

Abstract
Poly(ADP-ribosyl)ation, attaching the ADP-ribose polymer chain to the receptor protein, is a unique posttranslational modification. Poly(ADP-ribose) polymerase-1 (PARP-1) is a well-characterized member of the PARP family. In this review, we provide a general update on molecular structure and structure-based activity of this enzyme. However, we mainly focus on the roles of PARP-1 in inflammatory diseases. Specifically, we discuss the signaling pathway context that PARP-1 is involved in to regulate the pathogenesis of inflammation. PARP-1 facilitates diverse inflammatory responses by promoting inflammation-relevant gene expression, such as cytokines, oxidation-reduction–related enzymes, and adhesion molecules. Excessive activation of PARP-1 induces mitochondria-associated cell death in injured tissues and constitutes another mechanism for exacerbating inflammation.
There are many posttranslational protein modifications (eg, phosphorylation, acetylation, methylation, and ubiquitylation) that are involved in a wide scope of cellular processes, including chromatin remodeling, transcriptional regulation, and signal transmission response to extracellular stimulation. Poly(ADP-ribosyl)ation (PARylation) is one of such essential protein modifications, whereby polymers of ADP-ribose (PARs) are formed from donor NAD+ molecules and covalently attached via an ester linkage to glutamic acid and less commonly to aspartic acid or lysine of target proteins.1–3 The target proteins generally contain a PAR-binding consensus motif that frequently overlaps with a functional domain, such as a protein- or DNA-binding domain (DBD), and thus accounts for PAR modification, altering the functional properties of the targets.4
The process of PARylation is catalyzed by the poly(ADP-ribose) polymerase (PARP) family of enzymes that consists of 18 members.5 The PARPs, historically known as poly(ADP-ribose) synthases and poly(ADP-ribose) transferases,6,7 show different structure, cellular location, and functions.8,9 Only two members of this family (ie, PARP-1 and PARP-2) are DNA damage related; and PARP-1, the best-understood member, is an abundant nuclear enzyme and accounts for at least 85% of the cellular PARP activity.10
Since the discovery of PAR synthesis and PARP-1 decades ago,11,12 new discoveries have been consistently published related to their structure, property, and functions. PARP-1 is a multifunctional enzyme and has a key role in the spatial and temporal organization of DNA repair, thus maintaining genome integrity and facilitating cell survival. PARP-1 catalyzes the synthesis and attachment of highly negatively charged PARs to target proteins, including histones, topoisomerases, DNA helicases, and single-strand break repair and base excision repair factors; and facilitates relaxation of the chromatin superstructure, protein-protein interaction, and DNA-binding ability of the members of the DNA repair machinery. A recently published article13 reviews the role of PARP-1 in DNA repair. In addition, the importance of poly(ADP-ribose) synthesis has been established in many other cellular processes, such as chromatin replication, transcriptional regulation, and cell death, some of which will compose the main content of the present review. We will address the significant role of PARP-1 in inflammatory disorders and the precise signaling mechanisms by which PARP-1 regulates the pathogenesis of inflammation. We hope that positioning PARP-1 in the context of a well-described signaling pathway provides directions for new strategies to the PARP-1–targeted therapy of inflammation.
PARP-1 Structure, Properties, and Activation
PARP-1 is a large protein composed of 1014 amino acids (mol. wt., 116 kDa). It has three main domains: the DBD, the automodification domain (AMD), and the catalytic domain (NAD+-binding domain).14 The N-terminal 46-kDa DBD contains three zinc-finger motifs, of which the first two were described as “nick sensors” because early studies found that these zinc fingers recognize DNA damage and direct PARP-1 binding to the damaged DNA.10,15 More recently, the third zinc-finger motif in the DBD facilitated interdomain contact and assembly of the DNA-activated conformation of PARP-116; therefore, it was considered essential for DNA-dependent PARP-1 activity.16–18
The central AMD (mol. wt., 22 kDa) serves as a regulatory segment and consists of a breast cancer susceptibility protein C-terminus motif that is found in many other DNA repair and cell cycle proteins. There are several glutamic acid residues in this domain that are suggested to be the site for covalent binding with poly(ADP-ribose) on PARP-1 activation.19,20 However, other groups argue that individual lysine residues, not glutamic acid, serve as acceptor sites for ADP ribosylation in the AMD.21 The PAR modification of glutamic residues in the NAD+-binding domain and the DBD has also been noted.19,20 Irrespective of the site, automodification is accepted as a mechanism for regulating PARP-1 activity and control PAR synthesis19; however, the exact impact and mechanisms of automodification on enzyme function need further investigation.
The AMD is considered important for protein-protein interaction between PARP-1 and the members of the DNA repair and gene transcriptional machinery, as well as for PARP-1 homodimerization or heterodimerization with PARP-2.22–24 Dimerization is assumed to be a prerequisite for PARP-1 activation25; however, the AMD deletion mutant is catalytically active, indicating that this segment is not indispensable for PARP-1 activity.20 Other studies24,25 have suggested that interaction in the N-terminal region is required for PARP-1 dimerization and activation. The structural basis and significance of PARP-1's dimerization for its catalytic activity are not yet clear. Further investigation is still necessary because the blocking of PARP-1 dimerization might become a candidate strategy for inhibition of PARP-1 activation.
The C-terminal region (mol. wt., 54 kDa) is the most conserved part of the enzyme. It consists of an NAD+-binding domain and executes the catalytic function of PARP-1, synthesizing PARs by using NAD+ as a substrate. The highly conserved 50–amino acid “PARP signature” motif (NAD+-binding site) has been found in all PARP family members discovered thus far.26,27 The influence of the automodification of the glutamic acid residues within this domain on PARP-1's catalytic activity has not yet been fully addressed.19,20
In addition to DNA breaks, DNA hairpins, cruciform, and stably unpaired regions have all been considered effective determinants of PARP-1 activation.28,29 After its binding to DNA, PARP-1 catalyzes the formation of PARs. The most abundant poly(ADP-ribosyl)ated protein in the cell is PARP-1 itself, and the accumulation of PAR on PARP-1 leads to its repulsion and dissociation from DNA strands.30,31 The poly(ADP-ribose) glycohydrolase (PARG) recycles the PAR formed on PARP-1 and thereby allows PARP-1 to enter the next action round.13 The PARs are rapidly degraded by PARG,32,33 and PARG has both endoglycosidase and exoglycosidase activities (endoglycosidase being greater than exoglycosidase), producing free PAR and mono(ADP-ribose).34,35 The amount of PAR formation and its attachment to other proteins are controlled by PARP-1 and PARG. Thus, a balance between the activation of PARP-1 and PARG determines cell fate, both by influencing the level of energetic substrates (NAD+ and ATP) and PAR amount and has been investigated in recent studies.36–38
Involvement of PARP-1 in Inflammatory Diseases
Several studies have shown the simultaneous activation of inflammatory responses and PARP-1 in various disease models and noted that PARP-1 is rapidly activated; in addition, its activation is prolonged and sustained in pathophysiological conditions. PARP-1 may play an important role by up-regulating the expression of proinflammatory genes and/or inducing cell death in the injured tissues.39,40 We discuss PARP-1 activation in infectious and noninfectious diseases.
Studies in experimental models reflect the central role of PARP-1 in various diseases. For example, in a septic rat model, up-regulated PARP-1 expression colocalized with DNA breaks and correlated with sepsis-induced inflammation and early and late stages of myocardial dysfunction.41 In pulmonary inflammation models induced by intratracheal administration of lipopolysaccharide (LPS), PARP-1 suppression by genetic deletion or pharmacological inhibitors was beneficial in reducing the inflammatory cell recruitment to mouse airways.42,43 Likewise, the absence of PARP-1 in a mouse model of enterocolitis induced by Salmonella typhimurium decreased NF-κB–mediated proinflammatory gene expression and was associated with delayed gut inflammation.44 Others showed that PARP-1 inhibition protected the mouse brain from LPS-evoked systemic inflammation and other confounding factors, including the lowering of NAD+ concentration, mitochondrial biogenesis defects, translocation of apoptosis-inducing factor (AIF) to the nucleus, and enhanced lipid peroxidation.45 The role of PARP-1 in septic peritonitis was reviewed by Liaudet and Oddo.46
In addition to bacteria, PARP-1's involvement in the infection induced by other pathogenic agents, such as viruses, fungi, and parasites, has not yet been addressed. Recently, it was shown in an in vitro parasite infection model that PARP-1 induced inflammatory cytokine (IL-1) and tumor necrosis factor (TNF) α production in cardiomyocytes infected by Trypanosoma cruzi47 and provided a new direction for the mechanisms involved in Chagasic heart disease pathogenicity.
