

Abstract
Melanoma is the most deadly tumor of the skin, and systemic therapies for the advanced stage are still limited. Recent genetic analyses have revealed the molecular diversity of melanoma and potential therapeutic targets. By screening a cohort of 142 primary nonepithelial tumors, we discovered that about 10% of melanoma cases (4/39) harbored an IDH1 or IDH2 mutation. These mutations were found to coexist with BRAF or KIT mutation, and all IDH1 mutations were detected in metastatic lesions. BRAF-mutated melanoma cells, additionally expressing the cancer-related IDH1 mutant, acquired increased colony-forming and in vivo growth activities and showed enhanced activation of the MAPK and STAT3 pathways. Genome-wide gene expression profiling demonstrated that mutant IDH1 affected the expression of a set of genes. Especially, it caused the induction of growth-related transcriptional regulators (Jun, N-myc, Atf3) and the reduction of Rassf1 and two dehydrogenase genes (Dhrs1 and Adh5), which may be involved in the carcinogenesis of IDH1-mutated tumors. Our analyses demonstrate that IDH1 mutation works with other oncogenic mutations and could contribute to the metastasis in melanoma.
Melanoma is the most malignant tumor of the skin, and the median survival rate of patients with metastatic tumors is less than 1 year.1 Although the incidence of melanoma has been increasing around the world, systemic therapies for the advanced stage are still limited.2 Recent studies have provided a clearer picture of the molecular events leading to melanoma development and progression.3,4 Since the identification of prevalent activating mutations of BRAF kinase,5 further molecular studies have clarified the role of this pathway and others in melanomagenesis.6 Recent genetic investigations have also demonstrated specific genotype–phenotype correlations that would be potentially informative in the context of the molecular subclassification of melanoma and therapeutic target molecules.7 For example, the c-kit gene mutations have been frequently reported in acral lentiginous/mucosal melanomas and are associated with better responsiveness to the inhibitor, imatinib.8–10
Recently, unbiased whole-exon resequencing analysis of glioblastoma multiforme has revealed recurrent mutation of the two IDH (isocitrate dehydrogenase) isoforms, IDH1 and IDH2.11 Subsequent analysis showed that these mutations are frequent in glioma and associated with better prognosis12,13; furthermore, they have also been detected in a subset (about 8% to 16%) of acute myeloid leukemia (AML).14–17 These enzymes convert isocitrate to α-ketoglutarate (α-KG) with concurrent reduction of NADPH, but IDH1 is localized in the cytosol18 whereas IDH2 is localized in mitochondria.19 Mutations of the two genes affect the residues responsible for hydrophilic interactions with the substrate, and have been shown to impair the enzymatic activity, and therefore they are considered to be loss-of-function alleles.12 However, because the mutations are clustered in specific residues and only detected as heterozygous alleles, it could also be hypothesized that they are gain-of-function mutations. Recent milestone studies have revealed that mutant IDH1 or IDH2 acquires a new gain-of-function activity that results in reduction of α-ketoglutarate to 2-hydroxyglutarate (2HG) in glioma and leukemia, suggesting that IDH1/2 mutations could be gain-of-function alterations.20–22 Although accumulation of 2HG is associated with a risk of brain tumors including glioma,23,24 the significance of such metabolic change in carcinogenesis remains largely unknown.
Materials and Methods
Clinical Samples and DNA Extraction
Surgical or autopsied specimens (92 cases of sarcoma, 39 cases of melanoma, and 11 cases of mesothelioma) were obtained from patients who were diagnosed and underwent surgery at the National Cancer Center Hospital, Tokyo, Japan. Tumor cells and corresponding lymphocytes or normal skin tissue were dissected out under a microscope from methanol-fixed paraffin-embedded tissues, and the DNA was extracted. High molecular weight DNA was extracted from 13 melanoma cell lines as described previously.25 The study protocol for analysis of clinical samples was approved by the institutional review board of the National Cancer Center.
PCR and Sequence Analysis
We amplified exon 4 of the IDH1 gene, exon 4 of the IDH2 gene, exon 15 of the BRAF gene, exons 2 and 3 (covering codons 12, 13, and 61) of the NRAS gene, exon 3 of the CTNNB1 gene, and exons 11, 13, and 17 of the KIT gene by PCR using High Fidelity Taq polymerase (Roche Diagnostic, Basel, Switzerland) as described.26 The primers used in this study are IDH1-EX4F: 5′-AGAGAATCGTGATGCCACCAACG-3′, IDH1-EX4R: 5′-GCATAATGTTGGCGTCAAATGTGC-3′, IDH2-EX4F: 5′-ACATGCAAAATCACATTATTGCC-3′, IDH2-EX4R: 5′-CAAGTTGGAAATTTCTGGGCCATG-3′, BRAF-EX15F: 5′-AAACTCTTCATAATGCTTGCTCTG-3′, BRAF-EX15R: 5′-TAGCCTCAATTCTTACCATCCAC-3′, NRAS-EX2F: 5′-GATGTGGCTCGCCAATTAACCCTG-3′, NRAS-EX2R: 5′-GACAAGTGAGAGACAGGATCAGG-3′, NRAS-EX3F: 5′-TTACCCTCCACACCCCCAGGATTC-3′, NRAS-EX3R: 5′-AATGCTCCTAGTACCTGTAGAGG-3′, KIT-EX11F: 5′-CCAGAGTGCTCTAATGACTGAGAC-3′, KIT-EX11R: 5′-AAAGGTGACATGGAAAGCCCCTG-3′, KIT-EX13F: 5′-AGATGCTCAAGCGTAAGTTCCTG-3′, KIT-EX13R: 5′-AATAAAAGGCAGCTTGGACACGGC-3′, KIT-EX17F: 5′-GGTTTTCTTTTCTCCTCCAACCT-3′, KIT-EX17R: 5′-GTGATATCCCTAGACAGGATTTAC-3′, CTNNB1-EX3F: 5′-TATAGCTGATTTGATGGAGTTGG-3′, CTNNB1-EX3R: 5′-GCTACTTGTTCTTGAGTGAAGGAC-3′. All PCR products were purified (QIAquick PCR purification kit; QIAGEN, Hamburg, Germany) and analyzed by sequencing (Big Dye sequencing kit; Applied Biosystems, Carlsbad, CA).
