

Abstract
The role of endogenous galectin-1 (Gal-1) in acute inflammation has been poorly investigated. We therefore performed the carrageenan-induced paw edema model in wild-type and Gal-1−/− mice. On subplantar injection of carrageenan, Gal-1−/− mice displayed a similar first phase of edema (≤24 hours) to wild-type mice; however, a much less pronounced second phase (48 to 96 hours) was evident in this genotype. This reduced inflammation was associated with lower paw expression of inflammatory genes and cell infiltrates. Analysis of galectin protein and mRNA expression revealed high expression of Gal-1 in wild-type paws during resolution (≥48 hours), with some expression of galectin-9 (Gal-9). Administration of stable Gal-1 to wild-type mice completely ablated the first phase of edema but was ineffective when administered therapeutically at the 24-hour time point. Conversely, Gal-9 administration did not alter the first phase of edema but significantly reduced the second phase when administered therapeutically. This suggests anti-inflammatory actions for both proteins in this model albeit at different phases of the inflammatory response. Collectively, these data indicate that the absence of endogenous Gal-1 results in an abrogated response during the second phase of the edema reaction.
The process of leukocyte egress from the blood stream toward the site of tissue injury or infection is a life-saving process that is fundamental to survival of the host. Hence, it is governed by a series of cellular responses, the result of several fine-tuned molecular events, which assure the migration of the right type of leukocytes, in the correct vascular bed, ready to attack and dispose of the inflammatory insult.1 However, this response can potentially be disruptive due to the dangerous factors (reactive oxygen species and enzymes) released from emigrated white blood cells (eg, PMN) such that several mechanisms are activated in the host, not only to wall off production of lethal factors, but also to control the time- and spatial-dependency of the process of migration.2 Moreover, the life span of the emigrated leukocyte is also tightly regulated promoting safe/programmed cell death and removal.3 Therefore, besides classes of pro-inflammatory mediators and factors (eg, adhesion molecules) there exists an equally ever increasing list of endogenous anti-inflammatory mediators that promote the resolution of inflammation.2,4,5
Galectin-1 (Gal-1) is emerging as one of these effectors of endogenous anti-inflammation. In brief, several studies have indicated the inhibitory properties of Gal-1 on T cells, through its ability to induce apoptosis via interactions with several cell-surface glycoprotein receptors including CD7, CD2, CD43, and CD45.6–8 In the context of innate immunity, Gal-1 inhibits PMN chemotaxis,9 promotes phosphatidylserine (PS) exposure and subsequent phagocytosis of activated human neutrophils10 and reduces PMN interaction with endothelial cell monolayers.11 More importantly, pharmacological administration of Gal-1 inhibits several facets of acute inflammation including edema formation12 and tissue recovery of infiltrated PMN.9 However, not many studies have investigated the role of endogenous Gal-1 on specific aspects of the innate immune response. For instance, Gal-1−/− mice displayed increased levels of leukocyte recruitment in a model of peritonitis (http://www.functionalglycomics.org/) and increased vascular events such as leukocyte adhesion and emigration in the inflamed cremasteric microcirculation.11
Gal-1 is not the only galectin endowed with anti-inflammatory activities.13 Recent work has indicated Gal-9 as a novel and potent anti-inflammatory protein, largely due to its ability to induce T-cell apoptosis in models of experimental allergic encephalomyelitis14 and nephritis15 and inhibition of T-cell infiltration in a murine model of mite allergen-induced asthma.16 Moreover, Gal-9−/− mice develop spontaneous episodes of inflammatory arthritis and administration of this protein exerts inhibitory properties during the active phases of the collagen-induced arthritis model.17,18 PMN has also been identified as a target cell for Gal-9: in a murine model of the Schwartzman reaction, neutropenic mice were no longer protected by administration of recombinant Gal-9.19
The present study was undertaken to further our knowledge on the role of endogenous Gal-1 on the multiple facets of the inflammatory response, and the mouse carrageenan (CG) paw edema model was applied to Gal-1−/− mice. In the mouse paw, injection of CG elicits a complex and long-lasting response (compared to the more widely used rat edema model which lasts essentially 24 hours20) characterized by an acute (<24 hours) and a delayed (>24 hours) phase, with edema formation, marked influx of PMN, and the involvement of several mediators.21,22
Materials and Methods
Animals
Male C57BL/6J (WT) and Gal-1−/− mice were obtained from B&K (Hull, UK). Original breeding pairs of Gal-1−/− (Lgals1 null) animals were generously provided by the Consortium for Functional Glycomics (http://www.functionalglycomics.org). In all cases, animals (6–8 weeks old) were used 7 days after arrival according to guidelines laid down by the Local Ethical Committee for the use of Animals (Barts and The London School of Medicine). Moreover, animal work was performed according to Home Office regulations (Guidance on the Operation of Animals, Scientific Procedures Act, 1986).
Stable Forms of Gal-1 and Gal-9
We were kindly provided with stable forms of Gal-1 (sGal-1) and Gal-9 (sGal-9) from GalPharma (Takamatsu, Kagawa, Japan). Details relating to the generation, structure, and function of both sGal-1 and -9 can be found in previous reports.23,24 Briefly, Gal-1 is highly susceptible to oxidation, specifically at its cysteine residues, which renders the molecule inactive. sGal-1 is a cysteine-less mutant of Gal-1 which is resistant to oxidation and retains all of the known activities of WT Gal-1. In the case of Gal-9 it is highly susceptible to proteolytic degradation at the linker peptide. sGal-9 is a linker-less mutant of Gal-9 which is resistant to proteolytic degradation and again retains all known activities of WT Gal-9.
CG Model of Paw Edema
Paw edema was induced as previously described.21,25,26 Briefly, animals received a subplantar (s.pl) injection of 50 μl carrageenan (lambda species) 1% (w/v) in saline into the right paw with an equivalent volume of saline administered to the contralateral paw used as control. Paw volume was measured with a hydro-plethysmometer with mice paw adaptors (Ugo Basile, Milan, Italy) immediately before subplantar injection and up to 144 hours thereafter. Changes in paw volume were calculated by subtracting the paw volume of CG and saline (control) injected paws at each time point. Several experiments were conducted comparing responses in WT and Gal-1−/− mice. To assess the effects of the exogenous protein, three different regimes were followed: i) sGal-1 or sGal-9 (generous gifts of GalPharma, Takamatsu, Kagawa, Japan) were coadministered with CG, ii) administration of sGal-1 or sGal-9 intravenously (tail vein) 30 minutes before CG, and iii) administration of sGal-1 or sGal-9 intravenously (tail vein) 24 hours after induction with CG.