Recently, new evidence implicated PARP-1 in cytotoxic agent-induced inflammation. Asbestos is toxic to human mesothelial cells, and asbestos exposure activates human mesothelial cell PARP-1 associated with H2O2 secretion, ATP depletion, and translocation of high-mobility group box 1 protein from the nucleus to the cytoplasm and into the extracellular space.48 Experimental studies in mice and hamsters injected with asbestos validated the release of high-mobility group box 1 protein in the extracellular space of mesothelial cells and inflammatory cells around asbestos deposits. A contact hypersensitivity reaction is a form of a delayed type of hypersensitivity caused by allergens. Brunyanszki et al49 reported that PARP-1 inhibition reduces the extent of inflammation by modulating oxidative stress and impairing the activation of NF-κB and activator protein (AP) 1 in an oxazolone-induced contact hypersensitivity model.
The role of PARP-1 in inflammation associated with tissue injuries in stroke and trauma has been studied in detail. For example, in a spinal cord trauma model, the infiltration of neutrophils in spinal cord tissue was associated with a marked increase in immunoreactivity for PARs, an index of PARP activation; and treatment with PARP-1 inhibitors reduced the tissue inflammation and injury events associated with spinal cord trauma.50 In another study,51 PARP-1–deficient or wild-type mice treated with PARP-1 inhibitors [PJ34 (N-[6-oxo-5,6-dihydro-phenanthridin-2-yl]-N,N-dimethylactamide) or 3AB (3-aminobenzaminde)] were subjected to heat exposure as a model to study heat stroke. The PARP-1 inhibition increased the expression of 27- and 70-kDA heat shock proteins, and heat stroke–induced liver injury was attenuated in PARP-1–deficient mice when compared with findings in wild-type mice. The role of PARP-1 in sports injury was demonstrated by the observations that eccentric exercise-induced skeletal muscle damage was associated with protein oxidative and nitrosative adducts, leukocyte infiltration, PARP-1 up-regulation, and increased gene expression for inflammatory mediators (ie, cyclooxygenase-2, IL-1β, IL-6, TNF-α, and monocyte chemotactic protein-1).52 These symptoms were ameliorated by treatment with a Chinese herb (honokiol) that suppressed the PARP-1 activity and inflammation-mediated damage to muscle cells.52
The involvement of PARP-1 in noninfectious inflammation was noticed in the murine colitis and ischemia-reperfusion model in the 1990s.53,54 In succession, PARP-1 activation accompanied with depressed left ventricular function was demonstrated in chronic heart failure models.55,56 The key role of PARP-1 in many immune-abnormal conditions (eg, atherosclerosis, arthritis, lung injury, nephritis, diabetic complications, and spinal inflammation) attracted intensive attention. Atherosclerosis, a chronic inflammatory disease, is the leading cause of death in Western societies. PARP-1 enhances the expression of adhesion molecules and activates endothelial cells; it also contributes to the infiltration of inflammatory cells, inducing features of plaque vulnerability.57 An imbalance of tissue inhibitor of metalloproteinases and matrix metalloproteinases in the extracellular matrix may constitute a critical contributing factor to atherogenesis. Boulares and coworkers58,59 found that PARP-1 inhibition provides stability to atherosclerotic plaques associated with increased expression of tissue inhibitor of metalloproteinases 2, decreased activity of matrix metalloproteinase 9, and extracellular matrix degradation in a high-fat, diet-induced, dyslipidemic, dilated cardiomyopathy model. Rheumatoid arthritis is characterized as inflammation with synovial hyperplasia, pannus formation, and progressive destruction of cartilage and bone. PARP-1–deficient mice showed decreased transcription and expression of IL-1β, monocyte chemotactic protein-1, and TNF-α cytokines, providing evidence for the contribution of PARP-1 to the progression of arthritis.60 Inflammation plays a key role in lung injury and in the pathogenesis of asthma. In ovalbumin-challenged mice models, PARP-1 protein expression and its activity were greatly increased. Pharmaceutical inhibition and gene deletion of PARP-1 reduced inflammation by preventing eosinophilic infiltration into the airways of ovalbumin-challenged mice. In addition, the production of IL-5, IL-10, IL-13, and granulocyte macrophage colony-stimulating factor was completely inhibited in ex vivo ovalbumin-challenged lung cells derived from these animals, implying the role of PARP-1 in allergy-associated inflammation.61,62 Endothelial dysfunction leads to diabetic patients experiencing retinopathy, nephropathy, neuropathy, and accelerated atherosclerosis (so-called diabetic complications). PARP-1 is an important factor in the pathogenesis of endothelial dysfunction in diabetes63,64 and regulates the progression of autoimmune nephritis65 and spinal inflammation,64 indicative of its participation in autoimmune disorders.66 In summary, the studies presented in this section highlight the participation of PARP-1 in activation or sustenance of inflammatory processes in various diseases.
Role of PARP-1 in Inflammatory Gene Expression
PARP-1 facilitates diverse inflammatory responses by promoting inflammation-relevant gene expression. Various studies47,65,67–69 have shown that PARP-1 influences the expression of proinflammatory cytokines (eg, IL-1β, TNF-α, and monocyte chemotactic protein-1), enzymes involved in the deleterious effects of inflammation [eg, inducible nitric oxides synthase (iNOS), cyclo-oxygenase-2, and NAPDH oxidase],52,57,65,70 and inflammation-promoting adhesion molecules, such as intercellular adhesion molecule-1, P-selectin, and E-selectin.57,71 PARP-1 can influence inflammatory gene expression at three levels (ie, chromatin remodeling regulation, transcriptional activity regulation, and mRNA posttranscriptional stability regulation), as illustrated in Figure 1A.
Figure 1.
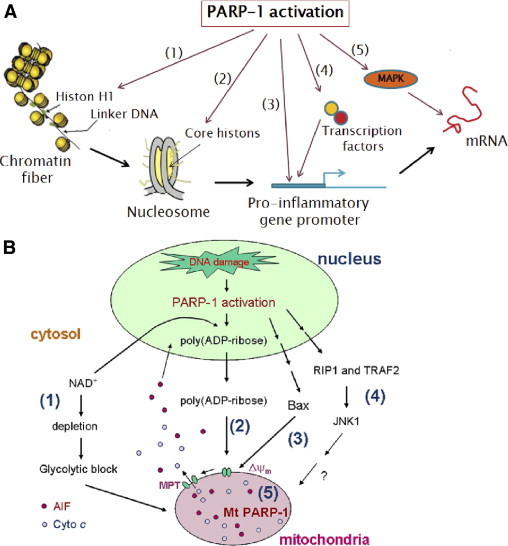
A: Potential routes of PARP-1 stimulation of inflammatory gene expression. First, at the level of chromatin remodeling, the modification of histone H1 by PARP-1 (1) relaxes the highly compacted chromatin fibers and facilitates transcription machinery access to the target genes (2). Second, at the level of transcription, activated PARP-1 either directly binds to the promoter region of target genes (3) or acts on transcription factors and regulates their transcriptional activity (4). Third, at the level of posttranscription, activated PARP-1 affects p38 (MAPK) and then regulates the posttranscriptional stability of mRNAs (5). B: Potential routes of nuclear-mitochondrial cross talk in PARP-1–dependent cell death pathways. 1: Extensive PARP-1 activation leads to depletion of NAD+ from the cytosol and accumulation of poly(ADP-ribose) in the nucleus. NAD+ depletion can block glycolysis and thereby block the delivery of glucose-derived substrates to the mitochondria. 2: Poly(ADP-ribose) formed in the nucleus might be transported out of the nucleus and act directly on mitochondrial membranes. 3: Pro-apoptosis protein Bcl-2–associated X protein (Bax) is activated and translocates to mitochondrial membranes. The actions of both poly(ADP-ribose) and Bax on mitochondrial membrane lead to cell death through a process involving mitochondrial depolarization (Δψm), MPT, and mitochondrial release of AIF and cytochrome c (Cyto c). 4: Blocking the receptor-interacting protein (RIP) 1–TNF receptor (TRAF)–associated factor 2–c-Jun NH2-terminal kinase (JNK) signal transduction pathway can also inhibit PARP-1–induced mitochondrial events and resultant cell death. It remains to be established whether these three pathways are interrelated or are independent of each other. 5: The localization of PARP-1 and the formation of PARs in mitochondria (mt) as mechanisms for impaired membrane potential (Ψm), NAD+ content, cellular respiration, and cell death factors (ie, Cyto c and AIF) remain to be examined in future studies.
