

Abstract
Epithelial neutrophil-activating peptide-78 (CXCL5), a member of the CXC chemokine family, has been shown to be involved in angiogenesis, tumor growth, and metastasis. The objective of this study was to determine the relationship between CXCL5 expression and tumor progression in human pancreatic cancer and to elucidate the mechanism underlying CXCL5-mediated tumor angiogenesis and cancer growth. We report herein that CXCL5 is overexpressed in human pancreatic cancer compared with paired normal pancreas tissue. Overexpression of CXCL5 is significantly correlated with poorer tumor differentiation, advanced clinical stage, and shorter patient survival. Patients with pancreatic cancer and CXCL5 overexpression who underwent resection of cancer had a mean survival time 25.5 months shorter than that of patients who did not overexpress CXCL5. Blockade of CXCL5 or its receptor CXCR2 by small-interfering RNA knockdown or antibody neutralization attenuated human pancreatic cancer growth in a nude mouse model. Finally, we demonstrated that CXCL5 mediates pancreatic cancer–derived angiogenesis through activation of several signaling pathways, including protein kinase B (Akt), extracellular signal–regulated kinase (ERK), and signal transducer and activator of transcription (STAT) in human endothelial cells. These data suggest that CXCL5 is an important mediator of tumor-derived angiogenesis and that it may serve as a survival factor for pancreatic cancer. Blockade of either CXCL5 or CXCR2 may be a critical adjunct antiangiogenic therapy against pancreatic cancer.
Pancreatic cancer is a highly lethal disease. By the time patients present to a physician with pancreatic cancer, 85% to 90% have locally advanced or metastatic disease. An estimated 42,470 Americans were diagnosed as having this disease in 2009, accounting for 3% of all new cancer diagnoses in that year.1 An estimated 35,240 Americans with pancreatic cancer died during the same year, making this type of cancer the fourth leading cause of cancer death overall. Five-year survival of patients with pancreatic cancer remains at 5%. Therapeutic options for patients with pancreatic cancer are limited. Only 10% to 15% of patients are candidates for surgical resection, and for those who are not, the current standard of care is chemotherapy.
Many potential mechanisms allow for maintenance of pancreatic cancer growth, including the imbalance of angiogenic and angiostatic factors. All solid tumor growth depends on the presence of neovascularization. In the absence of local capillary proliferation and delivery of oxygen and nutrients, neoplasms cannot grow beyond a volume of 2 mm3.2 A group of potent regulators of angiogenesis, CXC chemokines have been shown to be important in wound healing and in tumor growth.3–7 Epithelial neutrophil-activating peptide-78 (CXCL5), a member of the CXC chemokine family, was identified and isolated from a human alveolar epithelial cell line.8 It shares structural homologic qualities with and plays a similar role as IL-8 (CXCL8),8 another member of the CXC chemokine family, in inflammation and angiogenesis. CXCL5 is secreted by many types of cells, including neutrophils, monocytes, fibroblasts, and epithelial cells.9–12 Evidence is accumulating to suggest that CXCL5 is an important factor in cancer biology.7 CXCL5 has been found to directly induce endothelial cell proliferation and invasion in vitro,13,14 to promote tumor angiogenesis in vivo in non-small cell lung carcinoma, and to modulate tumor growth and metastasis.7 CXCL5 is overexpressed in gastric,15 prostate,16 endometrial,17 squamous cell,18 pancreatic,14 and colon cancer.19 Its expression is associated with advanced tumor stages, local invasion, and metastatic potential. Targeted down-regulation of CXCL5 by small-interfering RNA (siRNA) inhibited squamous carcinoma growth in vivo. In these studies, CXCL5 was found to directly stimulate cancer cell proliferation and invasion.16,18 Evidence suggested that CXCL5 may promote tumor growth and progression through direct stimulation of cancer cell growth and an indirect effect on angiogenesis. The direct effect of CXCL5 on cancer cell growth through the activation of mitogen-activated protein kinase (MAPK) and phosphoinositide-3 (PI3K) kinase signaling pathways has been proposed.16 We and others14,20,21 have previously reported the expression of CXCL5 in human pancreatic cancer. However, the precise role of CXCL5 in pancreatic cancer growth and the mechanism of CXCL5-mediated tumor angiogenesis in promoting tumor growth are not understood.
The purposes of this study were to determine the expression of CXCL5, the correlation between CXCL5 expression and clinical outcome in human pancreatic cancer, and the mechanisms of CXCL5 in promoting tumor growth and progression. We examined CXCL5 expression in cultured human pancreatic cancer cell lines and in human pancreatic adenocarcinoma specimens and the signaling pathways involved in CXCL5-mediated tumor angiogenesis. We found that CXCL5 is markedly expressed in human pancreatic cancer, and its expression is correlated with histopathologic grade, clinical stage, and patient survival. CXCL5 activated several signaling pathways in human endothelial cells and stimulated tumor angiogenesis. Targeted disruption of the CXCL5/CXCR2 axis impaired angiogenesis and slowed pancreatic cancer growth.
Materials and Methods
Cell Lines and Reagents
Human pancreatic cancer cell lines (AsPC-1, BxPC-3, CAPAN-2, HPAF-II, PANC-1, and MIA PaCa-2) and human umbilical vein endothelial cells (HUVECs) were obtained from American Type Culture Collection (Manassas, VA). These cell lines were maintained in culture as an adherent monolayer in RPMI 1640 medium or Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum or endothelial growth medium-2 supplemented with endothelial cell growth factors (Lonza Inc., Walkersville, MD). Recombinant human (rh) CXCL5, anti-CXCL5, anti-CXCR2 antibody, and phospho-kinase array were purchased from R&D Systems (Minneapolis, MN). Antihuman CD31 antibody was from Abcam (Cambridge, MA), and antimouse CD31 antibody was from Angio-Proteomie (Boston, MA). Antiserum to mouse CXCR2 was a gift from R.M.S.22 Phospho-specific antibody to protein kinase B (Akt) and extracellular signal–regulated kinase (ERK) and the inhibitors Wortmannin and PD98059 were obtained from Cell Signaling Technology (Danvers, MA).
Patient Specimens
A total of 153 archived (paraffin-embedded) pancreatic adenocarcinoma tissue samples (73 females and 80 males), 30 frozen pancreatic adenocarcinoma tissue samples with paired normal pancreas tissue, and 23 paraffin-embedded whole tumor tissue samples with paired normal pancreas tissue from 1989 to 2005 were obtained through the Departments of Surgery and Pathology at the University of California at Los Angeles. This study was approved by the Institutional Review Board at the University of California at Los Angeles School of Medicine.