Cellular and Biochemical Analyses
RNA Isolation and cDNA Synthesis
Mice were euthanized at selected time points by cervical dislocation and paws harvested. RNA isolation was performed; briefly, paws were cut and added to pre-weighed tubes containing ceramic beads (Stretton Scientific, Cepheid, Derbyshire, UK). Samples were weighed and a single volume of TRI reagent (1 ml TRI reagent/50–100 mg tissue) (Ambion, Warrington, UK) was added, followed by homogenization with the Bertin homogenizer (Bertin Technologies, Bordeaux, France) using the following parameters: four cycles at 6500 rpm for 30 seconds. Samples were centrifuged at 10,500 rpm for 2 minutes; supernatants were transferred to new tubes and subsequent steps were performed according to the manufacturers' guidelines to isolate the RNA. To remove all contaminating genomic DNA, the isolated RNA underwent treatment with TURBO DNase free kit (Ambion) following the manufacturer's “vigorous treatment” protocol. The concentration and purity of the RNA was analyzed using the Nanodrop ND-1000 (NanoDrop Technologies, Wilmington, DE). First strand cDNA synthesis was performed with the Superscript III reverse transcriptase system (Invitrogen, Carlsbad, CA) following the manufacturer's protocol on a total of 2.5 μg of RNA.
Real-Time PCR
Real-time PCR was performed with the ABI Prism 7900 Real-time PCR system (Applied Biosystems Inc, Foster City, CA). A standard thermal cycling amplification profile was applied: 95°C for 15 minutes; 35 cycles–94°C for 15 seconds, 55°C for 30 seconds, and 72°C for 30 seconds followed by a melt curve analysis. For each reaction, a total volume of 10 μl was used, which consisted of 2 μl of diluted cDNA (20 ng/μl), 5.2 μl of 1X Power SYBR Green mastermix (Applied Biosystems), 1 μl of gene specific primer (Qiagen, West Sussex, UK), and 1.8 μl of dH2O. mRNA data were normalized relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and then used to calculate expression levels. The comparative cycle threshold (Ct) method was used to measure the gene transcription in samples.27 The results are expressed as relative units based on calculation of 2−DDCt, which gives the relative amount of target gene normalized to endogenous control (GAPDH) and to the control (naïve) sample with the expression set as 1.
MPO Assay
The edema response was monitored for 12 hours, with some paws harvested at 4 hours for an MPO assay, which was performed as previously described.28
Western Blotting
Briefly, paws were perforated several times with a 19G needle followed by centrifugation at 5000 rpm for 5 minutes. Exudates were collected and a BCA protein assay performed. Protein levels were equalized and 6× hot sample buffer was added. Exudates were analyzed by standard SDS-PAGE and proteins transferred to PVDF membranes (Millipore, Watford, UK). Membranes were probed for galectin-1 (polyclonal goat anti-mouse galectin-1; 1:1000 dilution) or galectin-9 (polyclonal goat anti-mouse galectin-9; 1:1000 dilution) (R&D Systems, Abingdon, UK). Primary antibodies were diluted with 1% nonfat dried milk (w/v) made up in TBS containing 0.1% Tween20 and incubated with membranes overnight. Membranes underwent three 5-minute washes with TBS-T and were incubated with HRP-conjugated rabbit anti-goat (Dako, Cambridge, UK), and proteins were detected with enhanced chemiluminescence visualized on hyperfilm (GE Health Care, Little Chalfont, UK).
Histology and Immunohistochemistry
Paws were harvested from mice and fixed for 48 hours with 4% paraformaldehyde, decalcified in 4% EDTA for 21 days, and embedded in paraffin and 5-μm sections were cut and stained with H&E (Sigma-Aldrich, Gillingham, Dorset, UK). Specific immunostaining was performed on sections for Gal-1, Gal-9, and cleaved caspase-3. Briefly, paraffin sections were deparaffinized, followed by antigen retrieval with citrate buffered retrieval solution (Dako, Glostup, Denmark) for 10 minutes at 95°C. Endogenous avidin and biotin activity was blocked with a commercially available kit from Dako followed by incubation with serum-free protein block (Dako). Sections were then incubated with either polyclonal goat-anti-mouse galectin-1, polyclonal goat-anti-mouse galectin-9, or polyclonal rabbit anti-cleaved caspase-3 for 1 hour at RT. Sections were washed with TBS then incubated with rabbit-anti-goat biotin (1:100) or sheep-anti-rabbit biotin (1:100) (Dako) for 30 minutes at RT, followed by amplification with the Vectastain elite ABC kit (Vector Laboratories, Peterborough, UK). Sections were developed using DAB substrate kit (Dako), counterstained with hematoxylin, and mounted with DPX (VWR, Leicestershire, UK). In some sets of experiments, double immunofluorescence staining with anti-CD3 (Dako) and cleaved caspase-3 was performed and carried out as described above. In this instance secondary antibodies were conjugated to either an Alexa 488 or 555. DAPI (1:1000) (Dako) was used as a counterstain for cell nuclei. All primary and secondary antibodies were diluted in Dako antibody diluent. In all cases, a minimum ≥3 sections per animal were evaluated. Digital images were taken using Image Pro image analysis software (Media Cybernetics, Wokingham, Berkshire, UK).
The quantification of caspase-3 positive cells was carried out in a double-blinded manner. Six visual fields of the paw sections were counted at a 20× magnification. Two sections per paw were evaluated with the percentage of caspase-3 positive cells calculated from the total cell infiltrate per field.
Flow Cytometric Analysis
Cells recovered from the paws were washed with PBS and re-suspended in FACS buffer (PBS supplemented with 0.1% NaN3, 10% rat serum) containing CD16/CD32 FcγIIR blocking antibody (clone 93; eBioscience, Wembley, UK) for 30 minutes at 4°C. Specific leukocyte populations were identified by single or double staining with the following monoclonal conjugated antibodies; PE-conjugated rat-anti-mouse CD11b (Clone: M1/70.15; AbD Serotec, Kidlington, Oxford, UK), FITC-conjugated anti-mouse F4/80 (Clone Cl:A3-1; AbD Serotec), FITC-conjugated anti-mouse Ly6G (Gr1) (Clone RB6-8C5; eBioscience), PE-conjugated anti-mouse CCR7 (Clone 4B12; eBioscience), CY5-conjugated anti-mouse CD11c (Clone N418, eBioscience), and isotype controls (eBR2a, eBioscience) for 1 hour at 4°C. Cell populations were analyzed with FACScalibur flow cytometry using CellQuest software (BD Bioscience, Oxford, UK). A minimum of 10,000 events were acquired, with the percentage total events in each population compared to the total cell count to calculate the specific cell infiltrate per paw.
Statistical Tests
All data are reported as mean ± SEM of n observations, using at least three mice per group. Statistical evaluation was performed using one-way analysis of variance (analysis of variance) (Prism4 GraphPad Software, San Diego, CA) followed by Bonferroni test for posthoc analyses, taking a probability P < 0.05 as significant. Where two variables were analyzed, a Student's t-test was used.
Results
Exogenous and Endogenous Gal-1 in the Paw Edema Model
Before testing the potential role of endogenous Gal-1 in the CG-induced edema model, we determined how susceptible this inflammatory response was to the exogenous application of sGal-1. Treatment of mice with sGal-1 led to a complete ablation of the first phase of the edema response (Figure 1A) irrespective of whether it was administered intravenously as a pretreatment or co-injected into the paw with the CG. No effect was observed on the second phase of edema. In line with previous studies,9,11 the administration of exogenous sGal-1 also resulted in a reduced neutrophil recruitment, as assessed by quantifying myeloperoxidase activity at the 4-hour time point (Figure 1B).
