgements
This work was supported in part by NIH grants GM-43825 and CA-87584 to YAH and AG-016583, GM-062887, and a MERIT Award by the Office of Research and Development, Dept. of Veterans Affairs to LMO. Special thanks to Thomas D. Mullen and Cungui Mao for their helpful comments.
References
- 1.Mandon EC, et al. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J Biol Chem. 1992;267(16):11144–8. [PubMed] [Google Scholar]
- 2.Hornemann T, et al. Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem. 2006;281(49):37275–81. doi: 10.1074/jbc.M608066200. [DOI] [PubMed] [Google Scholar]
- 3.Yasuda S, Nishijima M, Hanada K. Localization, topology, and function of the LCB1 subunit of serine palmitoyltransferase in mammalian cells. J Biol Chem. 2003;278(6):4176–83. doi: 10.1074/jbc.M209602200. [DOI] [PubMed] [Google Scholar]
- 4.Hojjati MR, Li Z, Jiang XC. Serine palmitoyl-CoA transferase (SPT) deficiency and sphingolipid levels in mice. Biochim Biophys Acta. 2005;1737(1):44–51. doi: 10.1016/j.bbalip.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Gable K, et al. Tsc3p is an 80-amino acid protein associated with serine palmitoyltransferase and required for optimal enzyme activity. J Biol Chem. 2000;275(11):7597–603. doi: 10.1074/jbc.275.11.7597. [DOI] [PubMed] [Google Scholar]
- 6.Hanada K. Serine palmitoyltransferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta. 2003;1632(1-3):16–30. doi: 10.1016/s1388-1981(03)00059-3. [DOI] [PubMed] [Google Scholar]
- 7.Merrill AH., Jr. Characterization of serine palmitoyltransferase activity in Chinese hamster ovary cells. Biochim Biophys Acta. 1983;754(3):284–91. doi: 10.1016/0005-2760(83)90144-3. [DOI] [PubMed] [Google Scholar]
- 8.Dawkins JL, et al. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet. 2001;27(3):309–12. doi: 10.1038/85879. [DOI] [PubMed] [Google Scholar]
- 9.Spring PJ, et al. Autosomal dominant hereditary sensory neuropathy with chronic cough and gastro-oesophageal reflux: clinical features in two families linked to chromosome 3p22-p24. Brain. 2005;128(Pt 12):2797–810. doi: 10.1093/brain/awh653. [DOI] [PubMed] [Google Scholar]
- 10.Lindahl AJ, et al. Late-onset hereditary sensory neuropathy type I due to SPTLC1 mutation: autopsy findings. Clin Neurol Neurosurg. 2006;108(8):780–3. doi: 10.1016/j.clineuro.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Bejaoui K, et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet. 2001;27(3):261–2. doi: 10.1038/85817. [DOI] [PubMed] [Google Scholar]
- 12.McCampbell A, et al. Mutant SPTLC1 dominantly inhibits serine palmitoyltransferase activity in vivo and confers an age-dependent neuropathy. Hum Mol Genet. 2005;14(22):3507–21. doi: 10.1093/hmg/ddi380. [DOI] [PubMed] [Google Scholar]
- 13.Bejaoui K, et al. Hereditary sensory neuropathy type 1 mutations confer dominant negative effects on serine palmitoyltransferase, critical for sphingolipid synthesis. J Clin Invest. 2002;110(9):1301–8. doi: 10.1172/JCI16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawkins JL, et al. Exclusion of serine palmitoyltransferase long chain base subunit 2 (SPTLC2) as a common cause for hereditary sensory neuropathy. Neuromuscul Disord. 2002;12(7-8):656–8. doi: 10.1016/s0960-8966(02)00015-9. [DOI] [PubMed] [Google Scholar]
- 15.Beeler T, et al. The Saccharomyces cerevisiae TSC10/YBR265w gene encoding 3-ketosphinganine reductase is identified in a screen for temperature-sensitive suppressors of the Ca2+-sensitive csg2Delta mutant. J Biol Chem. 1998;273(46):30688–94. doi: 10.1074/jbc.273.46.30688. [DOI] [PubMed] [Google Scholar]
- 16.Kihara A, Igarashi Y. FVT-1 is a mammalian 3-ketodihydrosphingosine reductase with an active site that faces the cytosolic side of the endoplasmic reticulum membrane. J Biol Chem. 2004;279(47):49243–50. doi: 10.1074/jbc.M405915200. [DOI] [PubMed] [Google Scholar]