Real-Time PCR
Total cellular RNA was isolated from pancreatic cancer cell lines or patient tumor with paired normal pancreas using TRIzol reagent (Life Technologies Inc., Gaithersburg, MD). RNA isolation was performed from representative biopsies of surgically resected tumors and included stromal and cancer compartments. The percentage of cancer cells in a single specimen was not quantitated. RNA quality was measured using the Experion automated electrophoresis system (Bio-Rad, Hercules, CA). cDNAs were synthesized using 0.5 μg of RNA with the RNase-Free DNase Set (Qiagen Inc., Valencia, CA). Real-time PCR was performed using 0.5 μL of cDNA with SYBR Green and an iQ5 real-time PCR detection system (Bio-Rad) using a two-step PCR protocol. After the initial denaturing of the cDNA (3 minutes at 95°C), samples underwent 45 cycles at 95°C for 10 seconds and at 51.4°C for 20 seconds. Universal master mix as obtained from Applied Biosystems (Austin, TX) included all reagents, including Taq-polymerase, apart from specific primers and probes. PCR efficiencies for each primer pair were calculated from a standard dilution series. Amplification of unspecific products was excluded by means of melt curve analysis and agarose electrophoresis of PCR products. Gene expression was normalized to GAPDH as a housekeeping gene and was calculated using the dilution curve target method. The primers used for amplification of CXCL5 (GenBank accession number NM_002994) and GAPDH (GenBank accession number NM_002046) were as follows: CXCL5 sense, 5′-TGGACGGTGGAAACAAGG-3′; CXCL5 antisense, 5′-CTTCCCTGGGTTCAGAGAC-3′; GAPDH sense, 5′-CGCTCTCTGCTCCTCCTGTTCG-3′; and GAPDH antisense, 5′-CGGCTGGCGACGCAAAAGAAG-3′.
Tissue Microarray and Immunohistochemical Analysis
Two groups of archival paraffin-embedded patient pancreatic cancer or normal tissues were used. The first group of samples including 153 tumors was used for tissue microarray. A second group of samples containing 23 whole tumors paired with normal pancreas tissue was used for CXCL5 and CD31 immunohistochemical (IHC) analysis. The tissue microarray was constructed with tumors using multiple representative 1-mm cores from each tumor extracted from paraffin blocks and included on two arrays. Analysis by IHC on 4-μm-thick sections of tissue microarray was performed using CXCL5 antibody; IHC staining was performed as previously described.23 Briefly, sections (4 μmol/L) from FFPE tissues were deparaffinized in xylene. Unmasking of antigens was performed by incubating the sections in 0.01 mol/L citrate buffer, pH 6.0, in a microwave. Endogenous peroxidase activity was blocked by incubating sections with 3% H2O2. After blocking with serum and incubation with primary antibody overnight at 4°C, sections were washed with PBS and were incubated with biotinylated secondary antibody for 30 minutes at room temperature. Immunoreactivity was detected using an ABC Kit (Vector Laboratories, Burlingame, CA) or an EnVision Kit (Dako, Carpinteria, CA) for anti-mouse CD31. CXCL5 staining was scored as percentage of tumor cells staining positively for CXCL5. Microvascular density (MVD) was determined by counting CD31-positive blood vessels in the three areas with the highest blood vessel density.
Enzyme-Linked Immunosorbent Assay for Human CXCL5
Levels of CXCL5 in culture supernatant serum and tumor tissue were determined using the Quantikine Kit (R&D Systems) following the manufacturer's instructions. Briefly, 50 μL of sample or recombinant CXCL5 protein at different concentrations was added to each well and was incubated for 2 hours at room temperature. Then the plates were washed and incubated with CXCL5 conjugate for 2 hours. After washing, immunoreactivity was determined by adding substrate solution, and absorbance was determined using a Microplate Spectrophotometer (Bio-Rad). A curve of the absorbance versus the concentration of CXCL5 in the standard wells was plotted.
In Vitro Proliferation Assay
Endothelial cells were seeded in 96-well flat-bottom plates in triplicate and were allowed to adhere overnight. The cultures were then washed and incubated in control medium, pancreatic cancer cell conditioned medium (PCCM), or medium containing different concentrations of recombinant human (rh) CXCL5 with or without antibody to CXCL5 or CXCR2. After 48 or 72 hours of incubation, cell proliferation was determined by means of MTT assay. Percentage of growth stimulation was calculated as follows: growth stimulation = (A/B) − 1*100, where A is the number of viable cells in treated cultures and B is the number of viable cells in untreated control cultures.
In Vitro Angiogenesis
An in vitro capillary tube formation assay was performed as described earlier.24 Briefly, 104 HUVECs were plated in the wells of a Matrigel-coated (BD Biosciences, San Jose, CA) 48-well plate with medium alone or medium containing CXCL5, control IgG, or neutralizing antibody to CXCL5 or CXCR2. After 4 to 6 hours of incubation, the plate was examined for capillary tube formation and was photographed. Total tube length was determined using image analysis software (ImageJ; NIH, Bethesda, MD). The qualitative difference in tube formation was examined in treated and untreated cultures.
Western Blot Analysis and Phospho-Kinase Array
Cell extracts from treated and untreated HUVECs were quantitated. Equal amounts of proteins were separated on SDS–polyacrylamide gel electrophoresis and were electrotransferred to 0.45-μm nitrocellulose membranes. The membranes were blocked with 3% bovine serum albumin and were probed with primary antibody followed by horseradish peroxidase–conjugated secondary antibody. For phospho-kinase array, protein lysate was incubated with array membrane, and protein signal was visualized using a chemifluorescence detection system (Thermo Scientific, Waltham, MA) according to the manufacturer's protocol. Relative intensity of the specific protein expression was determined using Quantity One software (Bio-Rad).
Human Pancreatic Cancer Growth in Nude Mice
Nude mice (4 to 6 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and were maintained under pathogen-free conditions. All procedures were performed in accordance with institutional guidelines and were approved by the University of California at Los Angeles Institutional Animal Care and Use Committee. MIA PaCa-2 cells (4 × 106) were s.c. injected into nude mice. After the tumor reached 0.6 to 0.7 cm, intratumoral injection with 1 μg of CXLC5 or control siRNA was performed daily for 4 weeks. Tumor size was measured twice a week. Separate groups of nude mice received s.c. injection of BxPC3 cells. One week later, mice were treated with antiserum to CXCR2 or control serum i.p. at 0.5 ml per mouse daily for 6 weeks, and tumor size was measured. Tumors were fixed in 10% formalin, processed for histopathologic and IHC analyses, or kept at −70°C for further analysis. Tumor volume was calculated using the following formula: tumor volume = 4/3 π A × B × C, where A is the tumor's length, B is its width, and C is its height.
The gene sequences for CXCL5 (GenBank accession number NM_002994) and control siRNA (Applied Biosystems/Ambion, Austin, TX) were as follows: CXCL5 sense, 5′-CAAAGUGUCUUGAAUUGUA-3′; CXCL5 antisense, 5′-UACAAUUCAAGACACUUUG-3′; negative control sense, 5′-UAACGACGCGACGUAA-3′; and negative control antisense, 5′-UUACGUCGUCGCGUCGUUA-3′.
Statistical Analysis
Survival estimates were computed using the Kaplan-Meier method, and comparison of mean survival between groups was analyzed using an approximate normal test.25 The significance of the other data was determined using the t-test for continuous variables and the χ2 test for dichotomous variables. A P ≤ 0.05 was deemed significant.
