

Abstract
RhoB is an early-response gene whose expression is elevated by multiple cellular stresses; this gene plays an important role in cancer, macrophage motility, and apoptosis. These factors are essential for the onset of type 1 diabetes mellitus and related complications. This study explores the role of RhoB in β-cell depletion and hyperglycemia-associated complications and tests whether the pleiotropic effect of statins on glycemic control is RhoB dependent. We induced β-cell depletion in RhoB+/+, RhoB+/−, and RhoB−/− mice with streptozotocin (STZ). Diabetic status was assessed by glucose tolerance and pancreatic islet loss. RhoB−/− mice showed a significant reduction in the severity of STZ-induced diabetes; only 13% of the STZ-treated RhoB-null animals became hyperglycemic, as opposed to 61% of the wild-type controls. Diabetes-related complications, such as wound healing rate and onset of nephropathy, were also assessed. Hyperglycemic RhoB−/− mice had fewer signs of nephropathy and showed faster wound healing than RhoB+/+ animals. After assessing the diabetic status of mice treated simultaneously with STZ and simvastatin, we conclude that the effect of statins in improving glycemic control is RhoB independent. We propose that RhoB is a modifier of diabetes, important for the induction of β-cell loss. Suppression of RhoB expression may have potential application in the treatment of diabetes and associated complications.
RhoB is an early-response gene whose expression is elevated by cellular stresses, including UV irradiation and hypoxia.1 The RhoB gene encodes a small GTPase that regulates vascular sprouting and intracellular trafficking of v-akt murine thymoma viral oncogene homolog 1 (AKT) in endothelial cells.2 This protein has also been implicated in a variety of cellular processes, such as migration and adhesion in macrophages, endocytosis, epidermal growth factor signaling, actin organization, intracellular trafficking, and DNA damage–induced apoptosis.1,3–7 All of these are important processes for type 1 diabetes mellitus onset and for the complications associated with hyperglycemia, such as delayed wound healing and nephropathy. However, to our knowledge, RhoB has not been examined in the context of diabetes and hyperglycemia.
Rho GTPases can be found in two forms: the active GTP-bound form, generated by guanine nucleotide exchange factors; and the inactive form, bound to GDP by the GTPase-activating proteins. Both can be post-translationally modified by cysteine prenylation. Although RhoB can be prenylated by either a farnesyl group or a geranylgeranyl group, RhoA and RhoC can only be geranylgeranylated. The method of prenylation of the Rho GTPases determines its cellular localization.8,9
Statins have been shown in previous studies9,10 to prevent hyperglycemia in nonobese diabetic mice and streptozotocin (STZ)–treated mice. The mechanism by which this is achieved is still unclear. Statins are HMG-CoA reductase inhibitors broadly used to control cholesterol levels and mitigate cardiovascular disease. This inhibition affects the mevalonate pathway, inhibiting Rho prenylation by preventing the formation of farnesyl and geranylgeranyl isoprenoids. We tested the effect of the drug simvastatin in STZ-induced hyperglycemia in RhoB−/−, RhoB+/−, and RhoB+/+ mice to assess whether the effect of simvastatin was linked to RhoB.
Materials and Methods
Animals
RhoB−/−, RhoB+/−, and RhoB+/+ mice were kept in 12-hour night/day light cycles and fed at libitum. Equal numbers of female and male animals were used. Mice were aged 4 to 6 weeks. The Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee approved all animal protocols used in this study. The Principles of Laboratory Animal Care (National Institutes of Health publication 85-23, rev 1985) were followed.
Multiple Low-Dose STZ Treatment
Diabetes was induced in 8- to 10-week-old Balb/cJ, C57Bl/6J, RhoB+/+, RhoB+/−, and RhoB−/− mice by i.p. injection of STZ at 40 mg/kg in 10-mmol/L citrate buffer (pH 4.6) for five consecutive days. Animals fasted for 4 hours before each injection. Citrate buffer was injected as a control arm of the experiments. At 30 days after the STZ injection, diabetes was confirmed by blood glucose measurement using a blood glucose monitoring system (One Touch; Lifescan, Milpitas, NV). Animals were considered hyperglycemic when, after fasting for 4 hours, blood glucose levels were greater than 250 mg/dL.
Mice Islets Study
Islets from five STZ-treated and untreated mice were extracted using collagenase treatment, followed by the gradient centrifugation method.11 Real-time PCR was performed on them as described later.
Wound Healing Assay
Animals that had been hyperglycemic for 4 weeks were anesthetized with isoflurane; then, an 8-mm diameter skin punch was performed on the dorsal surface of the body. Every 2 days, pictures of the wound area were taken and the area of the wound was calculated.
Nephropathic Analysis
Kidneys were examined from animals that had been hyperglycemic for 3 to 4 months. Renal slices were fixed in 4% paraformaldehyde, embedded in paraffin, and deparaffinized in xylene. Then, 4-μm sections were stained with hematoxylin-eosin, PAS, and Masson trichrome. The extent of renal injury was assessed by morphometric analysis of the glomerular disease and interstitial fibrosis, as previously described.12 For glomerular damage, we evaluated mesangial expansion and enlargement of the glomeruli. A point counting method was used to quantify the mesangial matrix deposition according to a previous method, with some modifications.13 We analyzed 20 PAS-stained glomeruli from each mouse on a digital microscope screen grid containing 667 (29 × 23) points. To obtain the percentage of mesangial matrix deposition (mesangial matrix index) in a given glomerulus, the number of grid points that land on a pink or red mesangial matrix deposition was divided by the total number of points in the glomerulus. The relative interstitial volume was evaluated by morphometric analysis using a 10-μm graticule fitted into the eyepiece of the microscope. We evaluated 10 randomly selected cortical areas under ×200 magnification for each mouse.