The effects of PARP-1 on chromatin structure and transcriptional regulation were recently reviewed60 and discussed in the context of inflammatory gene expression herein. For function on chromatin-remodeling regulation, it is suggested that in the presence of saturating NAD+, PARP-1–dependent attachment of PARs to core histones and the linker histone H115,72 introduces massive negative charges and facilitates relaxation of the chromatin superstructure and recruitment of transcription machinery to the promoter or enhancer of target genes.73,74
The literature provides evidence that the effects of PARP-1 on transcription factors regulating inflammatory cytokine gene expression vary diversely, depending on the stimuli and cell and tissue types. For example, PARP-1 has been implicated in the activation of NF-κB, AP-1, and heat shock factor protein 1 transcription factors, classically known to signal inflammatory gene expression.47,67,75–77 Rel A (p65), the v-reticuloendotheliosis oncogene homolog A, an important subunit of activated NF-κB dimers (p65/p50, p65/p65, and p65/c-Rel), was poly(ADP-ribosyl)ated in an in vitro assay using recombinant protein as substrate.78 Poly(ADP-ribosyl)ation of p65 was a critical determinant for its interaction with nuclear export protein Crm1 (exportin 1), promoting nuclear retention of NF-κB and downstream events for cytokine gene expression.79 Others78,80–82 have reported that PARP-1 directly binds with NF-κB, and inhibition of PARP-1 attenuated the proinflammatory cytokine response via decreased assembly of transcription activation complex. However, it is not clear if enzymatically active PARP-1, nascent PARP-1, or both are capable of interacting with NF-κB and promoting DNA binding of NF-κB and cytokine gene expression.83 We did not detect either the interaction between PARP-1 and p65 or PARylation of p65 in cardiomyocytes infected by T. cruzi.47 Instead, PAR modification of p65-interacting nuclear proteins appeared to promote assembly of a transactivation-competent complex and cytokine gene expression in infected cardiomyocytes. In addition, PARP-1 may control inflammatory gene expression via regulation of cellular oxidation-reduction status. Nuclear respiratory factor (NRF) 1 is a key transcriptional activator of nuclear-encoded genes involved in mitochondrial biogenesis and function. Nuclear respiratory factor 1 participates in cellular oxidation-reduction signaling of inflammatory responses by increasing cytochrome c expression and mitochondrial respiratory capacity.84 The interactions of PARP-1 with NRF-1 and NRF-1 activation were regulated by automodification of PARP-1 and PARylation of NRF-1,84 suggesting that PARP-1 plays a regulatory role in NRF-1–dependent mitochondrial biogenesis, function, and cellular oxidation-reduction status. Conversely, PARP-1 may promote inflammation via negatively regulating other transcription factors. For example, the specificity protein 1 (Sp1) transcription factor mediates the transcriptional expression of hundreds of housekeeping genes whose products are anti-inflammatory mediators, and Sp1 activity is considered protective against systemic inflammation. A physical interaction of PARP-1 with Sp1 enhanced PARylation of Sp1 and resulted in its reduced binding to a consensus DNA target site.85 Interestingly, the AP-2α transcription factor is dually regulated by PARP-1 with distinct opposing outcomes.86 Separate regions of PARP-1 interact with AP-2α and independently control its transcriptional activation. The AMD domain of PARP-1 binds AP-2α with low affinity, enhancing the transcription rate. A high-affinity interaction of the catalytic domain of PARP-1 catalyzed poly-(ADP-ribosyl)ation of AP-2α and impaired its DNA-binding capacity and activity. The researchers propose that PARP-1 enhances the transcriptional activity of AP-2α in normal circumstances, whereas its enzymatic activity is used as a temporary shutoff mechanism during unfavorable conditions, to be validated in future studies. The complexity of PARP-1's regulation of various transcription factors in different cell types seems to indicate that use of PARP-1 inhibitors to block different types of inflammation should be tested on a case-by-case basis.
Another mechanism by which PARP-1 regulates cytokine gene expression is by direct binding to promoters. Poly(ADP-ribose) polymerase-1, in its inactive state, attaches to specific sequences in the C-X-C-motif chemokine ligand 1 promoter and prevents NF-κB (p65/p50) binding and transcription activation.87 In stress conditions, PARP-1 activation and PAR modification resulted in a loss of its binding to the CXC ligand 1 promoter, thus allowing NF-κB binding and enhanced CXC ligand 1 expression.87 Others88 showed that PARP-1 binding to the iNOS promoter enhances NO production, and s-nitrosylation of PARP-1 by NO negatively regulated the PARP-1 transactivation of the iNOS gene, likely by altering its binding and/or action at the iNOS promoter.
Although the study of the regulation of gene expression by PARP-1 has mainly focused on mechanisms that culminate in the control of transcription, a possible role of PARP-1 in the regulation of inflammatory gene expression at the posttranscriptional level has also been explored. A recent study68 indicated that PARP-1 might influence the posttranscriptional stability of mRNAs of inflammatory mediator interferon (IFN)–inducible protein 10. The PARP-1 deficiency in murine fibroblasts resulted in diminished IFN-γ–induced protein 10 expression that was associated with a defect in mitogen-activated protein kinase (MAPK) p38 activation. Whether PARP-1 regulation of mRNA stability constitutes a novel mechanism for tight regulation of chemokine expression in inflammatory diseases remains to be seen.
PARP-1 Activation and Cell Death
A second outcome resulting from PARP-1 activation is cell death, which triggers inflammatory reactions by multiple mechanisms. Necrosis, one type of cell death, incites an exudative inflammatory response in damaged tissue. Apoptosis and autophagy, two forms of programmed cell death, generally result in cell and nuclear shrinkage and fragmentation without colliquative cytolysis and inflammatory response. However, myocytes, probably because of their large size, elongated shape, and the presence of sarcomere, fail to manifest classic apoptotic morphological features and finally trigger the inflammatory process after undergoing apoptosis in injured hearts.89
PARP-1 acts on mitochondria and, depending on the extent of oxidative stress, DNA damage, and PARP-1 activation, different cell death pathways may be triggered (Figure 1B). A mild-to-moderate level of oxidative stress and PARP-1 activation may initiate cell death through a process involving mitochondrial depolarization/membrane permeability transition (MPT), resulting in the release of cytochrome c, second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low PI (Smac/Dablo), or AIF/endonuclease G from the mitochondrial intermembrane space into the cytosol.90,91 The AIF released from mitochondria moves to the nucleus and induces DNA fragmentation, which is considered an irreversible step in cell death and is caspase independent.92 Some studies93,94 have provided evidence for PARP-1–dependent cytochrome c and endonuclease G release from damaged mitochondria, inducing caspase-dependent apoptotic cell death. How PARP-1 activity is communicated to mitochondria is not known, and PARP-1 activation and the transport and binding of PARs to mitochondrial membranes catalyze mitochondrial MPT and initiate cytochrome c– or AIF/endonuclease G–dependent cell death pathways.95,96 However, this hypothesis should be experimentally corroborated in future studies. In most severe or sustained oxidative stress situations, excessive DNA damage resulting in hyperactivation of PARP-1 switches the mode of cell death from apoptosis to necrosis. This can possibly occur because of PARP-1–mediated excessive PARylation of apoptosis machinery and other essential proteins, resulting in their degradation and cell death. Alternatively, detachment of PAR from PARP-1 (by PARG) may allow the activated PARP-1 to bind DNA breaks again and use more NAD+ to continue the cycle. The NAD+ content is replenished by a nicotinic acid mononucleotide adenylyl transferase-1 enzyme that synthesizes NAD+ from nicotinamide mononucleotide and ATP.97 If not replenished or excessively used by hyperactive PARP-1, the depletion of NAD+ and the exhaustion of ATP result in impaired energy metabolism and, consequently, cell necrosis.98 Moreover, depletion of cytosolic NAD+ by PARP-1 activation blocks glycolysis at the NAD+-dependent glyceraldehyde-3-phosphate dehydrogenase step, thereby limiting glucose-derived substrate flow to the mitochondria.99,100 In summary, the cross talk between PARP-1 and mitochondria governs the fate of cells (ie, survival, apoptosis, or necrosis); and, depending on the extent of mitochondrial dysfunction and PARP-1 activation, inflammation and possibly other degenerative changes101,102 accrue in various diseases.
PARP-1–Related Signaling Pathways
Although accumulating data have indicated the important roles of PARP-1 in various inflammatory diseases, the signaling events that lead to PARP-1 activation and those modulated by PARP-1 have received attention in only the last decade. Herein, we aim at building up a framework to place PARP-1 in context to signaling pathways in inflammatory diseases. For those interested in the therapeutic application of PARP-1 inhibition, recent articles103–105 shed light on PARP-1 inhibitors and their use in human diseases.