Results
Expression of CXCL5 in Human Pancreatic Cancer Cell Lines
We examined the mRNA and protein expression of CXCL5 in six human pancreatic cancer cell lines: AsPC-1, BxPC3, CAPAN-2, HPAF, MIA PaCa-2, and PANC-1. The mRNA expression of CXCL5 was analyzed by means of real-time PCR using total RNA extracted from cultured cancer cell lines. We observed variable expression levels of CXCL5 mRNA (Figure 1A) in these cell lines. MIA PaCa-2 showed the highest mRNA expression, with mRNA levels 1000-fold higher than the other cell lines; the PANC-1 cell line had the lowest mRNA level. Protein expression of CXCL5 was examined by means of enzyme-linked immunosorbent assay using culture supernatants harvested from the six cancer cell lines (Figure 1B). Consistent with mRNA expression, we observed the highest CXCL5 protein level in MIA PaCa-2 cells but undetectable CXCL5 protein levels in AsPC-1 and PANC-1 cells. These data indicate that in cell lines, there is a difference in the pattern of expression of CXCL5.
Figure 1.
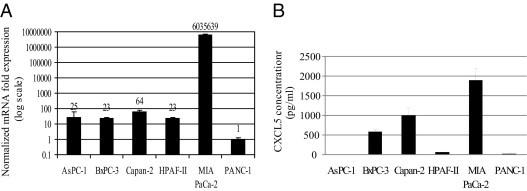
CXCL5 mRNA and protein expression in human pancreatic cancer cell lines. A: Total RNA extracted from pancreatic cancer cells was used for real-time PCR. Numbers represent the mean normalized mRNA expression index. Error bars represent SD. B: Supernatant from cultured pancreatic cancer cells was collected for enzyme-linked immunosorbent assay after 72 hours. The data represent the mean of three experiments performed in triplicate. Error bars represent SD.
Correlations between CXCL5 mRNA Expression and Clinical Variables in Human Pancreatic Cancer
We next examined the mRNA and protein expression of CXCL5 in paired human pancreatic cancer and normal pancreas specimens by means of real-time PCR and analyzed the relationships between mRNA expression of CXCL5 and histopathologic grade, clinical stage, tumor volume, and patient sex (Table 1). Twenty-three of 30 tumors (76.7%) showed increased CXCL5 mRNA expression compared with matched normal pancreas tissue. We further divided tumors into two groups based on the mRNA expression ratio of tumor to normal. Most tumors (69.2%) with low mRNA expression ratios (<4) were well to moderately differentiated, whereas 70.6% of tumors with high mRNA expression ratios were poorly differentiated. Most patients (82.4%) with high mRNA expression ratios had advanced clinical stage (T stages 3 and 4; P = 0.02). Mean tumor volume was larger in tumors with high rather than low mRNA expression ratios (13.2 versus 5.9 cm3). Data regarding adjuvant treatment for this series of patients were not available. We found that the expression of CXCL5 was significantly higher in male than in female patients (P = 0.02). These data further indicate that CXCL5 expression is associated with pancreatic cancer progression.
Table 1.
Correlations between CXCL5 mRNA Expression and Tumor Differentiation, T Stages, Tumor Volume, Node Status, and Sex in Pancreatic Cancer
| Ratio of tumor to normal | Tumor differentiation |
T stages |
Tumor volume (cm3) | Positive nodes | Sex |
Total | |||
|---|---|---|---|---|---|---|---|---|---|
| Well to moderate | Poor | 1, 2 | 3 | Female | Male | ||||
| <4 | 9 (69.2) | 4 (30.8) | 8 (61.5) | 5 (38.5) | 5.9 | 8 (61.5) | 10 (76.9) | 3 (23.1) | 13 |
| ≥4 | 5 (29.4) | 12 (70.6) | 3 (17.6) | 14 (82.4) | 13.2 | 10 (58.8) | 5 (29.4) | 12 (70.6) | 17 |
| P value | 0.06 | 0.02 | 0.12 | NS | 0.02 | ||||
Data are given as number (percentage) of tumors except for tumor volume.
NS, not significant.
Correlation between CXCL5 Protein Expression and Tumor Progression
To further determine CXCL5 protein expression and its relationship to tumor progression and MVD, we examined CXCL5 protein expression in 23 paraffin-embedded whole tumor tissues containing either cancer or pancreatic intraepithelial neoplasia (PanIN) lesions and matched normal pancreas by means of IHC analysis. CXCL5 staining is localized in apical cytoplasm in the tumor cells, and there is no CXCL5 immunoreactivity in acinar and duct epithelium in normal pancreas (Figure 2A). There was some scattered positivity in islet cells as well. The PanIN lesions are recognized as neoplastic precursors to pancreatic cancer. Pathologically, PanIN lesions are classified as dysregulated ductal epithelium progressing from PanIN-1 to PanIN-3.25,26 Both PanIN-2 and PanIN-3 are high-grade lesions representing the initial steps toward invasive carcinoma. CXCL5 immunoreactivity was occasionally observed in PanIN-1 but was increased in PanIN-2 and PanIN-3. The invasive carcinomas showed extensive staining of CXCL5 (Figure 2, B–F). Eleven of 12 cancers were positive for CXCL5, and 4 of 11 PanIN lesions showed positive CXCL5 immunoreactivity. Most cancer (66.7%) revealed strong CXCL5 positivity (Figure 2F). These data suggest that CXCL5 is constitutively overexpressed in human pancreatic cancer and that its expression correlates with tumor progression.
Figure 2.
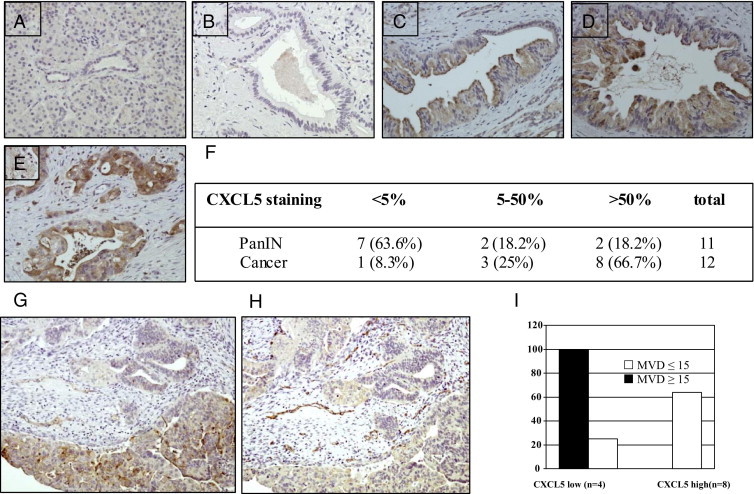
Correlations between CXCL5 expression and tumor progression and MVD. A: Normal human pancreas. B: PanIN-1. C: PanIN-2. D: PanIN-3. E: Invasive carcinoma. Original magnification, ×200. F: CXCL5 staining in PanIN lesions and cancer (P = 0.03). Tumor tissues stained with CXCL5 (G) and CD31 (H) were divided into CXCL5 low (≤50%) and CXCL5 high (>50%) groups (I). The MVD was subdivided into two groups (≤15 and >15). P = 0.03 by χ2 test.
