

Abstract
Many models of human tauopathies have been generated in mice by expression of a human mutant tau with maintained expression of mouse endogenous tau. Because murine tau might interfere with the toxic effects of human mutant tau, we generated a model in which a pathogenic human tau protein is expressed in the absence of wild-type tau protein, with the aim of facilitating the study of the pathogenic role of the mutant tau and to reproduce more faithfully a human tauopathy. The Tg30 line is a tau transgenic mouse model overexpressing human 1N4R double-mutant tau (P301S and G272V) that develops Alzheimer's disease-like neurofibrillary tangles in an age-dependent manner. By crossing Tg30 mice with mice invalidated for their endogenous tau gene, we obtained Tg30xtau−/− mice that express only exogenous human double-mutant 1N4R tau. Although Tg30xtau−/− mice express less tau protein compared with Tg30, they exhibit signs of decreased survival, increased proportion of sarkosyl-insoluble tau in the brain and in the spinal cord, increased number of Gallyas-positive neurofibrillary tangles in the hippocampus, increased number of inclusions in the spinal cord, and a more severe motor phenotype. Deletion of murine tau accelerated tau aggregation during aging of this mutant tau transgenic model, suggesting that murine tau could interfere with the development of tau pathology in transgenic models of human tauopathies.
Alzheimer's disease (AD) is defined by two neuropathological hallmarks: amyloid plaques and neurofibrillary tangles (NFTs). Amyloid plaques consist of an extracellular core of aggregated amyloid peptides cleaved from amyloid precursor protein (APP) by secretases. The NFTs are intraneuronal accumulation of abnormal filaments (paired helical filaments, PHFs). These PHFs are composed of highly and abnormally phosphorylated forms of the microtubule-associated protein tau; these abnormal tau proteins are called PHF-tau proteins. The mechanistic relationships between these lesions are under active investigation, with the aim of deciphering the basic mechanisms of AD. The amyloid peptide has been implicated as a primary upstream event leading to synaptic dysfunction, development of NFTs, and neuronal cell death,1 although neuronal dysfunction linked to tau pathology appears to be an essential element in the progression of AD and related tauopathies.2
In familial forms of AD, many pathogenic mutations have been identified in the APP and PSEN1 (alias PS1) genes. Expression of APP mutations or coexpression of APP and PSEN1 in transgenic models led to development of amyloid deposits in many transgenic models, but not of neurofibrillary tangles. Expression of PSEN1 mutations alone also did not lead to neurofibrillary tangles.
Although no mutations of the MAPT gene (on chromosome 17; alias FTDP-17) have been found to date in AD, ∼40 pathogenic mutations have been linked to this gene in families of hereditary frontotemporal dementia and parkinsonism patients (reviewed by van Swieten and Spillantini3). These tau mutations either promote tau aggregation, decrease the ability of tau to assemble microtubules or affect alternative splicing of tau mRNA.
Transgenic mice expressing mutant tau all demonstrate abnormal hyperphosphorylation and somatodendritic localization of tau. Most of the mutant tau transgenic mice develop NFTs and PHF-tau (reviewed by Denk and Wade-Martins,4) but they lack amyloid pathology. With the aim of analyzing the two pathological characteristics of AD in a single model, mice double-transgenic or triple-transgenic for Mapt, App, and Psen1 (ortholog to human MAPT, APP, and PSEN1) have been developed to mimic AD as experimental animal models.5–9
Wild-type mice do not spontaneously develop NFTs, and there are very few mouse models that demonstrate NFT formation in transgenic models expressing wild-type human tau, although all these models demonstrate hyperphosphorylation of tau.10–16 In a transgenic model that expressed six isoforms of non-mutant human tau and endogenous murine tau, there were no NFTs or PHF-tau observed.16 When these mice were crossed with tau knock-out animals, however, they exhibited AD-like NFTs and PHF-tau, as well as neuronal cell death.17,18 These findings suggest that endogenous murine tau might play a role by counteracting tau aggregation. In contrast, expression of 2N4R tau in the absence of murine tau did not lead to NFT development.19 It thus remains unclear to what extent murine tau interferes with NFT formation, and how this can affect modeling of tau pathology.
To further investigate this issue, we generated a transgenic mouse model expressing a human mutant tau protein but lacking endogenous murine tau. These mice exhibit a more severe motor phenotype and increased tau pathology, suggesting that the expression of murine tau might affect the development of tau pathology in transgenic models of Alzheimer's disease and other tauopathies.
Materials and Methods
Generation of Tg30xtau−/− Mice
Tg30 mice express a 1N4R human tau isoform mutated at positions G272V and P301S, under control of a thy-1 promoter.20,21 The tau−/− mouse line (Jackson Laboratories, Bar Harbor, ME) was generated by knock-in of the EGFP coding sequence into the first exon of the tau gene.22 Heterozygote Tg30 and tau−/− mice were maintained into a C57BL6 background. Tau+/− mice were generated by crossing tau−/− mice with C57BL6 mice. Tg30 and tau+/− mice were crossed to generate F1 Tg30xtau+/− animals. The F1 Tg30xtau+/− mice were then bred with tau+/− mice to generate F2 Tg30xtau−/− mice expressing only the human mutant tau G272V/P301S and no endogenous mouse tau, and littermates of the same genetic background. Genotyping was performed by three independent PCR amplifications of DNA extracted from tail DNA, using previously described primers for human mutant tau (forward 5′-ATGGCTGAGCCCCGCCAGGAG-3′, reverse 5′-TGGAGGTTCACCAGAGCTGGG-3′),23 mouse tau (forward 5′-CTCAGCATCCCACCTGTAAC-3′, reverse 5′-CCAGTTGTGTATGTCCACCC-3′), and the inserted EGFP cassette (forward 5′-AAGTTCATCTGCACCACCG-3′, reverse 5′-TCCTTGAAGAAGATGGTGCG) (Jackson Laboratories, Bar Harbor, ME).