Many intracellular and extracellular stimulators have been addressed as the signal “triggers” in different cell types. These include oxidative agents (eg, H2O2 and peroxynitrite),106,107 a DNA-alkylating agent (N-methyl-N′-nitro-N-nitrosoguanidine),107,108 excitotoxic injury (N-methyl-D-aspartic acid and glutamate),107,109 ethanol,110 immunological challenge (LPS and IL-1),67,75 Ca2+,111 angiotensin II,112 elevated extracellular glucose concentration,113 vitamin A depletion,114 and infection by the parasite T. cruzi.47 The activation of PARP-1 under most of these conditions directly results from final DNA damage by oxidants or genotoxicity; yet, how the DNA damage signal is transmitted to PARP-1 remains under further investigation. Duan et al109 recently elucidated a detailed pathway from an upstream stimulus to PARP-1 activation and mitochondrial release of AIF and cytochrome c in neurons. The researchers showed that glutamate excitotoxicity activates the N-methyl-D-aspartic acid receptor that leads to mitochondrial Ca2+ overload and increased reactive oxygen species (ROS) production and PARP-1 activation. Treatment with pharmacological inhibitors to block mitochondrial Ca2+ uptake or prevent the mitochondrial release of ROS, but not with inhibitors of cytosolic phospholipase A2 or xanthine oxidase (cytosolic ROS producers), inhibited mitochondrial ROS production, DNA damage, and PARP-1 activation. This study suggested that Ca2+ uptake and mitochondrial ROS production might be the early signaling events in the activation of PARP-1.109 Recent studies115 in an in vitro model of cardiomyocyte infection by T. cruzi support the previously described notion because we found that invasion by parasites triggered MPT and loss of membrane potential, which resulted in an inefficiency of the electron transport chain and increased ROS production. The ROS-induced DNA damage elicited PARP-1 activation; the latter, in turn, led to an increased formation of PARs.47 The T. cruzi attachment and invasion of host cells altered intracellular Ca2+,116,117 although whether Ca+2 flux by invading parasites was the key event initiating MPT/Δψ, electron leakage, and the superoxide anion formation that triggered PARP-1/PAR activation in cardiomyocytes remains to be determined.
In addition to ROS and DNA damage, phosphorylation of PARP-1 may result in its maximal activation. The observation of blockage of PARP-1 activity in extracellular signal–regulated kinase (ERK) 1/2-inhibited neuron cultures led Kauppinen et al107 to identify putative phosphorylation sites of PARP-1 by mass spectrometry. Transfection of PARP-1–deficient mouse embryonic fibroblasts with the mutant PARP-1 species (Ser372Ala and Thr373Ala) showed that a lack of phosphorylation at Ser372 and Thr373 impaired PARP-1 activation, whereas mutation to glutamate (S372E and T373E) to mimic constitutive phosphorylation increased PARP-1 activity and eliminated the effect of ERK1/2 inhibitors on PARP-1 activation.107 Others have shown that the Tec (Tyrosine kinase expressed in hepatocellular carcinoma) family tyrosine kinase, known to regulate type 1 helper T-cell cytokine production, also regulates PARP-1 phosphorylation. Maruyama et al118 demonstrated that Tec family tyrosine kinase phosphorylates PARP-1 and elongation factor-1 alpha (EF-1α) and that the resultant triprotein complex binds to the IFN-γ gene promoter in vitro. The N-terminal DBD of PARP-1 was important for triprotein complex formation. Several mutants of Tec family tyrosine kinase that lacked kinase activity were unable to form the trimolecular complex and stimulate cytokine expression.118 These data imply that phosphorylation of PARP-1 in response to different stimuli has diverse functional roles ranging from maximal PARP-1 activation/PAR formation to PARP-1 binding with DNA and proteins and modulation of gene expression of inflammatory mediators.
Few studies have examined the downstream signal events of PARP-1 in inflammatory disorders. Yet, it is widely accepted that PARP-1 signals the MAPK pathway by modulating the phosphorylation of ERK1/2, p38, and c-Jun NH2-terminal kinase.75,94,108 Andreone et al75 showed that PARP-1 signaled phosphorylation of c-Jun NH2-terminal kinase and c-Jun, leading to increased DNA binding of AP-1 transcription factor in murine fibroblasts. Receptor-interacting protein 1 and TNF receptor–associated factor 2 were essential for signal transfer from PARP-1 to c-Jun NH2-terminal kinase.108 Recently, it was reported that p38 MAPK is a downstream effector of PARP-1, accounting for mRNA stability of cytokine IFN-inducible protein 10.68 Conversely, others94 have shown that PARP-1 activation inhibited phosphorylation of ERK1/2 and Bcl-2–associated X protein and, thereby, promoted Bcl-2–associated X protein translocation to mitochondria and release of cytochrome c and AIF. Subsequently, it led to cell death. Thus far, the studies on the relationship between PARP-1 and MAPK suggest that PARP-1 and MAPK might stimulate each other in a positive feedback cycle to propagate the responses to the long-lasting stress signals. Overall, these studies show the complexity of PARP-1's role in interlinked signaling networks and require researchers interested in using PARP-1 inhibitors as a therapy for target inflammatory disorders to be attentive to the potential impact of PARP-1 blockage on different cellular processes.
PARP-1–Targeted Therapeutic Strategies
All PARP-1 inhibitors [eg, 3-AB, 5-aminoisoquinoline, 3,4-Dihydro-5-(4-[1-piperidinyl]butoxy)-1[2H]-isoquinolinone, PJ34, INO-1001, and GPI-21016] share a carboxamide group attached to an aromatic ring, like the normal PARP-1 substrate NAD+ or a carbamoyl group built into a polyaromatic heterocyclic skeleton; some of these inhibitors have been tested for efficacy against different diseases in clinical trials.104,119 Pharmacological inhibition of PARP-1 has provided benefits in rodent and large animal models of inflammatory disorders and is widely accepted as an approach in the therapy of inflammatory diseases.120 However, as discussed in a recent review,104 most of the inhibitors that have been in clinical trials for human use are aimed at cancer therapy. Accordingly, the therapeutic application of PARP-1 inhibitors in inflammatory disorders is short of clinical evidence.
Some non–PARP-1 inhibitor medicines have received attention for their PARP-1–targeted curative effects. For example, insulin, used for the treatment of hyperglycemia after exposure to LPS in a rat model of endotoxemia, blocked PARP-1 activation and blunted the LPS-induced TNF-α response.121 Oral administration of honokiol to rats 1 hour before eccentric exercise significantly ameliorated muscle damage and accelerated the recovery of limb function that was associated with suppression of PARP-1 up-regulation, cell fragmentation, protein nitrotyrosylation, and decline in lipid peroxidation and leukocyte infiltration.52 Recently, the effects of PARP-1 inhibition and the anti-inflammatory activity of some food-derived flavonoids were evaluated in a mouse model of LPS-induced acute pulmonary inflammation.122,123 Some of the flavonoids, such as fisetin, were efficacious in reducing pulmonary inflammation compared with the well-established anti-inflammatory glucocorticoid dexamethasone. Because the long-term intake of corticosteroids is accompanied by serious adverse effects, these results indicate that functional foods or nutraceuticals containing such food-derived flavonoids may be promising candidates for the treatment of chronic inflammatory diseases. In addition, the active metabolite of vitamin D acted as a PARP-1 inhibitor. The UV irradiation–mediated PARP activation in human keratinocytes was inhibited by treatment with vitamin D, 7-dehydrocholesterol, or 1α,25-dihydroxyvitamin D3. Vitamin D may protect keratinocytes against overactivation of PARP-1, resulting from exposure to sunlight, suggesting the pharmacological and anti-inflammatory effects of vitamin D.124
Conclusions
Because PARylation and PARP-1 were discovered a few decades ago, basic research showed more solicitude for PARP-1 structure, structure-based PARP-1 activity, and the molecular mechanisms by which PARP-1 participates in DNA repair, transcriptional regulation, and cell death. However, some contradictions remain to be clarified. In addition, further studies are necessary for an accurate understanding of the molecular basis for automodification, dimerization, and activation of PARP-1 to help in the design of specific strategies for blocking PARP-1 activation in disease therapy.