In Vitro Biological Assays
FLAG-tagged IDH1 full-length cDNA was amplified from human normal liver cDNA using reverse transcription (RT)-PCR and subcloned into a mammalian expression plasmid (Invitrogen, Carlsbad, CA). The R132H mutant was generated by site-directed mutagenesis (Quickchange; Stratagene, Santa Clara, CA). All plasmids were validated by sequencing. G361 cells were obtained from Japanese Collection of Research Bioresources (Sennan-shi, Japan) and maintained in DMEM supplemented with 10% fetal bovine serum. Linearized plasmid was transfected by lipofectamine (Invitrogen), and stable clones were isolated after Zeocin (Invitrogen) selection. Cell proliferation was measured using the 96-well plate format by MTS assay using Cell Titer 96 AQueous One Solution Reagent (Promega, Madison, WI).26 Colony formation assay and migration assay were performed as described.27 To measure ROS accumulation, cells were stained with 5- (and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Molecular Probes, Eugene, OR), and fluorescence-activated cell sorting (FACS) analysis was performed using FACScalibur (BD Biosciences, San Jose, CA) as instructed by the manufacturer.
In Vivo Tumorigenesis Analyses
For assessment of in vivo tumorigenicity, 1 × 106 cells were subcutaneously transplanted into the trunks of nude mice. After 12 weeks, the mice were sacrificed, and the number of subcutaneous tumors as well as metastasis in other organs was examined. The mice were kept at the Animal Care and Use Facilities of the National Cancer Center under specific pathogen-free conditions, and all experiments were approved by the institutional Animal Care and Ethics Committee.
Immunoblot Analysis
For protein extraction, we used a slightly modified buffer (10 mmol/L Tris-HCl [pH 7.5], 175 mmol/L NaCl, 5 mmol/L EDTA, 0.5% Triton-X, 0.5% NP-40) with a proteinase inhibitor cocktail (Roche), and the immunoblotting procedure was preformed as described previously.26 The antibodies used in this study are anti-FLAG peptide (clone M2; Sigma-Aldrich, St. Louis, MO), anti-MAPK, phospho-MAPK (pT202/pY204), AKT, phospho-AKT (pS473), phospho-STAT3 (pY705), p70S6K, phospho-p70S6K (pT389) antibodies (Cell Signaling Technologies, Danvers, MA), and anti-STAT3 antibody (BD Biosciences).
Gene Expression Profiling and Quantitative RT-PCR
From subconfluent G361 clones, total RNA was extracted using an RNAeasy kit (QIAGEN). Ten micrograms of total RNA was reverse-transcribed by MMLV-RT, and a Cy3-labeled cRNA probe was synthesized using T7 RNA polymerase and hybridized with a microarray covering the whole human genome (Whole Human Genome Oligo Microarray, G4112F; Agilent Technologies, Santa Clara, CA). All sample were analyzed in duplicate. After washing, the microarray was scanned by the DNA microarray scanner (Agilent Technologies). Data were normalized and statistical significance was measured by t-test with multiple testing correction (Benjamini and Hochberg false discovery rate) using GeneSpring software (Agilent Technologies).27 Quantitative RT-PCR was performed in triplicate and evaluated using universal probes for each amplicon and the LightCycler system (Roche). Primers designed by ProbeFinder (Version 2.45; Roche). The relative expression of each gene was determined by comparison with that of GAPDH.
Results
IDH1/2 Mutations in Melanoma
Since previous mutation analyses have reported that IDH1 mutation is rare in epithelial cancers in comparison to glioma and leukemia,28,29 we searched for the IDH1 gene mutation in a cohort of primary tumors of nonepithelial origin (92 sarcomas, 39 melanomas, and 11 malignant mesotheliomas). Our melanoma cohort included 17 metastatic cases. After screening these 142 tumors, we found 2 melanoma cases harboring heterozygous IDH1 mutation (R132C and R132H, 2/39) (Figure 1 and Table 1). These mutations affected exactly the same residue as that reported for glioma and AML.11,12,16 No IDH1 mutation was detected in sarcoma and mesothelioma cases. We then screened IDH2 mutation in the same cohort and found two heterozygous mutations (G171D and P158T) that affected well-conserved residues among species in two MM cases (Figure 1). Especially G171 is located next to the most frequently altered residue (R172), but its mutation in cancer has not been reported previously.12,14–17,28,29 These IDH1/2 mutations were not detected in the corresponding normal tissues, and in total, we detected four somatic IDH1/2 mutations out of 39 melanoma cases (10.3%). Three out of four IDH1/2 mutations occurred in either mucosal or acral lentiginous subtype, and IDH1 mutation was detected only in metastatic lesions (Table 1).
Figure 1.

IDH1 and IDH2 mutations in melanoma. Amino acid alignments of IDH1 and IDH2 proteins among human, mouse, rat, and chicken homologues are shown in the top panels. Sequence chromatographies of the IDH1 and IDH2 genes in primary melanomas (T) and corresponding normal (N) tissues are shown in the bottom panels. Arrows indicate the heterozygous mutation in tumor samples. Note that mutated amino acids residues are well conserved among species.
Table 1.