Figure 1.
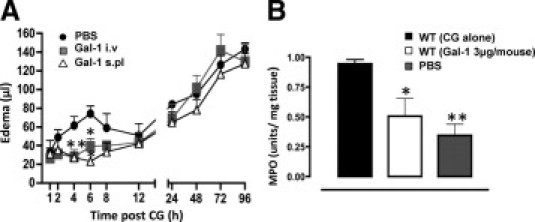
Gal-1 displays anti-inflammatory effects in the CG-induced paw edema model. A: sGal-1 (3 μg equivalent to 214 pmol) was either given intravenously (i.v.) 30 minutes before CG, or co-administered with CG subplantar (s.pl) into the right paw. An equal volume of saline was given s.pl into the left paw and the edema profile was monitored for 96 hours. Paw volume was assessed by plethysmography and edema calculated as the difference between CG and PBS injected paws. Data are expressed as mean ± SEM of six mice per treatment. *P < 0.05, **P < 0.01, as compared to WT (CG alone). B: Myeloperoxidase activity of paw tissue homogenates obtained 4 hours after injection with CG alone, CG+Gal-1, or PBS alone. Values are expressed as optical density units per mg of protein. Data are expressed as mean ± SEM of three mice per treatment. *P < 0.05, **P < 0.01, as compared to WT (CG alone).
We then compared the edema response in WT and Gal-1−/− mice throughout the full time course of this model.21,22 Injection of CG provoked an initial peak of edema (6–8 hours post-CG) followed by a delayed phase between the 48- and 96-hour time points (Figure 2A). In Gal-1−/− mice, the first phase followed the profile of WT mice; however, a blunted response was observed for the second phase with significantly lower edema values at 72 hours and 96 hours post-CG (Figure 2A). The extent of tissue pathology in the two genotypes is shown in representative images in Figure 2B (96-hour time point).
Figure 2.
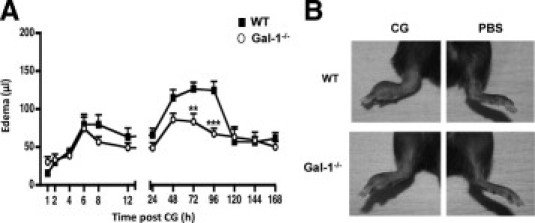
CG-induced paw edema profile in WT and Gal-1−/− mice. A: Paw edema was induced by s.pl injection of 1% lambda CG (50 μl) into the right hind paw of WT (black) and Gal-1−/− (white) mice. The left hind paw received the same volume of saline. Paw volume was assessed by plethysmography and edema calculated as the difference between CG and saline treated paws. Data are expressed as mean ± SEM of n = 15–25 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001 as compared to WT. B: Representative images of edema development in CG- (left) versus PBS-treated (right) paws from 48 hours time point in WT and Gal-1−/− mice.
The contrasting degree of edema observed in the Gal-1−/− mice at later time points instructed the next set of experiments where cell infiltrate and inflammatory mediator expression in the paw were determined. Table 1 reports the recovery of PMN (Gr1+ve cells), dendritic cells (CCR7 and CD11c double positive cells), and mono-macrophages (F4/80+ve cells). Whereas the injection of CG incremented the recovery of all three of these cell types at both 48 and 96 hours, absence of endogenous Gal-1 affected predominantly the number of PMN, with over 50% reduction being measured 48 hours post-CG. A trend toward an attenuation of both dendritic cell and macrophage infiltration was also evident at this time point, whereas no variation between genotype was detected at 96 hours post-CG (Table 1).
Table 1.
Characterization of Cellular Infiltrate in CG-Treated Paws of WT and Gal-1−/− Mice
| Marker | Time (hours) | Control (1 × 105/paw) | WT (1 × 105/paw) | Gal-1−/− (1 × 105/paw) |
|---|---|---|---|---|
| GR-1/CD11b | 48 | 0.29 ± 0.05 | 9.72 ± 1.43⁎† | 4.25 ± 1.23⁎ |
| 96 | 0.16 ± 0.02 | 0.53 ± 0.18 | 0.64 ± 0.10 | |
| CD11c/CCR7 | 48 | 0.04 ± 0.01 | 1.21 ± 0.24⁎ | 0.74 ± 0.15⁎ |
| 96 | 0.07 ± 0.03 | 5.95 ± 1.64 | 5.17 ± 2.21 | |
| F4/80 | 48 | 0.25 ± 0.02 | 3.7 ± 0.40⁎ | 2.31 ± 0.51⁎ |
| 96 | 0.22 ± 0.04 | 3.2 ± 1.41 | 2.29 ± 0.72 |
Flow cytometry was performed on cells recovered from CG treated paws of WT and Gal-1−/− mice at 48 hours (five mice per group) and 96 hours (three mice per group). Double and single staining was carried out for Gr1/CD11b (PMN), CD11c/CCR7 (migrating DC) and F4/80 (monocyte/ MØ). Data are expressed as mean ± SEM.
P < 0.05 as compared to WT naïve (control),
P < 0.05 as compared to Gal-1−/−.
Next, we measured gene expression of specific mediators in inflamed paw samples (96 hours post-GC). Absence of endogenous Gal-1 produced selective alterations such that both IL/1β mRNA and inductible nitric oxide synthase (iNOS) mRNA expression were markedly reduced in Gal-1−/− mice (Table 2) whereas no significant effect was observed for TNF-α, IL-6, and IL-10 mRNA. Interestingly, TGFβ mRNA was augmented in Gal-1−/− paws as compared to WT.
Table 2.
Cytokine mRNA Expression from CG-Treated Paws of WT and Gal-1−/− Mice
| Cytokine | WT (relative expression) | Gal-1−/− (relative expression) |
|---|---|---|
| IL-1β | 320 ± 61.79‡∥ | 23.47 ± 6.09 |
| TNF-α | 37.46 ± 7.03‡ | 23.58 ± 6.28⁎ |
| IL-6 | 41.5 ± 15.27 | 13.17 ± 3.40 |
| iNOS | 103.64 ± 27†¶ | 5.47 ± 1.57 |
| IL-10 | 34.04 ± 7.05† | 44.98 ± 7.67‡ |
| TGF-β | 12.21 ± 6.08⁎ | 25.47 ± 2.60‡⁎⁎ |
Real-time PCR analysis for TNF-α, IL-1β, IL-6, iNOS, IL-10, and TGF-β mRNA levels was performed on CG-treated paws of WT and Gal-1−/− mice collected at the 96-hour time-point. Data are expressed as mean ± SEM (8 per group).
§P < 0.05,
P < 0.05,
P < 0.01,
P < 0.001 as compared to control (WT Naïve),
P < 0.01,
P < 0.001 as compared to Gal-1−/−,
P < 0.01 as compared to WT.
