

Abstract
Cerebral malaria is the most severe neurologic complication in children and young adults infected with Plasmodium falciparum. T-cell activation is required for development of Plasmodium berghei ANKA (PbA)–induced experimental cerebral malaria (CM). To characterize the T-cell activation pathway involved, the role of protein kinase C-theta (PKC-θ) in experimental CM development was examined. PKC-θ–deficient mice are resistant to CM development. In the absence of PKC-θ, no neurologic sign of CM developed after blood stage PbA infection. Resistance of PKC-θ–deficient mice correlated with unaltered cerebral microcirculation and absence of ischemia, as documented by magnetic resonance imaging and magnetic resonance angiography, whereas wild-type mice developed distinct microvascular pathology. Recruitment and activation of CD8+ T cells, and ICAM-1 and CD69 expression were reduced in the brain of resistant mice; however, the pulmonary inflammation and edema associated with PbA infection were still present in the absence of functional PKC-θ. Resistant PKC-θ–deficient mice developed high parasitemia, and died at 3 weeks with severe anemia. Therefore, PKC-θ signaling is crucial for recruitment of CD8+ T cells and development of brain microvascular pathology resulting in fatal experimental CM, and may represent a novel therapeutic target of CM.
Malaria is 1 of the 5 most important infectious diseases worldwide, and cerebral malaria (CM) is a frequent cause of death in children and young adults infected with Plasmodium falciparum.1 Many aspects of the pathophysiology of CM are still poorly understood. Investigations in human beings and mice characterized the sequestration of erythrocytes (parasitized or not), platelets, and leukocytes in cerebral blood vessels,2,3 with increased expression of proinflammatory cytokines such as tumor necrosis factor4–6 and adhesion molecules.7 The specific role of T cells in the pathogenesis of CM has been difficult to address in human beings. In mice, however, T-cell sequestration and activation are crucial steps in the development of experimental CM after Plasmodium berghei ANKA (PbA) infection.8–10 In particular, brain sequestrated αβ CD8+ T cells have a pathogenic effector role in experimental CM development.10 T-cell activation is a complex process involving a cascade of intracellular enzymes. The present study addressed the role of protein kinase C (PKC) in the pathogenesis of CM in PbA infection.
PKC isozymes are important in cellular activation, differentiation, adhesion, motility, and survival. Among the PKC family, PKC-δ and PKC-θ are expressed in lymphocytes, and PKC-θ is also expressed in natural killer cells.11 PKC-θ is a critical regulator of T-cell receptor signaling and T-cell activation,12,13 and has been proposed as a therapeutic target for T-cell–mediated diseases.12 However, the role of PKC-θ in T-cell activation is complex, and PKC-θ is not equally involved in all T-cell responses. PKC-θ regulates Th2 helper cells that mediate immune response against helminth infection.12,13 It is involved in interferon I and II signaling in human T cells.14 PKC-θ is also important for T-cell survival and differentiation into functional cytotoxic T cells; however, it may be dispensable, compensated by other innate signals, for CD8+ T-cell proliferation and development.15 PKC-θ is not essential for mounting normal protective Th1 immune responses to clear some virus infections.16 Further, TLR9 ligand CpG was recently shown to directly act on T-cell proliferation and survival, bypassing the PKC-θ pathway.17 Therefore, the implication of PKC-θ in the T-cell pathway during development of experimental CM remains to be established.
In the present study, PKC-θ–deficient mice were used to identify PKC-θ as an obligatory pathway for development of CM in PbA infection. PKC-θ–deficient mice were protected from CM development in blood stage infection with PbA, and magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) confirmed the lack of ischemia and microvascular pathology and brain morphologic changes in these mice. Further, it was demonstrated that experimental CM protection is associated with reduced T-cell recruitment and activation in the brain, whereas lung edema is unaffected in the absence of functional PKC-θ.
Materials and Methods
Mice
Mice deficient in PKC-θ18 were obtained from Manfred Kopf (Molecular Biomedicine, ETH Zurich, Switzerland) and bred in our animal facility at the Transgenose Institute (CNRS, Orleans, France). Mice were of C57BL/6J genetic background (back-crossed at least 15 times), and wild-type C57BL/6J mice were used as controls. For experiments, adult (aged 8 to 10 weeks) mice were kept in filtered-cages in a P2 animal facility. All animal experiments complied with ethical and animal experiments regulations of the French government.
Experimental Malaria Infection
Recombinant PbA that constitutively expresses green fluorescent protein at a high level throughout the complete life cycle, from a transgene controlled by the strong promoter from a P berghei elongation factor-1α gene, was obtained from Dr. Andrew P. Waters (Glasgow Biomedical Research Center, University of Glasgow, Glasgow, United Kingdom).19 Mice were infected via intraperitoneal injection of 105 parasitized erythrocytes as described previously.20 Mice were observed daily, grip strength was evaluated during daily body weight measurement, and clinical neurologic signs of CM culminating in ataxia, paralysis, and coma were assessed.
Parasitemia
Parasitemia was assessed in 2 μL of blood collected from the tail vein at various time points after infection with EGFP-PbA. Blood was diluted in 3 ml of PBS containing 0.5% bovine serum albumin, and fluorescent cells were analyzed using a flow cytometer (FACScan LSR; Becton Dickinson, Grenoble, France)21 using FlowJo software (TreeAge Software, Inc, Williamstown, Massachusetts).
Hematologic Analysis
Blood was drawn with the mice under anesthesia with isofluorane (CSP, Fontenay sous Bois, France) into tubes containing EDTA (Vacutainer; Becton Dickinson) as indicated, and hematologic parameters were determined using a 5-part-differential hematology analyzer (MS 9.5; Melet Schloesing Laboratoires, Osny, France).
Assessment of Vascular Leak
Mice were injected intravenously with 0.2 ml of 325 mg/ml Evans blue solution (Sigma-Aldrich, St. Louis, MO) on day 7, shortly before the death of the wild-type mice. One hour later, mice were sacrificed, and the coloration of the brain was assessed. Brain extravasation was evaluated using Evans blue in formamide (Sigma-Aldrich) measured by absorbance at 610 nm of the tissue extracts as an indicator of increased capillary permeability.20 Similar mean (SD)baseline levels were observed in noninfected wild-type and PKC-θ−/− mice: 0.50 (0.21) μg/ml and 0.51 (0.26) μg/ml, respectively.