- 17.Krebs S, et al. A missense mutation in the 3-ketodihydrosphingosine reductase FVT1 as candidate causal mutation for bovine spinal muscular atrophy. Proc Natl Acad Sci U S A. 2007;104(16):6746–51. doi: 10.1073/pnas.0607721104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281(35):25001–5. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 19.Lahiri S, et al. Kinetic characterization of mammalian ceramide synthases: determination of K(m) values towards sphinganine. FEBS Lett. 2007;581(27):5289–94. doi: 10.1016/j.febslet.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 20.Venkataraman K, et al. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J Biol Chem. 2002;277(38):35642–9. doi: 10.1074/jbc.M205211200. [DOI] [PubMed] [Google Scholar]
- 21.Laviad EL, et al. Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem. 2008;283(9):5677–84. doi: 10.1074/jbc.M707386200. [DOI] [PubMed] [Google Scholar]
- 22.Riebeling C, et al. Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J Biol Chem. 2003;278(44):43452–9. doi: 10.1074/jbc.M307104200. [DOI] [PubMed] [Google Scholar]
- 23.Mizutani Y, Kihara A, Igarashi Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem J. 2005;390(Pt 1):263–71. doi: 10.1042/BJ20050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizutani Y, Kihara A, Igarashi Y. LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro)ceramide synthase with relatively broad substrate specificity. Biochem J. 2006;398(3):531–8. doi: 10.1042/BJ20060379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizutani Y, et al. 2-Hydroxy-ceramide synthesis by ceramide synthase family: enzymatic basis for the preference of FA chain length. J Lipid Res. 2008;49(11):2356–64. doi: 10.1194/jlr.M800158-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Bose R, et al. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82(3):405–14. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 27.Cadena DL, Kurten RC, Gill GN. The product of the MLD gene is a member of the membrane fatty acid desaturase family: overexpression of MLD inhibits EGF receptor biosynthesis. Biochemistry. 1997;36(23):6960–7. doi: 10.1021/bi970091l. [DOI] [PubMed] [Google Scholar]
- 28.Geeraert L, Mannaerts GP, van Veldhoven PP. Conversion of dihydroceramide into ceramide: involvement of a desaturase. Biochem J. 1997;327(Pt 1):125–32. doi: 10.1042/bj3270125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michel C, et al. Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide. J Biol Chem. 1997;272(36):22432–7. doi: 10.1074/jbc.272.36.22432. [DOI] [PubMed] [Google Scholar]
- 30.Savile CK, Fabrias G, Buist PH. Dihydroceramide delta(4) desaturase initiates substrate oxidation at C-4. J Am Chem Soc. 2001;123(19):4382–5. doi: 10.1021/ja010088w. [DOI] [PubMed] [Google Scholar]
- 31.Ternes P, et al. Identification and characterization of a sphingolipid delta 4-desaturase family. J Biol Chem. 2002;277(28):25512–8. doi: 10.1074/jbc.M202947200. [DOI] [PubMed] [Google Scholar]
- 32.Beauchamp E, et al. Myristic acid increases the activity of dihydroceramide Delta4-desaturase 1 through its N-terminal myristoylation. Biochimie. 2007;89(12):1553–61. doi: 10.1016/j.biochi.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 33.Mizutani Y, Kihara A, Igarashi Y. Identification of the human sphingolipid C4-hydroxylase, hDES2, and its up-regulation during keratinocyte differentiation. FEBS Lett. 2004;563(1-3):93–7. doi: 10.1016/S0014-5793(04)00274-1. [DOI] [PubMed] [Google Scholar]
- 34.Omae F, et al. DES2 protein is responsible for phytoceramide biosynthesis in the mouse small intestine. Biochem J. 2004;379(Pt 3):687–95. doi: 10.1042/BJ20031425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139–50. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 36.Holland WL, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5(3):167–79. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 37.Hanada K, et al. CERT and intracellular trafficking of ceramide. Biochim Biophys Acta. 2007;1771(6):644–53. doi: 10.1016/j.bbalip.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Kumagai K, et al. Interorganelle trafficking of ceramide is regulated by phosphorylation-dependent cooperativity between the PH and START domains of CERT. J Biol Chem. 2007;282(24):17758–66. doi: 10.1074/jbc.M702291200. [DOI] [PubMed] [Google Scholar]