Collectively, these unexpected results (reduced inflammatory response in Gal-1−/− mice) prompted the next set of experiments where expression of specific galectins was monitored.
Galectin Expression in Inflamed Paws
Gene product for Gal-1, Gal-3, and Gal-9 was modulated by the inflammatory response provoked by CG as measured in the whole paws collected from WT mice. Levels of mRNA for Gal-1 were found to be higher basally in the paw than was observed for Gal-3 and Gal-9 (Ct values of 20.80 ± 0.98, 27.48 ± 1.26, and 29.28 ± 1.10 for Lgals1, Lgals3, and Lgals9, respectively). Figure 3A shows that Gal-1 message increases in the inflamed paw at 48 and 96 hours post-CG, as compared to non-inflamed paws. Clearly no signal for Gal-1 message was observed in Gal-1−/− mice. The message for Gal-3 was delayed in its modulation, with no difference between genotypes (Figure 3B). In contrast, interesting observations were made for Gal-9 mRNA; in WT mice, Gal-9 mRNA increased steadily from the 24- to 96-hour time point, reaching at the later stage the highest degree of modulation as compared to the other galectins (approximately 40-fold increase) (Figure 3C). In Gal-1−/− mice, Gal-9 mRNA was already markedly elevated at the 24-hour time point and reached its peak at 48 hours post CG (Figure 3C).
Figure 3.
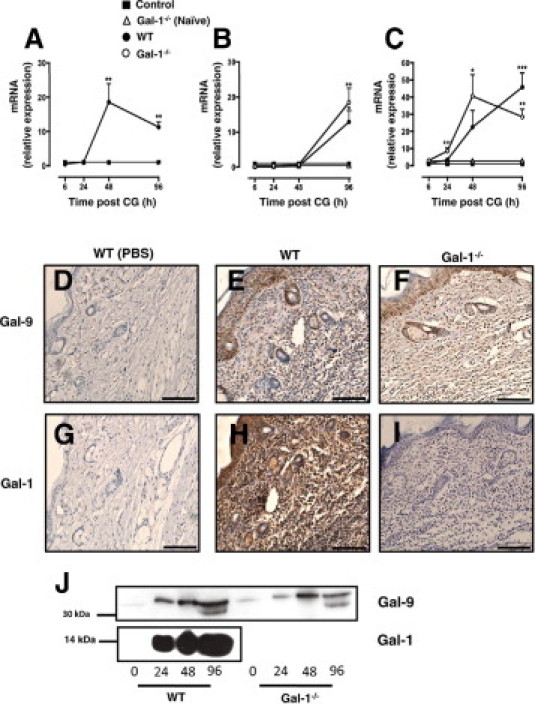
Galectin-mRNA expression profile in CG treated paws of WT and Gal-1−/− mice. Real-time PCR analysis for Gal-1 (A), Gal-3 (B), and Gal-9 (C) mRNA levels were assessed in CG-treated paws of WT (black circle) and Gal-1−/− (white circle) mice collected 6, 24, 48, 96 hours post-CG, respectively. Data are expressed as mean ± SEM (eight mice per group). *P < 0.05, **P < 0.01, ***P < 0.001 as compared to control (WT Naïve). Immunohistochemistry was performed on CG-treated paws of WT (E, H) and Gal-1−/− (F, I) mice from the 96-hour time point to examine Gal-9 and Gal-1 expression in the tissue. WT treated with saline were used as controls (D, G). Images shown are representative of five mice per group with similar results. Scale bar, 150 μm. J: Cell-free inflammatory exudates recovered from CG treated paws of WT and Gal-1−/− mice, taken from the indicated time points (24, 48, and 96 hours), were analyzed by Western blotting to determine soluble Gal-1 and Gal-9 protein levels in the inflamed paw.
Changes in Gal-1 and Gal-9 mRNA were then followed by analysis of protein expression in the inflamed paws of WT and Gal-1−/− mice. Fewer infiltrated cells were apparent in the Gal-1−/− mice compared to WT; of these cells, the degree of immunoreactivity against Gal-9 was comparable in both phenotypes (Figures 3E–F). A similar pattern of Gal-9 protein expression was also detected by Western blotting with comparable levels of Gal-9 observed at 48 hours post-CG in both WT and Gal-1−/− paw exudates (Figure 3J). Three isoforms of Gal-9 are known to exist in humans according to the length of the linker peptide that connects the two CRDs,29 while two isoforms have been identified to date in mice.30 As seen in Figure 3J, only one isoform of Gal-9 was detected within paw exudates at 24 and 48 hours while two isoforms were present at 96 hours. Gal-1 protein was also markedly expressed in the paws of WT mice with large amounts recovered both at 24, 48, and even more at 96 hours post CG (Figure 3J), a time point when resolution begins in WT animals (see Figure 2). When compared to Gal-9 the level of Gal-1 protein detected in both exudates and tissue was far greater with the peak in expression in paw exudates coinciding with the onset of resolution in WT mice.
Effect of Pharmacological Administration of sGal-9
The mRNA data prompted us to test the effect of exogenously administered sGal-9 in the last part of the study. Injection of different doses of sGal-9 intravenously 30 minutes before CG did not significantly reduce the first phase of edema (Figure 4A). Similarly, this early pharmacological treatment with sGal-9 did not affect the second phase of the edema response.
Figure 4.
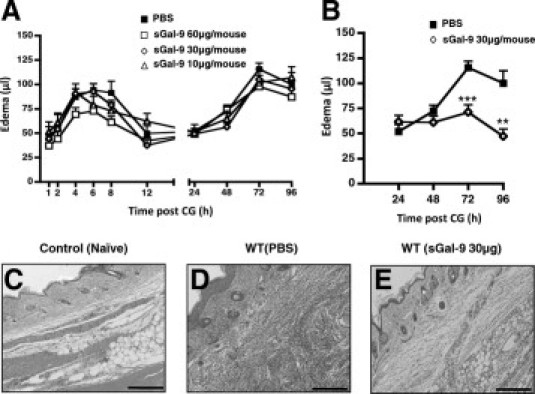
Administration of sGal-9 produces potent anti-edema effects. A: sGal-9 was administered intravenously (tail-vein) at three selected doses (10, 30, 60 μg per mouse) 30 minutes before edema induction with CG (right paw) and saline (left paw) in WT mice. PBS (100 μl) was given intravenously as a control. B: sGal-9 (30 μg) or PBS were administered intravenously 24 hours after induction with CG. In all cases, paw volume was assessed by plethysmography and edema calculated as the difference between CG and saline injected paws. Data are expressed as mean ± SEM of six mice per treatment. **P < 0.01, ***P < 0.001 as compared to control (PBS). C–E: Hematoxylin-eosin staining of cellular infiltrate was carried out on paws from the 96-hour time point, respectively, following intravenous administration 24 hours after CG induction with PBS (D) or sGal-9 (30 μg) (E). Control (WT Naïve) was injected with saline alone (C). Images shown are representative of six mice per group with similar results. Scale bar, 200 μm.