The MVD is a surrogate for tumor angiogenesis. We evaluated the relationship between CXCL5 expression and MVD by means of IHC staining of whole tumor tissue with CXCL5 and CD31 antibody on serial sections (Figure 2, G–I). In tumors with low CXCL5 staining (≤50%), 100% of tumors showed MVD of 15 or less, whereas more than 60% of tumors with strong CXCL5 staining (>50%) showed MVD greater than 15 (P = 0.02; Figure 2I), suggesting that CXCL5 may promote pancreatic cancer growth through neoangiogenesis.
Correlation between CXCL5 Expression and Patient Survival in Human Pancreatic Cancer
To determine the association between CXCL5 expression and patient survival, we performed an IHC analysis for CXCL5 on a tissue microarray containing 153 individual pancreatic cancer specimens (Figure 3). Mean ± SD postoperative survival for all the patients with pancreatic cancer in this series was 36.9 ± 3 months. For the cohort of 153 patients, the median age was 66 years, and 46.4% were female. All the patients underwent a pancreaticoduodenectomy. Based on the seventh edition of the American Joint Committee on Cancer criteria, there were 21 stage IA, 24 stage IB, 26 stage IIA, 79 stage IIB, and 3 stage IV tumors. The median tumor size was 2.9 cm, and 45.7% of the tumors were poorly differentiated. Of patients in the series, 12.4% had a positive margin on final analysis. Of the 153 specimens tested, 130 were positive for CXCL5 according to an independent pathologist. Using a 5.5% staining score as a cutoff point, a clear survival advantage (25.5 months) was identified for patients whose tumors had low CXCL5 expression (≤5.5%) compared with high CXCL5 expression (>5.5%) (P = 0.06 by log-rank test).
Figure 3.
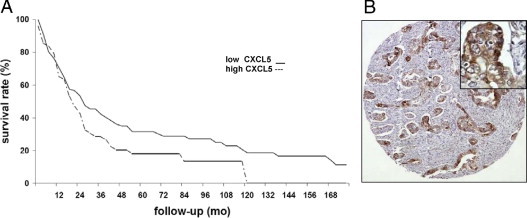
Kaplan-Meier analysis for patients with tumors that exhibited high (>5.5%) or low (≤5.5%) CXCL5 expression. A: Kaplan-Meier curve. High CXCL5 expression shortened patient survival time. Staining was scored based on the percentage of tumor cells staining positively. Two cores were evaluated per tumor. B: Representative tissue microarray of CXCL5 in patient tumor tissue. Original magnification: ×100 (B); ×400 (inset).
Mean ± SD survival was 64.0 ± 7.7 months for the low CXCL5 expression group but 38.5 ± 4.7 months for the high CXCL5 expression group. The mean difference in survival is significant (P = 0.002 by an approximate normal test). Median survival was 33 and 26 months for low and high CXCL5 expression, respectively. These data indicate that CXCL5 may be an important prognostic factor for patients with pancreatic cancer.
CXCL5 Mediates Pancreatic Cancer–Derived Angiogenesis
To evaluate the role of CXCL5 in pancreatic cancer–derived angiogenesis, we examined the effect of PCCM on human endothelial cell proliferation and in vitro angiogenesis. The PCCM was harvested from AsPC-1, BxPC3, or CAPAN-2 cell culture. The HUVECs grown on 96-well plates were cultured with PCCM for 48 hours. We observed significant increased cell proliferation in HUVECs cultured with PCCM (P < 0.05; Figure 4A). Neutralizing antibody to CXCL5 or its receptor CXCR2 significantly reduced pancreatic cancer cell–stimulated cell proliferation (P < 0.01). The HUVECs plated on Matrigel were treated with PCCM with or without neutralizing antibody to CXCL5 or CXCR2, and capillary tube formation was measured after 4 to 6 hours in an in vitro angiogenesis assay using Matrigel. The PCCM highly stimulated capillary tube formation, and neutralizing antibody to CXCL5 or CXCR2 significantly decreased PCCM-induced capillary tube formation (Figure 4, B and E; P < 0.001).
Figure 4.
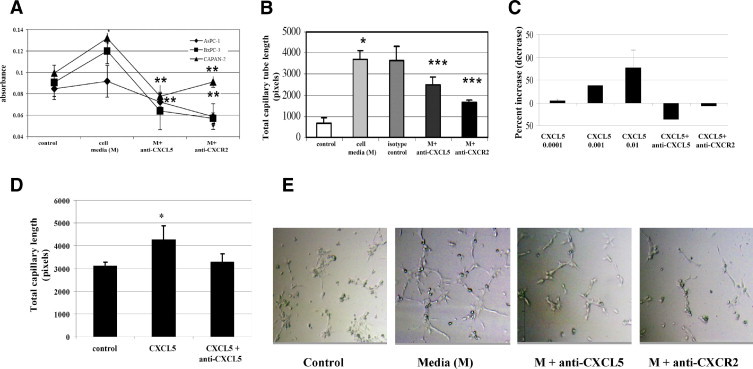
Mean growth and capillary tube formation of HUVECs in response to PCCM or rhCXCL5. A: Antibody to CXCL5 or CXCR2 reversed PCCM-stimulated cell growth. B: Antibody to CXCL5 or CXCR2 reduced PCCM-induced capillary tube formation. C: rhCXCL5 (0.001, 0.01, and 0.1 ng/ml) stimulated endothelial cell growth. Antibody to CXCL5 or CXCR2 reversed CXCL5-stimulated cell growth. D: rhCXCL5 induced capillary tube formation. Antibody to CXCL5 reversed the CXCL5 effect. E: rhCXCL5 induced capillary tube formation. Photograph of capillary tube formation in PCCM or PCCM combined with neutralizing antibody. *P < 0.05, **P < 0.01, ***P < 0.001. Error bars represent SD.
To further assess the role of CXCL5 in angiogenesis, we examined the effect of rhCXCL5 protein on HUVECs. We observed a dose-dependent increase in cell proliferation in response to rhCXCL5 treatment; neutralizing antibody to CXCL5 or CXCR2 completely blocked rhCXCL5-stimulated cell growth (Figure 4C). Recombinant human CXCL5 significantly induced, and neutralizing antibody reversed, CXCL5-mediated capillary tube formation (Figure 4D). These data suggest that CXCL5 plays an important role in pancreatic cancer–derived angiogenesis.
Mechanism of CXCL5-Mediated Angiogenesis
To analyze the signaling pathway involved in CXCL5-mediated angiogenesis, we performed a phospho-kinase array on HUVECs, which detects the activation of 46 kinases involved in several signaling pathways, including Akt, MAPK, signal transducer and activator of transcription (STAT) family, Src family kinase, focal adhesion kinase (FAK), cell cycle/check point protein and transcription factor, and others. CXCL5 activated most of the 46 kinases. Several pathways activated by CXCL5 are known to be involved in endothelial cell growth, differentiation, and migration. Quantitative data showed that four kinases in the Akt pathway are activated, with the highest level of phosphorylation for target of rapamycin (74.9% increase compared with control; P = 0.01) (Table 2). Phosphorylation of ERK and FAK was increased by 44.3% and 47.8%, respectively. More than a 40% increase was observed in six of the eight STAT family members. To validate the phospho-kinase array data, we performed Western blot analysis to detect Akt and ERK phosphorylation in response to CXCL5 stimulation. We reproduced the data observed in the phospho-array for Akt and ERK. Importantly, p-Akt or p-ERK specific inhibitor Wortmannin or PD98059 blocked CXCL5-stimulated Akt and ERK phosphorylation (Figure 5). The data suggest that CXCL5 activates multiple signaling pathways in endothelial cells.