- 39.Charruyer A, et al. Decreased ceramide transport protein (CERT) function alters sphingomyelin production following UVB irradiation. J Biol Chem. 2008;283(24):16682–92. doi: 10.1074/jbc.M800799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanada K, et al. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J Biol Chem. 1998;273(50):33787–94. doi: 10.1074/jbc.273.50.33787. [DOI] [PubMed] [Google Scholar]
- 41.Hanada K, et al. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426(6968):803–9. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- 42.Kudo N, et al. Structural basis for specific lipid recognition by CERT responsible for nonvesicular trafficking of ceramide. Proc Natl Acad Sci U S A. 2008;105(2):488–93. doi: 10.1073/pnas.0709191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watson P, Stephens DJ. ER-to-Golgi transport: form and formation of vesicular and tubular carriers. Biochim Biophys Acta. 2005;1744(3):304–15. doi: 10.1016/j.bbamcr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Stahl N, et al. Isolation, characterization, and expression of cDNA clones that encode rat UDP-galactose: ceramide galactosyltransferase. J Neurosci Res. 1994;38(2):234–42. doi: 10.1002/jnr.490380214. [DOI] [PubMed] [Google Scholar]
- 45.Coetzee T, et al. Molecular cloning, chromosomal mapping, and characterization of the mouse UDP-galactose:ceramide galactosyltransferase gene. Genomics. 1996;35(1):215–22. doi: 10.1006/geno.1996.0341. [DOI] [PubMed] [Google Scholar]
- 46.Fujimoto H, et al. Requirement of seminolipid in spermatogenesis revealed by UDP-galactose: Ceramide galactosyltransferase-deficient mice. J Biol Chem. 2000;275(30):22623–6. doi: 10.1074/jbc.C000200200. [DOI] [PubMed] [Google Scholar]
- 47.Zoller I, et al. Oligodendrocyte-specific ceramide galactosyltransferase (CGT) expression phenotypically rescues CGT-deficient mice and demonstrates that CGT activity does not limit brain galactosylceramide level. Glia. 2005;52(3):190–8. doi: 10.1002/glia.20230. [DOI] [PubMed] [Google Scholar]
- 48.Marcus J, et al. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53(4):372–81. doi: 10.1002/glia.20292. [DOI] [PubMed] [Google Scholar]
- 49.Ichikawa S, et al. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc Natl Acad Sci U S A. 1996;93(10):4638–43. doi: 10.1073/pnas.93.10.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeckel D, et al. Glucosylceramide is synthesized at the cytosolic surface of various Golgi subfractions. J Cell Biol. 1992;117(2):259–67. doi: 10.1083/jcb.117.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Angelo G, et al. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature. 2007;449(7158):62–7. doi: 10.1038/nature06097. [DOI] [PubMed] [Google Scholar]
- 52.Yamashita T, et al. A vital role for glycosphingolipid synthesis during development and differentiation. Proc Natl Acad Sci U S A. 1999;96(16):9142–7. doi: 10.1073/pnas.96.16.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jennemann R, et al. Integrity and barrier function of the epidermis critically depend on glucosylceramide synthesis. J Biol Chem. 2007;282(5):3083–94. doi: 10.1074/jbc.M610304200. [DOI] [PubMed] [Google Scholar]
- 54.Jennemann R, et al. Cell-specific deletion of glucosylceramide synthase in brain leads to severe neural defects after birth. Proc Natl Acad Sci U S A. 2005;102(35):12459–64. doi: 10.1073/pnas.0500893102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamashita T, et al. Conditional LoxP-flanked glucosylceramide synthase allele controlling glycosphingolipid synthesis. Genesis. 2005;43(4):175–80. doi: 10.1002/gene.20167. [DOI] [PubMed] [Google Scholar]
- 56.Tafesse FG, et al. Both sphingomyelin synthases SMS1 and SMS2 are required for sphingomyelin homeostasis and growth in human HeLa cells. J Biol Chem. 2007;282(24):17537–47. doi: 10.1074/jbc.M702423200. [DOI] [PubMed] [Google Scholar]
- 57.Huitema K, et al. Identification of a family of animal sphingomyelin synthases. Embo J. 2004;23(1):33–44. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tafesse FG, Ternes P, Holthuis JC. The multigenic sphingomyelin synthase family. J Biol Chem. 2006;281(40):29421–5. doi: 10.1074/jbc.R600021200. [DOI] [PubMed] [Google Scholar]