Clinicopathological and Mutation Profile of Melanoma Cases
| Case | Primary (P) or metastatis (M) site | Primary site | Subtype | IDH1 | IDH2 | BRAF | NRAS | KIT | CTNNB1 |
|---|---|---|---|---|---|---|---|---|---|
| MM-1 | M, left thigh | Face | nd | p.R132C, hetero | p.V600E, hetero | ||||
| MM-27 | M, liver | Anal | nd | p.R132H, hetero | p.K642E, homo | ||||
| MM-38 | P | Abdomen | nd | p.P158T, hetero | p.V600E, hetero | ||||
| MM-3 | P | Toe | NM | p.G171D, hetero | |||||
| MM-24 | M, pancreas | Finger | ALM | p.D594N, hetero | |||||
| MM-4 | P | Esophagus | nd | p.V600E, hetero | |||||
| MM-7 | P | Skin | ALM | p.V600E, hetero | |||||
| MM-12 | P | Esophagus | nd | p.V600E, hetero | |||||
| MM-14 | M, brain | Forearm | nd | p.V600E, hetero | |||||
| MM-16 | M, liver | Sole | ALM | p.V600E, hetero | |||||
| MM-25 | M, nd | Chest wall | NM | p.V600E, hetero | |||||
| MM-6 | P | Sole | ALM | p.V600E, homo | p.T41I, hetero | ||||
| MM-23 | M, LN | Skin | NM | p.V600E, homo | |||||
| MM-32 | P | Thigh | NM | p.V600E, homo | |||||
| MM-2 | P | Esophagus | nd | p.Q61H, hetero | |||||
| MM-10 | M, LN | Head | NM | p.G12S, hetero | |||||
| MM-15 | P | Esophagus | nd | p.G13R, hetero | |||||
| MM-26 | P | Pharyngeal | nd | p.Q61H, homo | |||||
| MM-28 | M, nd | Shoulder | NM | p.Q61R, homo | |||||
| MM-31 | P | Heel | ALM | p.G12S, hetero | |||||
| MM-33 | P | Sole | ALM | p.G12S, hetero | |||||
| MM-36 | P | Sole | ALM | p.Q61R, homo | |||||
| MM-20 | M, LN | Finger | ALM | p.K642E, homo | |||||
| MM-22 | M, LN | Skin | NM | p.N822K, hetero | |||||
| MM-30 | P | Sole | ALM | p.I817F, hetero | |||||
| MM-18 | M, nd | Conjunctiva | nd | p.T40I, hetero | |||||
| MM-19 | M, nd | Sole | ALM | p.P44L, hetero | |||||
| MM-37 | P | Sole | ALM | ||||||
| MM-39 | P | Leg | SSM | ||||||
| MM-5 | P | Face | ALM | ||||||
| MM-8 | P | Toe | ALM | ||||||
| MM-9 | M, skin | ND | nd | ||||||
| MM-11 | P | Rectum | nd | ||||||
| MM-13 | M, brain | Thigh | SSM | ||||||
| MM-17 | M, nd | Conjunctiva | nd | ||||||
| MM-21 | P | Abdomen | nd | ||||||
| MM-29 | M, nd | Forearm | nd | ||||||
| MM-34 | P | Conjunctiva | nd | ||||||
| MM-35 | P | Sole | ALM |
Clinicopathological (primary or metastasis, primary and metastatic organ site and histological subtype) and mutation (amino acid change and zygosity) data of the analyzed cases are shown.
nd, not determined; hetero, heterozygous mutation; homo, homozygous mutation; NM, nodular melanoma; ALM, acral lentigous melanoma; LN, lymph node; SSM, superficial spreading melanoma.
Association between IDH1/2 Mutation and Other Mutations in Melanoma
We next examined mutations of the melanoma-associated oncogenes (the BRAF, NRAS, KIT, and CTNNB1 genes) in our study cases. In this cohort, we detected 12 BRAF mutations (30.8%), 8 NRAS mutations (20.5%), 4 KIT mutations (10.2%), and 3 CTNNB1 mutations (7.7%) (Table 1). As reported previously,4,8 the existence of BRAF, NRAS, and KIT mutations is mutually exclusive, and one case contains both BRAF and CTNNB1 mutations. Among the IDH1/2-mutated cases, two had both BRAF and IDH1 or IDH2 mutations, and one had KIT and IDH1 mutations. We also screened IDH1, IDH2, BRAF, NRAS, and KIT mutations in 13 melanoma cell lines. We observed nine BRAF mutations (9/13, 69.2%) and one NRAS mutation (1/13, 7.7%) in these cell lines, but were unable to detect any IDH1 or IDH2 mutation (data not shown).
Mutant IDH1-Expressing Melanoma Cells Confer a Growth Advantage in Vivo
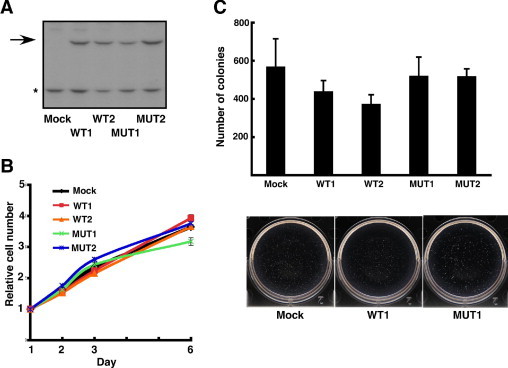
Previous studies have shown that BRAF mutation occurs at the early stage of melanoma development.30,31 Therefore, based on the above genetic analysis, we speculated that IDH mutation confers a growth advantage after acquiring BRAF or KIT mutation. Because the functional significance of IDH2 mutations detected in this study remains in need of further characterization, we focused on biological roles of a well-characterized IDH1 mutation (R132H) in melanoma. To examine the biological effect of mutant IDH1 in melanoma cells, we established clones expressing the wild or mutated (R132H) IDH1 gene from a BRAF-mutated (V600E) melanoma cell line (G361) (Figure 2A). We first compared the growth of these clones, but no significant difference was observed among control, wild-type, and mutant IDH1-expressing clones in vitro (Figure 2B). We then examined colony-forming activity of these clones and found that mutant IDH1-expressing clones formed significantly more colonies (Figure 2C). We also established wild or mutant IDH1-expressing clones from a BRAF wild-type melanoma cell line (GAK) (see Supplementary Figure S1A, http://ajp.amjpathol.org). In these clones, no significant difference in in vitro growth and colony-forming activities was detected (Supplementary Figure S1, B and C, at http://ajp.amjpathol.org).
