

Abstract
Introduction
Knowledge that antibodies of the IgG isotype have remarkably extended persistence in circulation and are able to pass through cell barriers has substantial implications. While is well-established that so-called neonatal Fc receptor, FcRn, acts throughout life to confer these unusual properties, its ramifications on clinical medicine and therapeutic uses are not broadly appreciated.
Scope
Here we discuss basic principles and gaps in understanding of FcRn, including its management of IgG antibodies and along with albumin, its impact on use and design of antibody-based therapeutics, and its genetics.
Keywords: Neonatal Fc receptor, FcRn, Fcgrt, β2 microglobulin, IgG, albumin, pharmokinetics, genetics, therapeutic antibodies
Novel properties of IgG antibodies
The humoral arm of adaptive immunity is comprised of IgA, IgD, IgE, IgM, and IgG antibodies. In all cases, the variable regions of the antibody (the Fab fragments), confer antigen specificity, while the Fc region of the heavy chain couples the Fabs to varying effector mechanisms, including complement fixation and cell-mediated mechanisms via Fc receptors. Among these antibody isotypes, IgG is regarded to play the most important role in protective long term humoral immunity against a wide range of pathogens and toxins.
Two properties of IgG maximize their effectiveness by increasing their bioavailability. The first is that they have a remarkably long half-life in circulation (~20d in humans) in comparison with a 1-2d for other antibodies classes [1]. This property, rather than more abundant plasma cells is why IgG is the most abundant antibody isotype in blood. The second is IgG’s biodistribution. Under non-inflammatory conditions, only IgG can efficiently traffic from its major repository in the circulation across endothelial cell barriers to permeate tissues, including the fetus. IgG can additionally pass through mucosal epithelial cells [2, 3]. These properties ensure that IgG antibodies equipped with potent effector domains are conserved and distributed for resolution of infections and are also made available to newborn animals for protective immunity. On the downside, these same properties can lead to deleterious effects by promoting the accumulation and distribution of pathological IgG antibodies that can cause a spectrum of humoral autoimmune disorders. Finally, both the extended persistence in circulation and the extravascular bioavailability of IgG afford substantial opportunities for treating a diversity of diseases utilizing therapeutic monoclonal antibodies (mAbs) and proteins fused to the Fc region of IgG.
The FcRn management system
Both the conservation and bioavailability of IgG at all stages of mammalian life are attributed to FcRn (reviewed in [4, 5]. The heavy chain of FcRn, also referred to as Fcgrt, is evolutionally distinct from all other Fc receptors [6] and is a novel member of the MHC class I protein family [7]. Like its prototypic family members, Fcgrt is a Type I membrane protein molecule comprised of 3 extracellular immunoglobulin superfamily domains (α1-3) and forms an obligate heterodimer with the β2 microglobulin (β2m) light chain [8-10] (Fig. 1a). Structurally, the FcRn heterodimer varies only subtly from conventional class I proteins by occlusion of the opposing α-helices that normally presents peptides to T cells and natural killer cells, and, most significantly, by the presence of localized glutamic and aspartic acid residues in the α2-3 domain junction [11] (Fig. 1b). These residues create an anionic pocket that allows easily protonated histidine residues unique to the hinge region of the CH2-CH3 IgG-Fc to engage in binding. However, binding only occurs in an acidic environment (pH ~6-6.5) with nanomolar affinity, and demonstrates a precipitous affinity drop at neutral pH [10-12]. These remarkable molecular adaptations are a prototypic illustration of how seemingly insignificant changes can give rise to novel, unpredicted protein functions that accommodate new physiological niches – control over the humoral arm of immunity in this case.
Figure 1. Structure features of FcRn and its binding partners and transcytotic pathways.

(A) FcRn is a heterodimer consisting of the FcRn heavy chain (green) and b2m (blue). FcRn binds at an acid pH to the CH2–CH3 hinge region of the Fc fragment of IgG antibodies. The structure is of rat FcRn complexed with the Fc fragment of rat IgG (PDB ID: 1I1A). (B) Close up view of the key human IgG1 and human FcRn amino acid residues that confer binding at an acid pH (PDB ID: 1I1A. (C) Structure of human FcRn (PDB ID: 3M17) indicating the histidine residue that is considered to engage in pH-dependent binding to human serum albumin. Depictions rendered in PyMol (DeLano, W.L. The PyMOL Molecular Graphics System (2002) DeLano Scientific). (D) Intracellular pathways by which FcRn rescues and transports IgG and albumin. In polarized cells, FcRn can recycle its cargo apically or transport it to the basolateral side, or from the basolateral to apical side (reverse transcytosis). In non-polarized cells, the directionality of transport is irrelevant; in such cells FcRn rescues its ligands from a catabolic fate.