Investigators have constantly noted PARP-1 activation in various diseases; however, the involvement of PARP-1 in inflammatory disorders received significant attention only in the last decade. The activation of PARP-1 results in an up-regulation of proinflammatory gene expression and mitochondria-associated cell death, accounting for the development of inflammation. The pathomechanism of inflammatory disorders, a consequence of integrated responses of the immune cells involved in the inflammatory foci and the local tissues/cells, is complex. The activation of PARP-1 showed different functions in different cell types, at least regarding signaling and activating transcription factors. Moreover, PARP-1 regulates the progression of autoimmune nephritis in males by inducing necrotic cell death and modulating inflammation65; PARP-1–mediated cell death is sexually dimorphic, participating in ischemic damage in the male, but not the female, brain.125 Because vitamin D can act as a PARP-1 inhibitor, and estrogen shares some structural properties with vitamin D, it is possible that protection from PARP-1 overactivation is afforded by estrogen in females (to be validated in future studies).
Current knowledge demonstrates the significance of PARP-1 in inflammatory diseases; however, the complexity of the signaling pathways linking PARP-1 in inflammation progression and the molecular mechanisms by which PARP-1 modulates the progression of inflammation remain to be elucidated. Yet, PARP-1 inhibitors are being tested in experimental models of various inflammatory diseases, with anticipation of regulating transcriptional pathways and rescuing cells from death. The assignment and testing of different PARP-1 inhibitors as therapy for specific cancers in clinical trials signifies that the application of PARP-1 inhibitors for inflammatory disorders requires caution. More important, investigators should analyze the signaling mechanisms affected by each PARP-1 inhibitor and weigh the beneficial-versus-harmful impact of each PARP-1 inhibitor before considering its application as therapy against inflammatory disorders.
Footnotes
Supported by grants from the NIH (AI053098 and HL094802 to N.J.G.) for work performed in the laboratory related to mitochondrial dysfunction and chronic inflammation.
CME Disclosure: The authors did not disclose any relevant financial relationships.
Current address of X.B.: Institute of Genetics and Cytology, Northeast Normal University, Changchun, People's Republic of China.
References
- 1.Riquelme P.T., Burzio L.O., Koide S.S. ADP ribosylation of rat liver lysine-rich histone in vitro. J Biol Chem. 1979;254:3018–3028. [PubMed] [Google Scholar]
- 2.Burzio L.O., Riquelme P.T., Koide S.S. ADP ribosylation of rat liver nucleosomal core histones. J Biol Chem. 1979;254:3029–3037. [PubMed] [Google Scholar]
- 3.Suzuki H., Quesada P., Farina B., Leone E. In vitro poly(ADP-ribosyl)ation of seminal ribonuclease. J Biol Chem. 1986;261:6048–6055. [PubMed] [Google Scholar]
- 4.Pleschke J.M., Kleczkowska H.E., Strohm M., Althaus F.R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000;275:40974–40980. doi: 10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 5.Otto H., Reche P.A., Bazan F., Dittmar K., Haag F., Koch-Nolte F. In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs) BMC Genomics. 2005;6:139. doi: 10.1186/1471-2164-6-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lautier D., Lagueux J., Thibodeau J., Menard L., Poirier G.G. Molecular and biochemical features of poly (ADP-ribose) metabolism. Mol Cell Biochem. 1993;122:171–193. doi: 10.1007/BF01076101. [DOI] [PubMed] [Google Scholar]
- 7.Lindahl T., Satoh M.S., Poirier G.G., Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 8.Nguewa P.A., Fuertes M.A., Valladares B., Alonso C., Perez J.M. Poly(ADP-ribose) polymerases: homology, structural domains and functions: novel therapeutical applications. Prog Biophys Mol Biol. 2005;88:143–172. doi: 10.1016/j.pbiomolbio.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber V., Dantzer F., Ame J.C., de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 10.Shieh W.M., Ame J.C., Wilson M.V., Wang Z.Q., Koh D.W., Jacobson M.K., Jacobson E.L. Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J Biol Chem. 1998;273:30069–30072. doi: 10.1074/jbc.273.46.30069. [DOI] [PubMed] [Google Scholar]
- 11.Chambon P., Weill J.D., Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. doi: 10.1016/0006-291x(63)90024-x. [DOI] [PubMed] [Google Scholar]
- 12.Slattery E., Dignam J.D., Matsui T., Roeder R.G. Purification and analysis of a factor which suppresses nick-induced transcription by RNA polymerase II and its identity with poly(ADP-ribose) polymerase. J Biol Chem. 1983;258:5955–5959. [PubMed] [Google Scholar]
- 13.Woodhouse B.C., Dianov G.L. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst) 2008;7:1077–1086. doi: 10.1016/j.dnarep.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Kameshita I., Matsuda Z., Taniguchi T., Shizuta Y. Poly (ADP-ribose) synthetase: separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J Biol Chem. 1984;259:4770–4776. [PubMed] [Google Scholar]
- 15.D'Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 16.Langelier M.F., Ruhl D.D., Planck J.L., Kraus W.L., Pascal J.M. The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. J Biol Chem. 2010;285:18877–18887. doi: 10.1074/jbc.M110.105668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tao Z., Gao P., Hoffman D.W., Liu H.W. Domain C of human poly(ADP-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry. 2008;47:5804–5813. doi: 10.1021/bi800018a. [DOI] [PubMed] [Google Scholar]
- 18.Langelier M.F., Servent K.M., Rogers E.E., Pascal J.M. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J Biol Chem. 2008;283:4105–4114. doi: 10.1074/jbc.M708558200. [DOI] [PubMed] [Google Scholar]
- 19.Desmarais Y., Menard L., Lagueux J., Poirier G.G. Enzymological properties of poly(ADP-ribose)polymerase: characterization of automodification sites and NADase activity. Biochim Biophys Acta. 1991;1078:179–186. doi: 10.1016/0167-4838(91)99007-f. [DOI] [PubMed] [Google Scholar]
- 20.Tao Z., Gao P., Liu H.W. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: analysis and implications. J Am Chem Soc. 2009;131:14258–14260. doi: 10.1021/ja906135d. [DOI] [PubMed] [Google Scholar]
- 21.Altmeyer M., Messner S., Hassa P.O., Fey M., Hottiger M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37:3723–3738. doi: 10.1093/nar/gkp229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caldecott K.W., Aoufouchi S., Johnson P., Shall S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Acids Res. 1996;24:4387–4394. doi: 10.1093/nar/24.22.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber V., Ame J.C., Dolle P., Schultz I., Rinaldi B., Fraulob V., Menissier-de Murcia J., de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 24.Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18:3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pion E., Ullmann G.M., Ame J.C., Gerard D., de Murcia G., Bombarda E. DNA-induced dimerization of poly(ADP-ribose) polymerase-1 triggers its activation. Biochemistry. 2005;44:14670–14681. doi: 10.1021/bi050755o. [DOI] [PubMed] [Google Scholar]
- 26.Simonin F., Hofferer L., Panzeter P.L., Muller S., de Murcia G., Althaus F.R. The carboxyl-terminal domain of human poly(ADP-ribose) polymerase: overproduction in Escherichia coli, large scale purification, and characterization. J Biol Chem. 1993;268:13454–13461. [PubMed] [Google Scholar]
- 27.Ruf A., Mennissier de Murcia J., de Murcia G., Schulz G.E. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc Natl Acad Sci U S A. 1996;93:7481–7485. doi: 10.1073/pnas.93.15.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lonskaya I., Potaman V.N., Shlyakhtenko L.S., Oussatcheva E.A., Lyubchenko Y.L., Soldatenkov V.A. Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. J Biol Chem. 2005;280:17076–17083. doi: 10.1074/jbc.M413483200. [DOI] [PubMed] [Google Scholar]