All studies on animals were performed in compliance with and after approval of the Ethical committee for the care and use of laboratory animals of the Medical School of the Free University of Brussels.
Motor Testing
Motor deficits in wild-type and Tg30 mice were evaluated by testing them on a rotarod apparatus (Ugo Basile), as described previously.24 Briefly, animals were first submitted to training sessions (three trials per day during 3 consecutive days) during which they were placed on the rod rotating with a progressive acceleration from 4 to 40 revolutions per minute. Animals were individually separated the day before the test and evaluated using the same experimental setting throughout 300 seconds. The latency to off the rotarod apparatus was recorded. Animals staying longer than 300 seconds were removed from the rotarod and their latency fall was recorded as 300 seconds.
Behavioral Testing
Mice were tested in the Y-maze for spontaneous alternation, as described previously.25 The number of total entries into the arms and the numbers of alternations were calculated. The percentage of spontaneous alternations was expressed as the number of alternations divided by the total number of arms minus 2. Repeated entries into the arms were accepted, resulting in a chance performance level of 22% alternation.26
Antibodies
The B19 antibody is a rabbit polyclonal antibody raised to adult bovine tau proteins. This antibody reacts with all known adult and fetal tau isoforms in bovine, rat, mouse, and human nervous tissue in a phosphorylation-independent manner.27
Two polyclonal antibodies (BR20 and BR21) against human tau were generated by immunizing rabbits with the synthetic peptide C-GTYGLGDRKDQGG conjugated to PPD, corresponding to residues 16–28 in the amino-terminal region of tau (in exon 1), with the addition of an amino-terminal cysteine. The BR10 tau antibody is specific for tau isoforms containing an amino-terminal insert of 58 residues (2N tau).28 Antibody JN-RF.5 mTau-5 (a generous gift from M. Mercken, Johnson & Johnson, Beerse, Belgium) reacts only with murine tau.29 The TP007 and TP70 rabbit polyclonal antibodies were raised against synthetic peptides mapping at the extreme N- and C- termini of human tau, respectively.30,31 The anti-cleaved tau (Asp421) mouse monoclonal antibody was purchased from Upstate Biotechnology Lake Placid, NY). The AT8, AT180, AT270, and AT100 mouse monoclonal antibodies (Innogenetics, Ghent, Belgium) are specific for tau phosphorylated at Ser202 and Thr205 (AT8),32 at Thr231 (AT180), and at Thr181 (AT270).33 The mouse monoclonal antibodies PHF-1 and CP13 (kindly provided by Drs. P. Davies and S. Greenberg, New York, NY) are specific for tau phosphorylated at Ser396/404 and Ser202/Thr205, respectively.34 The tau monoclonal antibody MC1 (kindly provided by Dr. P. Davies) recognizes a conformational epitope requiring both an N-terminal fragment and a C-terminal fragment.35 The phosphospecific rabbit polyclonal tau antibody to pSer262 was provided by BioSource (Nivelles, Belgium).
The following additional antibodies were also used: α-tubulin (clone DM1-A) and to β-actin (clone AC15) (Sigma-Aldrich, St. Louis, MO; Bornem, Belgium), to Cdk5 (clone DC17; Santa Cruz Biotechnology, Santa Cruz, CA), to β-catenin and to GSK-3β (TPK1) (BD Transduction Laboratories, San Diego, CA), and rabbit polyclonal antibodies to p35 (C-19) (Santa Cruz Biotechnology), to GSK-3 phosphorylated on Ser9 (Cell Signaling Technology, Danvers, MA), to GSK-3 phosphorylated on Tyr216/Tyr276 (BioSource), to neurofilament (NA1211; Affiniti Research Products, Exeter, UK), and to LC3 (Sigma-Aldrich).
Analysis of Sarkosyl-Insoluble and -Soluble Tau Fractions
The brains and spinal cords from sex-matched transgenic mice were dissected and homogenized in either a 10-fold (brain) or a 20-fold (spinal cord) volume of modified radioimmunoprecipitation assay buffer36: 50 mmol/L Tris-HCl (pH 7.4), 1% Nonidet P-40, 0.25% w/v Na-deoxycholate, 150 mmol/L NaCl, 1 mmol/L phenylmethyl sulfonyl fluoride, leupeptin 25 μg/ml, pepstatin 25 μg/ml, 20 mmol/L NaF, 1 mmol/L sodium orthovanadate, and 10 mmol/L sodium pyrophosphate. The homogenates were rotated at 4°C for 1 hour and centrifuged at 20,000 × g for 20 minutes at 4°C to obtain a pellet P1 and a supernatant S1. The protein concentrations in S1 fractions were equivalent for all mice used in this study. A same volume of S1 (2 ml for brain and 1 ml for spinal cord) was subjected to sarkosyl fractionation by incubation with 1% (w/v) N-lauroylsarcosine sodium salt (L-5125; Sigma-Aldrich) under mild rotation for 30 minutes at room temperature and centrifugation at 100,000 × g for 30 minutes at 4°C. The pellets (P2) containing the sarkosyl-insoluble material were resuspended in same volumes of 50 mmol/L Tris/HCl (pH 7.5). Sarkosyl-soluble (S2) and -insoluble (P2) (A68) fractions were analyzed by Western blotting.