Table 2.
Kinase Activation in Response to CXCL5 in HUVECs
| Phospho-Kinase | % increase | P value |
|---|---|---|
| Akt (S473) | 24.7 | 0.06 |
| Akt (T308) | 25.82 | 0.1 |
| TOR | 74.9 | 0.01 |
| GSK (3α/β) | 20.5 | 0.07 |
| p70 S6 kinase (T389) | 37.1 | 0.1 |
| p70 S6 kinase (T229) | 51.7 | 0.03 |
| p70 S6 kinase | 31.1 | 0.05 |
| ERK1/2 | 44.3 | 0.007 |
| MEK1/2 | 34.0 | 0.01 |
| p38α | 16.2 | 0.3 |
| JNK pan | 21.3 | 0.001 |
| RSK 1/2 | 36.5 | 0.002 |
| RSK 1/2/3 | 28.9 | 0.03 |
| FAK | 47.8 | 0.08 |
| β-catenin | 20.5 | 0.003 |
| Paxillin | 27.5 | 0.1 |
| Pyk-2 | 31.0 | 0.01 |
| Src | 59.4 | 0.002 |
| Fyn | 38.0 | 0.01 |
| Lyn | 43.8 | 0.05 |
| Yes | 35.1 | 0.02 |
| Lck | 39.7 | 0.001 |
| Hck | 38.2 | 0.02 |
| JNK pan | 21.3 | 0.1 |
| STAT1 | 60.1 | 0.007 |
| STAT2 | 36.3 | 0.01 |
| STAT3 | 43.5 | 0.0001 |
| STAT4 | 51.1 | 0.007 |
| STAT5a | 30.5 | 0.001 |
| STAT5a/b | 61.3 | 0.01 |
| STAT5b | 43.8 | 0.005 |
| STAT6 | 46.4 | 0.01 |
| p53 (S15) | 14.0 | 0.004 |
| p53 (S392) | 10.1 | 0.01 |
| p53 (S46) | 10.8 | 0.003 |
| p27 (T157) | 29.7 | 0.01 |
| P27 (T198) | 33.5 | 0.02 |
| CHK2 | 40.3 | 0.005 |
| C-Jun | 6.3 | 0.03 |
| CREB | 34.0 | 0.006 |
| eNOS | 3.06 | 0.09 |
| HSP27 | 36.4 | 0.03 |
| AMPKα1 | 22.7 | 0.01 |
| AMPKα2 | 31.4 | 0.08 |
| MSK1/2 | 24.1 | 0.01 |
| PLCg-1 | 29.9 | 0.03 |
Cell lysate from CXCL5-treated (0.5 ng/ml) HUVECs were used for phospho-kinase array.
Figure 5.
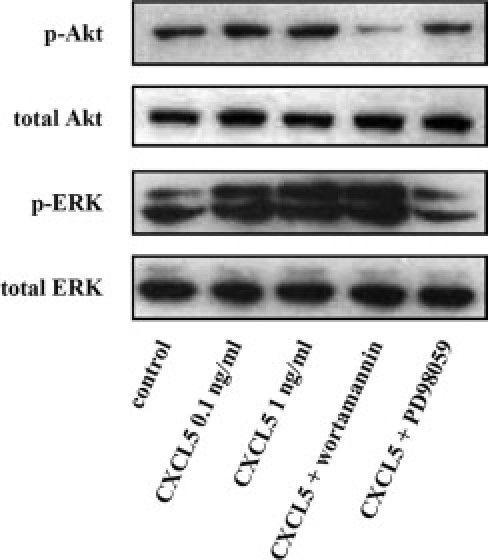
Effect of CXCL5 on Akt or ERK phosphorylation by means of Western blot analysis. HUVECs treated with CXCL5 with or without preblock with Akt (S473) inhibitor (Wortmannin, 1 mmol/L) or ERK inhibitor (PD98059, 20 mmol/L).
Knockdown of CXCL5 or Neutralization of CXCR2 Attenuated Human Pancreatic Cancer Growth in Nude Mice
To further address the functional significance of CXCL5 in human pancreatic cancer in vivo, we used two models to block CXCL5 effect: knockdown of CXCL5 by siRNA in high–CXCL5-expressing MIA PaCa-2 cells or neutralization of its receptor CXCR2 by antiserum treatment in relatively low–CXCL5-expressing BxPC3 cells. MIA PaCa-2 cells s.c. injected into nude mice were allowed to grow to 0.6 to 0.7 cm, and CXCL5 siRNA or control siRNA was then injected daily (intratumorally) for 4 weeks. A 44% reduction in tumor growth was observed in CXCL5 siRNA–treated mice compared with control mice (3.0 versus 5.3 cm3, P < 0.01; Figure 6A). Using CXCL5 enzyme-linked immunosorbent assay, we detected a reduction in CXCL5 protein by 47% (P = 0.002) in serum and 57% (P = 0.001) in tumor tissue of CXCL5 siRNA–treated mice. No change in MVD in treated and control mice was observed (data not shown). These data suggest that CXCL5 may promote tumor growth partially through an angiogenesis-independent mechanism.
Figure 6.