The functions of FcRn are facilitated by its intracellular transport behavior. The c-terminus of Fcgrt carries an endosomal targeting motif that directs FcRn to its steady state locatoin in the early acidic endosomes [13-15]. In this compartment, FcRn is thought to gain access to extracellular IgG mainly as the result of non-specific uptake of soluble extracellular material by processes, including fluid phase endocytosis. Fusion of the endocytic vesicles with the early endosomes exposes the cargo to the acidic environment, which permits the binding of FcRn to IgG [16, 17] (Fig. 1D). This complex is then sequestered as microvesicles that exclude other proteins and is transported back to the cell membrane, whereupon fusion with the plasma membrane leads to exposure to a neutral pH and the release IgG to the extracellular environment [18-20]. In contrast, proteins that are not rescued by FcRn (or other possible protein-specific rescue receptors) are destined for catabolism through the lysosomal pathway.
Albumin, the most abundant of all proteins in circulation is FcRn’s second ligand. While not as well-characterized as is IgG/FcRn, a preponderance of evidence supports a model in which albumin undergoes pH-dependent binding to FcRn and is trafficked intracellularly in manner that is similar to IgG [21-25]. Mice deficient in FcRn demonstrate a ~40% reduction in serum albumin concentrations and an abbreviated half-life in blood [21]. In humans, FcRn-mediated management results in a plasma half-life of albumin that is equivalent to that of IgG [26]. Notably, IgG and albumin bind FcRn in a non-competitive manner [27]. Mutational analysis of human FcRn has identified a key conserved histidine residue at p166 that is necessary for binding to yet to be defined residues of albumin at an acid but not neutral pH [28]. The ability of FcRn to non-competitively protect and traffic IgG and albumin explains the extended persistence of both proteins in circulation, and further illustrates the adaptability of MHC class I protein family members.
Understanding the tissues and cell types that engage in IgG and albumin management is an issue of substantial importance. In polarized barrier cells, such as the vascular endothelium, FcRn that has acquired its cargo from the bloodstream can recycle its cargo apically, back to the bloodstream (Fig. 1D). It can also transport its cargo from the apical to basolateral surfaces; this transcytotic route is considered to be essential for the biodistribution of IgG from blood to extravascular sites. Furthermore, transcytosis can operate bidirectionally, as has been best documented in epithelial cell lines [2, 29, 30]. This bidirectional transport system is thought operate in the mucosa to deliver IgG to luminal sites where it can bind and potentially incapacitate pathogenic microorganisms and then transport luminal antigen/antibody complexes to the lamina propria to reinforce the immune response [31-33]. Finally, IgG, and potentially albumin management by FcRn does not appear to be restricted to polarized cells, because bone marrow reconstitution experiments argue persuasively that monocyte lineage cells robustly express FcRn and extend the serum persistence of IgG and albumin ([34, 35] and our unpublished data).
Are there tissues in which FcRn manages IgG and albumin differently? Most models note similarities in IgG and albumin homeostasis based the vascular endothelial cell paradigm. However, while maternal IgG is transferred efficiently to the fetus, albumin is excluded in both conventional mice and mice that transgenically express human FcRn (Al Kabbaz et al, manuscript in preparation). Differences in IgG and albumin management are also evidenced in the kidney, an organ that must filter an enormous volume of blood each day. In recent kidney transplantation studies, it was shown that the absence of renal FcRn rendered mice hypoalbulinemic through the loss of albumin in urine. However, the loss was limited to albumin since mice maintained comparatively normal serum concentrations of IgG [36]. At a minimum, these intriguing findings indicate that albumin and IgG are handled differently in the kidney and underscore the importance of gaining a greater understanding of the physiology of FcRn in that organ. Overall, as both of FcRn’s ligands are increasingly being exploited for improving the efficacy of biotherapeutic proteins (reviewed in [25, 37, 38], there is a need to better understand the similarities and differences in pharmacokinetic behavior of these two ligands of FcRn.
Saturability of the FcRn protection pathway and implications for biological therapies
A principle of considerable clinical relevance is the fact that the FcRn salvage pathway is saturable, both in regards to IgG and albumin. This well-documented phenomenon, referred to as the concentration-catabolism effect [16, 39], is caused by the fact that the pool of FcRn available in cells to recycle or transport its ligands can be limiting. Thus, when FcRn is fully occupied, the unbound ligand is cleared, primarily through lysosomal degradation. As modeled with the IgG ligand, at low to physiological serum IgG concentrations, there is a sufficient amount of FcRn to rescue IgG very efficiently. However, superphysiological IgG concentrations in circulation can overwhelm FcRn’s recycling function. In such cases, the FcRn protection pathway can be sufficiently weakened as to negate its effect. This property can be exploited with benefit in treatment of autoimmune diseases with humoral involvement. Thus high dose intravenous IgG (IVIg) therapy is thought to act at least partially through FcRn saturation, flushing the body of intact, endogenous IgG, including that which is pathogenic [40]. However, the benefits of high dose IVIg acting through FcRn saturation are predicted to diminish in patients experiencing hypergammaglobulinemia because FcRn is already fully occupied.