Histologic Analysis
Mice were euthanized and perfused with intracardiac sterile PBS/EDTA, 0.002 mmol/L, to remove circulating red blood cells and leukocytes from the brain and lung. The tissues were then fixed with a second perfusion of PBS/paraformaldehyde 4% (Sigma-Aldrich). The brains and lungs from PbA-infected mice were fixed in 3.6% PBS/formaldehyde for 72 hours as described previously.20 Longitudinal sections of 3 μm were stained with H&E. Brain microvascular disease with endothelial damage, accumulation of mononuclear cells, and extravasated erythrocytes in the Virchov-Robin space was assessed semiquantitatively from a whole brain section, typically between −5.64 and −6.84 from the bregma. Erythrocyte accumulation in alveoli and thickening of alveolar septae of the lung were quantified using a semiquantitative score with increasing severity of changes (0 to 5) by 2 independent observers including a trained pathologist (B.R.). Brain microvascular obstruction was evaluated by scoring the number of vessels containing sequestered blood cells on multiple fields corresponding to whole brain sections (0% microvascular obstruction = 0, 1% to 19% microvascular obstruction = 1, 20% to 39% microvascular obstruction = 2, 40% to 59% microvascular obstruction = 3, 60% to 79% microvascular obstruction = 4, and 80% microvascular obstruction = 5). Lung sections were scored on 10 fields per section for erythrocyte accumulation in alveoli (less than 1 cell per alveoli = 0, 1 cell per alveoli = 1, 2 to 5 cells per alveoli = 2, 5 to 10 cells per alveoli = 3, 10 to 30 cells per alveoli = 4, and 30 cells pre alveoli = 5); alveolar size (all normal alveoli = 0, mix of normal and medium-sized alveoli = 1, medium-sized alveoli only = 2, mix of medium-sized and small alveoli = 3, all small alveoli = 4, and full obstruction = 5 (not observed); and thickening of alveolar septae due to inflammatory cells and edema was scored with increasing severity from 0 to 5 as an estimate of interstitial inflammation.
Immunohistologic Staining
Macrophages and T-cell populations were analyzed in situ using immunohistologic staining. Euthanized mice were perfused as described above, and brains and lungs were harvested, imbedded in OCT media (Tissue-Tek; Sakura Finetek Japan Co, Ltd, Tokyo, Japan), and frozen rapidly. Longitudinal cryosections of 10 μm were fixed in acetone, washed in PBS/Tween 0.05% and PBS, and incubated for 30 minutes with wild-type mouse serum at 2% in PBS. All incubations were carried out in a wet chamber. Brain and lung sections were then incubated overnight at 4°C with biotinylated rat antibodies to CD3e (clone 145-2C11), CD8α (clone 53-6.7), CD4 (clone RM4-5), CD54 (clone 3E2,), CD11b (clone M1/70), or IA/IE (clone 2G9) (all from BD Pharmingen, Inc, San Diego, CA) as indicated. After washing, sections were incubated for 30 minutes with an avidin-biotin peroxydase complex (ABC; Vector Laboratories, Inc, Burlingame, CA), followed by the peroxydase substrate (diaminobenzidine; Vector Laboratories, Inc). Finally, sections were washed in PBS, mounted using Eukitt medium (O. Kindler GmbH & Co, Freiburg, Germany), and examined microscopically.
Brain Imaging Using MRI
MRI experiments were performed as described previously21 using a horizontal 9.4 T/20 Bruker Biospec MR system (Bruker BioSpin SA, Wissembourg, France). A 12-element linear birdcage coil (Bruker BioSpin SA) with inner diameter of 35 mm and length of 60 mm was used to achieve uniform excitation and reception. A custom-built stereotaxic head holder was used to fix the animals in the birdcage coil. The mice were anesthetized using 1.5% isoflurane in a 1:1 oxygen/nitrous oxide mixture administered using a face mask, allowing free breathing. Respiration was monitored using a balloon taped to the abdomen and connected to a pressure transducer (SA Instruments, Inc, Stony Brook, NY). Mean (SD) mouse body temperature was maintained at 37°C (0.5°C) throughout the experiment, using warm water circulation.
Preparatory anatomical sagittal MRI was performed using a multisection multiecho sequence with the following parameters: rapid acquisition with relaxation enhancement factor = 8, repetition time/effective echo time = 4000/46 ms, field of view = 20 × 20 mm2, matrix = 128 × 128, 1 section 1-mm thick, number of averages = 1, and experimental time = 1 minute. Brain lesions and global changes in tissue structure were accessed via T2-weighted MRI using a multisection multiecho sequence with the following parameters: rapid acquisition with relaxation enhancement factor = 8, repetition time/effective echo time = 5000/46 ms, field of view = 17 × 17 mm2, matrix = 256 × 192 (zero-filled to 256 × 256), 37 axial sections 0.5-mm thick, number of averages = 8, and experimental time = 16 minutes. Three measures were used to estimate morphologic changes of the brain: line 1 from the pituitary gland to the aqueduct of Sylvius, the median line crossing the medial cerebellar nucleus, and the medium line stemming from the cerebellar obex. For each group, a region of interest was drawn manually in the corpus callosum, and gray-level mean was measured using Mazda software (Mazda version 3.20, copyright 1998–2002 by Piotr Szczypinski).
Vascular cerebral blood flow was measured at MRA using a fast low-angle shot sequence with the following parameters: field of view = 14 × 14 mm2, matrix = 128 × 128, repetition time/echo time = 30/5 ms, 51 axial sections 0.3-mm thick, number of averages = 4, and experimental time = 13 minutes. Angiograms were produced by generating maximum intensity projections after interpolating raw data to obtain an isotropic resolution (109 μm3). Image analysis and processing were performed using the public domain software Image J.
Isolation of Brain-Sequestered Leukocytes
Mice were euthanized and perfused as described,10 on day 7, when all wild-type mice exhibited neurologic symptoms of CM. Brains were removed and homogenized gently (30 seconds at 4000 rpm) using a sterile disposable homogenization system (Dispomix; Medic Tools AG, Zug, Switzerland) in RPMI 1640 medium containing 2% fetal calf serum. Homogenates were passed through a 100-μm nylon cell strainer (Becton Dickinson France SAS, Grenoble, France), and cells were centrifuged at 400g for 10 minutes.21 The mononuclear cells were separated over a 35% Percoll gradient (Amersham Biosciences AB, Uppsala, Sweden).
Flow Cytometry Analysis
Brain-sequestered cells were collected, washed, and saturated with mouse serum before staining with fluorescence-labeled antibodies for 30 minutes. The cells were then phenotyped at flow cytometry using the following rat antibodies: anti-mouse CD3 conjugated to fluorescein isothiocyanate (clone 145-2C11), CD4 conjugated to peridinin-chlorophyll-protein complex (clone L3T4 RM4-5), CD8α conjugated to allophycocyanin cyanin7 (clone 53-6.7), CD69 (clone H1.2F3) conjugated to phycoerythrin, and isotype-matched control antibodies (all from BD Pharmingen Inc). For each sample, 10,000 cells from the mononuclear population were scored. Data were analyzed using a flow cytometer and FlowJo software (Tree Star Inc, Ashland, Oregon).
Statistical Analysis
Unless otherwise indicated, data are given as mean (SD), indicated by error bars. Statistical significance was determined using commercially available software (Prism version 4.0; GraphPad Software, Inc, San Diego, CA). Differences between multiple groups were analyzed for statistical significance via 1-way parametric analysis of variance followed by the Bonferroni posttest for analyzing large sample groups or by a nonparametric Mann-Whitney test for analysis of small groups. P < 0.05 was considered statistically significant.
Results
PKC-θ Deficiency Confers Protection Against CM Development
The role of PKC-θ was assessed by investigating the development of neurologic signs of CM, survival, and parasitemia in mice deficient in PKC-θ. After injection of PbA-infected erythrocytes, wild-type control mice developed typical neurologic symptoms of CM such as ataxia, loss of grip strength, progressive paralysis, and coma, and all died within 1 week, whereas 75% of PKC-θ–deficient mice survived longer than 10 days, and 60% of PKC-θ–deficient mice survived longer than 14 days with no sign of CM (Figure 1A).
Figure 1.