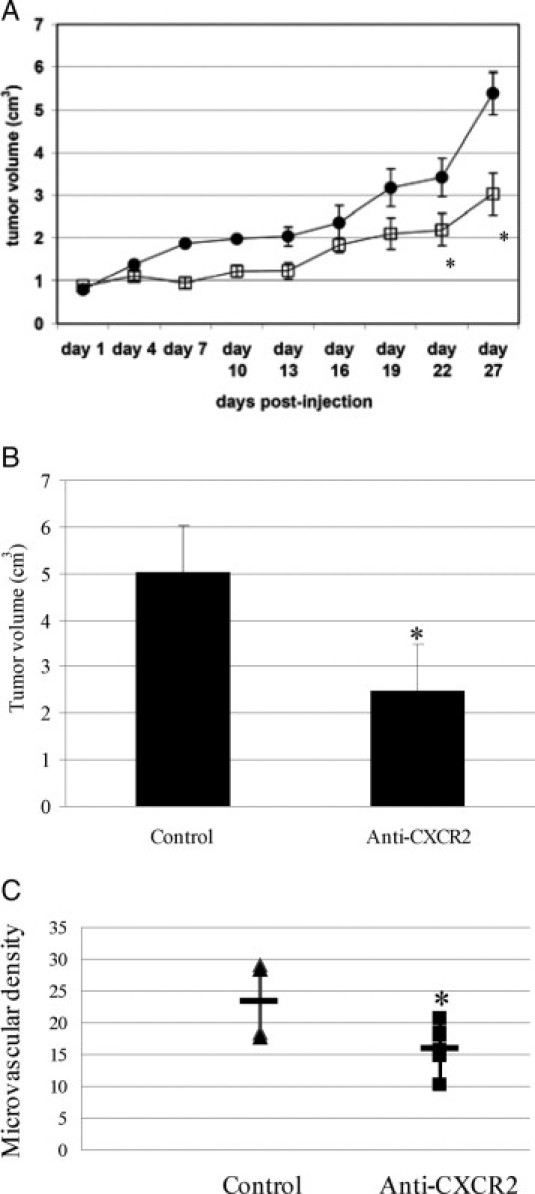
Blockade of CXCL5 or CXCR2 attenuated human pancreatic cancer growth. A: CXCL5 siRNA intratumoral treatment slowed MIA PaCa-2 cell growth in nude mice. Mice with intratumoral injection of CXCL5 (squares) or control (circles) siRNA were monitored for tumor growth twice weekly. Values are mean ± SD tumor volume. B: Antiserum to CXCR2 decreased BxPC3 cancer cell growth in nude mice. Mice were treated with antiserum to CXCR2 or control serum (i.p. injection) daily for 6 weeks. Values are mean ± SD tumor volume. C: CXCR2 antiserum treatment impaired angiogenesis in nude mice. Tumors from CXCR2 antiserum–treated or control mice were stained with antibody to CD31. The MVD was determined by counting blood vessels with positive CD31 staining. Horizontal bars indicate mean MVD. *P < 0.05.
One week after s.c. injection of BxPC3 cells, nude mice were treated with CXCR2 antiserum daily for 6 weeks. There was a 50% decrease in tumor growth in CXCR2 antiserum–treated mice compared with control mice (5.0 versus 2.4 cm3, P < 0.05; Figure 6B). To analyze the role of angiogenesis on tumor growth inhibition in these tumors, paraffin-embedded tumor tissues were stained with antibody to CD31, and MVD was determined by CD31-positive staining. We observed significantly decreased MVD in CXCR2 antiserum–treated tumor compared with control tumor, suggesting that blockade of CXCR2 impaired angiogenesis, thus inhibiting tumor growth.
Discussion
In the present study, we report that CXCL5 is markedly overexpressed in human pancreatic cancer. Its expression is progressively increased in premalignant PanIN lesions to invasive carcinoma and is associated with tumor progression. Expression of CXCL5 has a significant reverse correlation with tumor differentiation, clinical stage, and patient survival. Furthermore, we show that CXCL5 promotes pancreatic cancer growth directly through stimulation of cancer cell proliferation and indirectly through cancer cell–derived angiogenesis. Finally, we demonstrated that CXCL5 activates multiple signaling pathways in human endothelial cell.
Pancreatic adenocarcinoma is an extremely lethal disease with only approximately 23% of patients surviving 1 year after diagnosis. Pancreatic cancer has one of the lowest 5-year survival rates of all human malignancies.27 Even for those diagnosed as having localized disease, 5-year survival is only 15%. We observed a poor survival rate in CXCL5-overexpressing patients and a large 25.5-month shorter survival in high–CXCL5-expressing patients who underwent surgical resection. Consistent with the survival data, we observed that CXCL5 expression was correlated with large tumor volume, poor differentiation, and advanced clinical stage, indicating an important role of CXCL5 in pancreatic cancer progression. In contrast to a role in promoting tumor growth of CXCL5 in pancreatic cancer and other types of cancer,14–16,18 CXCL5 has been shown to have an opposite effect on colon cancer growth.19 Most tumors (70.6%) with a high CXCL5 expression ratio between tumor and normal pancreas were in male patients in this study. The cause of increased CXCL5 expression in male patients is not clear. Androgen receptor–sensitive and androgen receptor–insensitive prostate cancer cell lines are responsive to CXCL5 differentially, indicating a possible link between CXCL5 expression and sex hormone signaling.16
The PanINs are neoplastic precursors to human pancreatic cancer.25,26 Morphologic and molecular gene profiling suggested that PanINs represent a distinctive stepwise progression of the malignant transformation of pancreatic cancer. Several pancreatic cancer–associated gene alterations, including Ki-Ras, p16, and p53, are accumulated in high-grade PanIN-2 and PanIN-3 lesions but not in PanIN-1 lesions.28 Begley et al16 indicated that CXCL5 is up-regulated in prostate intraepithelial neoplasia, and it directly stimulated nontransformed and transformed prostate epithelial cell growth. A recent study by Singh et al29 demonstrated that overexpression of CXCR2 in nontumorigenic melanoma cells induced tumorigenicity. We found that CXCL5 expression is increased in PanIN-2 and PanIN-3 lesions (Figure 2) together with its receptor (CXCR2) expression in pancreatic cancer cells (data not shown), suggesting that CXCL5 may play an autocrine role in pancreatic cancer development.
Pancreatic cancer tissue with strong CXCL5 expression (>50%) revealed increased MVD (Figure 3; P = 0.03). Furthermore, conditioned media from cultured human pancreatic cancer significantly stimulated endothelial cell growth and in vitro angiogenesis (Figure 5). This stimulation is specific to CXCL5 because blockade of CXCL5 or its receptor, CXCR2, suppressed endothelial cell growth, and exogenous administration of rhCXCL5 induced endothelial cell growth and in vitro angiogenesis (Figure 5). We found in a previous work20 that conditioned media of these cells contains other pro-angiogenic factors, including vascular endothelial grow factor (VEGF) and the proangiogenic chemokines CXCL8 (IL-8) and CXCL1 (Gro). In CXCL5-negative AsPC-1 cells, no significant anti-CXCL5 effect is seen, whereas the anti-CXCR2 antibody significantly lowered endothelial cell growth. Other CXC ligands are activating growth and tubule formation because the CXCR2 antibody was most effective at blocking these. Taken together, these data suggest a critical role of CXCL5 in pancreatic cancer–mediated angiogenesis.
In response to CXCL5 stimulation, we demonstrated activation of several angiogenic signaling pathways in HUVECs, including AKT–mammalian target of rapamycin, mitogen-activated protein kinase, focal adhesion kinase, and the STAT family members and the Src family kinases that are known important regulators of endothelial cell function. The activation of Akt and ERK regulates multiple aspects of cellular function, including endothelial cell survival and growth.30,31 Activation of the JAK/STAT pathway stimulates cell proliferation, differentiation, migration, and apoptosis and is the principal signaling mechanism for cytokines.32 The JAK/STAT pathway has also been implicated in chemokine signaling.33 Recent studies demonstrated that STAT is involved in VEGF signaling34 and plays a pivotal role in hypoxia-induced angiogenesis.35 The Src family kinases are composed of nine structurally related molecules (Src, Blk, Fyn, Yes, Lyn, Lck, Hck, Fgr, and Yrk) with conserved peptide domains termed Src homology domains. Src family kinases interacting with VE-cadherin, VEGF, FAK, and caveolin signaling regulate endothelial cell permeability, contributing to tumor angiogenesis and metastasis.36–38 The multiple signaling pathways activated by CXCL5 play critical roles in endothelial function. After a phospho-kinase array screening assay, we further confirmed that CXCL5 induced Akt and ERK phosphorylation by Western blot (Figure 6B). The p-Akt–specific inhibitor Wortmannin blocked CXCL5-induced Akt activation, whereas the p-ERK–specific inhibitor PD98059 abolished CXCL5-stimulated ERK activation. These data suggest that CXCL5 is an important regulator of angiogenesis. Multiple signaling pathways are activated by CXCL5 in HUVECs; it would be interesting to see the interaction between CXCL5 and endothelial cell mitogenic VEGF signaling. A few studies have suggested a greater role of CXC chemokines, including CXCL5, than of VEGF in tumor angiogenesis in ovary, colon, and lung cancer.39–42 Ki-ras mutation increased expression of angiogenic CXC chemokines and decreased VEGF expression, particularly under hypoxic conditions,41 suggesting that CXC chemokines and VEGF may act through different mechanisms.