Proteins in tissue samples (100 μg protein/lane) were separated by 7.5% (w/v), 10% (w/v) or 15% (w/v) SDS-polyacrylamide gel electrophoresis, depending on the molecular weight of the analyzed proteins, and were transferred to nitrocellulose membrane using a liquid transfer system (Bio-Rad, Hercules, CA). For immunoblotting, the nitrocellulose sheets were blocked in semi-fat dry milk (10% w/v in Tris-buffered saline) for 1 hour at room temperature; they were then incubated with primary antibodies overnight, followed by anti-rabbit or anti-mouse immunoglobulins conjugated to alkaline phosphatase (Sigma-Aldrich). Finally, the membranes were incubated in developing buffer (0.1 mol/L Tris, 0.1 mol/L NaCl, 0.05 mol/L MgCl2; pH 9.5) containing nitro blue tetrazolium at a concentration of 0.33 mg/ml and 5-bromo-4-chloro-3-indolyl phosphate at a concentration of 0.175 mg/ml. The reaction was stopped by dipping the membranes in 10 mmol/L Tris, 1 mmol/L EDTA (pH 8). The levels of the signals were estimated by densitometry analysis using Image J software version 1.4 (NIH, Bethesda, MD), and adjusted for protein loading based on immunoblots performed with the anti-actin antibody and the B19 polyclonal tau antibody.
Histological Staining and Immunocytochemistry
Brains and spinal cords were fixed in 10% formalin for 24 hours before embedding in paraffin. Tissue sections (7 μm thick) were stained with the Gallyas silver staining method to identify neurofibrillary tangles. They were examined with a Zeiss Axioplan microscope and digital images acquired using an AxioCam HRc camera. The density of neurons with Gallyas-positive inclusions was estimated in the cortex and in the hippocampus on sagittal sections taken near the midline and in the cervical spinal cord on coronal sections (three adjacent sections at each level for each animal), as reported previously in Tg30 mice.24 The immunohistochemical labeling was performed using the ABC method. Briefly, tissue sections were treated with H2O2 to inhibit endogenous peroxidase and then were incubated with the blocking solution (10% (v/v) normal horse serum in TBS (0.01 mol/L Tris, 0.15 mol/L NaCl; pH 7.4). After an overnight incubation with the diluted primary antibody, the sections were sequentially incubated with either horse anti-mouse or goat anti-rabbit antibodies conjugated to biotin (Vector Laboratories, Burlingame, CA) followed by the ABC complex (Vector Laboratories). The peroxidase activity was developed using diaminobenzidine as chromogen.
Double immunolabeling was performed using fluorescent markers. The first antibody was detected using an anti-rabbit or an anti-mouse antibody conjugated to FITC (Jackson Laboratories) and the second antibody detected using an anti-rabbit or an anti-mouse antibody conjugated to biotin, followed by streptavidin conjugated to Alexa 594 (Molecular Probes, Eugene, OR). Double immunolabeling was followed by nuclear DAPI staining.
Sciatic Nerve Analysis
Sciatic nerves were prepared and analyzed as previously described.24 Semi-thin cross-sections (thickness of 1 μm) of the sciatic nerves were performed and stained with toluidine blue. The cross-sectional areas and the number of axons for each sciatic nerve were measured on digital images of these semi-thin sections with Image J software.
Ultrastructural Analysis in Electron Microscopy
Control and transgenic animals were anesthetized with ketamine hydrochloride and xylazine and were perfused intracardially with a solution of 2% (w/v) paraformaldehyde and 2% (v/v) glutaraldehyde in 0.1 mol/L phosphate buffer at pH 7.4. Tissue blocks of the spinal cord, sciatic nerves, hippocampus, and cerebral cortex were quickly dissected and further fixed by immersion in 4% (w/v) glutaraldehyde in 0.1 mol/L phosphate buffer at pH 7.4 for 90 minutes. After a washing in Millonig's buffer with 0.5% (w/v) sucrose for 24 hours, the tissue blocks were postfixed in 2% (w/v) OsO4 for 30 minutes, dehydrated and embedded in epoxy resin. Semi-thin sections were stained with toluidine blue. Ultrathin sections were counterstained with uranyl acetate and lead citrate and observed with a Zeiss EM 809 transmission electron microscope at 80 kV.
Immunolabeling of Tau Filaments in Electron Microscopy
The pellet containing the sarkosyl-insoluble material was resuspended in 50 mmol/L Tris/HCl (pH 7.5). This material was adsorbed onto Formvar carbon-coated electron microscope grids and then was immunolabeled and negatively stained with potassium phosphotungstate as previously described,37 before observation with a Zeiss EM 809 electron microscope at 80 kV.
Stereological Analysis with the Optical Fractionator
Stereological analysis was performed as previously reported,24 with slight modifications to the procedure. After dislocation, brains and spinal cord were dissected, fixed with 10% (v/v) formalin, embedded in paraffin, and serially cut in coronal sections (15 μm thickness). These sections were stained with Cresyl violet.
The total number of hippocampal pyramidal cells in the CA1, CA2, CA3, and CA4 sectors was estimated in the left hemispheres of 12-month-old wild-type mice (n = 4), tau−/− mice (n = 5), Tg30tau mice (n = 3), and Tg30xtau−/− mice (n = 3). For each animal, coronal sections at 225-μm intervals throughout the hippocampal pyramidal cell layer were used. An estimation of neuronal density in the anterior horn of the cervical spinal cord was also performed in the same animals, by analyzing 10 coronal sections at 75-μm intervals.