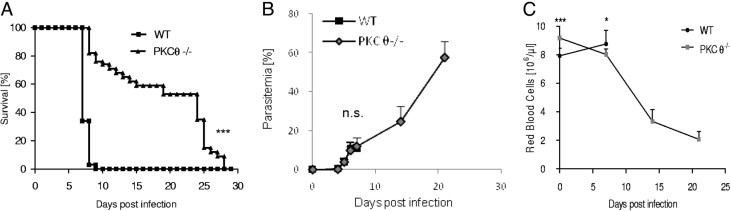
PKC-θ–deficient mice do not develop PbA-induced CM. A: Wild-type mice (WT) and PKC-θ–deficient mice were infected with 105Plasmodium berghei ANKA (PbA) parasitized red blood cells, and their survival was monitored daily. Values are given as mean (SD) of 25 PKC-θ–deficient and 29 WT mice, and are pooled from 5 independent experiments. B: Kinetics of parasitemia is shown up to day 7 for WT mice and day 21 after infection for CM-resistant PKC-θ–deficient mice. Values are given as mean (SD) from 5 independent experiments: 11 PKC-θ−/− and 14 WT mice on day 4; 7 PKC-θ−/− and 9 WT mice on day 5; 7 PKC-θ−/− and 9 WT mice on day 6; 38 PKC-θ−/− and 22 WT mice on day 7; and 19 WT and 16 PKC-θ−/− mice on days 14 and 21. C: Kinetics of peripheral red blood cell counts is shown up to day 7 for WT mice and day 21 after infection for CM-resistant PKC-θ–deficient mice. Values are given as mean (SD) from 4 independent experiments: 15 PKC-θ−/− and 21 WT mice on day 0; 21 PKC-θ−/− and 6 WT mice on day 7; and 4 WT and 3 PKC-θ−/− mice on days 14 and 21. NS, not significant. *P < 0.05. †P < 0.01. ‡P < 0.001.
Parasitemia was analyzed at flow cytometry using parasites transfected with green fluorescent protein.19 Although the onset of parasitemia was slightly lower in PKC-θ–deficient mice on day 4, parasitemia was similar in both groups on days 5 to 7, and PKC-θ–deficient mice failed to control parasite growth thereafter, with parasitemia increased to 25% at day 14 and reaching 58% at day 21 (Figure 1B). PKC-θ–deficient mice died after day 21 with a high parasite load (Figure 1, A and B) and severe anemia (Figure 1C) in the absence of neurologic signs. However, a retrospective analysis revealed that PKC-θ–deficient mice that died before day 15 without neurologic signs exhibited a slightly higher parasite load at day 7 compared with PKC-θ–deficient mice that survived beyond day 15 [mean (SD), 13.7 (3.8) and 9.6 (3.7%), respectively], indicating that early death in PKC-θ–deficient mice might be associated with a higher parasite load. Therefore, our data demonstrate a critical role of PKC-θ in CM development, but no drastic effect on parasite growth.
Reduced Cerebral Ischemia by Noninvasive Imaging in Absence of PKC-θ
Both MRI and MRA were used to explore neuropathologic disease in the brain of PbA-infected mice. These noninvasive techniques enable investigation of neurologic disease and are used in human beings to study brain changes during CM.21–25 In murine experimental CM, MRI and MRA enable semiquantitative analysis of swelling and edema, focal ischemia, morphologic changes, and vascular blood flow. This could be indicative of microvascular disease of the brain due to obstruction of small vessels with parasitized erythrocyte and leukocyte sequestration and damaging of endothelial cells.21,26–28 Wild-type and PKC-θ–deficient mice were examined at day 7, when sensitive mice start developing acute CM. Typical MRI and MRA images of uninfected and infected mice are shown in Figure 2,A and B, respectively.
Figure 2.
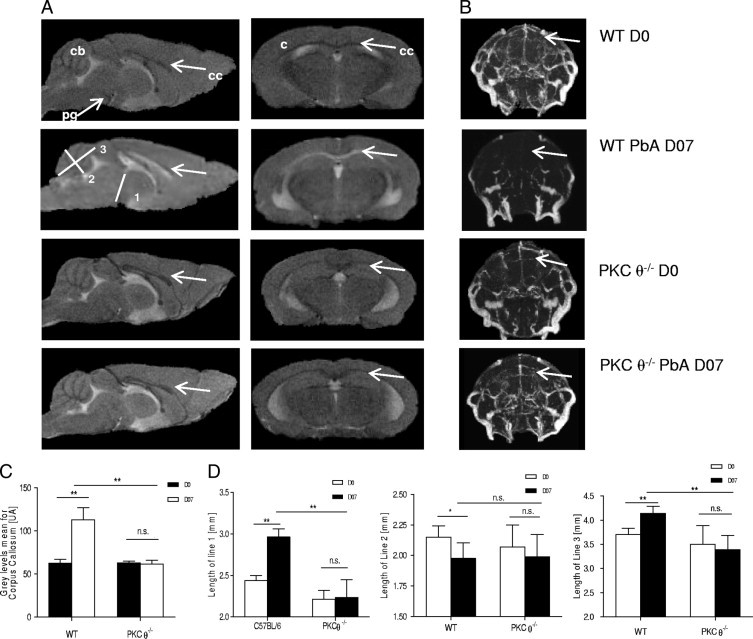
MRI analysis of brains of PbA-infected mice. Wild-type (WT) and PKC-θ–deficient mice were infected with 105PbA parasitized red blood cells as in Figure 1. A: MRI of uninfected and infected mice 7 days after infection. Left column: Mid-sagittal MRI. Right column: Typical axial MRI. Distinct anomalies are visible in WT infected mice; namely, edema formation is apparent as a bilateral hyperintense signal (arrows) at the corpus callosum, whereas WT uninfected mice and PKC-θ–deficient uninfected or infected mice do not show such alterations. MRIs are representative of 3 independent experiments: 6 PKC-θ−/− and 6 WT mice on day 0; and 6 PKC-θ−/− and 7 WT mice on day 7. B: At MRA, vascular blood flow perturbations are evident in WT mice 7 days after infection with 105PbA parasitized red blood cells, whereas WT uninfected mice and PKC-θ–deficient uninfected or infected mice seem unaffected. Arrows: Area demonstrating reduced brain vascularization in WT mice undergoing CM. Vascularization was not impaired in PbA-infected PKC-θ knockout mice. MRA images are representative of 3 independent experiments: 6 PKC-θ−/− and 6 WT mice on day 0; and 6 PKC-θ−/− and 7 WT mice on day 7. C: Corpus callosum mean gray level intensity for WT and PKC-θ–deficient uninfected mice (day 0, open bars) and infected mice (day 7, closed bars). Values are given as mean (SD) of 3 independent experiments: 6 PKC-θ−/− and 6 WT mice on day 0; and 6 PKC-θ−/− and 7 WT mice on day 7. D: Assessment of brain swelling in uninfected (day 0, open bars) and infected (day 7, closed bars) WT or PKC-θ–deficient mice in terms of measurement of distances 1 to 3 from the mid-sagittal MRIs: line 1 from the pituitary gland to the aqueduct of Sylvius; line 2, median line crossing the medial cerebellar nucleus; and line 3, medium line stemming from the cerebellar obex. Values are given as mean (SD) of 3 independent experiments: 6 PKC-θ−/− and 6 WT mice on day 0; and 6 PKC-θ−/− and 7 WT mice on day 7. c, cortex; cb, cerebellum; n.s., not significant; ml, corpus callosum; pg, pituary gland. *P < 0.05; **P < 0.01.