- 59.Villani M, et al. Sphingomyelin synthases regulate production of diacylglycerol at the Golgi. Biochem J. 2008;414(1):31–41. doi: 10.1042/BJ20071240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graf C, et al. Wild-type levels of ceramide and ceramide-1-phosphate in the retina of ceramide kinase-like-deficient mice. Biochem Biophys Res Commun. 2008;373(1):159–63. doi: 10.1016/j.bbrc.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 61.Wijesinghe DS, et al. Substrate specificity of human ceramide kinase. J Lipid Res. 2005;46(12):2706–16. doi: 10.1194/jlr.M500313-JLR200. [DOI] [PubMed] [Google Scholar]
- 62.Lamour NF, et al. Ceramide kinase uses ceramide provided by ceramide transport protein: localization to organelles of eicosanoid synthesis. J Lipid Res. 2007;48(6):1293–304. doi: 10.1194/jlr.M700083-JLR200. [DOI] [PubMed] [Google Scholar]
- 63.Kumagai K, et al. CERT mediates intermembrane transfer of various molecular species of ceramides. J Biol Chem. 2005;280(8):6488–95. doi: 10.1074/jbc.M409290200. [DOI] [PubMed] [Google Scholar]
- 64.Boath A, et al. Regulation and traffic of ceramide 1-phosphate produced by ceramide kinase: comparative analysis to glucosylceramide and sphingomyelin. J Biol Chem. 2008;283(13):8517–26. doi: 10.1074/jbc.M707107200. [DOI] [PubMed] [Google Scholar]
- 65.Sugiura M, et al. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem. 2002;277(26):23294–300. doi: 10.1074/jbc.M201535200. [DOI] [PubMed] [Google Scholar]
- 66.Mitsutake S, et al. The generation and behavioral analysis of ceramide kinase-null mice, indicating a function in cerebellar Purkinje cells. Biochem Biophys Res Commun. 2007;363(3):519–24. doi: 10.1016/j.bbrc.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 67.Graf C, et al. Neutropenia with impaired immune response to Streptococcus pneumoniae in ceramide kinase-deficient mice. J Immunol. 2008;180(5):3457–66. doi: 10.4049/jimmunol.180.5.3457. [DOI] [PubMed] [Google Scholar]
- 68.Bornancin F, et al. Characterization of a ceramide kinase-like protein. Biochim Biophys Acta. 2005;1687(1-3):31–43. doi: 10.1016/j.bbalip.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 69.Inagaki Y, Mitsutake S, Igarashi Y. Identification of a nuclear localization signal in the retinitis pigmentosa-mutated RP26 protein, ceramide kinase-like protein. Biochem Biophys Res Commun. 2006;343(3):982–7. doi: 10.1016/j.bbrc.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 70.Boudker O, Futerman AH. Detection and characterization of ceramide-1-phosphate phosphatase activity in rat liver plasma membrane. J Biol Chem. 1993;268(29):22150–5. [PubMed] [Google Scholar]
- 71.Shinghal R, Scheller RH, Bajjalieh SM. Ceramide 1-phosphate phosphatase activity in brain. J Neurochem. 1993;61(6):2279–85. doi: 10.1111/j.1471-4159.1993.tb07470.x. [DOI] [PubMed] [Google Scholar]
- 72.Brindley DN, et al. Analysis of ceramide 1-phosphate and sphingosine-1-phosphate phosphatase activities. Methods Enzymol. 2000;311:233–44. doi: 10.1016/s0076-6879(00)11086-9. [DOI] [PubMed] [Google Scholar]
- 73.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5(7):554–65. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 74.Sabourdy F, et al. Functions of sphingolipid metabolism in mammals--lessons from genetic defects. Biochim Biophys Acta. 2008;1781(4):145–83. doi: 10.1016/j.bbalip.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 75.Duan RD, et al. Purification, localization, and expression of human intestinal alkaline sphingomyelinase. J Lipid Res. 2003;44(6):1241–50. doi: 10.1194/jlr.M300037-JLR200. [DOI] [PubMed] [Google Scholar]
- 76.Schissel SL, et al. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J Biol Chem. 1998;273(29):18250–9. doi: 10.1074/jbc.273.29.18250. [DOI] [PubMed] [Google Scholar]
- 77.Schissel SL, et al. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271(31):18431–6. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- 78.Brady RO, et al. The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proc Natl Acad Sci U S A. 1966;55(2):366–9. doi: 10.1073/pnas.55.2.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Horinouchi K, et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet. 1995;10(3):288–93. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 80.Marathe S, et al. Creation of a mouse model for non-neurological (type B) Niemann-Pick disease by stable, low level expression of lysosomal sphingomyelinase in the absence of secretory sphingomyelinase: relationship between brain intra-lysosomal enzyme activity and central nervous system function. Hum Mol Genet. 2000;9(13):1967–76. doi: 10.1093/hmg/9.13.1967. [DOI] [PubMed] [Google Scholar]