Immunostaining
For insulin and glucagon labeling, 10- to 15-μm sections were prepared as previously described. Sections were blocked with 3% normal donkey serum for 30 minutes at room temperature and incubated at 4°C overnight with nonimmune serum, followed by a rabbit antiserum raised against glucagon (Linco Research Inc, St Charles, MO) at a dilution of 1:3000. After rinsing with PBS, slides were blocked again with 3% donkey serum for 10 minutes at room temperature and then localized with donkey anti–rabbit-conjugated biotin (Jackson ImmunoResearch Laboratories Inc, West Grove, PA) (1:500) for 1 hour at room temperature. Slides were then washed twice in PBS and incubated in conjugated antibody (Alexa Fluorescent; Molecular Probes, Eugene, OR; diluted 1:400 in PBS) for 1 hour at 37°C. Finally, slides were incubated in guinea pig anti–bovine insulin serum (1:200; Linco Research Inc) and localized with anti–guinea pig antibody (1:200) (Texas Red) for 1.5 hours at room temperature. Slides were mounted with fluorescent mounting medium (Vector Laboratories Inc, Burlingame, CA) and stored in the dark.
Glucose Challenge
After animals had fasted for 4 hours, their basal glucose level was measured using a blood glucose monitoring system (One Touch). They were then injected i.p. with 10-μL/g body weight of 10-mg/ml d-glucose in PBS. Blood glucose levels were measured at 15, 30, 60, and 120 minutes after injection.
Pancreatic Analysis
Pancreas specimens were surgically removed, fixed in 4% paraformaldehyde for 2 hours, and then placed in 30% sucrose and frozen in OCT. Sections of 10 μm were cut and placed in slides. Five nonconsecutive slides per animal were labeled for insulin and glucagon. The number of islets was then counted, and pictures were obtained. The location of β-cell apoptosis was identified using dual immunofluorescence for terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) and insulin staining. We used a labeling (TUNEL) kit (Roche; Molecular Biochemicals, Indianapolis, IN) with a modified protocol for immunofluorescence. The sections were examined using confocal microscopy. The TUNEL-positive β cells were identified by the presence of red nuclei and green cytoplasm. The area of the islets was calculated using software (ImageJ).
Statins and STZ
A total of 12 RhoB+/+, 21 RhoB+/−, and 34 RhoB−/− 8- to 10-week-old male and female mice were used for these experiments. The mice were divided into two treatment groups: one was treated with saline vehicle and multiple low-dose STZ treatment (MLDS), and the other was treated with simvastatin and MLDS. Simvastatin was dissolved in 100 μL of ethanol and 150 μL of 0.1 N sodium hydroxide, incubated at 50°C for 2 hours, pH adjusted to 7.0, and volume corrected to 1 ml. Once a day, for five consecutive days, the mice received an i.p. injection of either saline (0.2 ml) or freshly prepared STZ, as previously described. Two days before the STZ injections started and for 12 consecutive days, the mice received a daily i.p. injection of either vehicle (0.2 ml) or simvastatin (30-mg/kg body weight; 0.2 ml) dissolved in vehicle. When required, the STZ/saline injections were administered 30 minutes before the simvastatin/vehicle injections.
Statistical Analysis
Values are the mean ± SEM. The significance of the differences between the two groups was analyzed by the Student's t-test. Comparisons among three groups were performed by two-way analysis of variance, followed by Scheffé's test to evaluate the significance of the differences between any two groups. P ≤ 0.05 was defined as statistically significant.
Real-Time RT-PCR Analysis
RNA was extracted using reagent (Trizol; Invitrogen, Carlsbad, CA) following the manufacturer's instructions. All samples were treated with DNase I to prevent genomic DNA contamination. Reverse transcription was performed using a cDNA reverse transcription kit following the manufacturer's instructions (Applied Biosystems, Foster City, CA). The cDNA generated was stored at −80°C until required for quantification. An RT-PCR machine (ABI Prism 7000 Sequence Detection System; PE Applied Biosystems) was used to amplify cDNA and detect PCR amplicons using the following sequence-specific primers for glyceraldehyde-3-phosphate dehydrogenase (forward, 5′-AACTTTGGCATTGTGGAAGGGCTC-3′; reverse, 5′-TGGAAGAGTGGGAGTTGCTGTTGA-3′) and RhoB (forward, 5′-GCATCAACTGCTGCAAGGTGCTAT-3′; reverse, 5′-TAATTCTCAGCGTGACCAGCCAGA-3′). Because the RhoB gene contains no introns, tubes were added on every run containing RNA instead of cDNA. No significant genomic contamination was detected. PCR reactions were performed in 96-well optical reaction plates (PE Applied Biosystems). Quantitative real-time PCR was normalized to the copies of glyceraldehyde-3-phosphate dehydrogenase mRNA from the same sample. To compare relative levels of mRNAs and to reduce experimental error, from plate to plate, expression levels were compared with those of one sample that was run on every plate and used as an internal control. Results were given as relative quantization to one sample based on the following equation: ΔFC = 2−dCT, with dCT (ΔCT) calculated as the difference in CT values between the gene of interest and glyceraldehyde-3-phosphate dehydrogenase. Acquired data were analyzed by software (Sequence Detector Software; PE Applied Biosystems).
For all assays, controls and samples were analyzed in triplicate in a final reaction volume of 20 μL. PCR amplification included an initial phase of 2 minutes at 50°C, followed by 10 minutes at 95°C, and 40 cycles of 15 seconds at 95°C and 1 minute at 60°C.