While wild-type mice exhibited distinct signs of ischemic brain damage on blood stage PbA infection, PKC-θ–deficient mice demonstrated normal MRI parameters without any signs of edema or infection, similar to findings in uninfected mice. PbA-infected wild-type mice exhibited distinct changes at MRI, observable as a bilateral hyperintense signal at the corpus callosum (Figure 2A). The gray level intensity in the corpus callosum was significantly increased after PbA infection in wild-type mice, whereas no significant variation was observed in PKC-θ–infected or PKC-θ–noninfected mice (Figure 2C). Brain edema or swelling was documented in infected wild-type mice during acute CM by measuring 3 distances in the brain (Figure 2D): (1) line 1 from the pituitary gland to the aqueduct of Sylvius, (2) median line 2 crossing the medial cerebellar nucleus, and (3) medium line 3 stemming from the cerebellar obex.26 Distances 1 and 3 were significantly increased and distance 2 was significantly reduced in infected wild-type mice compared with noninfected mice (Figure 2D), in agreement with data from Penet et al.26 In contrast, these metric parameters were not significantly altered in infected PKC-θ–deficient mice compared with noninfected PKC-θ–deficient or wild-type mice (Figure 2D).
MRA analysis showed that while wild-type mice demonstrated distinct vascular blood flow perturbations on blood stage PbA infection, with a dramatic reduction of blood flow (Figure 2B), PKC-θ–deficient mice exhibited normal MRA parameters after infection, without any signs of microvascular obstruction, similar to findings in uninfected mice (Figure 2B). Therefore, PKC-θ signaling is critically involved in development of microvascular obstruction and ischemic brain damage consequent to PbA infection.
Microvascular Cerebral But Not Pulmonary Lesions Are Absent in PKC-θ–Deficient Mice on PbA Infection
To validate functionally the data obtained at MRI, microvascular lesions in the brain were investigated. Microvessel obstruction and disruption, which leads to increased protein extravasation from the capillary bed, was quantified via Evans blue leakage into tissue. PbA-infected wild-type mice demonstrated strong vascular leakage of Evans blue, whereas PKC-θ–deficient mice exhibited no discoloration of the brain parenchyma (Figure 3A), with nonsignificant blue extravasation (Figure 3B), compared with noninfected wild-type mice and noninfected PKC-θ–deficient mice (data not shown), demonstrating the integrity of the blood-brain barrier.
Figure 3.
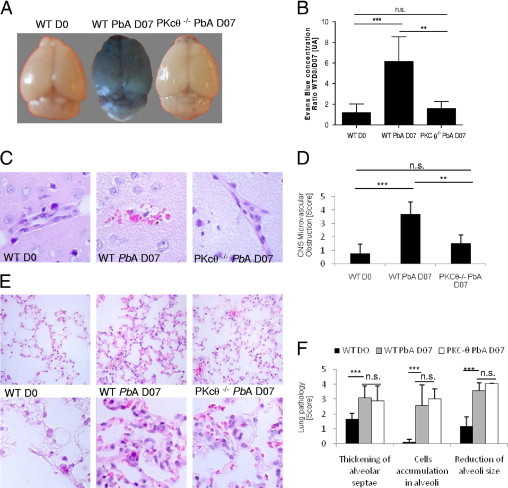
PKC-θ–deficient mice do not develop CM-associated brain vascular leakage. Mice were infected with 105PbA parasitized red blood cells as in Figure 1. A: Qualitative assessment of brain vascular leak using Evans blue extravasation. Wild-type (WT) mice with severe CM on day 7 after infection (WT PbA D07) exhibited blue discoloration of the brain, whereas uninfected control mice (WT D0) infected PKC-θ–deficient mice demonstrated no discoloration. Images are representative of 3 independent experiments: 12 WT mice on day 0; 7 WT mice on day 7; and 7 PKC-θ–deficient mice. B: Quantitative assessment of brain capillary permeability using Evans blue extravasation in formamide was measured via absorbance at 610 nm in brain extract. Values are given as mean (SD) from 3 different experiments as in A. C: Microvascular damage with mononuclear cell adhesion and perivascular hemorrhage is visible in the brain cortex of WT mice 7 days after infection but not in uninfected WT mice or PKC-θ–deficient mice. Microscopy sections (original magnification ×80) demonstrate representative H&E staining from 2 independent experiments: 8 WT mice on day 0; and 4 PKC-θ−/− and 7 WT mice on day 7 after infection. D: Severity of brain microvascular obstruction and local hemorrhage was assessed semiquantitatively from a whole-brain section between −5.64 and −6.84 from the bregma. Bar graphs show mean (SD) score from 2 independent experiments as in C. E: Lung alveolar damage of infected WT and PKC-θ–deficient mice on PbA infection. Magnifications are ×40 (upper panels) and ×100 (lower panels) of representative H&E staining from 2 independent experiments: 7 WT mice on day 0, and 4 PKC-θ−/− and 7 WT mice on day 7 after infection. F: Semiquantitative scores of thickening of alveolar septae, cell accumulation in alveoli, and reduction in size of alveoli. Bar graphs show mean (SD) scores from 2 independent experiments as in E. NS, not significant. *P < 0.05; **P < 0.01; ***P < 0.001.
Microscopically, the vascular blood flow perturbation was associated with microvascular lesions, with discrete damage to the small vessels and perivascular hemorrhage in the Virchov-Robin space and intravascular accumulation of mononuclear cells and infected and uninfected erythrocytes (Figure 3C), which was completely absent in PKC-θ–deficient mice. The severity of brain microvascular obstruction and local hemorrhage assessed semiquantitatively demonstrated a significant reduction in brain disease in PKC-θ–deficient mice (Figure 3D). Thus, data from the brain microscopic examination were in agreement with MRI results.
Pulmonary edema and acute respiratory distress are common complications in patients with CM.2 In the PbA CM model, mice exhibit signs of shock, most notably lung edema and inflammation, with vascular leakage.29 The pulmonary microvascular beds were investigated in PbA-infected wild-type and PKC-θ–deficient mice. The lungs of wild-type mice demonstrated congested capillaries of the alveolar septae, hemorrhage in alveoli, and interstitial edema after infection, similar to findings in PbA-infected PKC-θ–deficient mice (Figure 3E). Lung disease was investigated semiquantitatively, and no significant difference was observed after infection in wild-type and PKC-θ–deficient mice (Figure 3F), which suggests that the lung disease is PKC-θ–independent. Therefore, the data suggest that disruption of T-cell signaling in the absence of PKC-θ prevents brain microvascular disease, but not lung inflammation, induced by PbA blood stage infection.
Inactivation of PKC-θ Pathway Reduced Cerebral T-Cell Sequestration and Activation
Trafficking and recruitment of effector T lymphocytes into the brain are clearly necessary for development of disease associated with experimental CM.10,30 Leukocytes sequestered in the brain in wild-type and PKC-θ–deficient mice were quantified at a time point when sensitive mice develop neurologic symptoms of CM on PbA infection. Within CD3+ T cells, both CD4+ and CD8+ T-cell populations were strongly increased in the brain of PbA-infected wild-type mice compared with uninfected control mice (Figure 4, A–C). In contrast, T-cell recruitment was essentially absent in PbA-infected PKC-θ–deficient mice. Expression of CD69, a marker of T-cell activation, was up-regulated in CD8+ T cells on PbA infection in both groups (Figure 4E); however, marked CD69+ CD8+ T-cell recruitment was observed only in wild-type mice (Figure 4D). Thus, brain sequestration of activated effector T lymphocytes on PbA infection was drastically reduced in PKC-θ–deficient mice.