Counting of total numbers of neurons was carried out with a 100×/1.30 oil objective by using a random-sampling stereological counting tool, the optical fractionator38,39 implemented within a stereology workstation consisting of a light microscope (Axio Imager M1; Zeiss, Göttingen, Germany), Zeiss Plan-Neofluar objectives (10×/0.30; 100×/1.30 oil), a motorized specimen stage for automatic sampling (BioPrecision 2; Ludl Electronic Products, Hawthorne, NY), a focus measurement encoder, a charge-coupled device CCD color camera (Microfire; Optronics, Goleta, CA), and a microcomputer with stereology software (Stereoinvestigator version 8; MicroBrightField, Colchester, VT). Optical disectors were automatically randomly distributed throughout the delineated area, and each neuron whose nucleolus came into focus within an optical disector was counted. Estimated total numbers of pyramidal cells and the density of neurons in anterior spinal cord per specimen were hence calculated from the number of counted neurons, the volume of the pyramidal cell layer, or the volume of spinal gray matter and the sampling probability.40–42 Table 1 summarizes the parameters of the counting procedure.
Table.
Stereological Parameters Used for Cell Counting
| Area | Obj. | B (μm2) | H (μm) | Sampling grid (μm) | t (μm, mean ± SEM) | Σ OD | Σ CN | CE |
|---|---|---|---|---|---|---|---|---|
| Hippocampus | 100× | 900 | 9 | 125 × 90 | 19.9 ± 0.8 | 291 | 655 | 0.042 |
| Spinal cord | 100× | 2500 | 12 | 150 × 125 | 20.0 ± 0.3 | 183 | 157 | 0.082 |
B and H, base and height of the optical disectors; CE, average predicted coefficient of error of the estimated total numbers of neurons; Obj., objective lens power; t, measured average section thickness after histological processing; Σ CN, average number of counted neurons per specimen; Σ OD, average number of optical disectors used per specimen. For methodology, see Schmitz and Hof40,41 and Slomianka and West.42
Cavalieri Method to Estimate Brain Volume
The volume of the brain, the cortex, and the hippocampus was estimated using coronal sections (15 μm thickness) of the left hemispheres. The volume of a segment of the whole cervical spinal cord, the anterior horn, and total gray matter in this segment was estimated on coronal sections (15 μm thickness) of the left half-part of the cervical spinal cord. These volume estimations were performed using the Cavalieri estimator43 implemented in the Stereoinvestigator software. We used coronal sections at 600-μm intervals for the hippocampus and the cortex, at 225-μm intervals for the hippocampus, and at 75-μm intervals for the spinal cord. Neuroanatomical delineations were made according to the mouse brain atlas of Paxinos and Franklin.44
Statistical Analysis
Statistical analysis was performed using the GraphPad Prism software package version 5 (GraphPad Software, La Jolla, CA). Data were reported as mean ± SEM. Statistical comparisons were performed using one-way analysis of variance with Bonferroni post hoc test comparisons, unpaired two-tailed Student's t-test, or Mann-Whitney U-test. Values of P < 0.05 were considered significant.
Results
Generation of Tg30xtau−/− Mice
To study the relationship between tau pathology and endogenous murine tau, we generated a transgenic mouse that expresses human tau protein with double FTDP-17 mutations (P301S and G272V) in the absence of endogenous murine tau. To abolish the expression of murine tau protein, we crossed Tg30 mice with tau+/− to generate six distinct genotypes: tau+/+ (wild-type), tau+/−, tau−/−, Tg30xtau+/+ (Tg30), Tg30xtau+/−, and Tg30xtau−/−. PCR amplifications of genomic DNA allowed us to distinguish among these six genotypes (Figure 1). Tg30xtau−/− mice were identified by the presence of amplification products from the human tau transgene and the EGFP cassette, without any amplification product from the murine endogenous tau gene. Only the representative four genotypes (wild-type, tau−/−, Tg30, and Tg30x tau−/−) from the same littermates were analyzed further.
Figure 1.
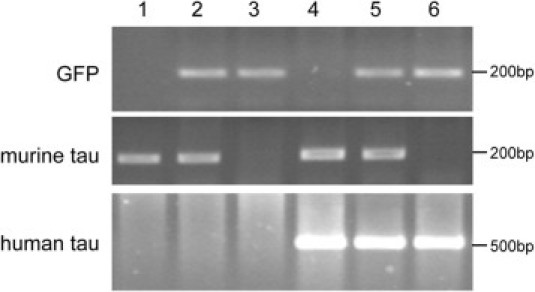
PCR genotyping of mice resulting from the crossing of Tg30xtau+/− mice with tau+/− mice. PCR amplification was performed with primers for GFP, murine tau, and human tau. Each lane represents a different genotype: lane 1, wild-type; lane 2, tau+/−; lane 3, tau−/−, lane 4, Tg30; lane 5, Tg30xtau+/−; and lane 6, g30Tg30xtau−/−. Only Tg30xtau−/− mice show amplification products for the human tau and GFP transgenes without amplification products of the murine tau gene. Molecular weight markers are indicated on the right.
Expression of Total Tau but Not of Mutant Human Tau Is Decreased in Tg30xtau−/− Mice
The level of human and murine tau expression was analyzed by Western blotting in 12-month-old mouse brain and spinal cord homogenates. A range of tau-positive bands was observed between 50 kDa and 64 kDa in wild-type, Tg30, and Tg30xtau−/− (but not in tau−/−) with the B19 tau antiserum, which recognizes both human and murine tau (Figure 2, A and B). With the human-specific BR21 tau antibody, tau bands were observed in Tg30 and Tg30xtau−/− mice, but not in wild-type and tau−/− mice. With the murine-specific mTau-5 tau antibody, tau bands were observed only in wild-type and in Tg30 mice (Figure 2). In Tg30xtau−/− mice, the total level of tau in brain estimated with the B19 antibody was significantly decreased, compared with Tg30 mice (Figure 2, C and D), but the total level of human tau estimated with the BR21 antibody was not different between these two genotypes (Figure 2, E and F). The immunocytochemical labeling (Figure 3) confirmed the presence of human tau only in Tg30 and Tg30xtau−/− mice (Figure 3, E–H) and of murine tau only in wild-type and Tg30 mice (Figure 3, I–L). A somatodendritic accumulation of tau was observed with the BR21 human tau antibody (and the B19 tau antibody) in Tg30 and Tg30xtau−/− mice (Figure 3, C, D, G, and H). An abnormal accumulation of endogenous murine tau was detected in Tg30 with the mTau-5 antibody but not in the other genotypes (Figure 3K).