Therefore, we next administered sGal-9 according to a therapeutic protocol, that is, 24 hours post-CG injection. This time point was chosen as it precedes the beginning of the bifurcation of the profile of the response between WT and Gal-1−/− mice (see Figure 2) and coincided also with the initial upregulation (between 8- and 10-fold) of Gal-9 mRNA measured in Gal-1−/− paw extracts (Figure 3C). When given according to this therapeutic protocol, sGal-9 blocked the edema response in WT mice so that no further increments were measured up to 96 hours (Figure 4B). This remarkable anti-inflammatory activity of sGal-9 was also confirmed when histological analysis of the inflamed paws was performed. Figure 4C-E shows the marked reduction in immune cell infiltrate after sGal-9 administration, as measured at 96 hours post-CG (that is 72 hours post-Gal-9), when compared to vehicle-treated mice. In contrast to sGal-9, administration of sGal-1 therapeutically had no effect on the second phase of edema (data not shown).
There is large body of literature linking galectin biology to cell apoptosis31; we attempted to determine the degree of apoptosis in the inflamed paws as at least one potential downstream mechanism responsible for the effects observed in Gal-1−/− mice and also, after sGal-9 administration. Figure 5 reports these results highlighting a higher degree of apoptosis, measured as expression of the active form of caspase-3 in Gal-1−/− mice compared to WT animals (96-hour tissue sections). Similarly, administration of sGal-9, according to the therapeutic pharmacological protocol, markedly incremented the percentage of immune cells positive for the caspase-3 staining (Figure 5F). Cumulative data, generated with ≥5 distinct animals, are presented in Figure 5G: increments varied from a twofold increase in active caspase-3+ve cells in Gal-1−/− mice versus WT animals to a ∼four-fold increase measured after therapeutic treatment with sGal-9.
Figure 5.
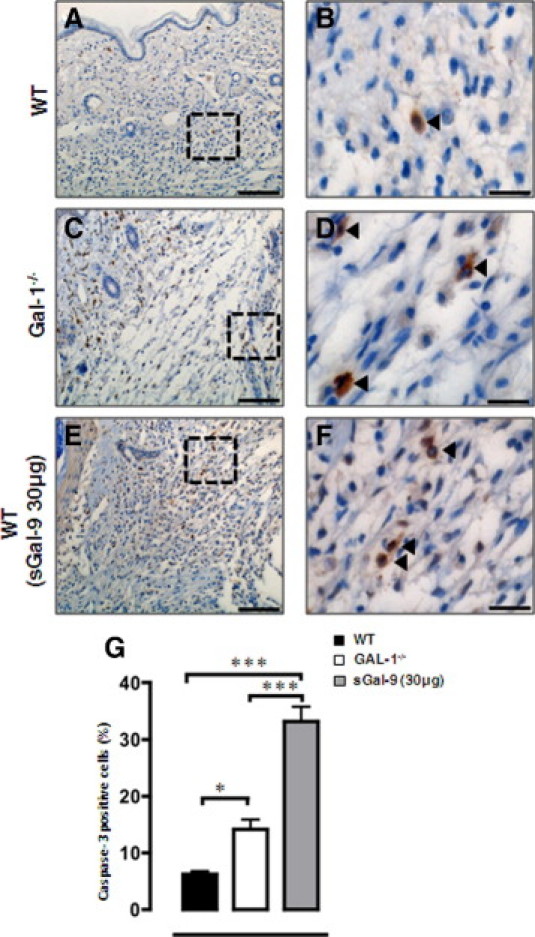
Cleaved caspase-3 expression as a marker of apoptosis. Immunohistochemistry was performed for cleaved caspase-3 expression on CG treated paws from WT (A, B), Gal-1−/− (C, D), and WT (E, F) (treated with sGal-9 30 μg, 24 hours after CG induction, see Figure 4B) mice from the 96-hour time point, respectively. Images shown are representative of six mice per group with similar results. Scale bar = 200 μm (A, C, E) 50 μm (B, D, F). G: Quantification of caspase-3 positive cells was carried out in a double-blinded manner, with six fields per section evaluated. *P < 0.05, ***P < 0.001 as compared to WT.
Finally, we attempted to identify the cells undergoing apoptosis as a result of treatment with sGal-9. To address this we performed double immunofluorescence staining with anti-CD3 (marker for T cells) with caspase-3. Figure 6 demonstrates strong co-localization between anti-CD3 and caspase-3, highlighting T cells as a specific cell type undergoing apoptosis during this phase of the edema response.
Figure 6.
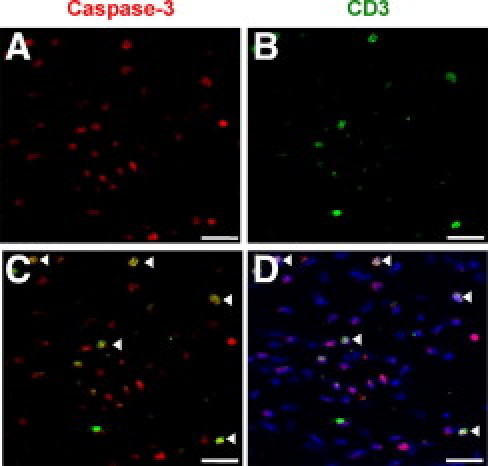
Cellular phenotype of caspase-3 positive cells. Double immunofluorescence was performed for cleaved caspase-3 (A) and anti-CD3 (B) on paw sections treated with sGal-9 30 μg, 24 hours after induction with CG. Panel C shows an overlay of the caspase-3 and anti-CD3 staining with co-localization indicated by arrowheads. D: DAPI was used as counterstain for cell nuclei. Scale bar = 150 μm.
Discussion
The novel findings of this study are that exogenous Gal-1 and -9 may act in concert to limit leukocyte recruitment and edema formation albeit at different stages of the inflammatory response. The study also highlights the differing functions of exogenous galectins and their endogenous counterparts.
The edema profiles obtained in both WT and Gal-1−/− mice followed a classical biphasic response as previously characterized.21 Our finding that administration of sGal-1 either as a pretreatment or concomitantly with CG abolished the first phase of edema and reduced the associated neutrophilic infiltrate correlates with the data of Rabinovich et al12 in which exogenous Gal-1 ameliorated phospholipase A2-induced paw edema in the rat. Exogenous Gal-1 has been shown previously to inhibit neutrophil recruitment in IL-1β induced peritonitis in mice9 and in vitro using a human system.11 The lack of any significant effect of Gal-9 on this first phase of edema suggests the anti-inflammatory properties of this protein are not through direct effects on neutrophil recruitment or inhibition of vascular permeability; indeed scant evidence exists for a role for Gal-9 in acute models of inflammation that are neutrophil driven. Data supporting any direct effects of Gal-9 on the trafficking of cells other than eosinophils is limited with conflicting evidence on whether it may also be chemotactic for neutrophils.19,32 The study by Tsuboi et al19 found Gal-9 to be chemotactic for PMN, although these PMN recruited by Gal-9 were shown to be anti-inflammatory PGE2-producing cells which led to a marked reduction in pro-inflammatory cytokines produced by recruited MØ. Their anti-inflammatory role was further emphasized by an enhanced inflammatory response when mice were rendered neutropenic in this murine model of the Schwartzman reaction.