- 29.Potaman V.N., Shlyakhtenko L.S., Oussatcheva E.A., Lyubchenko Y.L., Soldatenkov V.A. Specific binding of poly(ADP-ribose) polymerase-1 to cruciform hairpins. J Mol Biol. 2005;348:609–615. doi: 10.1016/j.jmb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 30.Kim M.Y., Mauro S., Gevry N., Lis J.T., Kraus W.L. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119:803–814. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Wacker D.A., Ruhl D.D., Balagamwala E.H., Hope K.M., Zhang T., Kraus W.L. The DNA binding and catalytic domains of poly(ADP-ribose) polymerase 1 cooperate in the regulation of chromatin structure and transcription. Mol Cell Biol. 2007;27:7475–7485. doi: 10.1128/MCB.01314-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidovic L., Vodenicharov M., Affar E.B., Poirier G.G. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 33.Van Wijk S.J., Hageman G.J. Poly(ADP-ribose) polymerase-1 mediated caspase-independent cell death after ischemia/reperfusion. Free Radic Biol Med. 2005;39:81–90. doi: 10.1016/j.freeradbiomed.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 34.Kim M.Y., Zhang T., Kraus W.L. Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 35.Heeres J.T., Hergenrother P.J. Poly(ADP-ribose) makes a date with death. Curr Opin Chem Biol. 2007;11:644–653. doi: 10.1016/j.cbpa.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 36.Andrabi S.A., Kim N.S., Yu S.W., Wang H., Koh D.W., Sasaki M., Klaus J.A., Otsuka T., Zhang Z., Koehler R.C., Hurn P.D., Poirier G.G., Dawson V.L., Dawson T.M. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohausz O., Blenn C., Malanga M., Althaus F.R. The roles of poly(ADP-ribose)-metabolizing enzymes in alkylation-induced cell death. Cell Mol Life Sci. 2008;65:644–655. doi: 10.1007/s00018-008-7516-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poitras M.F., Koh D.W., Yu S.W., Andrabi S.A., Mandir A.S., Poirier G.G., Dawson V.L., Dawson T.M. Spatial and functional relationship between poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in the brain. Neuroscience. 2007;148:198–211. doi: 10.1016/j.neuroscience.2007.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jagtap P., Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 40.Beneke S. Poly(ADP-ribose) polymerase activity in different pathologies: the link to inflammation and infarction. Exp Gerontol. 2008;43:605–614. doi: 10.1016/j.exger.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 41.Chopra M., Sharma A.C. Distinct cardiodynamic and molecular characteristics during early and late stages of sepsis-induced myocardial dysfunction. Life Sci. 2007;81:306–316. doi: 10.1016/j.lfs.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liaudet L., Pacher P., Mabley J.G., Virag L., Soriano F.G., Hasko G., Szabo C. Activation of poly(ADP-ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. Am J Respir Crit Care Med. 2002;165:372–377. doi: 10.1164/ajrccm.165.3.2106050. [DOI] [PubMed] [Google Scholar]
- 43.Zerfaoui M., Naura A.S., Errami Y., Hans C.P., Rezk B.M., Park J., Elsegeiny W., Kim H., Lord K., Kim J.G., Boulares A.H. Effects of PARP-1 deficiency on airway inflammatory cell recruitment in response to LPS or TNF: differential effects on CXCR2 ligands and Duffy antigen receptor for chemokines. J Leukoc Biol. 2009;86:1385–1392. doi: 10.1189/jlb.0309183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altmeyer M., Barthel M., Eberhard M., Rehrauer H., Hardt W.D., Hottiger M.O. Absence of poly(ADP-ribose) polymerase 1 delays the onset of Salmonella enterica serovar Typhimurium-induced gut inflammation. Infect Immun. 2010;78:3420–3431. doi: 10.1128/IAI.00211-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Czapski G.A., Cakala M., Gajkowska B., Strosznajder J.B. Poly(ADP-ribose) polymerase-1 inhibition protects the brain against systemic inflammation. Neurochem Int. 2006;49:751–755. doi: 10.1016/j.neuint.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Liaudet L., Oddo M. Role of poly(adenosine diphosphate-ribose) polymerase 1 in septic peritonitis. Curr Opin Crit Care. 2003;9:152–158. doi: 10.1097/00075198-200304000-00012. [DOI] [PubMed] [Google Scholar]
- 47.Ba X., Gupta S., Davidson M., Garg N.J. Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for up-regulation of cytokine expression in cardiomyocytes. J Biol Chem. 2010;285:11596–11606. doi: 10.1074/jbc.M109.076984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang H., Rivera Z., Jube S., Nasu M., Bertino P., Goparaju C., Franzoso G., Lotze M.T., Krausz T., Pass H.I., Bianchi M.E., Carbone M. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc Natl Acad Sci U S A. 2010;107:12611–12616. doi: 10.1073/pnas.1006542107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brunyanszki A., Hegedus C., Szanto M., Erdelyi K., Kovacs K., Schreiber V., Gergely S., Kiss B., Szabo E., Virag L., Bai P. Genetic ablation of PARP-1 protects against oxazolone-induced contact hypersensitivity by modulating oxidative stress. J Invest Dermatol. 2010;130:2629–2637. doi: 10.1038/jid.2010.190. [DOI] [PubMed] [Google Scholar]
- 50.Genovese T., Mazzon E., Muia C., Patel N.S., Threadgill M.D., Bramanti P., De Sarro A., Thiemermann C., Cuzzocrea S. Inhibitors of poly(ADP-ribose) polymerase modulate signal transduction pathways and secondary damage in experimental spinal cord trauma. J Pharmacol Exp Ther. 2005;312:449–457. doi: 10.1124/jpet.104.076711. [DOI] [PubMed] [Google Scholar]
- 51.Mota R.A., Hernandez-Espinosa D., Galbis-Martinez L., Ordonez A., Minano A., Parrilla P., Vicente V., Corral J., Yelamos J. Poly(ADP-ribose) polymerase-1 inhibition increases expression of heat shock proteins and attenuates heat stroke-induced liver injury. Crit Care Med. 2008;36:526–534. doi: 10.1097/01.CCM.0000299735.43699.E9. [DOI] [PubMed] [Google Scholar]
- 52.Chiang J., Shen Y.C., Wang Y.H., Hou Y.C., Chen C.C., Liao J.F., Yu M.C., Juan C.W., Liou K.T. Honokiol protects rats against eccentric exercise-induced skeletal muscle damage by inhibiting NF-kappaB induced oxidative stress and inflammation. Eur J Pharmacol. 2009;610:119–127. doi: 10.1016/j.ejphar.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 53.Thiemermann C., Bowes J., Myint F.P., Vane J.R. Inhibition of the activity of poly(ADP ribose) synthetase reduces ischemia-reperfusion injury in the heart and skeletal muscle. Proc Natl Acad Sci U S A. 1997;94:679–683. doi: 10.1073/pnas.94.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zingarelli B., Szabo C., Salzman A.L. Blockade of poly(ADP-ribose) synthetase inhibits neutrophil recruitment, oxidant generation, and mucosal injury in murine colitis. Gastroenterology. 1999;116:335–345. doi: 10.1016/s0016-5085(99)70130-7. [DOI] [PubMed] [Google Scholar]
- 55.Pacher P., Liaudet L., Mabley J., Komjati K., Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40:1006–1016. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 56.Molnar A., Toth A., Bagi Z., Papp Z., Edes I., Vaszily M., Galajda Z., Papp J.G., Varro A., Szuts V., Lacza Z., Gero D., Szabo C. Activation of the poly(ADP-ribose) polymerase pathway in human heart failure. Mol Med. 2006;12:143–152. doi: 10.2119/2006-00043.Molnar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Von Lukowicz T., Hassa P.O., Lohmann C., Boren J., Braunersreuther V., Mach F., Odermatt B., Gersbach M., Camici G.G., Stahli B.E., Tanner F.C., Hottiger M.O., Luscher T.F., Matter C.M. PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc Res. 2008;78:158–166. doi: 10.1093/cvr/cvm110. [DOI] [PubMed] [Google Scholar]
- 58.Hans C.P., Feng Y., Naura A.S., Troxclair D., Zerfaoui M., Siddiqui D., Jihang J., Kim H., Kaye A.D., Matrougui K., Lazartigues E., Boulares A.H. Opposing roles of PARP-1 in MMP-9 and TIMP-2 expression and mast cell degranulation in dyslipidemic dilated cardiomyopathy. Cardiovasc Pathol. 2010 doi: 10.1016/j.carpath.2010.03.007. [Epub ahead of press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oumouna-Benachour K., Hans C.P., Suzuki Y., Naura A., Datta R., Belmadani S., Fallon K., Woods C., Boulares A.H. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation. 2007;115:2442–2450. doi: 10.1161/CIRCULATIONAHA.106.668756. [DOI] [PubMed] [Google Scholar]