Figure 4.
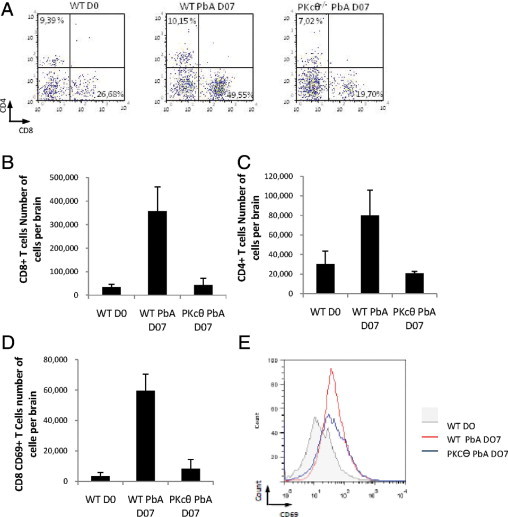
Reduced recruitment and activation of CD8+ lymphocytes in the brains of PbA-infected PKC-θ–deficient mice. Mice were infected with 105PbA parasitized red blood cells as in Figure 1. Flow cytometry analyses of brain-sequestered leukocytes from uninfected wild-type mice (WT NI), PbA-infected wild-type mice (WT PbA) with severe CM, and infected CM-resistant PKC-θ–deficient mice tested on the same day. A: Representative dot plots of CD4+ and CD8+ cells within the CD3+ cell populations, representative of 1 from 3 independent experiments. B and C: Bar graphs show absolute numbers of CD4+ and CD8+ T cells, respectively. Values are given as mean (SD) of 3 or 4 pools of 3 or 4 mice per group, and are representative of 1 from 3 independent experiments. D: Bar graph shows absolute numbers of CD8+CD69+ T cells representing the mean (SD) of 2 independent experiments: 3 to 4 pools of 3 to 4 mice per group. E: Representative histograms of CD69 staining in CD8+ T cells as in D, from 1 representative of 3 independent experiments. NS, not significant. *P < 0.01.
Topical distribution of the infiltrating T lymphocytes was determined via immunostaining brain sections (Figure 5A). CD3+, CD4+, and CD8+ T cells, present in brain of PbA-infected wild-type mice were largely absent in PbA-infected PKC-θ–deficient mice. The question then was whether adhesion molecule expression was influenced by the absence of PKC-θ signaling (Figure 5A). Expression of intercellular adhesion molecule-1 (CD54), as a marker of T-cell activation, was increased in PbA-infected wild-type mice, but less so in PKC-θ–deficient mice (Figure 5A). Intercellular adhesion molecule-1 expression was also up-regulated in the brain on PbA infection; however, the increase was much less marked in PKC-θ–deficient mice. Thus, microscopic examination confirmed the absence of sequestered activated effector T lymphocytes in the brain of PKC-θ–deficient mice on PbA infection.
Figure 5.
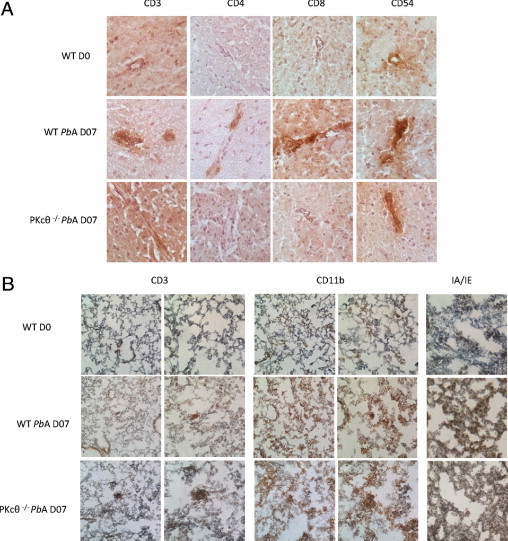
Immunohistologic staining shows reduced lymphocyte infiltration in the brains of PbA-infected PKC-θ–deficient mice. Mice were infected with 105PbA parasitized red blood cells as in Figure 1. A: Staining of CD3+, CD8+, CD4+, and CD54+ cells in brain vessels of infected wild-type (WT) mice that developed CM and in CM-resistant PKC-θ–deficient mice. Noninfected WT mice (D0) served as controls (micrograph ×40). Results are representative of 3 to 4 mice per group and are from 1 representative of 2 independent experiments. B: Staining of CD3+, CD11b+, and IA/IE+ cells in lung of infected WT mice and PKC-θ–deficient mice. Noninfected WT mice (D0) served as controls. Magnification ×20x (left panels) and ×40 (right panels) for CD3 and CD11b staining, and ×40 for IA/IE staining. Results are representative of 3 to 4 mice per group, and are from 1 representative of 2 independent experiments.
To further understand the absence of protection from lung inflammation in PKC-θ–deficient mice, immunohistologic staining of lung sections was performed (Figure 5B). Lung infiltration of CD3+ T cells was not increased 7 days after PbA infection in either wild-type or PKC-θ–deficient mice. In contrast, pulmonary infiltration of macrophages, demonstrated at CD11b staining, was largely increased during PbA infection, as was expression of major histocompatibility complex class II (IA and IE) (Figure 5B), indicating strong activation of lung tissue cells in both wild-type and PKC-θ–deficient mice. Therefore, PbA-induced lung disease is due to strong, PKC-θ–independent recruitment and activation of a non–T-cell leukocyte subset such as macrophages.
Hematologic Alterations in PKC-θ–Deficient Mice
Thrombocytopenia and anemia are hallmarks of the course of PbA infection in mice.21,31 Therefore, hematologic parameters were investigated in naïve and PbA-infected wild-type and PKC-θ–deficient mice. Hematologic analyses revealed similar reductions in platelet counts in wild-type and PKC-θ–deficient mice at 7 days after PbA infection (Figure 6A), which suggests that thrombocytopenia in PbA-infected PKC-θ–deficient mice is not an indicator of CM sensitivity in this model.
Figure 6.