Figure 2.
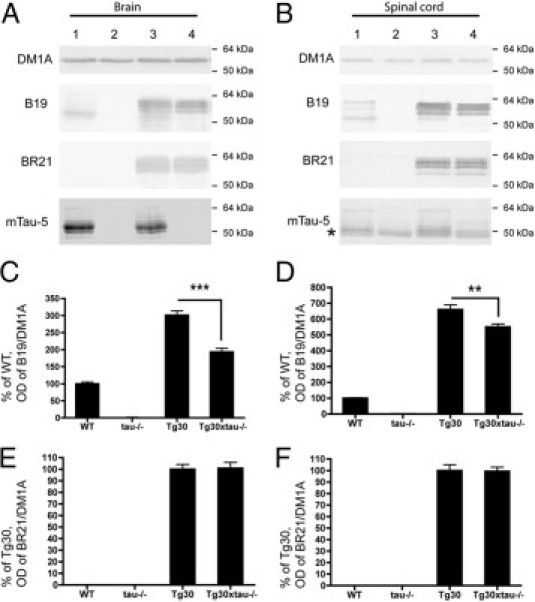
A, B: Levels of expression of tau proteins in wild-type mice (lane 1), in tau−/− mice (lane 2), in Tg30 mice (lane 3), and in Tg30xtau−/− mice (lane 4). Expression of tau was investigated in brain (A) and in spinal cord (B) with the B19 tau antibody (recognizing both human and mouse tau), the BR21 tau antibody (recognizing only human tau), and the mTau-5 antibody (recognizing only mouse tau). The asterisk in panel B indicates a nonspecific band of mouse immunoglobulin heavy chain detected by the secondary antibody in the soluble fraction of the spinal cord. Human tau was expressed only in Tg30 and Tg30xtau−/− mice and murine tau only in wild-type and Tg30 mice. C, D: The total level of tau (normalized to the level of α-tubulin detected with the DM1A antibody) was significantly decreased in the brain and in the spinal cord of Tg30xtau−/− mice, compared with Tg30 mice. E, F: The level of human tau (normalized to the level of a-tubulin) was not significantly different between Tg30 and Tg30xtau−/− mice. **P < 0.01 and ***P < 0.001 by one-way analysis of variance with Bonferroni post hoc tests [wild-type (WT), n = 4; tau−/−, n = 4; Tg30, n = 6; Tg30xtau−/−, n = 8].
Figure 3.
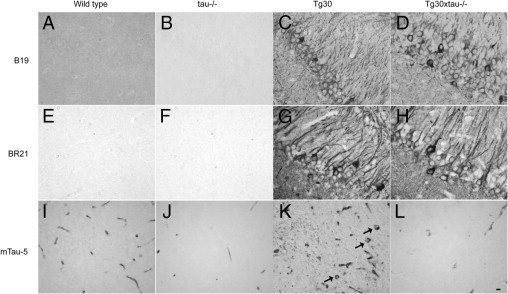
Tau immunohistochemical labeling of tissue sections of the hippocampus of 9-month-old wild-type, tau−/−, Tg30, and Tg30xtau−/− mice. A–D: The B19 tau antibody (reacting with mouse and human tau) shows the somatodendritic accumulation of tau in the CA1 sector of Tg30 and Tg30xtau−/− mouse hippocampus. E–H: The BR21 antibody specific for human tau also shows the somatodendritic accumulation of tau in Tg30 and in Tg30xtau−/− mice and does not show any immunoreactivity in wild-type mice. Some neurons containing neurofibrillary tangles show a stronger labeling in the CA1 sector of Tg30 and Tg30xtau−/− mouse hippocampus. I–L: The mTau-5 antibody specific for murine tau shows an abnormal somatodendritic accumulation of murine tau (arrows) in the subiculum of Tg30 mice, but not in the other genotypes. Blood vessels show a nonspecific labeling due to the use of an anti-mouse secondary antibody. Scale bar = 20 μm (A–L).
Survival of Tg30 and Tg30xtau−/− Mice Is Reduced
Kaplan-Meier survival curves were analyzed for wild-type, tau−/−, Tg30, and Tg30xtau−/− mice from postweaning age to 18 months of age (Figure 4A). The survival of Tg30 and Tg30xtau−/− mice was significantly reduced, compared with wild-type and tau−/− mice (P < 0.05 by log-rank test, using pairwise multiple comparison methods (Bonferroni-corrected threshold method). In addition, Tg30xtau−/− mice showed an accelerated mortality up to 3 months of age, compared with Tg30 mice.
Figure 4.