In Vitro Studies
Mouse insulinoma βTC3 cells were used for these studies. Cells were cultured in Dulbecco's modified Eagle's medium containing 25-mmol/L glucose supplemented with 10% fetal bovine serum, 100-IU/ml penicillin, 100-IU/ml streptomycin, and 2-mmol/L l-glutamine under 95% oxygen plus 5% carbon dioxide. The medium was changed twice a week, and cells were trypsinized and subcloned weekly. Cells were used between passages 20 and 65. The short hairpin (sh) RNAs for target genes used in this study were constructed in pLKO.1-puro vector (The RNAi Consortium at Broad Institute, Cambridge, MA). A plasmid carrying a nontargeting sequence was used to create the control cells. Lentiviral packaging and infection of the target cells were performed according to the The RNAi Consortium protocol, with some modifications. For virus packaging, 2.1 μg of the hairpin-pLKO.1 empty vector or vector containing the target gene-specific shRNA constructs was cotransfected with 3.7 μg of lentiviral packaging plasmid pCMVdR8.7psPAX2 containing gag, pol, and rev genes and 0.2 μg of envelope plasmid VSV-G into 293T cells. All plasmids were added to 400 μL of serum-free Dulbecco's modified Eagle's medium and 18 μL of polyethylenimine (1 mg/ml) (Polysciences, Inc, Warrington, PA). The mixture was vortex mixed for 10 seconds at high speed and incubated at room temperature for 15 minutes. The 293T cells were trypsinized. Then, the DNA mix was added to 3.5 × 106 293T cells. Forty-eight hours after transfection, the virus-containing media were collected, filtered, and added to trypsinized βTC3 cells in the presence of hexadimetherine bromide, 95% or greater (polybrene, 8 μg/ml) (Sigma-Aldrich, St. Louis, MO) for 24 hours. The infected cells were then subjected to selection with puromycin (Sigma-Aldrich) (2 μg/ml). For the cell proliferation study, plates were analyzed after 24, 48, 72, and 96 hours with cell proliferation reagent (WST-1; Roche Molecular Biochemicals) according to the manufacturer's instructions. For the STZ toxicity test, cells were either treated with media or STZ (5 mmol/L) for 6 hours. Viability was calculated by trypan blue dye exclusion.
Results
Hyperglycemia Induces RhoB Expression
The MLDS treatment used in our studies is one of the most common methods of inducing a type 1 diabetes–like pathological feature in vivo.14 The STZ is a nitrosourea compound that enters the β cell through a specific low-affinity Glut-2 glucose transporter; it causes massive methylation of the DNA and proteins, collapsing the DNA repair mechanism and leading to apoptosis and the subsequent attack of β cells by T cells and macrophages.9,10,15 We used this treatment with several different strains of mice (Balb/cJ, C57Bl/6J, and RhoB+/+), waited for 6 weeks for hyperglycemia to become well established, and measured RhoB expression in the kidney by Western blot analysis (Figure 1A). The results show an increase in RhoB protein levels in the diabetic kidney of each strain. We also used real-time RT-PCR to check whether RhoB expression levels changed in pancreatic islets after STZ treatment. Our results show that RhoB expression, in pancreatic islets of hyperglycemic mice, significantly increases 3 weeks after the last STZ injection when compared with those that only received the vehicle (Figure 1B).
Figure 1.
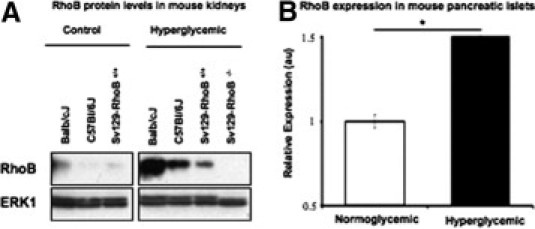
Expression of RhoB is induced by hyperglycemia. A: In all three tested strains of mice, the levels of RhoB protein in the kidney increased significantly 2 months after STZ injection. B: Results of RhoB expression in mouse pancreatic islets measured by RT-PCR before (white bar) and after (black bar) 3 weeks of hyperglycemia. The expression of RhoB in purified islets of RhoB+/+ mice is significantly higher than in those of normoglycemic control mice. Values are mean ± SE. The SE is shown as the error bar. Normoglycemic, n = 5; and hyperglycemic, n = 5. *P < 0.01 for normoglycemic versus hyperglycemic.
RhoB−/− Animals Are Protected against STZ-Induced Hyperglycemia
To study whether the association of RhoB expression and diabetes had functional consequences, we injected RhoB−/−, RhoB+/−, and RhoB+/+ control animals with STZ and measured the change in fasting glucose levels and the number and size of the pancreatic islets with time (Figure 2). Although 61% of the RhoB+/+ control animals became hyperglycemic (blood glucose >250 mg/dL) by 4 weeks after MLDS injections, only 13% of the RhoB−/−-treated animals became hyperglycemic (Figure 2A). Of those 13% of RhoB−/− animals that became diabetic, the level of hyperglycemia averaged values of approximately 350 mg/dL; in the RhoB+/+ controls, the fasting glucose levels averaged 450 to 500 mg/dL (Figure 2B). In both mice strains, there is an initial steep increase in fasting glucose levels 3 days after the last STZ injection. This increase corresponds to the initial wave of β-cell death. After a short plateau, the fasting glucose levels continued to increase in all hyperglycemic mice 24 days after the last injection.
Figure 2.