- 60.Garcia S., Bodano A., Gonzalez A., Forteza J., Gomez-Reino J.J., Conde C. Partial protection against collagen antibody-induced arthritis in PARP-1 deficient mice. Arthritis Res Ther. 2006;8:R14. doi: 10.1186/ar1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oumouna M., Datta R., Oumouna-Benachour K., Suzuki Y., Hans C., Matthews K., Fallon K., Boulares H. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. 2006;177:6489–6496. doi: 10.4049/jimmunol.177.9.6489. [DOI] [PubMed] [Google Scholar]
- 62.Havranek T., Aujla P.K., Nickola T.J., Rose M.C., Scavo L.M. Increased poly(ADP-ribose) polymerase (PARP)-1 expression and activity are associated with inflammation but not goblet cell metaplasia in murine models of allergen-induced airway inflammation. Exp Lung Res. 2010;36:381–389. doi: 10.3109/01902141003663360. [DOI] [PubMed] [Google Scholar]
- 63.Garcia Soriano F., Virag L., Jagtap P., Szabo E., Mabley J.G., Liaudet L., Marton A., Hoyt D.G., Murthy K.G., Salzman A.L., Southan G.J., Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 64.Szabo C., Zanchi A., Komjati K., Pacher P., Krolewski A.S., Quist W.C., LoGerfo F.W., Horton E.S., Veves A. Poly(ADP-ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002;106:2680–2686. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- 65.Jog N.R., Dinnall J.A., Gallucci S., Madaio M.P., Caricchio R. Poly(ADP-ribose) polymerase-1 regulates the progression of autoimmune nephritis in males by inducing necrotic cell death and modulating inflammation. J Immunol. 2009;182:7297–7306. doi: 10.4049/jimmunol.0803565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Genovese T., Cuzzocrea S. Role of free radicals and poly(ADP-ribose)polymerase-1 in the development of spinal cord injury: new potential therapeutic targets. Curr Med Chem. 2008;15:477–487. doi: 10.2174/092986708783503177. [DOI] [PubMed] [Google Scholar]
- 67.Veres B., Gallyas F., Jr, Varbiro G., Berente Z., Osz E., Szekeres G., Szabo C., Sumegi B. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochem Pharmacol. 2003;65:1373–1382. doi: 10.1016/s0006-2952(03)00077-7. [DOI] [PubMed] [Google Scholar]
- 68.Galbis-Martinez M., Saenz L., Ramirez P., Parrilla P., Yelamos J. Poly(ADP-ribose) polymerase-1 modulates interferon-gamma-inducible protein (IP)-10 expression in murine embryonic fibroblasts by stabilizing IP-10 mRNA. Mol Immunol. 2010;47:1492–1499. doi: 10.1016/j.molimm.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 69.Kaplin A.I., Deshpande D.M., Scott E., Krishnan C., Carmen J.S., Shats I., Martinez T., Drummond J., Dike S., Pletnikov M., Keswani S.C., Moran T.H., Pardo C.A., Calabresi P.A., Kerr D.A. IL-6 induces regionally selective spinal cord injury in patients with the neuroinflammatory disorder transverse myelitis. J Clin Invest. 2005;115:2731–2741. doi: 10.1172/JCI25141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park E.M., Cho S., Frys K., Racchumi G., Zhou P., Anrather J., Iadecola C. Interaction between inducible nitric oxide synthase and poly(ADP-ribose) polymerase in focal ischemic brain injury. Stroke. 2004;35:2896–2901. doi: 10.1161/01.STR.0000147042.53659.6c. [DOI] [PubMed] [Google Scholar]
- 71.Mazzon E., Genovese T., Di Paola R., Muia C., Crisafulli C., Malleo G., Esposito E., Meli R., Sessa E., Cuzzocrea S. Effects of 3-aminobenzamide, an inhibitor of poly (ADP-ribose) polymerase, in a mouse model of acute pancreatitis induced by cerulein. Eur J Pharmacol. 2006;549:149–156. doi: 10.1016/j.ejphar.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 72.Kraus W.L., Lis J.T. PARP goes transcription. Cell. 2003;113:677–683. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 73.Ogata N., Ueda K., Hayaishi O. ADP-ribosylation of histone H2B: identification of glutamic acid residue 2 as the modification site. J Biol Chem. 1980;255:7610–7615. [PubMed] [Google Scholar]
- 74.Aubin R.J., Dam V.T., Miclette J., Brousseau Y., Huletsky A., Poirier G.G. Hyper(ADP-ribosyl)ation of histone H1. Can J Biochem. 1982;60:1085–1094. doi: 10.1139/o82-139. [DOI] [PubMed] [Google Scholar]
- 75.Andreone T.L., O'Connor M., Denenberg A., Hake P.W., Zingarelli B. Poly(ADP-ribose) polymerase-1 regulates activation of activator protein-1 in murine fibroblasts. J Immunol. 2003;170:2113–2120. doi: 10.4049/jimmunol.170.4.2113. [DOI] [PubMed] [Google Scholar]
- 76.Zingarelli B., Hake P.W., Burroughs T.J., Piraino G., O'Connor M., Denenberg A. Activator protein-1 signalling pathway and apoptosis are modulated by poly(ADP-ribose) polymerase-1 in experimental colitis. Immunology. 2004;113:509–517. doi: 10.1111/j.1365-2567.2004.01991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hassa P.O., Haenni S.S., Buerki C., Meier N.I., Lane W.S., Owen H., Gersbach M., Imhof R., Hottiger M.O. Acetylation of poly(ADP-ribose) polymerase-1 by p300/CREB-binding protein regulates coactivation of NF-kappaB-dependent transcription. J Biol Chem. 2005;280:40450–40464. doi: 10.1074/jbc.M507553200. [DOI] [PubMed] [Google Scholar]
- 78.Kameoka M., Ota K., Tetsuka T., Tanaka Y., Itaya A., Okamoto T., Yoshihara K. Evidence for regulation of NF-kappaB by poly(ADP-ribose) polymerase. Biochem J. 2000;346(pt 3):641–649. [PMC free article] [PubMed] [Google Scholar]
- 79.Zerfaoui M., Errami Y., Naura A.S., Suzuki Y., Kim H., Ju J., Liu T., Hans C.P., Kim J.G., Abd Elmageed Z.Y., Koochekpour S., Catling A., Boulares A.H. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J Immunol. 2010;185:1894–1902. doi: 10.4049/jimmunol.1000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boulares A.H., Zoltoski A.J., Sherif Z.A., Jolly P., Massaro D., Smulson M.E. Gene knockout or pharmacological inhibition of poly(ADP-ribose) polymerase-1 prevents lung inflammation in a murine model of asthma. Am J Respir Cell Mol Biol. 2003;28:322–329. doi: 10.1165/rcmb.2001-0015OC. [DOI] [PubMed] [Google Scholar]
- 81.Mabley J.G., Jagtap P., Perretti M., Getting S.J., Salzman A.L., Virag L., Szabo E., Soriano F.G., Liaudet L., Abdelkarim G.E., Hasko G., Marton A., Southan G.J., Szabo C. Anti-inflammatory effects of a novel, potent inhibitor of poly (ADP-ribose) polymerase. Inflamm Res. 2001;50:561–569. doi: 10.1007/PL00000234. [DOI] [PubMed] [Google Scholar]
- 82.Adaikalakoteswari A., Rema M., Mohan V., Balasubramanyam M. Oxidative DNA damage and augmentation of poly(ADP-ribose) polymerase/nuclear factor-kappa B signaling in patients with type 2 diabetes and microangiopathy. Int J Biochem Cell Biol. 2007;39:1673–1684. doi: 10.1016/j.biocel.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 83.Chang W.J., Alvarez-Gonzalez R. The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J Biol Chem. 2001;276:47664–47670. doi: 10.1074/jbc.M104666200. [DOI] [PubMed] [Google Scholar]
- 84.Hossain M.B., Ji P., Anish R., Jacobson R.H., Takada S. Poly(ADP-ribose) polymerase 1 interacts with nuclear respiratory factor 1 (NRF-1) and plays a role in NRF-1 transcriptional regulation. J Biol Chem. 2009;284:8621–8632. doi: 10.1074/jbc.M807198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zaniolo K., Desnoyers S., Leclerc S., Guerin S.L. Regulation of poly(ADP-ribose) polymerase-1 (PARP-1) gene expression through the post-translational modification of Sp1: a nuclear target protein of PARP-1. BMC Mol Biol. 2007;8:96. doi: 10.1186/1471-2199-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li M., Naidu P., Yu Y., Berger N.A., Kannan P. Dual regulation of AP-2alpha transcriptional activation by poly(ADP-ribose) polymerase-1. Biochem J. 2004;382:323–329. doi: 10.1042/BJ20040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amiri K.I., Ha H.C., Smulson M.E., Richmond A. Differential regulation of CXC ligand 1 transcription in melanoma cell lines by poly(ADP-ribose) polymerase-1. Oncogene. 2006;25:7714–7722. doi: 10.1038/sj.onc.1209751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu Z., Kuncewicz T., Dubinsky W.P., Kone B.C. Nitric oxide-dependent negative feedback of PARP-1 trans-activation of the inducible nitric-oxide synthase gene. J Biol Chem. 2006;281:9101–9109. doi: 10.1074/jbc.M511049200. [DOI] [PubMed] [Google Scholar]