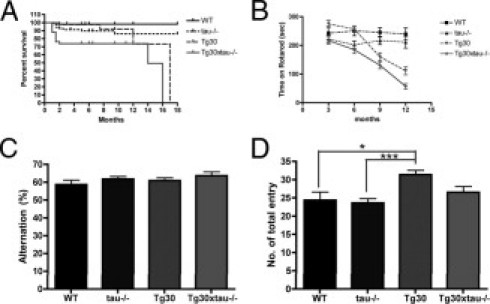
Survival and behavioural analysis of wild-type, tau−/−, Tg30 and Tg30xtau−/− mice. A: Kaplan-Meir survival curves. Tg30xtau−/− mice have a significantly reduced survival by comparison with other genotypes and an accelerated mortality during the first 3 months (P < 0.05, by log-rank test, using pairwise multiple comparison methods) (Bonferroni-corrected threshold method) (n = 178 for four genotypes). B: Rotarod testing. Tg30xtau−/− mice developed a motor deficit starting at 3 months and exhibited a more severe motor deficit than other genotypes (n > 11 for each age, for each genotype). C: Y maze test for alternations at 3 to 6 months. The percentage of alternations was not significantly different among all genotypes (wild-type: n = 11; tau−/−: n = 12; Tg30: n = 15; Tg30xtau−/−: n = 16). D: Number of entries in arms during Y-maze test at 3 to 6 months. The total number of entries was similar among genotypes, except that Tg30 mice had a higher number of entries than wild-type and tau−/− mice. *P < 0.05; ***P < 0.001 by one-way analysis of variance with Bonferonni post hoc tests.
Motor Deficit Is More Pronounced in Tg30xtau−/− Mice than in Tg30 Mice
We next compared the evolution of motor deficits in wild-type, tau−/−, Tg30, and Tg30xtau−/− mice by rotarod testing from 3 to 12 months of age. (Figure 4B). The tau−/− mice did not show a significant motor impairment up to 12 months. As reported previously,24 Tg30 mice showed a progressive motor deficit, significant at 9 and 12 months, compared with wild-type and tau−/− mice. Tg30xtau−/− mice also showed a progressive motor deficit that was significant from 6 to 12 months, compared with wild-type and tau−/− mice. At all ages, this motor impairment was significantly more severe in Tg30xtau−/− than in Tg30 mice.
Spatial Working Memory Is Not Significantly Different in Tg30 and Tg30xtau−/− Mice
Working memory was tested with the Y-maze, in experimental groups of 3- to 6-month-old mice (Figure 4C). The alternation scores of Tg30xtau−/− (63.96 ± 2.920%, mean ± SEM) and of Tg30 (60.28 ± 1.939%, mean ± SEM) mice were not significantly different between them or compared with wild-type and tau−/− mice. The number of total entries into the arms at 3 to 6 months did not differ among genotypes (except that it was significantly higher for Tg30 mice, compared with wild-type and tau−/− mice), indicating that locomotor activity associated with this exploratory behavior was not decreased in Tg30 and Tg30xtau−/− mice.
Increased Proportion of Insoluble Human Mutant Tau in the Absence of Murine Tau
To further investigate the effect of lack of endogenous murine tau in tau pathology, we analyzed the levels of soluble tau and insoluble PHF-tau by sarkosyl fractionation in Tg30 and Tg30xtau−/− mice (Figure 5). Sarkosyl-insoluble tau in both Tg30 and Tg30xtau−/− mice contained a major 64-kDa band immunoreactive with the human-specific tau antibody BR21. There were no tau-positive bands in sarkosyl-insoluble material from wild-type or tau−/− mice (data not shown). Soluble tau (S3) contained human and murine tau in Tg30 mice and only human tau in Tg30xtau−/− mice. The endogenous 110-kDa mouse big tau, immunoreactive with the B19 and the murine mTau-5 but not with the human BR21 tau antibody, was detected in the soluble spinal cord fractions of Tg30 mice but not in Tg30xtau−/− mice.
Figure 5.
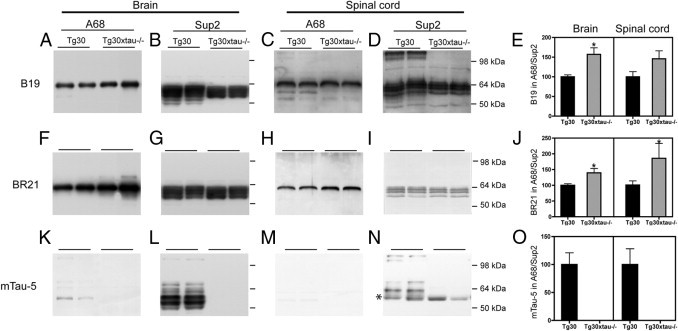
The proportion of sarkosyl-insoluble tau was increased in Tg30xtau−/− mice, compared with Tg30 mice. The levels of tau in the sarkosyl-insoluble (A68) and the soluble (Sup2) fractions were analyzed by immunoblotting in the brain and the spinal cord of Tg30 and Tg30xtau−/− mice. The panels show two representative blots for each condition. A–E: Panspecific B19 tau antibody. F–J: Human-specific tau antibody BR21. K–O: Mouse-specific tau antibody mTau5. The B19 and the BR21 antibodies recognize a major 64-kDa insoluble tau species in the brain (A and F) and the spinal cord (C and H) of Tg30 and Tg30xtau−/− mice. Additional minor bands in the sarkosyl-insoluble fraction were detected by the B19 antibody but not by the human-specific BR21 tau antibody. These minor bands are detected by the murine-specific tau antibody (K and M). The proportion of insoluble tau (ratio of insoluble tau to soluble tau) was increased in brain and spinal cord of Tg30xtau−/− mice, as estimated with the B19 antibody (E) and the human-specific BR21 antibody (J). The 110-kDa murine tau isoform (big tau) was detected only in Tg30 mice (D, M, and N). The asterisk in panel N indicates a nonspecific band of mouse immunoglobulin heavy chain detected by the secondary antibody in the soluble fraction of spinal cord (Tg30, n = 5; Tg30xtau−/−, n = 7). *P < 0.05 by unpaired Student's t-test.
The ratio of insoluble tau versus soluble tau was calculated, and we found that the proportion of sarkosyl-insoluble tau (estimated with both the B19 and the BR21 tau antibodies) was significantly increased in the brains of Tg30xtau−/− mice, compared with Tg30 mice (Figure 5, E and J).