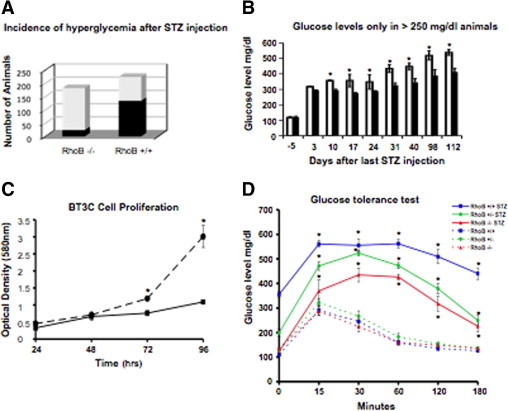
RhoB−/− animals are resistant to STZ-induced diabetes. A: Incidence of hyperglycemia in RhoB−/− and RhoB+/+ mice. When treated with multiple-dose STZ injections, after 4 weeks, only 13% of the RhoB−/− animals became hyperglycemic (left bar, black portion) (glucose level >250 mg/dL), whereas in the RhoB+/+ background control, 61% of the animals became hyperglycemic (right bar, black portion). RhoB−/−, n = 170; and RhoB+/+, n = 214. B: Fasting glucose levels in hyperglycemic animals. In animals with a fasting glucose level of greater than 250 mg/dL, the average glucose level of the hyperglycemic RhoB−/− animals (black bars) was lower than in the RhoB+/+ animals (white bars). Both groups showed a slow increase in their average fasting glucose levels with time. Values are mean ± SE. The SE is shown as the error bar (d = −5). RhoB+/+, n = 45; and RhoB−/−, n = 92 (any other point). RhoB+/+, n = 14; and RhoB−/−, n = 11. *P < 0.005 for RhoB+/+ versus RhoB−/−. C: Cell proliferation study on mouse insulinoma βTC3 cells. We used short-hairpin RNA to knock down RhoB in these cells. The solid line represents the proliferation rate of cells in which RhoB had been knocked down, whereas the dotted line represents cells infected with empty vector. Knocking down RhoB did not increase the proliferation rate of the cells. Indeed, it significantly slowed it down. *P < 0.001 for RhoB shRNA versus empty vector. D: Glucose tolerance test. All RhoB+/+, RhoB+/−, and RhoB−/− STZ-treated animals showed poor glycemic control after i.p. glucose injection (1 g/kg) (solid lines) when compared with the vehicle-injected controls (dashed lines). Still, glycemic control was worse in animals with one or more copies of RhoB. Values are mean ± SE. The SE is shown as the error bar. RhoB+/+, n = 6; RhoB+/−, n = 6; RhoB−/−, n = 5; STZ-RhoB+/+, n = 15; STZ-RhoB+/−, n = 8; and STZ-RhoB−/−, n = 21. *P < 0.01 for the STZ-injected versus the vehicle group.
The β cells can multiply in certain situations.16,17 To test whether β cells of RhoB-null animals are less sensitive to STZ or maintained function by repopulating islets after initial cell death, we knocked down RhoB in mouse insulinoma βTC3 cells and performed a cell proliferation study by observing them for 96 hours (Figure 2C). Our results show that knocking down RhoB in βTC3 cells does not increase the proliferation rate of the βTC3 cells and that it slows cell proliferation down, suggesting that the improved blood glucose levels are not because of β-cell recovery.
We also assessed the ability of β cells to produce insulin and mediate glucose intake after 4 weeks of STZ treatment by performing a glucose tolerance test (Figure 2D). All phenotypes showed signs of defective insulin production and delayed glucose uptake when compared with their non–STZ-injected controls. RhoB expression correlated with poor glycemic control during glucose challenge. Rather, when we looked at the pancreas of these animals (Figure 3, A–D), we found that 14 and 65 days after STZ injection, there were significantly more islets in the RhoB-null animals' pancreas than in the RhoB+/+ controls (Figure 3A), indicating that reduced islet damage may explain the better blood glucose levels in the RhoB−/− mice.
Figure 3.
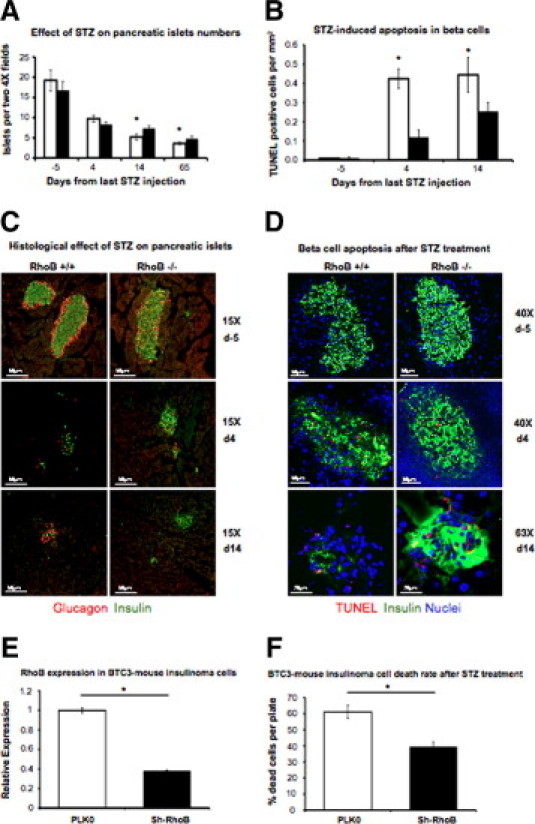
A: Effect of STZ on the number of pancreatic islets. In both strains, the number of pancreatic islets decreased 4, 14, and 65 days after the last STZ injection, but the number of islets is significantly higher in RhoB−/− (black bars) pancreas than in the RhoB+/+ (white bars) at days 14 and 65. Values are mean ± SE. The SE (n = 5 to 8) is shown as the error bar. *P < 0.05 for RhoB+/+ versus RhoB−/−. B: Quantitative analysis of apoptotic β cells in the pancreas after STZ treatment shows that 4 and 14 days after the last STZ injection, RhoB−/− mice have significantly fewer TUNEL-positive β cells than RhoB+/+ mice. Values are the mean ± SE. The SE (n = 5 to 8) is shown as the error bar. *P < 0.05 for RhoB+/+ versus RhoB−/−. C: Immunohistochemistry labeling of pancreatic β cells (insulin in green and glucagon in red). For both, RhoB+/+ and RhoB−/− show STZ-induced β-cell depletion and decrease in size and number of pancreatic islets. Still, the effect is more dramatic in RhoB+/+, in which, after 14 days of the last STZ injection, the ratio of insulin-producing cells/glucagon-producing cells decreased significantly while insulin-producing islets can still be found in RhoB−/− sections. D: Immunohistochemistry labeling of pancreatic β cells (insulin in green, TUNEL positive in red, and nuclei in blue). E:RhoB expression measured by RT-PCR in mouse insulinoma βTC3 cells. The white bar is the expression of cells infected with empty plasmid. The black bar represents RhoB expression after the cells have been infected with shRNA to knock down RhoB. The expression level decreased 60%. Values are the mean ± SE. The SE is shown as the error bar. PLK0, n = 5; and sh-RhoB, n = 5. *P < 0.01 for PLK0 versus sh-RhoB. F: Knocking down RhoB in βTC3 cells significantly reduced their sensitivity to STZ-induced cell death. Values are the mean ± SE. The SE is shown as the error bar. PLK0, n = 5; and sh-RhoB, n = 5. *P < 0.01 for PLK0 versus sh-RhoB.