Although Gal-1 administration abolished the first phase of edema it was ineffective against the second phase suggesting that the two phases are distinct from each other. Bucci et al33 found that both phases of the response were abolished in eNOS−/− mice as is the case for Akt1−/− mice34; changes in vascular permeability induced by histamine and bradykinin were also inhibited in these mice suggesting a direct effect on endothelial permeability. In contrast, Gal-1 administration has been shown to be ineffective against histamine induced-vascular permeability changes suggesting that its effects are mediated through limiting leukocyte recruitment rather than a direct effect on the vasculature.12
Against our prediction, during the second phase of the response the intensity of the edema was significantly lower in the Gal-1−/− mice compared to WT. A lower degree of edema and leukocyte infiltration in the Gal-1−/− mice appears to be at odds with the perceived anti-inflammatory properties of this protein.13 However, a majority of studies have focused on the role of the exogenous protein with relatively few investigations being carried out in the Gal-1−/− mice. The effectiveness of Gal-1 in many chronic models of inflammation and autoimmune disease is largely thought to be mediated through its ability to induce T-cell apoptosis. On the neutrophil, while it does not induce apoptosis Gal-1 induces PS exposure leading to increased clearance of these cells suggesting a pro-resolution function for this protein. Chronic inflammatory models that have been carried out in the Gal-1−/− mice suggest that the endogenous protein is indeed an important pro-apoptotic signal, at least for T cells.35 Our hypothesis was that lack of Gal-1 would exacerbate and prolong this second phase of edema. The fact that this was not the case suggests that either other pathways/mediators are at play in these experimental settings or that the endogenous protein functions in a different manner to the exogenous one. Indeed, there is a strong possibility that the differences observed in the edema response in the Gal-1−/− mice may be related to the role of intracellular rather than extracellular Gal-1. In the case of Gal-3 it has been shown that extracellular Gal-3 can induce apoptosis in activated T cells36 whereas intracellular Gal-3 can promote T-cell survival.37 A similar dual functionality may also exist for Gal-1. The potential function of Gal-1 as a pro-resolving factor in this model is supported, however, by its increased expression at both gene and protein level in WT mice at the later time points, with peak expression correlating with the onset of resolution.
The second phase of edema in this model is reminiscent of a delayed-type hypersensitivity reaction22 and is dependent on iNOS-derived NO. Levels of iNOS were significantly reduced in Gal-1−/− mice compared to WT mice and this may account for the decreased edema observed. iNOS expression and activity is significantly increased in WT mice from 24 hours onward in this model33,38 and inhibition of iNOS by L-NMMA significantly blunts the second phase of edema.22 Although in the study of Ianaro et al NOS inhibition blunted the edema response, cellular infiltrate was unaffected. This is in contrast to the findings in Gal-1−/− mice where both cellular infiltrate and edema are reduced suggesting a lack of iNOS induction is unlikely to be the only mechanism in these mice. As well as a decrease in iNOS expression, increased levels of transforming growth factor β (TGFβ) were detected in Gal-1−/− mice. TGFβ has been shown to inhibit delayed-type hypersensitivity responses in mice39 and more specifically to significantly inhibit CG-induced paw edema in mice40 through inhibition of cytokine generation by draining lymph node cells.
The mRNA expression profiles of Gal-1, Gal-3, and Gal-9 in the inflamed paws of WT and Gal-1−/− mice revealed a significant increase in Gal-9 gene expression in the Gal-1−/− mice which coincided with the onset of resolution in these mice. This prompted speculation that over-expressed Gal-9 may be functioning to reduce inflammation in Gal-1−/− mice. However, when monitoring Gal-9 protein expression by immunohistochemistry and Western blot in both tissue and exudates of inflamed paws, no apparent differences between phenotypes were observed. However, Gal-9 protein levels were increased during the inflammatory response and, interestingly, two isoforms of Gal-9 were detected by Western blot of paw exudates, suggesting that Gal-9-expressing cells are present within the inflamed paws and also that Gal-9 might be secreted into the extracellular milieu where it can presumably bind to target cells.
The complete abrogation of the second phase of edema following therapeutic administration of sGal-9 coupled with the significantly reduced leukocyte infiltration may be due to the induction of a subset of immunosuppressive MØ. A recent study from Arikawa et al41 demonstrated that Gal-9 significantly suppressed Trichosporon asahii-induced hypersensitivity pneumonitis in mice through induction of this subset of myeloid-derived suppressor cells. Expansion of these suppressor cells resulted in inhibition of T-cell proliferation and cytokine generation suggesting a possible mechanism of action for Gal-9 in this model. Gal-9 has also been found to directly promote TGFβ-induced Treg differentiation in vitro18 and sGal-9 increased Treg expansion in mice in models of CIA18 and contact hypersensitivity.42
Like Gal-1, Gal-9 is also known to induce apoptosis of various cell types including T cells14 as well as fibroblasts.17 We could unveil a strong modulatory function for exogenous Gal-9 and endogenous Gal-1 (possibly associated with augmented endogenous levels of Gal-9) on the viability of immune cells that had infiltrated the inflamed paws. Gal-9 induces apoptosis of Th1 cells through interaction with T-cell immunoglobulin domain and mucin domain 3 (Tim-3),14 as the second phase of edema is characterized by a Th1 response40; induction of apoptosis of T cells is a plausible mechanism of action for Gal-9 in this model. Evidence from the double staining performed with anti-CD3 and caspase-3 revealed co-localization between T cells and caspase-3, which adds further support to the notion that Gal-9 is endowed with pro-resolution properties in this model through induction of leukocyte apoptosis and increased phagocytic clearance. Clearly, additional studies will be required not only to reinforce the role of Gal-9 in this model but also the potential interlink between members of the galectin family.
The effects of galectins are complex and vary depending on local concentration, intracellular or extracellular localization, or differentiation status of the target cell. At low (30 nmol/L) concentrations Gal-1 inhibits Th1 responses43 and induces FcγRI on monocytes, an effect that is lost at high concentrations.44 Intracellular Gal-3 protects T cells from apoptosis while the extracellular protein induces apoptosis (reviewed in 45), while Gal-9 induces maturation of human dendritic cells46 and upregulates FcγRIIb on macrophages.47 It is therefore not inconceivable that galectins may cooperate during an inflammatory response.
In conclusion, loss of Gal-1 leads to an abrogation of CG induced second-phase edema in the mouse paw. This reduced inflammatory response is associated with decreased expression of iNOS and IL-1β, in conjunction with an increased expression of TGFβ and Gal-9 and increased apoptosis of infiltrated leukocytes. We wish to speculate that a Gal-1/Gal-9 cross talk may be operative during the course of a resolving model of acute inflammation, a feature that may also have repercussions in chronic settings, though future studies will address this hypothesis in detail.
Footnotes
Supported by a BBSRC-CASE PhD Studentship and by an Arthritis Research UK fellowship (18103) (D.C.). This work forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit, which is supported and funded by the National Institute of Health Research.