This same principle may be even more relevant to the use and efficacy of therapeutic antibodies and related Fc-based biologics. Efficacy, both in terms of persistence in circulation and bioavailability at the target site is predicted to be greatest in patients with low to normal level of serum IgG (~12 mg/ml) (Fig. 2) while the therapeutic benefit will likely to diminish in relationship to their higher serum IgG concentrations. Patients with >20 mg/ml of IgG may therefore be less responsive to treatment. Routine consideration of this factor in the decision for use and dosing of antibody-based therapeutics may have value.
Figure 2. Relationship of serum IgG concentrations to clearance in humans.
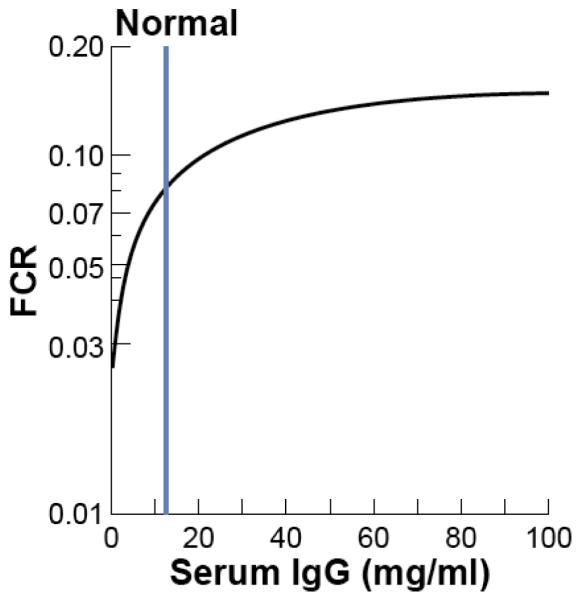
Representation of data from ref. [1]. FCR, fractional catabolic rate; the fraction of the serum IgG pool that disappears per day. The asymptote approaches that expected for complete loss of FcRn-mediated protection IgG and is expected to be similar to non-protected IgG isotypes.
Therapeutic monoclonal antibodies and ways to improve them
Exploitation of the FcRn interaction is proving to be a generalized way to extend pharmacokinetics. Originally, fully mouse mAbs (e.g., OKT3) tested in the clinic were found to have precipitous elimination kinetics making them only useful for short term therapy. It is now recognized that a primary the reason for this rapid clearance of mouse mAbs in humans was the failure of the Fc fragment of mouse IgG mAbs to interact efficiently with and be protected by human FcRn [41]. In the many cases where prolongation of the pharmacokinetics of therapeutic mAbs is desired, it has thus become standard to use the mouse Fc with human Fc, most commonly of the IgG1 subclass. Similarly, the incorporation of the human Fc into therapeutic proteins is becoming routine (e.g., the TNF receptor fusion protein, Enbrel®) as the pharmacokinetics of such fusion proteins benefit from the human FcRn interaction.
Further improving therapeutic mAb and Fc-fusion protein pharmacokinetics by augmenting the Fc/FcRn interaction is gaining considerable traction. Owing both to crystallization and mutagenesis studies, the topology of the Fc/FcRn binding site and the critical amino acid residues involved is increasingly well understood [6, 9, 11, 42-45] (Fig. 1B). This concept was pioneered by Sally Ward and her colleagues by their demonstration that certain amino acid substitutions in the CH2-CH3 Fc region of IgG increased mAb binding affinity at acidic pH and extended its serum persistence in standard mice [46]. Similarly, amino acid changes in the CH2-CH3 hinge of human IgG1 Fc improve the pH-dependent binding to human FcRn [43, 45, 47-51]. This gain in affinity may be explained by an increase in H-bonding and surface contact between Fc and FcRn [52]. However, such substitutions failed to increase their serum persistence in standard mice, raising questions regarding their utility for modeling of therapeutic mAbs [51, 53]. This point of confusion is resolved by the knowledge that mouse FcRn promiscuously binds IgGs from many species, including human, with such high affinity as to mask their pharmacokinetic behavior in humans [41, 53]. Analysis of mAbs modified in this manner showed much better correlations between IgG/FcRn affinity and serum half-lives in primates [47, 49, 50] and in mice expressing the human FcRn heavy chain [51, 54, 55]. This greatly strengthened the prospect that second “generation” mAbs that are engineered to enhance the FcRn interaction will improve their effectiveness beyond that observed with current mAb therapeutics. This key question was tackled recently by Zalevsky and colleagues at Xencor using substitutions in the CH2 CH3 Fc region (M428L/N434S) that were known to increase pH-dependent binding to human FcRn [55]. Substantially extended pharmokinetics and anti-tumor activity in immunodeficient human FCGRT-transgenic mice was realized when these amino acid substitutions were incorporated into the anti-VEGF mAb Cetuximab and the anti-EGFR mAb Bevacizumab. Therapeutic mAbs and fusion proteins engineered to enhance the FcRn interaction have multiple applications not only in treating cancer and autoimmune diseases but also for passive immunization, and are likely to assume widespread use in the future.