Fourteen days after the last STZ injection (Figure 3C), there are few insulin-producing cells left in the RhoB+/+ mice and most of the cells remaining in the islets are glucagon-producing α cells. We used double staining with TUNEL and insulin to detect apoptosis and determine the rate of cell death after STZ injection (Figure 3, B and D). Three days after STZ treatment, the RhoB+/+ mice showed five times more TUNEL-positive β cells than the RhoB−/− mice. The difference is still significant 14 days after treatment, with more TUNEL-positive nuclei in the pancreatic islets of RhoB+/+ mice.
To determine that the cause of the resistance to the drug of RhoB−/− animals is indeed caused by different sensitivity of their β cells and not different reasons, such as different permeability of STZ, we performed an in vitro study using βTC3 cells. For this study, we checked the survival rate of these cells to STZ after knocking down RhoB. We show that RhoB can be efficiently knocked down in these cells (Figure 3E) and that reduction of RhoB expression significantly reduces the death rate caused by STZ in βTC3 cells (Figure 3F). We concluded from these studies that RhoB functions in hyperglycemic stress to exacerbate islet damage and metabolic dysfunction. This could be related to the already established function of RhoB in promoting DNA damage–induced apoptosis; in this case, this occurs in the pancreatic islets after STZ exposure.
Diabetic RhoB−/− Animals Show Fewer Signs of Nephropathy
In part because we had observed RhoB induction in the kidney of diabetic mice (Figure 1A), and because we had previously shown RhoB to be important for microvascular function in other organs,2 we investigated whether RhoB expression affected secondary damage from hyperglycemia to organs other than the pancreas. Nephropathy is a common complication of diabetes, characterized by accumulation of extracellular matrix in the glomeruli. This leads to thickening of the capillary and Bowman's capsule basement membrane and mesangium in the glomerulus. The result is glomerulosclerosis and eventually proteinuria and renal failure. Mice show early signs of nephropathy in the mesangial matrix after 3 months of becoming hyperglycemic, allowing us to observe early onset of nephropathy in the diabetic animals. The STZ-treated animals used in this experiment were diabetic for at least 6 months before the analysis; and the fasting blood glucose levels of RhoB−/− and RhoB+/+ mice were matched as closely as possible, considering that in most cases RhoB−/− mice had significantly lower fasting glucose levels than RhoB+/+ mice.
We looked at glomerular size and mesangial matrix accumulation in STZ-treated and nontreated RhoB−/− and RhoB+/+ controls (Figure 4). In both groups of STZ-treated animals, there was a significant increase in nephropathy compared with non–STZ-treated animals, represented by an increase of glomerular surface area (Figure 4, A and B) and accumulation of mesangial matrix (Figure 4, A and C) when compared with the non–STZ-injected animals. When we examined the glomeruli, we observed no difference between the RhoB−/− and RhoB+/+ control or the MLDS-treated groups (Figure 4B). However, with a threefold increase in the RhoB+/+ strain and a twofold increase in the RhoB−/− animals, there was a significant difference in the mesangial matrix index between the STZ-treated animals and their normoglycemic counterparts. Still, the diabetic RhoB−/− animals show significantly less nephropathy than the diabetic wild-type mice (Figure 4C). RhoB−/− mice showed significantly less matrix accumulation around the capillaries of the glomeruli. In all, long-term hyperglycemia in the absence of RhoB resulted in reductions in kidney damage.
Figure 4.
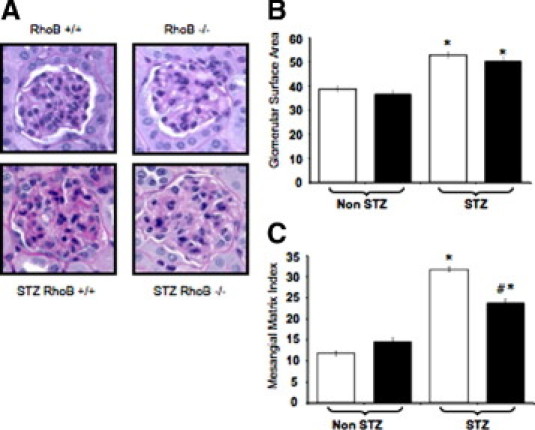
Hyperglycemic RhoB−/− kidneys showed fewer signs of early nephropathy compared with those of the wild-type control group. A: Hematoxylin-eosin staining of the glomeruli. Both diabetic groups (bottom) had an enlarged glomerular surface area and accumulation of mesangial matrix compared with the nondiabetic groups (top) at 6 months of diabetes duration. B: Glomerular size increases with hyperglycemia in both RhoB+/+ (white bars) and RhoB−/− (black bars) groups. Values are the mean ± SE. The SE is shown as the error bar. RhoB+/+, n = 79; RhoB−/−, n = 60; STZ-RhoB+/+, n = 80; and STZ-RhoB−/−, n = 60. *P < 0.01 versus non-STZ. C: The STZ-treated RhoB−/− mice (black bar, right) showed less severe mesangial matrix accumulation compared with diabetic RhoB+/+ (white bar, right). Values are the mean ± SE. The SE is shown as the error bar. #P < 0.01 versus STZ RhoB+/+, and *P < 0.01 versus non-STZ. RhoB+/+, n = 79; RhoB−/−, n = 60; STZ-RhoB+/+, n = 80; and STZ-RhoB−/−, n = 60.