Current address of F.M., University of Naples “Federico II”, Naples, Italy; of A.L.F.S., Ensino e Inovação, CTM - Far-Manguinhos, Brazil.
Contributor Information
Mauro Perretti, Email: m.perretti@qmul.ac.uk.
Dianne Cooper, Email: d.cooper@qmul.ac.uk.
References
- 1.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 2.Serhan C.N., Brain S.D., Buckley C.D., Gilroy D.W., Haslett C., O'Neill L.A., Perretti M., Rossi A.G., Wallace J.L. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hallett J.M., Leitch A.E., Riley N.A., Duffin R., Haslett C., Rossi A.G. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol Sci. 2008;29:250–257. doi: 10.1016/j.tips.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Gilroy D.W., Lawrence T., Perretti M., Rossi A.G. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 5.Perretti M., D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9:62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 6.Pace K.E., Hahn H.P., Pang M., Nguyen J.T., Baum L.G. CD7 delivers a pro-apoptotic signal during galectin-1-induced T cell death. J Immunol. 2000;165:2331–2334. doi: 10.4049/jimmunol.165.5.2331. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez J.D., Nguyen J.T., He J., Wang W., Ardman B., Green J.M., Fukuda M., Baum L.G. Galectin-1 binds different CD43 glycoforms to cluster CD43 and regulate T cell death. J Immunol. 2006;177:5328–5336. doi: 10.4049/jimmunol.177.8.5328. [DOI] [PubMed] [Google Scholar]
- 8.Perillo N.L., Pace K.E., Seilhamer J.J., Baum L.G. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 9.La M., Cao T.V., Cerchiaro G., Chilton K., Hirabayashi J., Kasai K., Oliani S.M., Chernajovsky Y., Perretti M. A novel biological activity for galectin-1: inhibition of leukocyte-endothelial cell interactions in experimental inflammation. Am J Pathol. 2003;163:1505–1515. doi: 10.1016/s0002-9440(10)63507-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dias-Baruffi M., Zhu H., Cho M., Karmakar S., McEver R.P., Cummings R.D. Dimeric galectin-1 induces surface exposure of phosphatidylserine and phagocytic recognition of leukocytes without inducing apoptosis. J Biol Chem. 2003;278:41282–41293. doi: 10.1074/jbc.M306624200. [DOI] [PubMed] [Google Scholar]
- 11.Cooper D., Norling L.V., Perretti M. Novel insights into the inhibitory effects of Galectin-1 on neutrophil recruitment under flow. J Leukoc Biol. 2008;83:1459–1466. doi: 10.1189/jlb.1207831. [DOI] [PubMed] [Google Scholar]
- 12.Rabinovich G.A., Sotomayor C.E., Riera C.M., Bianco I., Correa S.G. Evidence of a role for galectin-1 in acute inflammation. Eur J Immunol. 2000;30:1331–1339. doi: 10.1002/(SICI)1521-4141(200005)30:5<1331::AID-IMMU1331>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 13.Norling L.V., Perretti M., Cooper D. Endogenous galectins and the control of the host inflammatory response. J Endocrinol. 2009;201:169–184. doi: 10.1677/JOE-08-0512. [DOI] [PubMed] [Google Scholar]
- 14.Zhu C., Anderson A.C., Schubart A., Xiong H., Imitola J., Khoury S.J., Zheng X.X., Strom T.B., Kuchroo V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 15.Tsuchiyama Y., Wada J., Zhang H., Morita Y., Hiragushi K., Hida K., Shikata K., Yamamura M., Kanwar Y.S., Makino H. Efficacy of galectins in the amelioration of nephrotoxic serum nephritis in Wistar Kyoto rats. Kidney Int. 2000;58:1941–1952. doi: 10.1111/j.1523-1755.2000.00366.x. [DOI] [PubMed] [Google Scholar]
- 16.Katoh S., Ishii N., Nobumoto A., Takeshita K., Dai S.Y., Shinonaga R., Niki T., Nishi N., Tominaga A., Yamauchi A., Hirashima M. Galectin-9 inhibits CD44-hyaluronan interaction and suppresses a murine model of allergic asthma. Am J Respir Crit Care Med. 2007;176:27–35. doi: 10.1164/rccm.200608-1243OC. [DOI] [PubMed] [Google Scholar]
- 17.Seki M., Sakata K.M., Oomizu S., Arikawa T., Sakata A., Ueno M., Nobumoto A., Niki T., Saita N., Ito K., Dai S.Y., Katoh S., Nishi N., Tsukano M., Ishikawa K., Yamauchi A., Kuchroo V., Hirashima M. Beneficial effect of galectin 9 on rheumatoid arthritis by induction of apoptosis of synovial fibroblasts. Arthritis Rheum. 2007;56:3968–3976. doi: 10.1002/art.23076. [DOI] [PubMed] [Google Scholar]
- 18.Seki M., Oomizu S., Sakata K.M., Sakata A., Arikawa T., Watanabe K., Ito K., Takeshita K., Niki T., Saita N., Nishi N., Yamauchi A., Katoh S., Matsukawa A., Kuchroo V., Hirashima M. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008;127:78–88. doi: 10.1016/j.clim.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Tsuboi Y., Abe H., Nakagawa R., Oomizu S., Watanabe K., Nishi N., Nakamura T., Yamauchi A., Hirashima M. Galectin-9 protects mice from the Shwartzman reaction by attracting prostaglandin E2-producing polymorphonuclear leukocytes. Clin Immunol. 2007;124:221–233. doi: 10.1016/j.clim.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 20.Di Rosa M., Giroud J.P., Willoughby D.A. Studies on the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J Pathol. 1971;104:15–29. doi: 10.1002/path.1711040103. [DOI] [PubMed] [Google Scholar]
- 21.Henriques M.G., Silva P.M., Martins M.A., Flores C.A., Cunha F.Q., Assreuy-Filho J., Cordeiro R.S. Mouse paw edema: A new model for inflammation? Braz J Med Biol Res. 1987;20:243–249. [PubMed] [Google Scholar]
- 22.Ianaro A., O'Donnell C.A., Di Rosa M., Liew F.Y. A nitric oxide synthase inhibitor reduces inflammation, down-regulates inflammatory cytokines and enhances interleukin-10 production in carrageenin-induced oedema in mice. Immunology. 1994;82:370–375. [PMC free article] [PubMed] [Google Scholar]
- 23.Nishi N., Abe A., Iwaki J., Yoshida H., Itoh A., Shoji H., Kamitori S., Hirabayashi J., Nakamura T. Functional and structural bases of a cysteine-less mutant as a long-lasting substitute for galectin-1. Glycobiology. 2008;18:1065–1073. doi: 10.1093/glycob/cwn089. [DOI] [PubMed] [Google Scholar]