- 81.Sawai H, et al. Function of the cloned putative neutral sphingomyelinase as lyso-platelet activating factor-phospholipase C. J Biol Chem. 1999;274(53):38131–9. doi: 10.1074/jbc.274.53.38131. [DOI] [PubMed] [Google Scholar]
- 82.Zumbansen M, Stoffel W. Neutral sphingomyelinase 1 deficiency in the mouse causes no lipid storage disease. Mol Cell Biol. 2002;22(11):3633–8. doi: 10.1128/MCB.22.11.3633-3638.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tani M, Hannun YA. Neutral sphingomyelinase 2 is palmitoylated on multiple cysteine residues. Role of palmitoylation in subcellular localization. J Biol Chem. 2007;282(13):10047–56. doi: 10.1074/jbc.M611249200. [DOI] [PubMed] [Google Scholar]
- 84.Marchesini N, et al. Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J Biol Chem. 2004;279(24):25101–11. doi: 10.1074/jbc.M313662200. [DOI] [PubMed] [Google Scholar]
- 85.Stoffel W, et al. Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc Natl Acad Sci U S A. 2005;102(12):4554–9. doi: 10.1073/pnas.0406380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stoffel W, et al. Neutral sphingomyelinase (SMPD3) deficiency causes a novel form of chondrodysplasia and dwarfism that is rescued by Col2A1-driven smpd3 transgene expression. Am J Pathol. 2007;171(1):153–61. doi: 10.2353/ajpath.2007.061285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krut O, et al. Novel tumor necrosis factor-responsive mammalian neutral sphingomyelinase-3 is a C-tail-anchored protein. J Biol Chem. 2006;281(19):13784–93. doi: 10.1074/jbc.M511306200. [DOI] [PubMed] [Google Scholar]
- 88.Corcoran CA, et al. Neutral sphingomyelinase-3 is a DNA damage and nongenotoxic stress-regulated gene that is deregulated in human malignancies. Mol Cancer Res. 2008;6(5):795–807. doi: 10.1158/1541-7786.MCR-07-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Momoi T, Ben-Yoseph Y, Nadler HL. Substrate-specificities of acid and alkaline ceramidases in fibroblasts from patients with Farber disease and controls. Biochem J. 1982;205(2):419–25. doi: 10.1042/bj2050419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li CM, et al. Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics. 2002;79(2):218–24. doi: 10.1006/geno.2002.6686. [DOI] [PubMed] [Google Scholar]
- 91.Tani M, Iida H, Ito M. O-glycosylation of mucin-like domain retains the neutral ceramidase on the plasma membranes as a type II integral membrane protein. J Biol Chem. 2003;278(12):10523–30. doi: 10.1074/jbc.M207932200. [DOI] [PubMed] [Google Scholar]
- 92.Tani M, Igarashi Y, Ito M. Involvement of neutral ceramidase in ceramide metabolism at the plasma membrane and in extracellular milieu. J Biol Chem. 2005;280(44):36592–600. doi: 10.1074/jbc.M506827200. [DOI] [PubMed] [Google Scholar]
- 93.Kono M, et al. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J Biol Chem. 2006;281(11):7324–31. doi: 10.1074/jbc.M508382200. [DOI] [PubMed] [Google Scholar]
- 94.Ohlsson L, et al. Purification and characterization of human intestinal neutral ceramidase. Biochimie. 2007;89(8):950–60. doi: 10.1016/j.biochi.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 95.Sun W, et al. Upregulation of the human alkaline ceramidase 1 and acid ceramidase mediates calcium-induced differentiation of epidermal keratinocytes. J Invest Dermatol. 2008;128(2):389–97. doi: 10.1038/sj.jid.5701025. [DOI] [PubMed] [Google Scholar]
- 96.Xu R, et al. Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. Faseb J. 2006;20(11):1813–25. doi: 10.1096/fj.05-5689com. [DOI] [PubMed] [Google Scholar]
- 97.Mao C, et al. Cloning and characterization of a novel human alkaline ceramidase. A mammalian enzyme that hydrolyzes phytoceramide. J Biol Chem. 2001;276(28):26577–88. doi: 10.1074/jbc.M102818200. [DOI] [PubMed] [Google Scholar]