Finally, we showed that disruption of CXCL5/CXCR2 signaling in vivo abolished human pancreatic cancer growth in a nude mouse model (Figure 6A). Small-interfering RNA against human CXCL5 slowed tumor growth, with no effect on MVD. These data further suggest an autocrine effect of CXCL5 on pancreatic cancer cell growth. This may be due to other ligands of CXCR2 that were not abrogated in this experiment. In contrast to CXCL5 siRNA–treated tumor, antiserum to mouse CXCR2 suppressed tumor growth with decreased MVD. These data indicate a dual effect of CXCL5 on cancer cells and endothelial cells. CXCL5 may promote tumor growth partially through a receptor-mediated angiogenesis and partially through an autocrine growth effect on cancer cells.
In conclusion, we demonstrate that CXCL5 is an important survival factor for human pancreatic cancer. CXCL5 stimulated human endothelial cell growth and differentiation through multiple signaling pathways, and CXCL5- induced angiogenesis is critical for tumor growth and progression. Targeting CXCL5/CXCR2 signaling may be a powerful therapeutic strategy for the treatment of pancreatic cancer.
Footnotes
Supported by grants R21 CA124609 and PO1CA003960 from the National Institutes of Health and by the Hirshberg Foundation.
References
- 1.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J., Cotran R. Relation of vascular proliferation to tumor growth. Int Rev Exp Pathol. 1976;16:207–248. [PubMed] [Google Scholar]
- 3.Devalaraja R.M., Nanney L.B., Du J., Qian Q., Yu Y., Devalaraja M.N., Richmond A. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115:234–244. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh S., Sadanandam A., Singh R.K. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev. 2007;26:453–467. doi: 10.1007/s10555-007-9068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider G.P., Salcedo R., Welniak L.A., Howard O.M., Murphy W.J. The diverse role of chemokines in tumor progression: prospects for intervention (review) Int J Mol Med. 2001;8:235–244. doi: 10.3892/ijmm.8.3.235. [DOI] [PubMed] [Google Scholar]
- 6.Wang J.M., Deng X., Gong W., Su S. Chemokines and their role in tumor growth and metastasis. J Immunol Methods. 1998;220:1–17. doi: 10.1016/s0022-1759(98)00128-8. [DOI] [PubMed] [Google Scholar]
- 7.Arenberg D.A., Keane M.P., DiGiovine B., Kunkel S.L., Morris S.B., Xue Y.Y., Burdick M.D., Glass M.C., Iannettoni M.D., Strieter R.M. Epithelial-neutrophil activating peptide (ENA-78) is an important angiogenic factor in non-small cell lung cancer. J Clin Invest. 1998;102:465–472. doi: 10.1172/JCI3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walz A., Burgener R., Car B., Baggiolini M., Kunkel S.L., Strieter R.M. Structure and neutrophil-activating properties of a novel inflammatory peptide (ENA-78) with homology to interleukin 8. J Exp Med. 1991;174:1355–1362. doi: 10.1084/jem.174.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imaizumi T., Albertine K.H., Jicha D.L., McIntyre T.M., Prescott S.M., Zimmerman G.A. Human endothelial cells synthesize ENA-78: relationship to IL-8 and to signaling of PMN adhesion. Am J Respir Cell Mol Biol. 1997;17:181–192. doi: 10.1165/ajrcmb.17.2.2818. [DOI] [PubMed] [Google Scholar]
- 10.Schnyder-Candrian S., Strieter R.M., Kunkel S.L., Walz A. Interferon-α and interferon-γ down-regulate the production of interleukin-8 and ENA-78 in human monocytes. J Leukoc Biol. 1995;57:929–935. doi: 10.1002/jlb.57.6.929. [DOI] [PubMed] [Google Scholar]
- 11.Z'Graggen K., Walz A., Mazzucchelli L., Strieter R.M., Mueller C. The C-X-C chemokine ENA-78 is preferentially expressed in intestinal epithelium in inflammatory bowel disease. Gastroenterology. 1997;113:808–816. doi: 10.1016/s0016-5085(97)70175-6. [DOI] [PubMed] [Google Scholar]
- 12.Koch A.E., Kunkel S.L., Harlow L.A., Mazarakis D.D., Haines G.K., Burdick M.D., Pope R.M., Walz A., Strieter R.M. Epithelial neutrophil activating peptide-78: a novel chemotactic cytokine for neutrophils in arthritis. J Clin Invest. 1994;94:1012–1018. doi: 10.1172/JCI117414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi H., Numasaki M., Lotze M.T., Sasaki H. Interleukin-17 enhances bFGF-, HGF- and VEGF-induced growth of vascular endothelial cells. Immunol Lett. 2005;98:189–193. doi: 10.1016/j.imlet.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Matsuo Y., Raimondo M., Woodward T.A., Wallace M.B., Gill K.R., Tong Z., Burdick M.D., Yang Z., Strieter R.M., Hoffman R.M., Guha S. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int J Cancer. 2009;125:1027–1037. doi: 10.1002/ijc.24383. [DOI] [PubMed] [Google Scholar]
- 15.Park J.Y., Park K.H., Bang S., Kim M.H., Lee J.E., Gang J., Koh S.S., Song S.Y. CXCL5 overexpression is associated with late stage gastric cancer. J Cancer Res Clin Oncol. 2007;133:835–840. doi: 10.1007/s00432-007-0225-x. [DOI] [PubMed] [Google Scholar]
- 16.Begley L.A., Kasina S., Mehra R., Adsule S., Admon A.J., Lonigro R.J., Chinnaiyan A.M., Macoska J.A. CXCL5 promotes prostate cancer progression. Neoplasia. 2008;10:244–254. doi: 10.1593/neo.07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong Y.F., Cheung T.H., Lo K.W., Yim S.F., Siu N.S., Chan S.C., Ho T.W., Wong K.W., Yu M.Y., Wang V.W., Li C., Gardner G.J., Bonome T., Johnson W.B., Smith D.I., Chung T.K., Birrer M.J. Identification of molecular markers and signaling pathway in endometrial cancer in Hong Kong Chinese women by genome-wide gene expression profiling. Oncogene. 2007;26:1971–1982. doi: 10.1038/sj.onc.1209986. [DOI] [PubMed] [Google Scholar]
- 18.Miyazaki H., Patel V., Wang H., Edmunds R.K., Gutkind J.S., Yeudall W.A. Down-regulation of CXCL5 inhibits squamous carcinogenesis. Cancer Res. 2006;66:4279–4284. doi: 10.1158/0008-5472.CAN-05-4398. [DOI] [PubMed] [Google Scholar]