- 24.Nishi N., Itoh A., Fujiyama A., Yoshida N., Araya S., Hirashima M., Shoji H., Nakamura T. Development of highly stable galectins: truncation of the linker peptide confers protease-resistance on tandem-repeat type galectins. FEBS Lett. 2005;579:2058–2064. doi: 10.1016/j.febslet.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 25.Calhoun W., Chang J., Carlson R.P. Effect of selected antiinflammatory agents and other drugs on zymosan, arachidonic acid: PAF and carrageenan induced paw edema in the mouse. Agents Actions. 1987;21:306–309. doi: 10.1007/BF01966499. [DOI] [PubMed] [Google Scholar]
- 26.Morris C.J. Carrageenan-induced paw edema in the rat and mouse. Methods Mol Biol. 2003;225:115–121. doi: 10.1385/1-59259-374-7:115. [DOI] [PubMed] [Google Scholar]
- 27.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maione F., Paschalidis N., Mascolo N., Dufton N., Perretti M., D'Acquisto F. Interleukin 17 sustains rather than induces inflammation. Biochem Pharmacol. 2009;77:878–887. doi: 10.1016/j.bcp.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 29.Chabot S., Kashio Y., Seki M., Shirato Y., Nakamura K., Nishi N., Nakamura T., Matsumoto R., Hirashima M. Regulation of galectin-9 expression and release in Jurkat T cell line cells. Glycobiology. 2002;12:111–118. doi: 10.1093/glycob/12.2.111. [DOI] [PubMed] [Google Scholar]
- 30.Wada J., Kanwar Y.S. Identification and characterization of galectin-9, a novel beta-galactoside-binding mammalian lectin. J Biol Chem. 1997;272:6078–6086. doi: 10.1074/jbc.272.9.6078. [DOI] [PubMed] [Google Scholar]
- 31.Liu FT, Rabinovich GA: Galectins: regulators of acute and chronic inflammation. Ann NY Acad Sci 1183:158–182 [DOI] [PubMed]
- 32.Matsumoto R., Matsumoto H., Seki M., Hata M., Asano Y., Kanegasaki S., Stevens R.L., Hirashima M. Human ecalectin, a variant of human galectin-9, is a novel eosinophil chemoattractant produced by T lymphocytes. J Biol Chem. 1998;273:16976–16984. doi: 10.1074/jbc.273.27.16976. [DOI] [PubMed] [Google Scholar]
- 33.Bucci M., Roviezzo F., Posadas I., Yu J., Parente L., Sessa W.C., Ignarro L.J., Cirino G. Endothelial nitric oxide synthase activation is critical for vascular leakage during acute inflammation in vivo. Proc Natl Acad Sci USA. 2005;102:904–908. doi: 10.1073/pnas.0408906102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Lorenzo A., Fernandez-Hernando C., Cirino G., Sessa W.C. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci USA. 2009;106:14552–14557. doi: 10.1073/pnas.0904073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toscano M.A., Bianco G.A., Ilarregui J.M., Croci D.O., Correale J., Hernandez J.D., Zwirner N.W., Poirier F., Riley E.M., Baum L.G., Rabinovich G.A. Differential glycosylation of TH1: TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–834. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 36.Fukumori T., Takenaka Y., Yoshii T., Kim H.R., Hogan V., Inohara H., Kagawa S., Raz A. CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 2003;63:8302–8311. [PubMed] [Google Scholar]
- 37.Yang R.Y., Hsu D.K., Liu F.T. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci USA. 1996;93:6737–6742. doi: 10.1073/pnas.93.13.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Posadas I., Bucci M., Roviezzo F., Rossi A., Parente L., Sautebin L., Cirino G. Carrageenan-induced mouse paw oedema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase-2 expression. Br J Pharmacol. 2004;142:331–338. doi: 10.1038/sj.bjp.0705650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meade R., Askenase P.W., Geba G.P., Neddermann K., Jacoby R.O., Pasternak R.D. Transforming growth factor-beta 1 inhibits murine immediate and delayed type hypersensitivity. J Immunol. 1992;149:521–528. [PubMed] [Google Scholar]
- 40.Ianaro A., Xu D., O'Donnell C.A., Di Rosa M., Liew F.Y. Expression of TGF-beta in attenuated Salmonella typhimurium: oral administration leads to the reduction of inflammation: IL-2 and IFN-gamma, but enhancement of IL-10, in carrageenin-induced oedema in mice. Immunology. 1995;84:8–15. [PMC free article] [PubMed] [Google Scholar]
- 41.Arikawa T., Saita N., Oomizu S., Ueno M., Matsukawa A., Katoh S., Kojima K., Nagahara K., Miyake M., Yamauchi A., Kohrogi H., Hirashima M. Galectin-9 expands immunosuppressive macrophages to ameliorate T-cell-mediated lung inflammation. Eur J Immunol. 2010;40:548–558. doi: 10.1002/eji.200939886. [DOI] [PubMed] [Google Scholar]
- 42.Niwa H., Satoh T., Matsushima Y., Hosoya K., Saeki K., Niki T., Hirashima M., Yokozeki H. Stable form of galectin-9, a Tim-3 ligand, inhibits contact hypersensitivity and psoriatic reactions: a potent therapeutic tool for Th1- and/or Th17-mediated skin inflammation. Clin Immunol. 2009;132:184–194. doi: 10.1016/j.clim.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 43.Rabinovich G.A., Daly G., Dreja H., Tailor H., Riera C.M., Hirabayashi J., Chernajovsky Y. Recombinant galectin-1 and its genetic delivery suppress collagen-induced arthritis via T cell apoptosis. J Exp Med. 1999;190:385–398. doi: 10.1084/jem.190.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrionuevo P., Beigier-Bompadre M., Ilarregui J.M., Toscano M.A., Bianco G.A., Isturiz M.A., Rabinovich G.A. A novel function for galectin-1 at the crossroad of innate and adaptive immunity: galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J Immunol. 2007;178:436–445. doi: 10.4049/jimmunol.178.1.436. [DOI] [PubMed] [Google Scholar]
- 45.Hsu D.K., Chen H.Y., Liu F.T. Galectin-3 regulates T-cell functions. Immunol Rev. 2009;230:114–127. doi: 10.1111/j.1600-065X.2009.00798.x. [DOI] [PubMed] [Google Scholar]
- 46.Dai S.Y., Nakagawa R., Itoh A., Murakami H., Kashio Y., Abe H., Katoh S., Kontani K., Kihara M., Zhang S.L., Hata T., Nakamura T., Yamauchi A., Hirashima M. Galectin-9 induces maturation of human monocyte-derived dendritic cells. J Immunol. 2005;175:2974–2981. doi: 10.4049/jimmunol.175.5.2974. [DOI] [PubMed] [Google Scholar]
- 47.Arikawa T., Watanabe K., Seki M., Matsukawa A., Oomizu S., Sakata K.M., Sakata A., Ueno M., Saita N., Niki T., Yamauchi A., Hirashima M. Galectin-9 ameliorates immune complex-induced arthritis by regulating Fc gamma R expression on macrophages. Clin Immunol. 2009;133:382–392. doi: 10.1016/j.clim.2009.09.004. [DOI] [PubMed] [Google Scholar]