This result suggests that RhoB knockout inhibits the accumulation of mesangial matrix without decreasing the size of enlarged glomeruli in those with diabetes.
Diabetic RhoB−/− Animals Show Fewer Signs of Retarded Wound Healing
Because the STZ-treated hyperglycemic RhoB−/− mice showed fewer signs of nephropathy than the RhoB+/+ controls, we decided to assess another diabetic complication: delayed wound healing. Hyperglycemia induced by STZ treatment of mice was previously reported to cause delayed wound healing.18 The STZ and non–STZ-treated RhoB−/− and RhoB+/+ animals were wounded with an 8-mm-diameter full-thickness dorsal skin punch. No significant differences were observed comparing the normoglycemic groups (RhoB+/+ versus RhoB−/−) (Figure 5, A–C). Yet, there was a significant difference in STZ-treated animals of both groups (Figure 5, A–C). The hyperglycemic RhoB+/+ animals experienced delayed wound healing. Notably, after 14 days, they still had a large wound area, with 30% of the initial punch still healing. On the other hand, the RhoB−/− animals showed little delay in the healing process. In fact, the healing was similar to their non–STZ-treated (nondiabetic) peers. Microscopic analysis of the wounds (Figure 5, B and C) showed delayed healing in the RhoB+/+ STZ-treated animals with abundant granulating tissue, edema, and scabbing 14 days after wounding. At the same point, the RhoB−/− STZ-treated animals had almost completely healed, showing less granulated tissue around the site of the wound and overall better organization of the epidermal layer.
Figure 5.
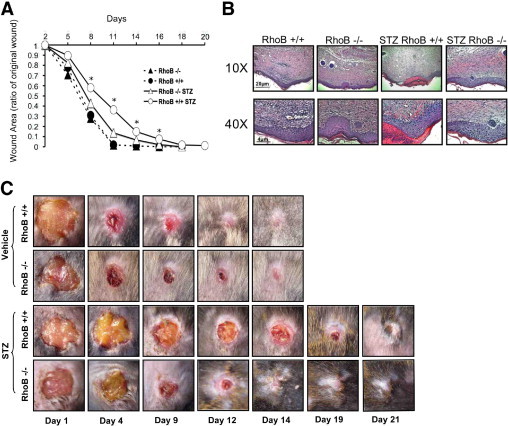
RhoB loss improves wound healing in hyperglycemic mice. A: There was no difference in the wound-healing rates of the non–STZ-treated RhoB+/+ and RhoB−/− animals. When the same experiment was performed in STZ-treated RhoB−/− and RhoB+/+ hyperglycemic mice, the RhoB+/+ animals showed significantly delayed wound healing compared with the non–STZ-treated controls and the hyperglycemic RhoB−/− mice (triangles, solid line). STZ treated RhoB−/− mice healed significantly faster than their wild-type peers. Values are the mean ± SE. The SE is shown as the error bar. RhoB+/+, n = 18; RhoB−/−, n = 20; STZ-RhoB+/+, n = 33; and STZ-RhoB−/−, n = 20. *P < 0.05 versus diabetic RhoB−/−. B: Histological examination of the wounds at day 14. No difference was observed in the wounds of RhoB−/− and RhoB+/+ untreated mice. On the other hand, the STZ-treated RhoB+/+ mice showed a significant delay in their wound-healing process when compared with the STZ-treated RhoB−/− mice. The STZ-treated RhoB+/+ unhealed wounds showed extended granulated tissue, edema, significant scabbing, and lack of a good layer definition. On the other hand, the wounds of hyperglycemic RhoB−/− mice showed histological features similar to those of the untreated mice. C: Representative image of the wound-healing process showing the delayed wound healing in RhoB+/+ hyperglycemic animals.
Statin Amelioration of Hyperglycemia Further Improves Outcome in RhoB-Null Mice
Statins ameliorate hyperglycemia in nonobese diabetic and STZ-treated mice.9 Because RhoB becomes prenylated on cysteines and the prenylation pathways are downstream of statins, we surmised that the pleiotropic effect of statins on glycemic control might be a consequence of the inhibition of RhoB prenylation. To test whether those two events were related, we injected RhoB−/−, RhoB+/−, and RhoB+/+ animals with simvastatin for 14 days. On day 2 after the first simvastatin injection, some of the animals were also injected with low-dose STZ. Simvastatin decreased the average fasting blood glucose level (Figure 6, dashed lines), when compared with animals that only received STZ treatment and vehicle (Figure 6, solid lines), and also decreased the percentage of animals developing diabetes in all groups. These experiments showed that glycemic control levels of RhoB and simvastatin are independent of each other and can be an added benefit in fighting diabetes.
Figure 6.