- 98.Mao C, Obeid LM. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta. 2008;1781(9):424–34. doi: 10.1016/j.bbalip.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mao C, et al. Cloning and characterization of a mouse endoplasmic reticulum alkaline ceramidase: an enzyme that preferentially regulates metabolism of very long chain ceramides. J Biol Chem. 2003;278(33):31184–91. doi: 10.1074/jbc.M303875200. [DOI] [PubMed] [Google Scholar]
- 100.Wattenberg BW, Pitson SM, Raben DM. The sphingosine and diacylglycerol kinase superfamily of signaling kinases: localization as a key to signaling function. J Lipid Res. 2006;47(6):1128–39. doi: 10.1194/jlr.R600003-JLR200. [DOI] [PubMed] [Google Scholar]
- 101.Johnson KR, et al. PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA) J Biol Chem. 2002;277(38):35257–62. doi: 10.1074/jbc.M203033200. [DOI] [PubMed] [Google Scholar]
- 102.Pitson SM, et al. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. Embo J. 2003;22(20):5491–500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buehrer BM, Bardes ES, Bell RM. Protein kinase C-dependent regulation of human erythroleukemia (HEL) cell sphingosine kinase activity. Biochim Biophys Acta. 1996;1303(3):233–42. doi: 10.1016/0005-2760(96)00092-6. [DOI] [PubMed] [Google Scholar]
- 104.Yamanaka M, et al. Sphingosine kinase 1 (SPHK1) is induced by transforming growth factor-beta and mediates TIMP-1 up-regulation. J Biol Chem. 2004;279(52):53994–4001. doi: 10.1074/jbc.M410144200. [DOI] [PubMed] [Google Scholar]
- 105.Nakade Y, et al. Regulation of sphingosine kinase 1 gene expression by protein kinase C in a human leukemia cell line, MEG-O1. Biochim Biophys Acta. 2003;1635(2-3):104–16. doi: 10.1016/j.bbalip.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 106.Olivera A, et al. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol. 1999;147(3):545–58. doi: 10.1083/jcb.147.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ancellin N, et al. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J Biol Chem. 2002;277(8):6667–75. doi: 10.1074/jbc.M102841200. [DOI] [PubMed] [Google Scholar]
- 108.Inagaki Y, et al. Identification of functional nuclear export sequences in human sphingosine kinase 1. Biochem Biophys Res Commun. 2003;311(1):168–73. doi: 10.1016/j.bbrc.2003.09.194. [DOI] [PubMed] [Google Scholar]
- 109.Thompson CR, et al. Sphingosine kinase 1 (SK1) is recruited to nascent phagosomes in human macrophages: inhibition of SK1 translocation by Mycobacterium tuberculosis. J Immunol. 2005;174(6):3551–61. doi: 10.4049/jimmunol.174.6.3551. [DOI] [PubMed] [Google Scholar]
- 110.Stahelin RV, et al. The mechanism of membrane targeting of human sphingosine kinase 1. J Biol Chem. 2005 doi: 10.1074/jbc.M507574200. [DOI] [PubMed] [Google Scholar]
- 111.Pitson SM, et al. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J Exp Med. 2005;201(1):49–54. doi: 10.1084/jem.20040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365(6446):557–60. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- 113.Edsall LC, Pirianov GG, Spiegel S. Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J Neurosci. 1997;17(18):6952–60. doi: 10.1523/JNEUROSCI.17-18-06952.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shu X, et al. Sphingosine kinase mediates vascular endothelial growth factor-induced activation of ras and mitogen-activated protein kinases. Mol Cell Biol. 2002;22(22):7758–68. doi: 10.1128/MCB.22.22.7758-7768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sarkar S, et al. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005;579(24):5313–7. doi: 10.1016/j.febslet.2005.08.055. [DOI] [PubMed] [Google Scholar]
- 116.El-Shewy HM, et al. Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine-1-phosphate receptors. J Biol Chem. 2006 doi: 10.1074/jbc.M605339200. [DOI] [PubMed] [Google Scholar]
- 117.Ma MM, et al. Sphingosine kinase 1 participates in insulin signalling and regulates glucose metabolism and homeostasis in KK/Ay diabetic mice. Diabetologia. 2007 doi: 10.1007/s00125-006-0589-5. [DOI] [PubMed] [Google Scholar]