- 19.Speetjens F.M., Kuppen P.J., Sandel M.H., Menon A.G., Burg D., van de Velde C.J., Tollenaar R.A., de Bont H.J., Nagelkerke J.F. Disrupted expression of CXCL5 in colorectal cancer is associated with rapid tumor formation in rats and poor prognosis in patients. Clin Cancer Res. 2008;14:2276–2284. doi: 10.1158/1078-0432.CCR-07-4045. [DOI] [PubMed] [Google Scholar]
- 20.Wente M.N., Keane M.P., Burdick M.D., Friess H., Buchler M.W., Ceyhan G.O., Reber H.A., Strieter R.M., Hines O.J. Blockade of the chemokine receptor CXCR2 inhibits pancreatic cancer cell-induced angiogenesis. Cancer Lett. 2006;241:221–227. doi: 10.1016/j.canlet.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 21.Frick V.O., Rubie C., Wagner M., Graeber S., Grimm H., Kopp B., Rau B.M., Schilling M.K. Enhanced ENA-78 and IL-8 expression in patients with malignant pancreatic diseases. Pancreatology. 2008;8:488–497. doi: 10.1159/000151776. [DOI] [PubMed] [Google Scholar]
- 22.Addison C.L., Daniel T.O., Burdick M.D., Liu H., Ehlert J.E., Xue Y.Y., Buechi L., Walz A., Richmond A., Strieter R.M. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165:5269–5277. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- 23.Li A., Hasui K., Yonezawa S., Tanaka S., Sato E. Immunohistochemical analysis of pericryptal fibroblast sheath and proliferating epithelial cells in human colorectal adenomas and carcinomas with adenoma components. Pathol Int. 1999;49:426–434. doi: 10.1046/j.1440-1827.1999.00891.x. [DOI] [PubMed] [Google Scholar]
- 24.Li A., Varney M.L., Valasek J., Godfrey M., Dave B.J., Singh R.K. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8:63–71. doi: 10.1007/s10456-005-5208-4. [DOI] [PubMed] [Google Scholar]
- 25.Hruban R.H., Adsay N.V., Albores-Saavedra J., Compton C., Garrett E.S., Goodman S.N., Kern S.E., Klimstra D.S., Kloppel G., Longnecker D.S., Luttges J., Offerhaus G.J. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 26.Takaori K., Kobashi Y., Matsusue S., Matsui K., Yamamoto T. Clinicopathological features of pancreatic intraepithelial neoplasias and their relationship to intraductal papillary-mucinous tumors. J Hepatobiliary Pancreat Surg. 2003;10:125–136. doi: 10.1007/s00534-003-0756-8. [DOI] [PubMed] [Google Scholar]
- 27.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M.J. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 28.Sipos B., Frank S., Gress T., Hahn S., Kloppel G. Pancreatic intraepithelial neoplasia revisited and updated. Pancreatology. 2009;9:45–54. doi: 10.1159/000178874. [DOI] [PubMed] [Google Scholar]
- 29.Singh S., Nannuru K.C., Sadanandam A., Varney M.L., Singh R.K. CXCR1 and CXCR2 enhances human melanoma tumourigenesis, growth and invasion. Br J Cancer. 2009;100:1638–1646. doi: 10.1038/sj.bjc.6605055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J., Somanath P.R., Razorenova O., Chen W.S., Hay N., Bornstein P., Byzova T.V. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alavi A., Hood J.D., Frausto R., Stupack D.G., Cheresh D.A. Role of Raf in vascular protection from distinct apoptotic stimuli. Science. 2003;301:94–96. doi: 10.1126/science.1082015. [DOI] [PubMed] [Google Scholar]
- 32.Rawlings J.S., Rosler K.M., Harrison D.A. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 33.Mellado M., Rodriguez-Frade J.M., Manes S., Martinez A.C. Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu Rev Immunol. 2001;19:397–421. doi: 10.1146/annurev.immunol.19.1.397. [DOI] [PubMed] [Google Scholar]
- 34.Yahata Y., Shirakata Y., Tokumaru S., Yamasaki K., Sayama K., Hanakawa Y., Detmar M., Hashimoto K. Nuclear translocation of phosphorylated STAT3 is essential for vascular endothelial growth factor-induced human dermal microvascular endothelial cell migration and tube formation. J Biol Chem. 2003;278:40026–40031. doi: 10.1074/jbc.M301866200. [DOI] [PubMed] [Google Scholar]
- 35.Dudley A.C., Thomas D., Best J., Jenkins A. A VEGF/JAK2/STAT5 axis may partially mediate endothelial cell tolerance to hypoxia. Biochem J. 2005;390:427–436. doi: 10.1042/BJ20050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim M.P., Park S.I., Kopetz S., Gallick G.E. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335:249–259. doi: 10.1007/s00441-008-0682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eliceiri B.P., Paul R., Schwartzberg P.L., Hood J.D., Leng J., Cheresh D.A. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 38.Werdich X.Q., Penn J.S. Src, Fyn and Yes play differential roles in VEGF-mediated endothelial cell events. Angiogenesis. 2005;8:315–326. doi: 10.1007/s10456-005-9021-x. [DOI] [PubMed] [Google Scholar]
- 39.Yoneda J., Kuniyasu H., Crispens M.A., Price J.E., Bucana C.D., Fidler I.J. Expression of angiogenesis-related genes and progression of human ovarian carcinomas in nude mice. J Natl Cancer Inst. 1998;90:447–454. doi: 10.1093/jnci/90.6.447. [DOI] [PubMed] [Google Scholar]
- 40.Belperio J.A., Keane M.P., Arenberg D.A., Addison C.L., Ehlert J.E., Burdick M.D., Strieter R.M. CXC chemokines in angiogenesis. J Leukoc Biol. 2000;68:1–8. [PubMed] [Google Scholar]
- 41.Mizukami Y., Jo W.S., Duerr E.M., Gala M., Li J., Zhang X., Zimmer M.A., Iliopoulos O., Zukerberg L.R., Kohgo Y., Lynch M.P., Rueda B.R., Chung D.C. Induction of interleukin-8 preserves the angiogenic response in HIF-1α–deficient colon cancer cells. Nat Med. 2005;11:992–997. doi: 10.1038/nm1294. [DOI] [PubMed] [Google Scholar]
- 42.McClelland M.R., Carskadon S.L., Zhao L., White E.S., Beer D.G., Orringer M.B., Pickens A., Chang A.C., Arenberg D.A. Diversity of the angiogenic phenotype in non-small cell lung cancer. Am J Respir Cell Mol Biol. 2007;36:343–350. doi: 10.1165/rcmb.2006-0311OC. [DOI] [PMC free article] [PubMed] [Google Scholar]