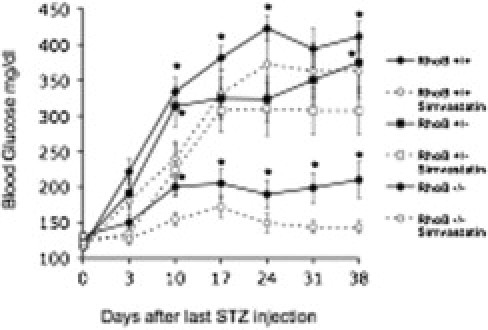
Simvastatin improves glycemic control. RhoB−/−, RhoB+/−, and RhoB+/+ animals were injected with simvastatin 2 days before the first STZ injection and for 14 consecutive days. Simvastatin succeeded in bringing the level of hyperglycemia down in all groups (dashed lines, animals treated with simvastatin and STZ; solid lines, animals treated with STZ and vehicle). Values are the mean ± SE. The SE (n = 16 to 30) is shown as the error bar. RhoB+/+, n = 10; RhoB+/−, n = 16; RhoB−/−, n = 11; simvastatin-RhoB+/+, n = 7; simvastatin-RhoB+/−, n = 13; simvastatin-RhoB−/−, n = 21. *P < 0.05 versus the same genotype injected with simvastatin.
Discussion
The data we have collected provide strong evidence for RhoB as a potential target to ameliorate diabetes by improving glycemic control and reducing complications. Extensive work1,2,4,19–23 has been performed in the cancer field on RhoB's proapoptotic properties, its capacity to regulate angiogenesis, and its role as a tumor suppressor. RhoB is also important for intracellular transport, actin regulation, macrophage motility, and activation.3 Perhaps what is common in these various roles of RhoB is that they all reflect functions for RhoB in pathological settings or states of cellular activation that follow RhoB's induction because of multiple types of cellular stress. Indeed, we observed that there is an increase in RhoB expression both in the kidneys of mice that have been diabetic for 3 months and in pancreatic islets of hyperglycemic mice 3 weeks after the last STZ injection.
Several studies have shown different approaches for protecting β cells from STZ-induced apoptosis, including using antioxidants24,25 and recombinant gene therapy.26,27 Our study shows that loss of RhoB protects both pancreatic β cells from destruction by inhibiting apoptosis and organs from secondary complications associated with hyperglycemia. Specifically, we examined nephropathy and delayed wound healing in STZ-treated animals. Both diabetic nephropathy and diabetic wound healing have been related to diabetic microvascular disease. In the kidney, microvascular damage and leak leads to the accumulation of extracellular basement membrane, which exacerbates the failure of filtration in the glomeruli. In wound healing, angiogenesis and lymphangiogenesis are rate limiting for healing and clearing of tissue damage. Thus, the additional benefit gained by loss of RhoB on the microvasculature could participate in the improvement of diabetic complications in RhoB-null animals.
In our experiments, only hyperglycemic animals were included in the STZ-treated groups. However, we observed that hyperglycemic RhoB−/− mice had lower glucose levels than STZ-treated wild-type mice. Consequently, it is difficult to determine whether the improved complications are because of the lack of RhoB in affected tissues or simply the more favorable glycemic levels in the STZ-treated RhoB−/− mice. Because we used a constitutive knockout model, it is also difficult to conclude whether the improvements in wound healing and nephropathy are because of improved glycemic control or loss of RhoB in the microvasculature of those affected tissues. Further studies using a model that allows tissue-specific deletion of RhoB could identify a specific mechanism for the milder diabetic complications observed in the RhoB−/− mice. In summary, RhoB presents itself as an attractive cellular target for diabetic therapy.
Previous studies have used statins to reduce the STZ-induced hyperglycemia in control animals and in nonobese diabetic mice.9 Our experiments show that simvastatin injections reduced the number of animals that developed hyperglycemia and the average fasting glucose level, regardless of the number of copies of RhoB in the genome. Therefore, the effect of simvastatin on protecting the islets is not solely dependent on RhoB. Rydgren and Sandler28 found that the beneficial effect of statins in glycemic control after STZ treatment was independent of HMG-CoA reductase inhibition in their models. However, because RhoB function is strongly associated with prenylation status, we cannot rule out the possibility that the positive effects of simvastatin in RhoB wild-type settings are benefited by loss of RhoB prenylation. Nevertheless, we propose that RhoB-targeted therapy could be used as an alternative or in combination with statins to improve the lives of diabetic patients.
Footnotes
Supported by the American Diabetes Association (grant 705RA10) and the National Institutes of Health (grant HL071049 to L.E.B).
Current address of L.E.B.: ImClone Systems, New York, New York.