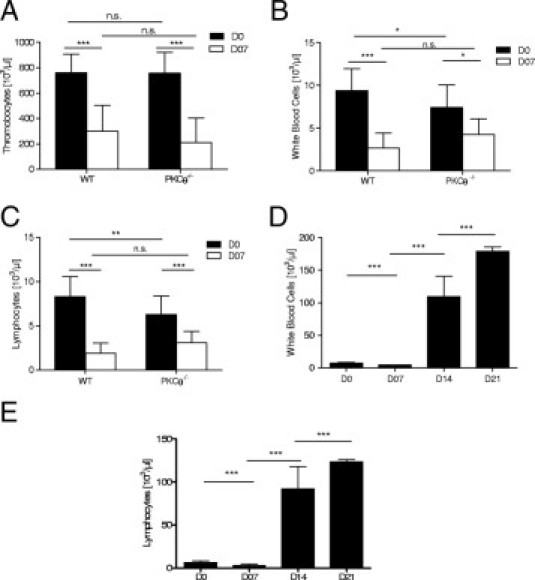
Hematologic alterations in PKC-θ–deficient mice. Mice were infected with 105PbA parasitized red blood cells as in Figure 1. Platelet (A), total white blood cell (B), and lymphocyte (C) counts were determined in peripheral blood of naïve and 7-day infected wilt-type (WT) and PKC-θ–deficient mice. Results are mean (SD) from 4 independent experiments: 15 PKC-θ−/− and 21 WT mice on day 0; and 21 PKC-θ−/− and 6 WT mice on day 7. Kinetics of total white blood cell (D) and lymphocyte (E) counts are shown up to day 7 for WT mice and up to day 21 after infection for CM-resistant PKC-θ–deficient mice. Values are given as mean (SD) from 4 independent experiments: 15 PKC-θ−/− and 21 WT mice on day 0; 21 PKC-θ−/− and 6 WT mice on day 7; and 4 and 3 PKC-θ−/− mice on days 14 and 21. NS, not significant. *P < 0.05; **P < 0.01; ***P < 0.001.
After PbA infection for 7 days, wild-type mice show a reduction of circulating white blood cells (Figure 6B), largely attributed to a decrease in lymphocytes (Figure 6D). Although the total white blood cell count was slightly lower in naïve PKC-θ–deficient mice compared with wild-type mice, there was a decrease in white blood cell and lymphocyte counts in PKC-θ–deficient mice at 7 days after PbA infection (Figure 6, B and D). However, white blood cell and lymphocyte counts increased dramatically to greater than 100 × 103/μL in CM-resistant PKC-θ–deficient mice on days 14 to 21 after infection (Figure 6, C and E). Although no significant anemia developed in wild-type mice at 7 days after infection (Figure 6F), PKC-θ–deficient mice developed dramatic anemia at 2 and 3 weeks after PbA infection, reaching a mean (SD) of 2.07 (0.54) × 106 red blood cells per microliter (Figure 6G).
Therefore, resistance of PKC-θ–deficient mice to CM development was not associated with thrombocytopenia. The PbA-infected PKC-θ–deficient mice that were resistant to CM and developed a high degree of parasitemia died at 3 weeks with severe anemia and drastic leukocytosis.
Discussion
PKC-θ signaling is critical for development of fatal experimental CM. Using PKC-θ–deficient mice, it was demonstrated that disruption of PKC-θ signaling abrogated both the T-cell recruitment into the brain and the cerebral microvascular disease that lead to fatal CM. However, PKC-θ signaling has little effect on the associated pulmonary inflammation and edema.
T-cell sequestration and activation are crucial steps in development of CM after PbA infection.8–10 Suidan et al30 and Baier et al32 demonstrated the role of Th1 response in initiation of central nervous system vascular permeability as a CD8+ T cell perforin-dependent process. CD8+ T cells may mediate circulatory shock, vascular permeability changes, and edema in the brain during PbA-infection.29 CD8+ T cells are the primary effector cells of CM in the PbA mouse model, although their precise mechanism of action is not known.8 PKC-θ is expressed in T cells and is a key component of T-cell receptor signaling and activation.12,13,33,34 However, PKC-θ–dependent pathways are differently required in T-cell activation and are especially critical for development of Th2 but not Th1 cells.12,13 PKC-θ is not essential for T-cell activation leading to viral clearance.16 PKC-θ–deficient mice develop less severe neurologic signs in T-cell–dependent experimental autoimmune encephalomyelitis models, with delayed CD4+ T-cell recruitment and modulation of Th1 and Th17 cytokine profiles.35,36 Moreover, they developed less severe Th1-dependent responses in antigen-induced arthritis.37
To address the role of PKC-θ in CM pathogenesis, PKC-θ–deficient mice were investigated for several aspects of the pathophysiology of CM development. The present study demonstrated protection of PKC-θ–deficient mice against CM after infection with PbA, in agreement with the recent study of Ohayon et al,38 who also demonstrated that PKC-θ has an effect on CM development; however, dissociated were the CM and the lung disease that are not affected by PKC-θ deficiency. Most PKC-θ–deficient mice survived beyond day 15 without signs of CM. The early death observed in 40% of PKC-θ–deficient mice was not associated with any of the classic visible CM symptoms observed in wild-type mice such as loss of gripping reflex, and might be related to a higher parasite load rather than to experimental CM development, although the explanation for the cause of early death remains speculative. However, the propagation of PbA infection in erythrocytes was PKC-θ–independent. Indeed, CM-resistant PKC-θ–deficient mice developed a high parasite load together with severe anemia and leukocytosis, which were fatal after 3 weeks of infection, whereas wild-type mice developed fatal CM at 7 days after PbA infection. This result was associated with unaffected cerebral perfusion as assessed at MRA. There was a correlation of MRA data and microscopic observations. In vivo imaging data correlated with absence of microvascular disease, accumulation of mononuclear cells, vascular leak, hemorrhage, and inflammation in the brain of PbA-infected PKC-θ–deficient mice.
However, PKC-θ expression is not restricted to T cells. It is also expressed in natural killer cells39; in platelets, where it may modulate activation and thrombus formation40; in skeletal muscle cells41; in the endothelium of muscle resistance arteries, where it modulates vascular tone through insulin-induced vasoconstriction42; and in hypothalamus tissue, where it contributes to insulin signaling.43 PKC-θ could, therefore, exert multifold influence on development of CM, through platelet activation and vascular tone modulation, beyond a direct effect on T cells.
Specifically investigated was whether absence of PKC-θ affected T-cell recruitment during PbA-induced CM. Flow cytometric analysis of thymus, spleen and lymph node cells revealed no abnormalities in PKC-θ–deficient mice.18 However, after PbA infection, analysis of brain-sequestered cells clearly showed a sharp reduction in brain-infiltrating CD4+, and more drastically of CD8+ and CD8+ CD69+ T cells in CM-resistant PKC-θ–deficient mice. These results are in agreement with those of Saibil et al,44 who observed that the absence of PKC-θ strongly affected the primary activated CD8+ T cells, whereas CD4+ T cells exhibited only a moderate survival defect. Immunohistologic analysis confirmed reduced recruitment and activation of CD4+ and CD8+ T cells in the brains of PKC-θ–deficient mice. The reduced T-cell sequestration was associated with a slight reduction in expression of the activation marker CD54 in the brain vessels of PKC-θ–deficient mice. Defective T-cell activation and recruitment to the brain may, therefore, be responsible for protection of PKC-θ–deficient mice from CM development.
CD8+ T cells were central not only to CM8–10,21 but also to the CM-associated “shock” features including vascular leakage of lung, kidney, and brain; metabolic acidosis; and lowered mean arterial pressure.29 CD8+ T-cell depletion completely prevented neurologic signs of CM, significantly reduced the symptoms of circulating shock including lung vascular leakage, and restored cardiac output.29 In the present study, however, there was little CD3+ T-cell infiltration in the lungs from infected wild-type or PKC-θ–deficient mice, whereas there was a strong CD11b+ macrophage infiltration with strong major histocompatibility complex class II expression in both wild-type and PKC-θ–deficient mice. Furthermore, the pulmonary microvascular damage with alveolitis, and plasma and erythrocyte leakage in the alveolar space was similar in wild-type and PKC-θ–deficient mice. Thus, PbA-induced brain microvascular damage and inflammation are primarily PKC-θ–dependent, whereas PbA-induced lung microvascular damage, edema, and alveolitis are largely PKC-θ–independent.