Increased Number of Gallyas-Positive NFTs in the Hippocampus of Tg30xtau−/− Mice Compared with Tg30 Mice
Because the proportion of insoluble tau increased in the absence of murine tau, we became interested in the formation of NFTs in our Tg30xtau−/− mice. We performed Gallyas silver staining (which detects NFTs) in the brains of 12-month-old mice (Figure 6). As reported previously in Tg30 mice,24 Gallyas-positive NFTs were observed in the hippocampus, the cortex, subcortical areas, brainstem, and spinal cord of Tg30xtau−/− mice but not in wild-type and tau−/− mice. We measured the density of Gallyas-positive NFTs in the cortex, the hippocampus (subiculum and hippocampal CA1 sector), and the cervical spinal cord; the density of Gallyas-positive NFTs was significantly increased in the hippocampus of Tg30xtau−/−, compared with Tg30 mice, but was similar in the spinal cord of these two genotypes (Figure 6). Although Tg30xtau−/− mice showed a trend for increased NFT density in the cortex, this increase was not significant: 5.2 ± 0.67 NFT/mm2 for Tg30xtau−/− and 3.2 ± 0.85 NFT/mm2 for Tg30: (mean ± SEM; P = 0.097 by t-test).
Figure 6.
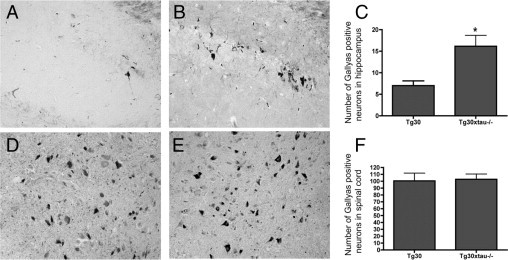
The load of neurofibrillary tangles estimated with Gallyas staining was increased in Tg30xtau−/− mice, compared with Tg30 mice (12-month-old mice). A–C: Hippocampus (CA1 sector). The density of NFTs was significantly higher in Tg30xtau−/− than in Tg30 mice (C). *P <0.05 by unpaired t-test (P = 0.032) (Tg30, n = 5; Tg30xtau−/−, n = 6). D–F: Lumbar spinal cord, anterior horn. The density of NFTs was not significantly different between Tg30xtau−/− andTg30 mice (F) (P = 0.8670). The density of NFTs is expressed as the number of NFTs per slice counted in the hippocampus and subiculum in three adjacent sagittal sections (Tg30, n = 6; Tg30xtau−/−, n = 8).
Decreased Phosphorylation of Soluble Tau in Tg30xtau−/− Compared with Tg30 Littermates
We compared the phosphorylation of sarkosyl-soluble tau in Tg30 and Tg30xtau−/− mice using a panel of tau antibodies to phosphorylated epitopes or to conformational epitopes (Figure 7, A–E). The phosphorylation levels of soluble tau detected by the AT270, CP13, AT180, PHF-1, and MC1 antibodies, normalized to total tau detected by the B19 antibody (Figure 7F), were significantly decreased in Tg30xtau−/− mice, compared with Tg30 mice (Figure 7G).
Figure 7.
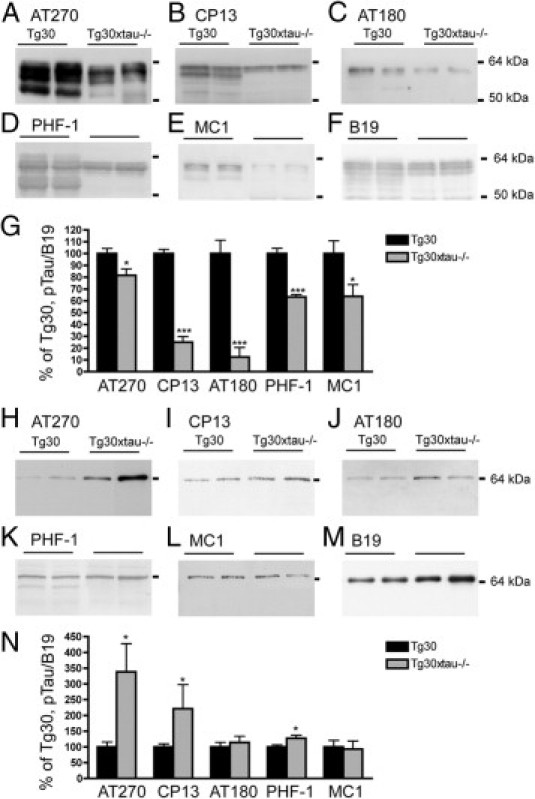
Highly phosphorylated soluble tau was rapidly aggregated into sarkosyl-insoluble fraction in Tg30xtau−/− mouse brain. The phosphorylation of tau in the sarkosyl-soluble fraction S2 (A–F) and sarkosyl-insoluble A68 fraction (H–M) was analyzed by immunoblotting with phosphotau antibodies AT270 (A and H), CP13 (B and I), AT180 (C and J), PHF-1 (D and K), and the conformational antibodies MC1 (E and L) and the polyclonal tau antibody B19 (F and M). The mean relative levels of each phosphotau species estimated by densitometry analysis (normalized to total tau estimated with the B19 antibody) are shown in panels G and N. The majority of pathological phosphotau species were reduced in sarkosyl-soluble fraction, whereas the levels of certain phosphotau species were increased in sarkosyl-insoluble fraction of Tg30xtau−/− mice. *P < 0.05 and ***P < 0.001 by Student's t-test (Tg30, n = 5; Tg30xtau−/−, n = 7).