Is there functionally-relevant genetic variation in FcRn?
Monogenic Disorders
Given the key role for FcRn in IgG and albumin homeostasis and transcytosis, it is reasonable to consider the possibility that there are naturally occurring allelic variants of FcRn that have functional consequence. Identification of individuals with compromised FcRn function by routine clinical measures – low serum IgG and albumin and hyperlipidemia (as the result of hypoalbulinemia) – are confounded by the overlap of these abnormal parameters with numerous other disorders, especially those that compromise renal function. Evidence for a true monogenic disorder in FcRn, Familial Idiopathic Hypercatabolic Proteinemia, emanated from the remarkable early studies by Waldman and Strober in their evaluations of immunoglobulin metabolism [56]. They identified 2 siblings from a consanguineous marriage that had abnormally low serum levels of IgG and albumin while maintaining normal concentrations of other antibody isotypes. These siblings showed no evidence for a defect in IgG synthesis or leakage through the renal or gastrointestinal routes. Most telling was a substantial increase in the fractional catabolic rate (FCR) of both proteins based on infusion of radiolabeled IgG and albumin. Trangulation of this disorder to FcRn dysfunction awaited the advent of molecular tools many years later that allowed Anderson and colleagues to identify a loss-of-function allele of β2 microglobulin (B2M) as the probable cause of [56]. Given the fact that B2M encodes is the obligate light chain of numerous MHC class I family proteins, these individuals would likely have numerous immunological and non-immunological abnormalities. A Mendelian genetic disorder caused by a mutation in FCGRT, thus affecting only FcRn’s functions, is yet to be described in humans.
Secondary effects by other monogenic disorders?
Another intriguing, but unexplained observation again originally made by Waldman and Strober concerns patients with the dominantly-acting neuromuscular wasting disorder, Myotonic Dystrophy (MD) [1, 57]. The majority of patients with this disorder demonstrate a reduction in serum IgG concentrations while maintaining normal concentration of other antibody subclasses and albumin [57, 58]. Attribution of this specific IgG reduction to increased catabolism was made by the finding of a substantially increased fractional catabolic rate (FCR) for IgG in these patients while maintaining normal FCRs for other antibody isotypes and albumin [57, 59]. The failure to observe this defect in patients with other degenerative neuromuscular disorders lent credence to this increased catabolism being specific for MD. While no subsequent studies have evaluated FCRs, many other studies have confirmed the selective lowering of serum IgG in such patients [59, 60]. Trinucleotide CTG expansions in the 3′ untranslated region of dystrophia myotonica-protein kinase gene (DMPK) on chromosome 19 are responsible for the more severe form (MD1) and accounts for the great majority of the cases. Such explanations can be massive (up to 4,000) and correlate with disease severity and age of onset [61]. Of note is that FCGRT maps ~ 4 Mb distal to DMPK. This fact and evidence that such repeats in DMPK can reduce the activity of the closely-linked homeobox gene, SIX1 [62] and with possible effects on more distal 3′ genes [63] lead to the intriguing notion that DMPK RNA containing these repeats acts in cis to compromise the expression of FCGRT [64]. However, the predominance of current evidence supports trans-acting effects in which the transcribed repeats act dominantly by binding key nucleoproteins, CUG-binding protein 1 and muscleblind-like 1, which then affect alterations of mRNA splicing, translation, and stability of multiple gene products, and by doing so contribute to varied manifestations of MD syndromes [61, 65]. Even under this scenario, FCGRT (or B2M) would have to be one of the target genes affected if this gene was directly involved in the hyper-IgG catabolic trait. While one study of Japanese MD1 patients reported a strong correlation between the number of DMPK CTG expansions and serum concentrations of IgG, analysis of substantially larger Swedish cohorts failed to confirm this finding and furthermore failed to find obvious correlations between numbers of DMPK trinucleotide repeats, serum IgG concentrations, and FCGRT transcription of muscle and lymphocytes as detected by quantitative PCR techniques [60]. Moreover, as noted by Pan-Hammarstrom et al, patients with the alternative form of MD (MD2), caused by tetranucleotide repeat expansions zinc finger 9 (ZNF9) (unlinked to FCGRT), also show a selective reduction in serum IgG [60]. The selectivity for IgG while not affecting FcRn’s other ligand, albumin, would not be predicted by a global influence on FCGRT expression. However, as mentioned in a preceding section, selectivity for IgG could be explained by the tissue affected: if the kidney is most critical for albumin but not for IgG homeostasis and tissues more generally abnormal in MD (skeletal muscle and its microvasculature) are more critical for IgG homeostasis, one would expect to observe changes in IgG but not albumin homeostasis. Thus, given the facts that the fractional catabolic rates of IgM and IgA are not altered in MD, it is still plausible that the IgG/FcRn recycling pathway is compromised by this disease in affected tissues. Trans-acting effects caused by either DMPK or ZNF9 trinucleotide repeat expansions and acting through CUGBP1 and MBNL1 could interfere with FCGRT mRNA splicing and/or translation. Alternatively, these expansions could negatively impact the IgG/FcRn recycling pathway more generally by interfering with elements of endosomal trafficking that are needed for FcRn to perform its functions. A test of these possibilities could be through analysis of mice that carry a CTG expanded, expressed human DMPK transgenes that recapitulate MD1 pathophysiology (reviewed in [61]).