Therefore, PKC-θ seems to be a critical enzyme that regulates selective T-cell functions and may represent an attractive drug target for treatment of early CM. In conclusion, data from the present study suggest that accumulation of pathogenic CD8+ T cells induced by PbA infection is PKC-θ–dependent and that PKC-θ signaling is required and nonredundant for development of microvascular disease resulting in fatal CM. However, the lung disease can be dissociated from CM. PKC-θ deficiency does not protect against PbA-induced lung microvascular damage, edema, and alveolitis.
Acknowledgments
We thank Manfred Kopf (Molecular Biomedicine, ETH Zurich, Switzerland) for the kind gift of the PKC-θ–deficient mice, and Drs. Maitre and Kerdraon, pathologists at the Regional Hospital Center of Orléans (CHRO, Orléans, France), for expert evaluation of lung and brain histologic analyses.
Footnotes
Supported by CNRS, the interdisciplinary program MIE from the French Research Minister, and European FP6 MPCM project.
References
- 1.Miller L.H., Good M.F., Milon G. Malaria pathogenesis. Science. 1994;264:1878–1883. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 2.Idro R., Jenkins N.E., Newton C.R. Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol. 2005;4:827–840. doi: 10.1016/S1474-4422(05)70247-7. [DOI] [PubMed] [Google Scholar]
- 3.Hunt N.H., Grau G.E. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 4.Grau G.E., Taylor T.E., Molyneux M.E., Wirima J.J., Vassalli P., Hommel M., Lambert P.H. Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med. 1989;320:1586–1591. doi: 10.1056/NEJM198906153202404. [DOI] [PubMed] [Google Scholar]
- 5.Kern P., Hemmer C.J., Van Damme J., Gruss H.J., Dietrich M. Elevated tumor necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. Am J Med. 1989;87:139–143. doi: 10.1016/s0002-9343(89)80688-6. [DOI] [PubMed] [Google Scholar]
- 6.Kwiatkowski D., Hill A.V., Sambou I., Twumasi P., Castracane J., Manogue K.R., Cerami A., Brewster D.R., Greenwood B.M. TNF concentration in fatal cerebral, nonfatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336:1201–1204. doi: 10.1016/0140-6736(90)92827-5. [DOI] [PubMed] [Google Scholar]
- 7.Fry A.E., Auburn S., Diakite M., Green A., Richardson A., Wilson J., Jallow M., Sisay-Joof F., Pinder M., Griffiths M.J., Peshu N., Williams T.N., Marsh K., Molyneux M.E., Taylor T.E., Rockett K.A., Kwiatkowski D.P. Variation in the ICAM1 gene is not associated with severe malaria phenotypes. Genes Immun. 2008;9:462–469. doi: 10.1038/gene.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Renia L., Potter S.M., Mauduit M., Rosa D.S., Kayibanda M., Deschemin J.C., Snounou G., Gruner A.C. Pathogenic T cells in cerebral malaria. Int J Parasitol. 2006;36:547–554. doi: 10.1016/j.ijpara.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 9.Schofield L., Grau G.E. Immunological processes in malaria pathogenesis. Nat Rev Immunol. 2005;5:722–735. doi: 10.1038/nri1686. [DOI] [PubMed] [Google Scholar]
- 10.Belnoue E., Kayibanda M., Vigario A.M., Deschemin J.C., van Rooijen N., Viguier M., Snounou G., Renia L. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol. 2002;169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 11.Vyas Y.M., Maniar H., Dupont B. Cutting edge: differential segregation of the SRC homology 2-containing protein tyrosine phosphatase-1 within the early NK cell immune synapse distinguishes noncytolytic from cytolytic interactions. J Immunol. 2002;168:3150–3154. doi: 10.4049/jimmunol.168.7.3150. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhary D., Kasaian M. PKC-theta: a potential therapeutic target for T-cell-mediated diseases. Curr Opin Invest Drugs. 2006;7:432–437. [PubMed] [Google Scholar]
- 13.Marsland B.J., Soos T.J., Spath G., Littman D.R., Kopf M. Protein kinase C theta is critical for the development of in vivo T helper (Th) 2 cell but not Th1 cell responses. J Exp Med. 2004;200:181–189. doi: 10.1084/jem.20032229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava K.K., Batra S., Sassano A., Li Y., Majchrzak B., Kiyokawa H., Altman A., Fish E.N., Platanias L.C. Engagement of protein kinase C-theta in interferon signaling in T-cells. J Biol Chem. 2004;279:29911–29920. doi: 10.1074/jbc.M401997200. [DOI] [PubMed] [Google Scholar]
- 15.Marsland B.J., Nembrini C., Schmitz N., Abel B., Krautwald S., Bachmann M.F., Kopf M. Innate signals compensate for the absence of PKC-(theta) during in vivo CD8(+) T cell effector and memory responses. Proc Natl Acad Sci USA. 2005;102:14374–14379. doi: 10.1073/pnas.0506250102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannoni F., Lyon A.B., Wareing M.D., Dias P.B., Sarawar S.R. Protein kinase C theta is not essential for T-cell–mediated clearance of murine gammaherpesvirus 68. J Virol. 2005;79:6808–6813. doi: 10.1128/JVI.79.11.6808-6813.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsland B.J., Nembrini C., Grun K., Reissmann R., Kurrer M., Leipner C., Kopf M. TLR ligands act directly upon T cells to restore proliferation in the absence of protein kinase C-theta signaling and promote autoimmune myocarditis. J Immunol. 2007;178:3466–3473. doi: 10.4049/jimmunol.178.6.3466. [DOI] [PubMed] [Google Scholar]
- 18.Sun Z., Arendt C.W., Ellmeier W., Schaeffer E.M., Sunshine M.J., Gandhi L., Annes J., Petrzilka D., Kupfer A., Schwartzberg P.L., Littman D.R. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature. 2000;404:402–407. doi: 10.1038/35006090. [DOI] [PubMed] [Google Scholar]
- 19.Franke-Fayard B., Trueman H., Ramesar J., Mendoza J., van der Keur M., van der Linden R., Sinden R.E., Waters A.P., Janse C.J. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 20.Rudin W., Eugster H.P., Bordmann G., Bonato J., Muller M., Yamage M., Ryffel B. Resistance to cerebral malaria in tumor necrosis factor-alpha/beta–deficient mice is associated with a reduction of intercellular adhesion molecule-1 up-regulation and T helper type 1 response. Am J Pathol. 1997;150:257–266. [PMC free article] [PubMed] [Google Scholar]