- 89.Buja L.M., Vela D. Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc Pathol. 2008;17:349–374. doi: 10.1016/j.carpath.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 90.Yu S.W., Wang H., Poitras M.F., Coombs C., Bowers W.J., Federoff H.J., Poirier G.G., Dawson T.M., Dawson V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 91.Yu S.W., Andrabi S.A., Wang H., Kim N.S., Poirier G.G., Dawson T.M., Dawson V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Modjtahedi N., Giordanetto F., Madeo F., Kroemer G. Apoptosis-inducing factor: vital and lethal. Trends Cell Biol. 2006;16:264–272. doi: 10.1016/j.tcb.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 93.Tanaka S., Takehashi M., Iida S., Kitajima T., Kamanaka Y., Stedeford T., Banasik M., Ueda K. Mitochondrial impairment induced by poly(ADP-ribose) polymerase-1 activation in cortical neurons after oxygen and glucose deprivation. J Neurochem. 2005;95:179–190. doi: 10.1111/j.1471-4159.2005.03353.x. [DOI] [PubMed] [Google Scholar]
- 94.Ethier C., Labelle Y., Poirier G.G. PARP-1-induced cell death through inhibition of the MEK/ERK pathway in MNNG-treated HeLa cells. Apoptosis. 2007;12:2037–2049. doi: 10.1007/s10495-007-0127-z. [DOI] [PubMed] [Google Scholar]
- 95.Cipriani G., Rapizzi E., Vannacci A., Rizzuto R., Moroni F., Chiarugi A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J Biol Chem. 2005;280:17227–17234. doi: 10.1074/jbc.M414526200. [DOI] [PubMed] [Google Scholar]
- 96.Blenn C., Althaus F.R., Malanga M. Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochem J. 2006;396:419–429. doi: 10.1042/BJ20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Belenky P., Bogan K.L., Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 98.Alano C.C., Garnier P., Ying W., Higashi Y., Kauppinen T.M., Swanson R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alano C.C., Ying W., Swanson R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- 100.Ying W., Alano C.C., Garnier P., Swanson R.A. NAD+ as a metabolic link between DNA damage and cell death. J Neurosci Res. 2005;79:216–223. doi: 10.1002/jnr.20289. [DOI] [PubMed] [Google Scholar]
- 101.Peralta-Leal A., Rodriguez M.I., Oliver F.J. Poly(ADP-ribose)polymerase-1 (PARP-1) in carcinogenesis: potential role of PARP inhibitors in cancer treatment. Clin Transl Oncol. 2008;10:318–323. doi: 10.1007/s12094-008-0207-8. [DOI] [PubMed] [Google Scholar]
- 102.Moncada S., Bolanos J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J Neurochem. 2006;97:1676–1689. doi: 10.1111/j.1471-4159.2006.03988.x. [DOI] [PubMed] [Google Scholar]
- 103.Pacher P., Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peralta-Leal A., Rodriguez-Vargas J.M., Aguilar-Quesada R., Rodriguez M.I., Linares J.L., de Almodovar M.R., Oliver F.J. PARP inhibitors: new partners in the therapy of cancer and inflammatory diseases. Free Radic Biol Med. 2009;47:13–26. doi: 10.1016/j.freeradbiomed.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 105.Krishnakumar R., Kraus W.L. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tapodi A., Debreceni B., Hanto K., Bognar Z., Wittmann I., Gallyas F., Jr, Varbiro G., Sumegi B. Pivotal role of Akt activation in mitochondrial protection and cell survival by poly(ADP-ribose)polymerase-1 inhibition in oxidative stress. J Biol Chem. 2005;280:35767–35775. doi: 10.1074/jbc.M507075200. [DOI] [PubMed] [Google Scholar]
- 107.Kauppinen T.M., Chan W.Y., Suh S.W., Wiggins A.K., Huang E.J., Swanson R.A. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci U S A. 2006;103:7136–7141. doi: 10.1073/pnas.0508606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu Y., Huang S., Liu Z.G., Han J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem. 2006;281:8788–8795. doi: 10.1074/jbc.M508135200. [DOI] [PubMed] [Google Scholar]
- 109.Duan Y., Gross R.A., Sheu S.S. Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. J Physiol. 2007;585:741–758. doi: 10.1113/jphysiol.2007.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maffi S.K., Rathinam M.L., Cherian P.P., Pate W., Hamby-Mason R., Schenker S., Henderson G.I. Glutathione content as a potential mediator of the vulnerability of cultured fetal cortical neurons to ethanol-induced apoptosis. J Neurosci Res. 2008;86:1064–1076. doi: 10.1002/jnr.21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yakovlev A.G., Wang G., Stoica B.A., Boulares H.A., Spoonde A.Y., Yoshihara K., Smulson M.E. A role of the Ca2+/Mg2+-dependent endonuclease in apoptosis and its inhibition by poly(ADP-ribose) polymerase. J Biol Chem. 2000;275:21302–21308. doi: 10.1074/jbc.M001087200. [DOI] [PubMed] [Google Scholar]
- 112.Szabo C., Pacher P., Zsengeller Z., Vaslin A., Komjati K., Benko R., Chen M., Mabley J.G., Kollai M. Angiotensin II-mediated endothelial dysfunction: role of poly(ADP-ribose) polymerase activation. Mol Med. 2004;10:28–35. doi: 10.2119/2004-00001.szabo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Du X., Matsumura T., Edelstein D., Rossetti L., Zsengeller Z., Szabo C., Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chiu H.J., Fischman D.A., Hammerling U. Vitamin A depletion causes oxidative stress, mitochondrial dysfunction, and PARP-1-dependent energy deprivation. FASEB J. 2008;22:3878–3887. doi: 10.1096/fj.08-112375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gupta S., Bhatia V., Wen J.J., Wu Y., Huang M.H., Garg N.J. Trypanosoma cruzi infection disturbs mitochondrial membrane potential and ROS production rate in cardiomyocytes. Free Radic Biol Med. 2009;47:1414–1421. doi: 10.1016/j.freeradbiomed.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Caler E.V., Morty R.E., Burleigh B.A., Andrews N.W. Dual role of signaling pathways leading to Ca(2+) and cyclic AMP elevation in host cell invasion by Trypanosoma cruzi. Infect Immun. 2000;68:6602–6610. doi: 10.1128/iai.68.12.6602-6610.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Burleigh B.A., Caler E.V., Webster P., Andrews N.W. A cytosolic serine endopeptidase from Trypanosoma cruzi is required for the generation of Ca2+ signaling in mammalian cells. J Cell Biol. 1997;136:609–620. doi: 10.1083/jcb.136.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maruyama T., Nara K., Yoshikawa H., Suzuki N. Txk, a member of the non-receptor tyrosine kinase of the Tec family, forms a complex with poly(ADP-ribose) polymerase 1 and elongation factor 1alpha and regulates interferon-gamma gene transcription in Th1 cells. Clin Exp Immunol. 2007;147:164–175. doi: 10.1111/j.1365-2249.2006.03249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Esposito E., Cuzzocrea S. Superoxide: NO, peroxynitrite and PARP in circulatory shock and inflammation. Front Biosci. 2009;14:263–296. doi: 10.2741/3244. [DOI] [PubMed] [Google Scholar]
- 120.Giansanti V., Donà F., Tillhon M., Scovassi A.I. PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol. 2010;80:1869–1877. doi: 10.1016/j.bcp.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 121.Horvath E.M., Benko R., Gero D., Kiss L., Szabo C. Treatment with insulin inhibits poly(ADP-ribose)polymerase activation in a rat model of endotoxemia. Life Sci. 2008;82:205–209. doi: 10.1016/j.lfs.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Geraets L., Haegens A., Brauers K., Haydock J.A., Vernooy J.H., Wouters E.F., Bast A., Hageman G.J. Inhibition of LPS-induced pulmonary inflammation by specific flavonoids. Biochem Biophys Res Commun. 2009;382:598–603. doi: 10.1016/j.bbrc.2009.03.071. [DOI] [PubMed] [Google Scholar]
- 123.Geraets L., Moonen H.J., Brauers K., Wouters E.F., Bast A., Hageman G.J. Dietary flavones and flavonoles are inhibitors of poly(ADP-ribose)polymerase-1 in pulmonary epithelial cells. J Nutr. 2007;137:2190–2195. doi: 10.1093/jn/137.10.2190. [DOI] [PubMed] [Google Scholar]
- 124.Mabley J.G., Wallace R., Pacher P., Murphy K., Szabo C. Inhibition of poly(adenosine diphosphate-ribose) polymerase by the active form of vitamin D. Int J Mol Med. 2007;19:947–952. [PMC free article] [PubMed] [Google Scholar]
- 125.Vagnerova K., Liu K., Ardeshiri A., Cheng J., Murphy S.J., Hurn P.D., Herson P.S. Poly (ADP-ribose) polymerase-1 initiated neuronal cell death pathway: do androgens matter? Neuroscience. 2010;166:476–481. doi: 10.1016/j.neuroscience.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]