Allelic variation
Even with the very rare case of Familial Idiopathic Hypercatabolic Proteinemia, there is minimal support for functionally relevant allelic variants of FcRn that segregate in the human population. Individuals with hypomorphic alleles that broadly compromise FcRn’s function (either by regulatory or protein-coding changes) would be expected to show moderate hypergammaglobulinemia and hypoalbuminemia. It is less certain whether such individuals would be immune compromised to any substantial extent. Unfortunately, these symptoms overlap with gastrointestinal and renal disorders that leak IgG and albumin, and therefore, would only be distinguished by the absence of other clinical abnormalities. The efficiency of maternofetal transfer of IgG, as measured by the ratios of mother’s IgG to newborn’s IgG, is another possibility that would not be as confounded by such disorders.
Individuals with hypermorphic alleles of FcRn would be expected to support higher concentrations of IgG (and albumin) in circulation by raising FcRn’s saturation threshold. This would potentially increase the severity of autoimmune diseases in which pathogenic IgG autoantibodies play an important role. However, while limited, our studies in mice, including those genetically prone to develop autoimmune disease, failed to identify functionally conspicuous allelic variants in FcRn that are indicated by abnormally shortened or increased serum half-lives of IgG [66]. There is evidence that FcRn-mediated transport in mammary glands of cattle contributes to the supply of IgG in colostrum [67]. While a limited study, single sequence polymorphisms (SNP) in bovine FCGRT were associated with concentrations of colostral IgG [68]. Studies in humans identified variable number of tandem repeat (VNTR) polymorphisms within the human FCGRT promotor that alter the transcriptional activity of this gene in monocytes [69]. Another study investigated whether such VNTR were associated with risk for glomerular nephritis in Chinese and found no such correlation [70]. Finally, a more recent study that evaluated whether these VNTRs influence maternofetal transfer of IgG and found no such effect [71]. Genome wide association studies provide an unbiased strategy to evaluate whether allelic variations contribute meaningfully to the disease studied. To the best of our knowledge, no associations with human FCGRT have been revealed with this approach to date and it seems unlikely that polymorphisms of substantial disease relevance will emerge. Overall, the possibility of functionally relevant allelic variants in the FcRn heavy chain remain to be firmly established.
Acknowledgements
This work was supported by NIH R01 DK56597 (DCR) and T31 DK07449 (VZS).