- 21.Togbe D., de Sousa P.L., Fauconnier M., Boissay V., Fick L., Scheu S., Pfeffer K., Menard R., Grau G.E., Doan B.T., Beloeil J.C., Renia L., Hansen A.M., Ball H.J., Hunt N.H., Ryffel B., Quesniaux V.F. Both functional LTbeta receptor and TNF receptor 2 are required for the development of experimental cerebral malaria. PLoS ONE. 2008;3:e2608. doi: 10.1371/journal.pone.0002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kampfl A.W., Birbamer G.G., Pfausler B.E., Haring H.P., Schmutzhard E. Isolated pontine lesion in algid cerebral malaria: clinical features, management, and magnetic resonance imaging findings. Am J Trop Med Hygiene. 1993;48:818–822. doi: 10.4269/ajtmh.1993.48.818. [DOI] [PubMed] [Google Scholar]
- 23.Looareesuwan S., Wilairatana P., Krishna S., Kendall B., Vannaphan S., Viravan C., White N.J. Magnetic resonance imaging of the brain in patients with cerebral malaria. Clin Infect Dis. 1995;21:300–309. doi: 10.1093/clinids/21.2.300. [DOI] [PubMed] [Google Scholar]
- 24.Cordoliani Y.S., Sarrazin J.L., Leveque C., Goasdoue P., Sabbah P. Imaging of encephalic parasitic diseases [in French] J Neuroradiol. 1998;25:290–305. [PubMed] [Google Scholar]
- 25.Patankar T.F., Karnad D.R., Shetty P.G., Desai A.P., Prasad S.R. Adult cerebral malaria: prognostic importance of imaging findings and correlation with postmortem findings. Radiology. 2002;224:811–816. doi: 10.1148/radiol.2243010588. [DOI] [PubMed] [Google Scholar]
- 26.Penet M.F., Viola A., Confort-Gouny S., Le Fur Y., Duhamel G., Kober F., Ibarrola D., Izquierdo M., Coltel N., Gharib B., Grau G.E., Cozzone P.J. Imaging experimental cerebral malaria in vivo: significant role of ischemic brain edema. J Neurosci. 2005;25:7352–7358. doi: 10.1523/JNEUROSCI.1002-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoelcker B., Hehlgans T., Weigl K., Bluethmann H., Grau G.E., Mannel D.N. Requirement for tumor necrosis factor receptor 2 expression on vascular cells to induce experimental cerebral malaria. Infect Immun. 2002;70:5857–5859. doi: 10.1128/IAI.70.10.5857-5859.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engwerda C.R., Mynott T.L., Sawhney S., De Souza J.B., Bickle Q.D., Kaye P.M. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J Exp Med. 2002;195:1371–1377. doi: 10.1084/jem.20020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang W.L., Jones S.P., Lefer D.J., Welbourne T., Sun G., Yin L., Suzuki H., Huang J., Granger D.N., van der Heyde H.C. CD8(+)-T-cell depletion ameliorates circulatory shock in Plasmodium berghei-infected mice. Infect Immun. 2001;69:7341–7348. doi: 10.1128/IAI.69.12.7341-7348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nitcheu J.B.O., Combadiere C., Tefit M., Seilhean D., Mazier D., Combadiere B. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol. 2003;170:22221–22228. doi: 10.4049/jimmunol.170.4.2221. [DOI] [PubMed] [Google Scholar]
- 31.Grau GE P.P., Gretener D., Vesin C., Lambert P.H. Immunopathology of thrombocytopenia in experimental malaria. Immunology. 1988;65:501–506. [PMC free article] [PubMed] [Google Scholar]
- 32.Suidan G.L., McDole J.R., Chen Y., Pirko I., Johnson A.J. Induction of blood brain barrier tight junction protein alterations by CD8 T cells. PLoS ONE. 2008;3:e3037. doi: 10.1371/journal.pone.0003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baier G., Telford D., Giampa L., Coggeshall K.M., Baier-Bitterlich G., Isakov N., Altman A. Molecular cloning and characterization of PKC theta, a novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J Biol Chem. 1993;268:4997–5004. [PubMed] [Google Scholar]
- 34.Isakov N., Altman A. Protein kinase C(theta) in T cell activation. Annu Rev Immunol. 2002;20:761–794. doi: 10.1146/annurev.immunol.20.100301.064807. [DOI] [PubMed] [Google Scholar]
- 35.Salek-Ardakani S., So T., Halteman B.S., Altman A., Croft M. Protein kinase Ctheta controls Th1 cells in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:7635–7641. doi: 10.4049/jimmunol.175.11.7635. [DOI] [PubMed] [Google Scholar]
- 36.Tan S.L., Zhao J., Bi C., Chen X.C., Hepburn D.L., Wang J., Sedgwick J.D., Chintalacharuvu S.R., Na S. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase C theta–deficient mice. J Immunol. 2006;176:2872–2879. doi: 10.4049/jimmunol.176.5.2872. [DOI] [PubMed] [Google Scholar]
- 37.Healy A.M., Izmailova E., Fitzgerald M., Walker R., Hattersley M., Silva M., Siebert E., Terkelsen J., Picarella D., Pickard M.D., LeClair B., Chandra S., Jaffee B. PKC-theta–deficient mice are protected from Th1-dependent antigen-induced arthritis. J Immunol. 2006;177:1886–1893. doi: 10.4049/jimmunol.177.3.1886. [DOI] [PubMed] [Google Scholar]
- 38.Ohayon A., Golenser J., Sinay R., Tamir A., Altman A., Pollack Y., Isakov N. PKC(theta) deficiency increases resistance of C57BL/6J mice to Plasmodium berghei infection-induced cerebral malaria. Infect Immun. 2010;78:4195–420578. doi: 10.1128/IAI.00465-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vyas Y.M., Maniar H., Dupont B. Cutting edge: differential segregation of the SRC homology 2–containing protein tyrosine phosphatase–1 within the early NK cell immune synapse distinguishes noncytolytic from cytolytic interactions. J Immunol. 2002;168:3150–3154. doi: 10.4049/jimmunol.168.7.3150. [DOI] [PubMed] [Google Scholar]
- 40.Harper M.T., Poole A.W. Diverse functions of protein kinase C isoforms in platelet activation and thrombus formation. J Thromb Haemost. 2010;8:454–462. doi: 10.1111/j.1538-7836.2009.03722.x. [DOI] [PubMed] [Google Scholar]
- 41.Hilgenberg L., Miles K. Developmental regulation of a protein kinase C isoform localized in the neuromuscular junction. J Cell Sci. 1995;108(pt 1):51–61. doi: 10.1242/jcs.108.1.51. [DOI] [PubMed] [Google Scholar]
- 42.Bakker W., Sipkema P., Stehouwer C.D., Serne E.H., Smulders Y.M., van Hinsbergh V.W., Eringa E.C. Protein kinase C theta activation induces insulin-mediated constriction of muscle resistance arteries. Diabetes. 2008;57:706–713. doi: 10.2337/db07-0792. [DOI] [PubMed] [Google Scholar]
- 43.Benoit S.C., Kemp C.J., Elias C.F., Abplanalp W., Herman J.P., Migrenne S., Lefevre A.L., Cruciani-Guglielmacci C., Magnan C., Yu F., Niswender K., Irani B.G., Holland W.L., Clegg D.J. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J Clin Invest. 2009;119:2577–2589. doi: 10.1172/JCI36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saibil S.D., Jones R.G., Deenick E.K., Liadis N., Elford A.R., Vainberg M.G., Baerg H., Woodgett J.R., Gerondakis S., Ohashi P.S. CD4+ and CD8+ T cell survival is regulated differentially by protein kinase Ctheta, c-Rel, and protein kinase B. J Immunol. 2007;178:2932–2939. doi: 10.4049/jimmunol.178.5.2932. [DOI] [PubMed] [Google Scholar]