Figure 2.

Growth advantage by mutant IDH1 in BRAF-mutated melanoma cells. A: Immunoblot analysis of mock, wild-type IDH1 (WT1 and 2), and mutant IDH1 (MUT1 and 2). The FLAG-tagged wild or mutant (R132H) IDH1 genes were introduced in BRAF-mutated G361 cells. The cell lysates were electrophoresed and immunoblotted with anti-flag antibody. Asterisk indicates nonspecific signal. B: In vitro cell proliferation of mock, wild-type (WT1 and 2), and mutant (MUT1 and 2) IDH1-expressing clones under two culture conditions (DMEM supplemented with 10% or with 1% serum). C: Colony-forming activity of mock, wild-type IDH1 (WT1 and 2), and mutant IDH1 (MUT1 and 2). Representative plate of each clone is shown at the bottom. D: Migration activity of mock, wild-type IDH1 (WT1 and 2) and mutant IDH1 (MUT1 and 2). Representative picture of migrated cells in each clone is shown at the bottom. E: The weight of in vivo tumors produced by mock, wild-type (WT1), and mutant (MUT1) IDH1-expressing clones (Mock: n = 5, WT1: n = 5, MUT: n = 8) (left). Histological appearance (hematoxylin-eosin staining) of tumors formed by mock and mutant IDH1-expressing clones (right). Scale bar = 100 μm.
Because IDH1 mutation was detected only in metastatic lesions, we next examined migration activity in these clones, but no difference was observed (Figure 2D). We finally tested the in vivo tumorigenicity of these clones. Mutant IDH1-expressing clones induced more frequently (8/8) than mock (5/8) and wild (5/8) IDH1-expressing clones. Moreover, mutant IDH1-expressing clones produced larger tumors than control clones (Figure 2E). Therefore, this IDH1 mutation appears to confer a growth advantage in vivo. Histologically, IDH1-mutated clones showed spindle morphology in vivo compared to the tumors of mock and wild IDH1-expressing clones (Figure 2E).
No Significant ROS Accumulation in IDH1 Mutant Expressing Melanoma Cells
Recent metabolome analysis has shown that mutant IDH1 proteins, including the R132H mutant, specifically produce 2HG,20–22 which may cause oxidative stress and induce DNA damage in affected cells. Therefore, we examined the accumulation of reactive oxygen species (ROS) in the mutant IDH1 clones in comparison to clones expressing wild-type IDH1 or the control. We did not detect any significant increase of ROS accumulation in mutant IDH1-expressing clones relative to the parental and wild-type IDH1-expressing clones (Figure 3).
Figure 3.
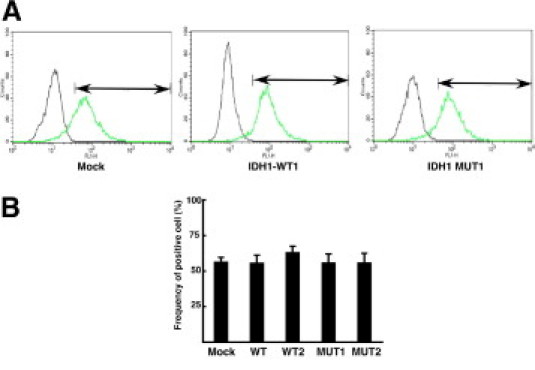
No significant accumulation of reactive oxygen species by mutant IDH1 in melanoma cells. A: Measurement of reactive oxygen species (ROS) in mock and IDH1-expressing clones by fluorescence-activated cell sorting analysis. Representative data of ROS accumulation (right peak) and control signal (left peak) are indicated. Positive cell fraction is shown by arrows. B: Frequency of fluorescence positive cells in mock, wild-type IDH1-, and mutant IDH1-expressing clones (n = 3).
Downstream Signal Pathways Affected by Mutant IDH1 in Melanoma Cells
To examine whether mutant IDH1 affects any known oncogenic pathways, we examined the activation of node proteins in the various molecular pathways (MAPK, AKT, S6K, and STAT3) in the control, wild IDH1-, or mutant IDH1-expressing clones and found that phosphorylation of MAPK and STAT3 were specifically increased in mutant IDH1-expressing clones (Figure 4A).
Figure 4.

Downstream signal pathways of mutant IDH1 in melanoma cells. A: Detection of phosphorylated and non-phosphorylated forms of MAPK, AKT, STAT3, and S6K in mock, wild-type IDH1-, and mutant IDH1-expressing clones. β-actin expression was a loading control. B and D: Quantitative RT-PCR analysis of the candidate genes regulated by mutant IDH1.C: In vitro cell proliferation of mock, wild-type (WT1 and 2), and mutant (MUT1 and 2) IDH1-expressing clones under hypoxic condition (1% O2).
Finally, to further elucidate the molecular changes induced by mutant IDH1, we conducted a genome-wide gene expression profiling of control, wild-type IDH1-, and mutant IDH1-expressing clones. The only reported downstream target of mutant IDH1 is activation of the hypoxia pathway through prevention of HIF1α protein degradation by prolyl hydroxylase (PHD) activity,32 but we did not observe any change in HIF1α targets such as Glut-1 and Pgk1 in melanoma cells (Figure 4B). We also examined the growth activity of these clones under hypoxic condition (1% O2), but no significant difference was observed (Figure 4C). Eight genes increased significantly in mutant IDH1-expressing clones compared to the mock and wild IDH1-expressing clones. Remarkably, they include four transcriptional factors (ATF3, JUN, MYCN, and SOX8) (Table 2, Figure 4B). The expression of 47 genes decreased in mutant IDH1-expressing clones compared with the mock and wild IDH1-expressing clones. We note that mutant IDH1 reduced the expression of RASSF1, a negative regulator of RAS signaling,33 and two dehydrogenases (DHRS1 and ADH5) in melanoma cells (Figure 4D, top 20 genes are shown in Table 2).
Table 2.