References Cited
- 1.Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1–110. doi: 10.1159/000385919. [DOI] [PubMed] [Google Scholar]
- 2.Dickinson BL, Badizadegan K, Wu Z, Ahouse JC, Zhu X, Simister NE, Blumberg RS, Lencer WI. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J Clin Invest. 1999;104:903–11. doi: 10.1172/JCI6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masuda A, Yoshida M, Shiomi H, Morita Y, Kutsumi H, Inokuchi H, Mizuno S, Nakamura A, Takai T, Blumberg RS, Azuma T. Role of Fc Receptors as a therapeutic target. Inflamm Allergy Drug Targets. 2009;8:80–6. doi: 10.2174/187152809787582525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 5.Ward ES, Ober RJ. Chapter 4: Multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv Immunol. 2009;103:77–115. doi: 10.1016/S0065-2776(09)03004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raghavan M, Bjorkman PJ. Fc receptors and their interactions with immunoglobulins. Annu Rev Cell Dev Biol. 1996;12:181–220. doi: 10.1146/annurev.cellbio.12.1.181. [DOI] [PubMed] [Google Scholar]
- 7.Simister NE, Ahouse JC. The structure and evolution of FcRn. Res Immunol. 1996;147:333–7. doi: 10.1016/0923-2494(96)89647-7. discussion 53. [DOI] [PubMed] [Google Scholar]
- 8.Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature. 1994;372:336–43. doi: 10.1038/372336a0. [DOI] [PubMed] [Google Scholar]
- 9.Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. 1994;372:379–83. doi: 10.1038/372379a0. [DOI] [PubMed] [Google Scholar]
- 10.West AP, Jr., Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,) Biochemistry. 2000;39:9698–708. doi: 10.1021/bi000749m. [DOI] [PubMed] [Google Scholar]
- 11.Martin WL, West AP, Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7:867–77. doi: 10.1016/s1097-2765(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 12.Vaughn DE, Bjorkman PJ. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure. 1998;6:63–73. doi: 10.1016/s0969-2126(98)00008-2. [DOI] [PubMed] [Google Scholar]
- 13.McCarthy KM, Yoong Y, Simister NE. Bidirectional transcytosis of IgG by the rat neonatal Fc receptor expressed in a rat kidney cell line: a system to study protein transport across epithelia. J Cell Sci. 2000;113(Pt 7):1277–85. doi: 10.1242/jcs.113.7.1277. [DOI] [PubMed] [Google Scholar]
- 14.Wu Z, Simister NE. Tryptophan- and dileucine-based endocytosis signals in the neonatal Fc receptor. J Biol Chem. 2001;276:5240–7. doi: 10.1074/jbc.M006684200. [DOI] [PubMed] [Google Scholar]
- 15.Newton EE, Wu Z, Simister NE. Characterization of basolateral-targeting signals in the neonatal Fc receptor. J Cell Sci. 2005;118:2461–9. doi: 10.1242/jcs.02367. [DOI] [PubMed] [Google Scholar]
- 16.Brambell FW, Hemmings WA, Morris IG. A Theoretical Model of Gamma-Globulin Catabolism. Nature. 1964;203:1352–4. doi: 10.1038/2031352a0. [DOI] [PubMed] [Google Scholar]
- 17.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93:5512–6. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ober RJ, Martinez C, Lai X, Zhou J, Ward ES. Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci U S A. 2004;101:11076–81. doi: 10.1073/pnas.0402970101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward ES, Martinez C, Vaccaro C, Zhou J, Tang Q, Ober RJ. From sorting endosomes to exocytosis: association of Rab4 and Rab11 GTPases with the Fc receptor, FcRn, during recycling. Mol Biol Cell. 2005;16:2028–38. doi: 10.1091/mbc.E04-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan Z, Ram S, Vaccaro C, Ober RJ, Ward ES. Analyses of the Recycling Receptor, FcRn, in Live Cells Reveal Novel Pathways for Lysosomal Delivery. Traffic. 2009;10:600–14. doi: 10.1111/j.1600-0854.2009.00887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, Anderson CL. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197:315–22. doi: 10.1084/jem.20021829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson CL, Chaudhury C, Kim J, Bronson CL, Wani MA, Mohanty S. Perspective--FcRn transports albumin: relevance to immunology and medicine. Trends Immunol. 2006;27:343–8. doi: 10.1016/j.it.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Bronson CL, Hayton WL, Radmacher MD, Roopenian DC, Robinson JM, Anderson CL. Albumin turnover: FcRn-mediated recycling saves as much albumin from degradation as the liver produces. Am J Physiol Gastrointest Liver Physiol. 2006;290:G352–60. doi: 10.1152/ajpgi.00286.2005. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn-mediated recycling of IgG and albumin in human: Pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin Immunol. 2006 doi: 10.1016/j.clim.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersen JT, Sandlie I. The versatile MHC class I-related FcRn protects IgG and albumin from degradation: implications for development of new diagnostics and therapeutics. Drug Metab Pharmacokinet. 2009;24:318–32. doi: 10.2133/dmpk.24.318. [DOI] [PubMed] [Google Scholar]