- 118.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4(5):397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 119.Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol. 2006;39(2):113–31. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- 120.Igarashi N, et al. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem. 2003;278(47):46832–9. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- 121.Tani M, Ito M, Igarashi Y. Ceramide/sphingosine/sphingosine 1-phosphate metabolism on the cell surface and in the extracellular space. Cell Signal. 2007;19(2):229–37. doi: 10.1016/j.cellsig.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 122.Venkataraman K, et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem J. 2006;397(3):461–71. doi: 10.1042/BJ20060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cuvillier O. Sphingosine kinase-1 - a potential therapeutic target in cancer. Anticancer Drugs. 2007;18(2):105–10. doi: 10.1097/CAD.0b013e328011334d. [DOI] [PubMed] [Google Scholar]
- 124.Bonhoure E, et al. Sphingosine kinase-1 is a downstream regulator of imatinib-induced apoptosis in chronic myeloid leukemia cells. Leukemia. 2008 doi: 10.1038/leu.2008.95. [DOI] [PubMed] [Google Scholar]
- 125.Taha TA, et al. Down-regulation of sphingosine kinase-1 by DNA damage: dependence on proteases and p53. J Biol Chem. 2004;279(19):20546–54. doi: 10.1074/jbc.M401259200. [DOI] [PubMed] [Google Scholar]
- 126.Taha TA, et al. Tumor necrosis factor induces the loss of sphingosine kinase-1 by a cathepsin B-dependent mechanism. J Biol Chem. 2005;280(17):17196–202. doi: 10.1074/jbc.M413744200. [DOI] [PubMed] [Google Scholar]
- 127.Taha TA, et al. Sphingosine kinase-1 is cleaved by cathepsin B in vitro: identification of the initial cleavage sites for the protease. FEBS Lett. 2006;580(26):6047–54. doi: 10.1016/j.febslet.2006.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maceyka M, et al. Sphk1 and Sphk2: Sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005 doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 129.Mitra P, et al. Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci U S A. 2006;103(44):16394–9. doi: 10.1073/pnas.0603734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Le Stunff H, et al. Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J Biol Chem. 2007;282(47):34372–80. doi: 10.1074/jbc.M703329200. [DOI] [PubMed] [Google Scholar]
- 131.Allende ML, et al. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279(50):52487–92. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- 132.Kohno M, et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26(19):7211–23. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kawamori T, et al. Role for sphingosine kinase 1 in colon carcinogenesis. Faseb J. 2008 doi: 10.1096/fj.08-117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Snider AJ, et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. Faseb J. 2008 doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mizugishi K, et al. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25(24):11113–21. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liu Y, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106(8):951–61. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mizugishi K, et al. Maternal disturbance in activated sphingolipid metabolism causes pregnancy loss in mice. J Clin Invest. 2007;117(10):2993–3006. doi: 10.1172/JCI30674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pyne S, Pyne NJ. Sphingosine 1-phosphate signalling and termination at lipid phosphate receptors. Biochim Biophys Acta. 2002;1582(1-3):121–31. doi: 10.1016/s1388-1981(02)00146-4. [DOI] [PubMed] [Google Scholar]
- 139.Pyne S, et al. Lipid phosphate phosphatases and lipid phosphate signalling. Biochem Soc Trans. 2005;33(Pt 6):1370–4. doi: 10.1042/BST0331370. [DOI] [PubMed] [Google Scholar]
- 140.Alderton F, et al. G-protein-coupled receptor stimulation of the p42/p44 mitogen-activated protein kinase pathway is attenuated by lipid phosphate phosphatases 1, 1a, and 2 in human embryonic kidney 293 cells. J Biol Chem. 2001;276(16):13452–60. doi: 10.1074/jbc.M006582200. [DOI] [PubMed] [Google Scholar]