Genes Aberrantly Regulated in Mutant IDH1 Expressing Melanoma Cells
| Ratio (Mut/Mock) | Gene name | Annotation |
|---|---|---|
| 5.56 | PRKCA | Kinase |
| 4.47 | ATF3 | TF |
| 4.12 | EIF5 | Translation |
| 4.12 | JUN | TF |
| 4.02 | LYPD3 | Membranous |
| 3.7 | MYCN | TF |
| 3.31 | ANKDD1A | |
| 3.26 | SOX8 | TF |
| 0.0839 | BCL11A | ZF |
| 0.159 | KIF5C | Transport |
| 0.169 | RASSF1 | Ras signal |
| 0.178 | HEY1 | TF |
| 0.184 | SDC2 | Membranous |
| 0.219 | RANBP5 | Nuclear import |
| 0.224 | NR2F2 | TF |
| 0.231 | TTC26 | |
| 0.235 | PPP4R1 | Phosphatase |
| 0.239 | ZDHHC23 | |
| 0.25 | JAG2 | Membranous |
| 0.261 | AHNAK2 | |
| 0.273 | C9orf40 | |
| 0.273 | CA13 | |
| 0.274 | LEF1 | TF |
| 0.276 | DHRS1 | Oxidative stress |
| 0.279 | DOCK9 | Adaptor |
| 0.288 | MCTP2 | Membranous |
| 0.289 | ZNF222 | ZF |
| 0.297 | ADH5 | Oxidative stress |
Ratio of average expression (mutant IDH1 clones [Mut]/mock [Mock] clones), gene name, and annotation are shown. False discovery rate is <0.15.
TF, transcription factor; ZF, zinc finger protein.
Discussion
IDH1/2 belong to a novel family of cancer-related genes that were discovered by unbiased genome-wide exon sequence analysis.11 Since the IDH1/2 genes encode ubiquitous metabolic enzymes that convert isocitrate to α-KG, it remains unclear why genetic alterations of these genes occur specifically and frequently in glioma and leukemia, and are rarely detected in epithelial tumors. In the present study, we conducted focused sequence analysis of nonepithelial tumors to determine the prevalence of IDH1/2 mutation in tumors other than glioma and leukemia. No previous studies have analyzed IDH1/2 mutations in sarcomas, and our present analysis revealed that they are not frequent. Our analysis revealed that about 10% of melanomas (4/39) in a Japanese population harbored an IDH1 or IDH2 mutation. Bleeker et al analyzed 23 cases of melanoma, but did not detect any mutation.28 Very recently, Lopez et al analyzed 78 melanoma cases and reported IDH1 mutation in one case.34 Interestingly, this positive case is a lung metastasis. Because our analysis also detected IDH1 mutation exclusively in metastatic lesions, this alteration may occur in metastatic or advanced melanomas that might not have been extensively analyzed in the previous studies. It is also possible that there are ethnic or histological differences in the frequency of IDH1/2 mutations in melanoma. The frequency of each histological subtype varies among different ethnic groups,35 and our cohort contains a rather high percentage of the acral lentiginous subtype (15/39, 38.5%), which occurs dominantly in the Asian ethnic group. It should also be noted that three out of four IDH1/2 mutations occurred in either the mucosal or acral lentiginous subtype. Validation analysis of an additional larger cohort should be performed to determine the exact frequency of IDH1/2 mutations in melanoma and to see whether IDH1-mutated melanoma can be classified as a specific molecular or histopathological entity as recently reported in glioma and AML cases.16,36 IDH1 mutation has also been identified in 11% of anaplastic thyroid cancer, which is a very aggressive type of thyroid tumor and harbors BRAF, NRAS, and CTNNB1 mutations.37 Because melanocyte, thyroid, and glia originate from neural crest cells, it could be possible that the IDH1 gene alteration might confer some advantageous characteristics to tumors of specific developmental origin. It is also important to characterize the biological significance of rare substitutions such as G171D and P158T of the IDH2 gene since rare IDH2 mutations (R140Q and R140G) are discovered as germline variations of D-2-hydroxyglutaric aciduria.38
Our analysis has also revealed that IDH1 or IDH2 mutation coexists with other prevailing mutations such as BRAF or KIT mutations in melanoma. A previously reported IDH1-mutated melanoma case also harbored a BRAF mutation (p.V600E).34 Because BRAF, NRAS, and KIT mutations converge to the activation of RAS/RAF/MAPK signaling and they occur mutually exclusively in melanoma,4,8 it can be hypothesized that mutant IDH1/2 have a biological function distinct from this signaling pathway. Chou et al have reported that IDH1 mutation is significantly associated with NPM1 mutation in AML.15 It has also been reported that some AML cases harbor both IDH1 and NRAS mutations.14,15 To evaluate the biological significance of IDH1 mutation in melanoma, we established stable clones expressing mutant IDH1 with the common BRAF mutation. We found that they acquired growth activity especially in vivo and, surprisingly, enhanced activation of the MAPK pathway relative to the parental or wild IDH1-expressing cells. To uncover the molecular signature by mutant IDH1, we then conducted genome-wide gene expression profiling of mutant IDH1-expressing cells and found that the expression of RASSF1 mRNA was specifically down-regulated. It has been reported that RASSF1 associates with and negatively regulates the RAS signaling,33,39 and reduced RASSF1 expression could partly explain the MAPK activation in mutant IDH1-expressing melanoma cells. The expression of RASSF1 gene is epigenetically silenced in a wide range of cancers including melanoma,40 and recently, IDH1 mutation has been shown to associate with the CpG island methylator phenotype in glioma.41 Further study should address whether IDH1/2 mutations have any relation to the epigenetic alterations in other tumors including melanoma. Additionally, mutant IDH1 induced the expression of other growth-related transcriptional factors, including MYCN and JUN oncoproteins and ATF3.42 These molecular signatures could provide a key to understanding how mutant IDH1 modulates cellular signaling in cancer.