- 26.Peters T., Jr. Serum albumin. Adv Protein Chem. 1985;37:161–245. doi: 10.1016/s0065-3233(08)60065-0. [DOI] [PubMed] [Google Scholar]
- 27.Chaudhury C, Brooks CL, Carter DC, Robinson JM, Anderson CL. Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry. 2006;45:4983–90. doi: 10.1021/bi052628y. [DOI] [PubMed] [Google Scholar]
- 28.Andersen JT, Dee Qian J, Sandlie I. The conserved histidine 166 residue of the human neonatal Fc receptor heavy chain is critical for the pH-dependent binding to albumin. Eur J Immunol. 2006;36:3044–51. doi: 10.1002/eji.200636556. [DOI] [PubMed] [Google Scholar]
- 29.Ramalingam TS, Detmer SA, Martin WL, Bjorkman PJ. IgG transcytosis and recycling by FcRn expressed in MDCK cells reveals ligand-induced redistribution. Embo J. 2002;21:590–601. doi: 10.1093/emboj/21.4.590. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Tzaban S, Massol RH, Yen E, Hamman W, Frank SR, Lapierre LA, Hansen SH, Goldenring JR, Blumberg RS, Lencer WI. The recycling and transcytotic pathways for IgG transport by FcRn are distinct and display an inherent polarity. J Cell Biol. 2009;185:673–84. doi: 10.1083/jcb.200809122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, Lencer WI, Blumberg RS. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity. 2004;20:769–83. doi: 10.1016/j.immuni.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida M, Masuda A, Kuo TT, Kobayashi K, Claypool SM, Takagawa T, Kutsumi H, Azuma T, Lencer WI, Blumberg RS. IgG transport across mucosal barriers by neonatal Fc receptor for IgG and mucosal immunity. Springer Semin Immunopathol. 2006;28:397–403. doi: 10.1007/s00281-006-0054-z. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi K, Qiao SW, Yoshida M, Baker K, Lencer WI, Blumberg RS. An FcRn-dependent role for anti-flagellin immunoglobulin G in pathogenesis of colitis in mice. Gastroenterology. 2009;137:1746–56 e1. doi: 10.1053/j.gastro.2009.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, Wang Y, Robert C, Wu B, Smith PD, Lencer WI, Blumberg RS. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol. 2001;166:3266–76. doi: 10.4049/jimmunol.166.5.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179:4580–8. doi: 10.4049/jimmunol.179.7.4580. [DOI] [PubMed] [Google Scholar]
- 36.Sarav M, Wang Y, Hack BK, Chang A, Jensen M, Bao L, Quigg RJ. Renal FcRn reclaims albumin but facilitates elimination of IgG. J Am Soc Nephrol. 2009;20:1941–52. doi: 10.1681/ASN.2008090976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Presta LG. Molecular engineering and design of therapeutic antibodies. Curr Opin Immunol. 2008;20:460–70. doi: 10.1016/j.coi.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 38.Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol. 2009;20:685–91. doi: 10.1016/j.copbio.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 39.Humphrey JH, Fahey JL. The Metabolism of Normal Plasma Proteins and Gamma-Myeloma Protein in Mice Bearing Plasma-Cell Tumors. J Clin Invest. 1961;40:1696–705. doi: 10.1172/JCI104392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin F, Balthasar JP. Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Hum Immunol. 2005;66:403–10. doi: 10.1016/j.humimm.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 41.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13:1551–9. doi: 10.1093/intimm/13.12.1551. [DOI] [PubMed] [Google Scholar]
- 42.Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu Rev Immunol. 2000;18:739–66. doi: 10.1146/annurev.immunol.18.1.739. [DOI] [PubMed] [Google Scholar]
- 43.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, Fox JA, Presta LG. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 44.Zhou J, Johnson JE, Ghetie V, Ober RJ, Ward ES. Generation of mutated variants of the human form of the MHC class I-related receptor, FcRn, with increased affinity for mouse immunoglobulin G. J Mol Biol. 2003;332:901–13. doi: 10.1016/s0022-2836(03)00952-5. [DOI] [PubMed] [Google Scholar]
- 45.Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23:1283–8. doi: 10.1038/nbt1143. [DOI] [PubMed] [Google Scholar]
- 46.Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. 1997;15:637–40. doi: 10.1038/nbt0797-637. [DOI] [PubMed] [Google Scholar]
- 47.Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, Keller S, Tang MT, Tso JY, Vasquez M, Tsurushita N. Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem. 2004;279:6213–6. doi: 10.1074/jbc.C300470200. [DOI] [PubMed] [Google Scholar]
- 48.Kamei DT, Lao BJ, Ricci MS, Deshpande R, Xu H, Tidor B, Lauffenburger DA. Quantitative methods for developing Fc mutants with extended half-lives. Biotechnol Bioeng. 2005;92:748–60. doi: 10.1002/bit.20624. [DOI] [PubMed] [Google Scholar]