- 141.Long J, et al. Regulation of cell survival by lipid phosphate phosphatases involves the modulation of intracellular phosphatidic acid and sphingosine 1-phosphate pools. Biochem J. 2005;391(Pt 1):25–32. doi: 10.1042/BJ20050342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Delon C, et al. Sphingosine kinase 1 is an intracellular effector of phosphatidic acid. J Biol Chem. 2004;279(43):44763–74. doi: 10.1074/jbc.M405771200. [DOI] [PubMed] [Google Scholar]
- 143.Boujaoude LC, et al. Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J Biol Chem. 2001;276(38):35258–64. doi: 10.1074/jbc.M105442200. [DOI] [PubMed] [Google Scholar]
- 144.Pyne S, Kong KC, Darroch PI. Lysophosphatidic acid and sphingosine 1-phosphate biology: the role of lipid phosphate phosphatases. Semin Cell Dev Biol. 2004;15(5):491–501. doi: 10.1016/j.semcdb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 145.Mandala SM, et al. Sphingoid base 1-phosphate phosphatase: a key regulator of sphingolipid metabolism and stress response. Proc Natl Acad Sci U S A. 1998;95(1):150–5. doi: 10.1073/pnas.95.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mandala SM, et al. Molecular cloning and characterization of a lipid phosphohydrolase that degrades sphingosine-1- phosphate and induces cell death. Proc Natl Acad Sci U S A. 2000;97(14):7859–64. doi: 10.1073/pnas.120146897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ogawa C, et al. Identification and characterization of a novel human sphingosine-1-phosphate phosphohydrolase, hSPP2. J Biol Chem. 2003;278(2):1268–72. doi: 10.1074/jbc.M209514200. [DOI] [PubMed] [Google Scholar]
- 148.Le Stunff H, et al. Sphingosine-1-phosphate phosphohydrolase in regulation of sphingolipid metabolism and apoptosis. J Cell Biol. 2002;158(6):1039–49. doi: 10.1083/jcb.200203123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kihara A, et al. Transmembrane topology of sphingoid long-chain base-1-phosphate phosphatase, Lcb3p. Genes Cells. 2003;8(6):525–35. doi: 10.1046/j.1365-2443.2003.00653.x. [DOI] [PubMed] [Google Scholar]
- 150.Johnson KR, et al. Role of human sphingosine-1-phosphate phosphatase 1 in the regulation of intra- and extracellular sphingosine-1-phosphate levels and cell viability. J Biol Chem. 2003;278(36):34541–7. doi: 10.1074/jbc.M301741200. [DOI] [PubMed] [Google Scholar]
- 151.Le Stunff H, et al. Sphingosine-1-phosphate and lipid phosphohydrolases. Biochim Biophys Acta. 2002;1582(1-3):8–17. doi: 10.1016/s1388-1981(02)00132-4. [DOI] [PubMed] [Google Scholar]
- 152.Mechtcheriakova D, et al. Sphingosine 1-phosphate phosphatase 2 is induced during inflammatory responses. Cell Signal. 2006 doi: 10.1016/j.cellsig.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 153.Ikeda M, Kihara A, Igarashi Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem Biophys Res Commun. 2004;325(1):338–43. doi: 10.1016/j.bbrc.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 154.Stoffel W, LeKim D, Sticht G. Distribution and properties of dihydrosphingosine-1-phosphate aldolase (sphinganine-1-phosphate alkanal-lyase) Hoppe Seylers Z Physiol Chem. 1969;350(10):1233–41. doi: 10.1515/bchm2.1969.350.2.1233. [DOI] [PubMed] [Google Scholar]
- 155.Schwab SR, et al. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309(5741):1735–9. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 156.Fyrst H, Saba JD. Sphingosine-1-phosphate lyase in development and disease: sphingolipid metabolism takes flight. Biochim Biophys Acta. 2008;1781(9):448–58. doi: 10.1016/j.bbalip.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mendel J, et al. Sphingosine phosphate lyase expression is essential for normal development in Caenorhabditis elegans. J Biol Chem. 2003;278(25):22341–9. doi: 10.1074/jbc.M302857200. [DOI] [PubMed] [Google Scholar]
- 158.Li G, et al. Sphingosine-1-phosphate lyase has a central role in the development of Dictyostelium discoideum. Development. 2001;128(18):3473–83. doi: 10.1242/dev.128.18.3473. [DOI] [PubMed] [Google Scholar]
- 159.Schmahl J, Raymond CS, Soriano P. PDGF signaling specificity is mediated through multiple immediate early genes. Nat Genet. 2007;39(1):52–60. doi: 10.1038/ng1922. [DOI] [PubMed] [Google Scholar]