Since the discovery of IDH1/2 mutations, their biological significance has been debated. The first comprehensive study of IDH1/2 mutation in glioma also demonstrated that the mutant proteins lose their original enzymatic activity,11 but the mutation profile (accumulation in specific residues and occurring only heterozygously) cast doubt on the idea that they might be tumor suppressor genes. Zhao et al have reported that mutant IDH1 heterodimerizes with wild-type IDH1 protein and diminishes the production of α-KG by a dominant-negative fashion.32 Since α-KG is required for PHD activity, which promotes HIF1α degradation, mutant IDH1 induces HIF1α accumulation and confers resistance to hypoxia. To determine whether mutant IDH1 activates the hypoxia-responsive signaling in melanoma, we examined the expression of HIF1α target genes. However, we were unable to detect any increase in the expression of well-characterized HIF1α-regulated genes (Glut-1 and Pgk1) by mutant IDH1 in melanoma cells and no growth advantage of mutant IDH1-expressing clones was observed under hypoxic condition.
Recent studies have identified that mutant IDH1/2 can convert α-ketoglutarate to 2-hydroxyglutarate, which is a completely different function from that of the wild-type enzyme.20–22 These findings suggest that mutant IDH1/2 may have a gain-of-function effect in tumors, although the biological significance of 2HG production in human carcinogenesis remains unclear. D-2-hydroxyglutaric aciduria, caused by germline mutations of the D-2-hydroxyglutarate dehydrogenase or IDH2 genes,38 shows accumulation of 2HG, and its symptoms include encephalopathy and some brain tumors.23,24 Accumulation of 2HG has been suggested to cause oxidative stress that could be associated with increased DNA damage or epigenetic alteration,43,44 thus driving carcinogenesis. Interestingly, our gene expression profiling revealed that melanoma cells harboring mutant IDH1 significantly reduced the expression of two dehydrogenases. Although the underlining molecular mechanisms for their transcriptional regulation remain unknown, mutant IDH1 seems to affect redox status of the cell through multiple ways (accumulation of 2HG and reduction of dehydrogenases), which might increase the mutation frequency. It has been reported that STAT3 is activated by oxidative stress.45 To evaluate the oxidative stress induced by mutant IDH1, we measured ROS accumulation in mutant IDH-expressing clones, but were unable to detect any significant increase of ROS. It is possible that 2HG may modulate the oxidative state of specific metabolites, which were not detectable under the conditions we used. Further metabolomic approach would be helpful to resolve this question and also to test whether 2HG could be used as a new diagnostic marker for melanoma as proposed in AML.21
In conclusion, we have identified IDH1/2 mutations in a small subset of melanoma. IDH1/2 mutation coexists with BRAF or KIT mutations and mutant IDH1 confers an in vivo growth advantage in BRAF-mutated melanoma cells partly through transcriptional regulation of growth-associated and dehydrogenase genes and the MAPK/STAT3 pathway activation. Further analysis to clarify the biological roles and clinical significance of mutant IDH1/2 and underlining molecular mechanisms in melanoma is warranted.
Acknowledgments
We thank Dr. Toshikazu Ushijima (Carcinogenesis Division, National Cancer Center Research Institute) for providing genomic DNA of melanoma cell lines.
Footnotes
Supported in part by Grant-in-Aid for Cancer Research from the Ministry of Health, Labor and Welfare of Japan.
Supplemental material for this manuscript can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.12.011.
Current address of M.M., Division of Gastroenterologic and General Surgery, Department of Surgery, Asahikawa Medical University, Asahikawa, Japan.
Supplementary data
Supplementary Figure S1.
GAK cells were obtained from Japanese Collection of Research Bioresources (Sennan-shi, Japan) and maintained in DMEM supplemented with 10% fetal bovine serum. The FLAG-tagged wild or mutant (R132H) IDH1 genes were introduced in BRAF-wild GAK cells. A: Immunoblot analysis of mock, wild-type IDH1 (WT1 and 2), and mutant IDH1 (MUT1 and 2). The cell lysates were electrophoresed and immunoblotted with anti-flag antibody. Arrow indicates expected IDH1 protein. Asterisk indicates nonspecific signal. B: In vitro cell proliferation of mock, wild-type (WT1 and 2), and mutant (MUT1 and 2) IDHI-expressing clones. The culture condition is DMEM supplemented with 10% serum. C: Colony-forming activity of mock, wild-type IDH1 (WT1 and 2), and mutant IDH1 (MUT1 and 2). Representative plate of each clone is shown at the bottom.