- 49.Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn) J Biol Chem. 2006;281:23514–24. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 50.Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. An engineered human IgG1 antibody with longer serum half-life. J Immunol. 2006;176:346–56. doi: 10.4049/jimmunol.176.1.346. [DOI] [PubMed] [Google Scholar]
- 51.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–69. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- 52.Oganesyan V, Damschroder MM, Woods RM, Cook KE, Wu H, Dall’acqua WF. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol Immunol. 2009;46:1750–5. doi: 10.1016/j.molimm.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 53.Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103:18709–14. doi: 10.1073/pnas.0606304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roopenian DC, Christianson GJ, Sproule TJ. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol Biol. 602:93–104. doi: 10.1007/978-1-60761-058-8_6. [DOI] [PubMed] [Google Scholar]
- 55.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wani MA, Haynes LD, Kim J, Bronson CL, Chaudhury C, Mohanty S, Waldmann TA, Robinson JM, Anderson CL. Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant beta2-microglobulin gene. Proc Natl Acad Sci U S A. 2006;103:5084–9. doi: 10.1073/pnas.0600548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wochner RD, Drews G, Strober W, Waldmann TA. Accelerated breakdown of immunoglobulin G (IgG) in myotonic dystrophy: a hereditary error of immunoglobulin catabolism. J Clin Invest. 1966;45:321–9. doi: 10.1172/JCI105346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zinneman HH, Rotstein J. A study of gamma globulins in dystrophia myotonica. J Lab Clin Med. 1956;47:907–16. [PubMed] [Google Scholar]
- 59.Roberts DF, Bradley WG. Immunoglobulin levels in dystrophia myotonica. J Med Genet. 1977;14:16–9. doi: 10.1136/jmg.14.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan-Hammarstrom Q, Wen S, Ghanaat-Pour H, Solders G, Forsberg H, Hammarstrom L. Lack of correlation between the reduction of serum immunoglobulin concentration and the CTG repeat expansion in patients with type 1 dystrophia [correction of Dystrofia] myotonica. J Neuroimmunol. 2003;144:100–4. doi: 10.1016/s0165-5728(03)00271-6. [DOI] [PubMed] [Google Scholar]
- 61.Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans. 2009;37:1281–6. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klesert TR, Cho DH, Clark JI, Maylie J, Adelman J, Snider L, Yuen EC, Soriano P, Tapscott SJ. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat Genet. 2000;25:105–9. doi: 10.1038/75490. [DOI] [PubMed] [Google Scholar]
- 63.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–7. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 64.Junghans RP, Ebralidze A, Tiwari B. Does (CUG)n repeat in DMPK mRNA ‘paint’ chromosome 19 to suppress distant genes to create the diverse phenotype of myotonic dystrophy?: A new hypothesis of long-range cis autosomal inactivation. Neurogenetics. 2001;3:59–67. doi: 10.1007/s100480000103. [DOI] [PubMed] [Google Scholar]
- 65.Junghans RP. Dystrophia myotonia: why focus on foci? Eur J Hum Genet. 2009;17:543–53. doi: 10.1038/ejhg.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Christianson GJ, Brooks W, Vekasi S, Manolfi EA, Niles J, Roopenian SL, Roths JB, Rothlein R, Roopenian DC. Beta 2-microglobulin-deficient mice are protected from hypergammaglobulinemia and have defective antibody responses because of increased IgG catabolism. J Immunol. 1997;159:4781–92. [PubMed] [Google Scholar]
- 67.Mayer B, Doleschall M, Bender B, Bartyik J, Bosze Z, Frenyo LV, Kacskovics I. Expression of the neonatal Fc receptor (FcRn) in the bovine mammary gland. J Dairy Res. 2005;72:107–12. doi: 10.1017/s0022029905001135. Spec No. [DOI] [PubMed] [Google Scholar]
- 68.Zhang R, Zhao Z, Zhao Y, Kacskovics I, Eijk M, Groot N, Li N, Hammarstrom L. Association of FcRn Heavy Chain Encoding Gene (FCGRT) Polymorphisms with IgG Content in Bovine Colostrum. Anim Biotechnol. 2009;20:242–6. doi: 10.1080/10495390903196448. [DOI] [PubMed] [Google Scholar]
- 69.Sachs UJ, Socher I, Braeunlich CG, Kroll H, Bein G, Santoso S. A variable number of tandem repeats polymorphism influences the transcriptional activity of the neonatal Fc receptor alpha-chain promoter. Immunology. 2006;119:83–9. doi: 10.1111/j.1365-2567.2006.02408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou XJ, Yu L, Zhu L, Hou P, Lv JC, Yu F, Zhang H. Association between polymorphisms in the FCGRT gene and lupus nephritis in Chinese patients. Clin Exp Rheumatol. 2009;27:609–14. [PubMed] [Google Scholar]
- 71.Freiberger T, Ravcukova B, Grodecka L, Kurecova B, Jarkovsky J, Bartonkova D, Thon V, Litzman J. No association of FCRN promoter VNTR polymorphism with the rate of maternal-fetal IgG transfer. J Reprod Immunol. 85:193–7. doi: 10.1016/j.jri.2010.04.002. [DOI] [PubMed] [Google Scholar]