References
- 1.Huang M., Prendergast G.C. RhoB in cancer suppression. Histol Histopathol. 2006;21:213–218. doi: 10.14670/HH-21.213. [DOI] [PubMed] [Google Scholar]
- 2.Adini I., Rabinovitz I., Sun J.F., Prendergast G.C., Benjamin L.E. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003;17:2721–2732. doi: 10.1101/gad.1134603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wheeler A.P., Ridley A.J. RhoB affects macrophage adhesion, integrin expression and migration. Exp Cell Res. 2007;313:3505–3516. doi: 10.1016/j.yexcr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Fritz G., Kaina B. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets. 2006;6:1–14. [PubMed] [Google Scholar]
- 5.Mazieres J., Tovar D., He B., Nieto-Acosta J., Marty-Detraves C., Clanet C., Pradines A., Jablons D., Favre G. Epigenetic regulation of RhoB loss of expression in lung cancer. BMC Cancer. 2007;7:220. doi: 10.1186/1471-2407-7-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S., Yan-Neale Y., Fischer D., Zeremski M., Cai R., Zhu J., Asselbergs F., Hampton G., Cohen D. Histone deacetylase 1 represses the small GTPase RhoB expression in human nonsmall lung carcinoma cell line. Oncogene. 2003;22:6204–6213. doi: 10.1038/sj.onc.1206653. [DOI] [PubMed] [Google Scholar]
- 7.Hackam D.J., Rotstein O.D., Schreiber A., Zhang W., Grinstein S. Rho is required for the initiation of calcium signaling and phagocytosis by Fcgamma receptors in macrophages. J Exp Med. 1997;186:955–966. doi: 10.1084/jem.186.6.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adamson P., Marshall C., Hall A., Tilbrook P. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 9.Rydgren T., Vaarala O., Sandler S. Simvastatin protects against multiple low-dose streptozotocin-induced type 1 diabetes in CD-1 mice and recurrence of disease in nonobese diabetic mice. J Pharmacol Exp Ther. 2007;323:180–185. doi: 10.1124/jpet.107.122655. [DOI] [PubMed] [Google Scholar]
- 10.Zhu B., Shen H., Zhou J., Lin F., Hu Y. Effects of simvastatin on oxidative stress in streptozotocin-induced diabetic rats: a role for glomeruli protection. Nephron Exp Nephrol. 2005;101:e1–e8. doi: 10.1159/000085712. [DOI] [PubMed] [Google Scholar]
- 11.Gotoh M., Maki T., Kiyoizumi T., Satomi S., Monaco A.P. An improved method for isolation of mouse pancreatic islets. Transplantation. 1985;40:437–438. doi: 10.1097/00007890-198510000-00018. [DOI] [PubMed] [Google Scholar]
- 12.Zeisberg M., Hanai J.-i., Sugimoto H., Mammoto T., Charytan D., Strutz F., Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 13.Brocco E., Fioretto P., Mauer M., Saller A., Carraro A., Frigato F., Chiesura-Corona M., Bianchi L., Baggio B., Maioli M., Abaterusso C., Velussi M., Sambataro M., Virgili F., Ossi E., Nosadini R. Renal structure and function in non-insulin dependent diabetic patients with microalbuminuria. Kidney Int Suppl. 1997;63:40–44. [PubMed] [Google Scholar]
- 14.Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50:537–546. [PubMed] [Google Scholar]
- 15.Murata M., Takahashi A., Saito I., Kawanishi S. Site-specific DNA methylation and apoptosis: induction by diabetogenic streptozotocin. Biochem Pharmacol. 1999;57:881–887. doi: 10.1016/s0006-2952(98)00370-0. [DOI] [PubMed] [Google Scholar]
- 16.Dhawan S., Tschen S.I., Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009;23:906–911. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tschen S.I., Dhawan S., Gurlo T., Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009;58:1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Covington D.S., Xue H., Pizzini R., Lally K.P., Andrassy R.J. Streptozotocin and alloxan are comparable agents in the diabetic model of impaired wound healing. Diabetes Res. 1993;23:47–53. [PubMed] [Google Scholar]
- 19.Laufs U., Liao J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 20.Turner S.J., Zhuang S., Zhang T., Boss G.R., Pilz R.B. Effects of lovastatin on Rho isoform expression, activity, and association with guanine nucleotide dissociation inhibitors. Biochem Pharmacol. 2008;75:405–413. doi: 10.1016/j.bcp.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazieres J., Antonia T., Daste G., Muro-Cacho C., Berchery D., Tillement V., Pradines A., Sebti S., Favre G. Loss of RhoB expression in human lung cancer progression. Clin Cancer Res. 2004;10:2742–2750. doi: 10.1158/1078-0432.ccr-03-0149. [DOI] [PubMed] [Google Scholar]
- 22.Marlow L.A., Reynolds L.A., Cleland A.S., Cooper S.J., Gumz M.L., Kurakata S., Fujiwara K., Zhang Y., Sebo T., Grant C., McIver B., Wadsworth J.T., Radisky D.C., Smallridge R.C., Copland J.A. Reactivation of suppressed RhoB is a critical step for the inhibition of anaplastic thyroid cancer growth. Cancer Res. 2009;69:1536–1544. doi: 10.1158/0008-5472.CAN-08-3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couderc B., Pradines A., Rafii A., Golzio M., Deviers A., Allal C., Berg D., Penary M., Teissie J., Favre G. In vivo restoration of RhoB expression leads to ovarian tumor regression. Cancer Gene Ther. 2008;15:456–464. doi: 10.1038/cgt.2008.12. [DOI] [PubMed] [Google Scholar]
- 24.Cemek M., Kaga S., Simsek N., Buyukokuroglu M.E., Konuk M. Antihyperglycemic and antioxidative potential of Matricaria chamomilla L. in streptozotocin-induced diabetic rats. J Nat Med. 2008;62:284–293. doi: 10.1007/s11418-008-0228-1. [DOI] [PubMed] [Google Scholar]
- 25.Coskun O., Kanter M., Korkmaz A., Oter S. Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and beta-cell damage in rat pancreas. Pharmacol Res. 2005;51:117–123. doi: 10.1016/j.phrs.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Schnedl W.J., Ferber S., Johnson J.H., Newgard C.B. STZ transport and cytotoxicity: specific enhancement in GLUT2-expressing cells. Diabetes. 1994;43:1326–1333. doi: 10.2337/diab.43.11.1326. [DOI] [PubMed] [Google Scholar]
- 27.Mori H., Shichita T., Yu Q., Yoshida R., Hashimoto M., Okamoto F., Torisu T., Nakaya M., Kobayashi T., Takaesu G., Yoshimura A. Suppression of SOCS3 expression in the pancreatic beta-cell leads to resistance to type 1 diabetes. Biochem Biophys Res Commun. 2007;359:952–958. doi: 10.1016/j.bbrc.2007.05.198. [DOI] [PubMed] [Google Scholar]
- 28.Rydgren T., Sandler S. The protective effect of simvastatin against low dose streptozotocin induced type 1 diabetes in mice is independent of inhibition of HMG-CoA reductase. Biochem Biophys Res Commun. 2009;379:1076–1079. doi: 10.1016/j.bbrc.2009.01.017. [DOI] [PubMed] [Google Scholar]