References
- 1.Tsao H., Atkins M.B., Sober A.J. Management of cutaneous melanoma. N Engl J Med. 2004;351:998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 2.Verma S., Quirt I., McCready D., Bak K., Charette M., Iscoe N. Systematic review of systemic adjuvant therapy for patients at high risk for recurrent melanoma. Cancer. 2006;106:1431–1442. doi: 10.1002/cncr.21760. [DOI] [PubMed] [Google Scholar]
- 3.Kuphal S., Bosserhoff A. Recent progress in understanding the pathology of malignant melanoma. J Pathol. 2009;219:400–409. doi: 10.1002/path.2617. [DOI] [PubMed] [Google Scholar]
- 4.Curtin J.A., Fridlyand J., Kageshita T., Patel H.N., Busam K.J., Kutzner H., Cho K.H., Aiba S., Bröcker E.B., LeBoit P.E., Pinkel D., Bastian B.C. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 5.Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M.J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B.A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G.J., Bigner D.D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J.W., Leung S.Y., Yuen S.T., Weber B.L., Seigler H.F., Darrow T.L., Paterson H., Marais R., Marshall C.J., Wooster R., Stratton M.R., Futreal P.A. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 6.Singh M., Lin J., Hocker T.L., Tsao H. Genetics of melanoma tumorigenesis. Br J Dermatol. 2008;158:15–21. doi: 10.1111/j.1365-2133.2007.08316.x. [DOI] [PubMed] [Google Scholar]
- 7.Fecher L.A., Cummings S.D., Keefe M.J., Alani R.M. Toward a molecular classification of melanoma. J Clin Oncol. 2007;25:1606–1620. doi: 10.1200/JCO.2006.06.0442. [DOI] [PubMed] [Google Scholar]
- 8.Curtin J.A., Busam K., Pinkel D., Bastian B.C. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 9.Beadling C., Jacobson-Dunlop E., Hodi F.S., Le C., Warrick A., Patterson J., Town A., Harlow A., Cruz F., 3rd, Azar S., Rubin B.P., Muller S., West R., Heinrich M.C., Corless C.L. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14:6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 10.Handolias D., Hamilton A.L., Salemi R., Tan A., Moodie K., Kerr L., Dobrovic A., McArthur G.A. Clinical responses observed with imatinib or sorafenib in melanoma patients expressing mutations in KIT. Br J Cancer. 2010;102:1219–1223. doi: 10.1038/sj.bjc.6605635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parsons D.W., Jones S., Zhang X., Lin J.C., Leary R.J., Angenendt P., Mankoo P., Carter H., Siu I.M., Gallia G.L., Olivi A., McLendon R., Rasheed B.A., Keir S., Nikolskaya T., Nikolsky Y., Busam D.A., Tekleab H., Diaz L.A., Jr., Hartigan J., Smith D.R., Strausberg R.L., Marie S.K., Shinjo S.M., Yan H., Riggins G.J., Bigner D.D., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V.E., Kinzler K.W. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan H., Parsons D.W., Jin G., McLendon R., Rasheed B.A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G.J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K.W., Velculescu V.E., Vogelstein B., Bigner D.D. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanson M., Marie Y., Paris S., Idbaih A., Laffaire J., Ducray F., El Hallani S., Boisselier B., Mokhtari K., Hoang-Xuan K., Delattre J.Y. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- 14.Mardis E.R., Ding L., Dooling D.J., Larson D.E., McLellan M.D., Chen K., Koboldt D.C., Fulton R.S., Delehaunty K.D., McGrath S.D., Fulton L.A., Locke D.P., Magrini V.J., Abbott R.M., Vickery T.L., Reed J.S., Robinson J.S., Wylie T., Smith S.M., Carmichael L., Eldred J.M., Harris C.C., Walker J., Peck J.B., Du F., Dukes A.F., Sanderson G.E., Brummett A.M., Clark E., McMichael J.F., Meyer R.J., Schindler J.K., Pohl C.S., Wallis J.W., Shi X., Lin L., Schmidt H., Tang Y., Haipek C., Wiechert M.E., Ivy J.V., Kalicki J., Elliott G., Ries R.E., Payton J.E., Westervelt P., Tomasson M.H., Watson M.A., Baty J., Heath S., Shannon W.D., Nagarajan R., Link D.C., Walter M.J., Graubert T.A., DiPersio J.F., Wilson R.K., Ley T.J. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou W.C., Hou H.A., Chen C.Y., Tang J.L., Yao M., Tsay W., Ko B.S., Wu S.J., Huang S.Y., Hsu S.C., Chen Y.C., Huang Y.N., Chang Y.C., Lee F.Y., Liu M.C., Liu C.W., Tseng M.H., Huang C.F., Tien H.F. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115:2749–2754. doi: 10.1182/blood-2009-11-253070. [DOI] [PubMed] [Google Scholar]
- 16.Abbas S., Lugthart S., Kavelaars F.G., Schelen A., Koenders J., Zeilemaker A., van Putten W.J., Rijneveld A., Löwenberg B., Valk P.J. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;16:2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- 17.Paschka P., Schlenk R.F., Gaidzik V.I., Habdank M., Krönke J., Bullinger L., Späth D., Kayser S., Zucknick M., Götze K., Horst H.A., Germing U., Döhner H., Döhner K. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–3643. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 18.Geisbrecht B.V., Gould S.J. The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem. 1999;274:30527–30533. doi: 10.1074/jbc.274.43.30527. [DOI] [PubMed] [Google Scholar]
- 19.Nekrutenko A., Hillis D.M., Patton J.C., Bradley R.D., Baker R.J. Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol Biol Evol. 1998;15:1674–1684. doi: 10.1093/oxfordjournals.molbev.a025894. [DOI] [PubMed] [Google Scholar]
- 20.Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C., Marks K.M., Prins R.M., Ward P.S., Yen K.E., Liau L.M., Rabinowitz J.D., Cantley L.C., Thompson C.B., Vander Heiden M.G., Su S.M. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross S., Cairns R.A., Minden M.D., Driggers E.M., Bittinger M.A., Jang H.G., Sasaki M., Jin S., Schenkein D.P., Su S.M., Dang L., Fantin V.R., Mak T.W. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward P.S., Patel J., Wise D.R., Abdel-Wahab O., Bennett B.D., Coller H.A., Cross J.R., Fantin V.R., Hedvat C.V., Perl A.E., Rabinowitz J.D., Carroll M., Su S.M., Sharp K.A., Levine R.L., Thompson C.B. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haliloglu G., Jobard F., Oguz K.K., Anlar B., Akalan N., Coskun T., Sass J.O., Fischer J., Topcu M. L-2-hydroxyglutaric aciduria and brain tumors in children with mutations in the L2HGDH gene: neuroimaging findings. Neuropediatrics. 2008;39:119–122. doi: 10.1055/s-2008-1081217. [DOI] [PubMed] [Google Scholar]
- 24.Aghili M., Zahedi F., Rafiee E. Hydroxyglutaric aciduria and malignant brain tumor: a case report and literature review. J Neurooncol. 2009;91:233–236. doi: 10.1007/s11060-008-9706-2. [DOI] [PubMed] [Google Scholar]
- 25.Furuta J., Nobeyama Y., Umebayashi Y., Otsuka F., Kikuchi K., Ushijima T. Silencing of Peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006;66:6080–6086. doi: 10.1158/0008-5472.CAN-06-0157. [DOI] [PubMed] [Google Scholar]
- 26.Shibata T., Kokubu A., Gotoh M., Ojima H., Ohta T., Yamamoto M., Hirohashi S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–1368. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 27.Shibata T., Kokubu A., Miyamoto M., Hosoda F., Gotoh M., Tsuta K., Asamura H., Matsuno Y., Kondo T., Imoto I., Inazawa J., Hirohashi S. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene. 2010;29:4671–4681. doi: 10.1038/onc.2010.217. [DOI] [PubMed] [Google Scholar]
- 28.Bleeker F.E., Lamba S., Leenstra S., Troost D., Hulsebos T., Vandertop W.P., Frattini M., Molinari F., Knowles M., Cerrato A., Rodolfo M., Scarpa A., Felicioni L., Buttitta F., Malatesta S., Marchetti A., Bardelli A. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009;30:7–11. doi: 10.1002/humu.20937. [DOI] [PubMed] [Google Scholar]
- 29.Kang M.R., Kim M.S., Oh J.E., Kim Y.R., Song S.Y., Seo S.I., Lee J.Y., Yoo N.J., Lee S.H. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125:353–355. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- 30.Papp T., Schipper H., Kumar K., Schiffmann D., Zimmermann R. Mutational analysis of the BRAF gene in human congenital and dysplastic melanocytic naevi. Melanoma Res. 2005;15:401–407. doi: 10.1097/00008390-200510000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Poynter J.N., Elder J.T., Fullen D.R., Nair R.P., Soengas M.S., Johnson T.M., Redman B., Thomas N.E., Gruber S.B. BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res. 2006;16:267–273. doi: 10.1097/01.cmr.0000222600.73179.f3. [DOI] [PubMed] [Google Scholar]
- 32.Zhao S., Lin Y., Xu W., Jiang W., Zha Z., Wang P., Yu W., Li Z., Gong L., Peng Y., Ding J., Lei Q., Guan K.L., Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armesilla A.L., Williams J.C., Buch M.H., Pickard A., Emerson M., Cartwright E.J., Oceandy D., Vos M.D., Gillies S., Clark G.J., Neyses L. Novel functional interaction between the plasma membrane Ca2+ pump 4b and the proapoptotic tumor suppressor Ras-associated factor 1 (RASSF1) J Biol Chem. 2004;279:31318–31328. doi: 10.1074/jbc.M307557200. [DOI] [PubMed] [Google Scholar]
- 34.Lopez G.Y., Reitman Z.J., Solomon D., Waldman T., Bigner D.D., McLendon R.E., Samuels Y., Yan H. IDH1 (R132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem Biophys Res Commun. 2010;398:585–587. doi: 10.1016/j.bbrc.2010.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Byrd-Miles K., Toombs E.L., Peck G.L. Skin cancer in individuals of African: Asian, Latin-American, and American-Indian descent: differences in incidence, clinical presentation, and survival compared to Caucasians. J Drugs Dermatol. 2007;6:10–16. [PubMed] [Google Scholar]
- 36.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., Alexe G., Lawrence M., O'Kelly M., Tamayo P., Weir B.A., Gabriel S., Winckler W., Gupta S., Jakkula L., Feiler H.S., Hodgson J.G., James C.D., Sarkaria J.N., Brennan C., Kahn A., Spellman P.T., Wilson R.K., Speed T.P., Gray J.W., Meyerson M., Getz G., Perou C.M., Hayes D.N., Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murugan A.K., Bojdani E., Xing M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun. 2010;393:555–559. doi: 10.1016/j.bbrc.2010.02.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kranendijk M., Struys E.A., van Schaftingen E., Gibson K.M., Kanhai W.A., van der Knaap M.S., Amiel J., Buist N.R., Das A.M., de Klerk J.B., Feigenbaum A.S., Grange D.K., Hofstede F.C., Holme E., Kirk E.P., Korman S.H., Morava E., Morris A., Smeitink J., Sukhai R.N., Vallance H., Jakobs C., Salomons G.S. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330:336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- 39.Ortiz-Vega S., Khokhlatchev A., Nedwidek M., Zhang X.F., Dammann R., Pfeifer G.P., Avruch J. The putative tumor suppressor RASSF1A homodimerizes and heterodimerizes with the Ras-GTP binding protein Nore1. Oncogene. 2002;21:1381–1390. doi: 10.1038/sj.onc.1205192. [DOI] [PubMed] [Google Scholar]
- 40.Spugnardi M., Tommasi S., Dammann R., Pfeifer G.P., Hoon D.S. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003;63:1639–1643. [PubMed] [Google Scholar]
- 41.Noushmehr H., Weisenberger D.J., Diefes K., Phillips H.S., Pujara K., Berman B.P., Pan F., Pelloski C.E., Sulman E.P., Bhat K.P., Verhaak R.G., Hoadley K.A., Hayes D.N., Perou C.M., Schmidt H.K., Ding L., Wilson R.K., Van Den Berg D., Shen H., Bengtsson H., Neuvial P., Cope L.M., Buckley J., Herman J.G., Baylin S.B., Laird P.W., Aldape K. Cancer Genome Atlas Research Network: Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yin X., Dewille J.W., Hai T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene. 2008;27:2118–2127. doi: 10.1038/sj.onc.1210861. [DOI] [PubMed] [Google Scholar]
- 43.Latini A., Scussiato K., Rosa R.B., Llesuy S., Belló-Klein A., Dutra-Filho C.S., Wajner M. D-2-hydroxyglutaric acid induces oxidative stress in cerebral cortex of young rats. Eur J Neurosci. 2003;17:2017–2022. doi: 10.1046/j.1460-9568.2003.02639.x. [DOI] [PubMed] [Google Scholar]
- 44.Lim S.O., Gu J.M., Kim M.S., Kim H.S., Park Y.N., Park C.K., Cho J.W., Park Y.M., Jung G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135:2128–2140. doi: 10.1053/j.gastro.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 45.Uckun F.M., Qazi S., Ma H., Tuel-Ahlgren L., Ozer Z. STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress. Proc Natl Acad Sci USA. 2010;107:2902–2907. doi: 10.1073/pnas.0909086107. [DOI] [PMC free article] [PubMed] [Google Scholar]