Increased Phosphorylation of Sarkosyl-Insoluble Tau in Tg30xtau−/− Mouse Brain
We then compared the phosphorylation levels of sarkosyl-insoluble tau (Figure 7, H–M). The levels of phosphotau recognized by the AT270, CP13, and PHF-1 antibodies was significantly increased in the sarkosyl-insoluble fraction of Tg30xtau−/−, compared with Tg30 mouse brains (Figure 7N).
Increased Levels of Y216 GSK-3β in Tg30xtau−/− Mice
We analyzed the levels of GSK-3β, pS9 GSK-3β, and pY216 GSK-3β and of the p35 and p25 activators of Cdk5 (Figure 8A). The total levels of GSK-3β and of pS9 GSK-3β (Figure 8B) were similar in Tg30 and Tg30xtau−/− mice, but the levels of Y216 GSK-3β were significantly increased in the brain of Tg30xtau−/− mice, compared with all the other genotypes (Figure 8C). Both GSK-3α and -β can be cleaved by calpain at the N-terminus, and the truncation increases kinase activity.45 Calpain-cleaved GSK-3 immunoreactive with Y216 GSK-3 antibody (arrow in Figure 8A) was generally increased in Tg30 and Tg30xtau−/− mice (Figure 8D). The levels of p25 were not significantly different between Tg30 and Tg30xtau−/− mice (Figure 8E). The level of Cdk5 was similar and not significantly different among four genotypes (data not shown).
Figure 8.
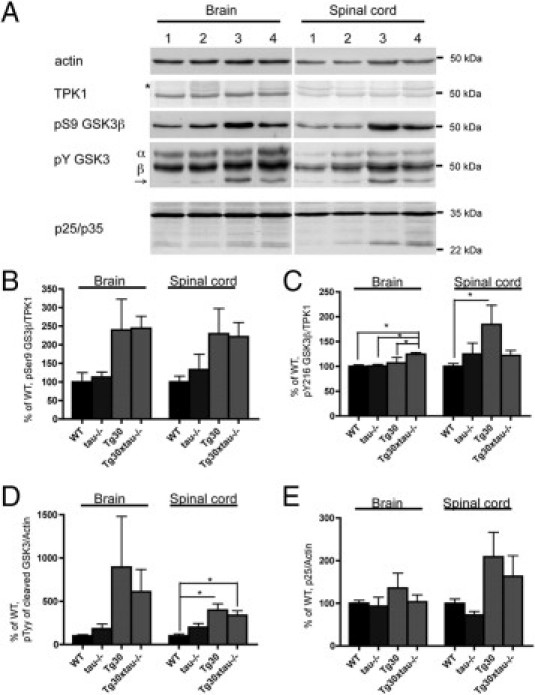
Kinase activation was analyzed by Western blotting for GSK-3 and p35. A: Immunoblotting for actin, TPK1, pSer9 GSK-3β, pTyr GSK-3, and p25/p35 in brain and spinal cord of wild-type mice (lane 1), tau−/− mice (lane 2), Tg30 mice (lane 3), and Tg30xtau−/− mice (lane 4). The asterisk at TPK1 indicates nonspecific bands of mouse IgG heavy chain. The arrow points to the calpain-cleaved GSK-3. B: The level of pSer9 GSK-3β normalized to TPK1 was generally (but not significantly) increased in the presence of tau pathology. C: The level of pTyr 216 GSK-3β normalized to TPK1 was significantly increased in the brain of Tg30xtau−/− mice and in the spinal cord of Tg30. D: pTyr GSK-3-positive calpain-cleaved GSK-3 was significantly increased in spinal cord of Tg30 and Tg30xtau−/−. E: The level of p25 normalized to actin was generally (but not significantly) increased in Tg30 mice (wild-type, n = 4; tau−/−, n = 4; Tg30, n = 4; Tg30xtau−/−, n = 6).
Endogenous Mouse Tau Is Recruited at Low Level into PHFs
We analyzed whether endogenous mouse tau isoforms could be recruited in NFTs by analyzing the tau isoform composition and their immunoreactivity with mouse and human-specific tau antibodies in Tg30 and Tg30xtau−/− mice. In addition to the major 64-kDa band, two minor bands of 63 kDa and 67 kDa were detected with the B19 and PHF-1 tau antibodies in sarkosyl-insoluble fraction from Tg30 mice (Figure 5), accounting for 5% of the total tau immunoreactivity in this fraction, as judged by densitometry analysis. The endogenous 110-kDa mouse big tau was also detected with the PHF-1 antibody in the sarkosyl-insoluble spinal cord fraction of Tg30 mice. These minor bands were not immunoreactive with the BR21 human tau antibody and were absent in sarkosyl-insoluble material from Tg30xtau−/− mice, which suggests that they corresponded to endogenous murine tau proteins.
The presence at low level of mouse tau proteins in PHFs from Tg30 mice was confirmed by immunolabeling in electron microscopy of isolated PHFs present in sarkosyl-insoluble fractions (Figure 9, A–D). These PHFs were strongly labeled by the BR21 human tau antibody (Figure 9A) and weakly by the murine tau antibody (Figure 9B), with both human and mouse tau detected in the same PHF. In Tg30xtau−/− mice, the PHFs were immunoreactive only to human tau (Figure 9C) and not to murine tau (Figure 9D). Finally, we looked for mouse tau in NFTs in brain tissue sections by using the BR10 tau antibody to 2N tau isoforms. The BR10 antibody, which could react only with endogenous mouse 2N4R tau isoform present in adult mouse brain, labeled a subset of NFTs in Tg30 mice but never in Tg30xtau−/− mice (not shown). BR10-positive NFTs inTg30 mice were much more frequently observed in the brainstem and the spinal cord than in the hippocampus or the cortex.
Figure 9.
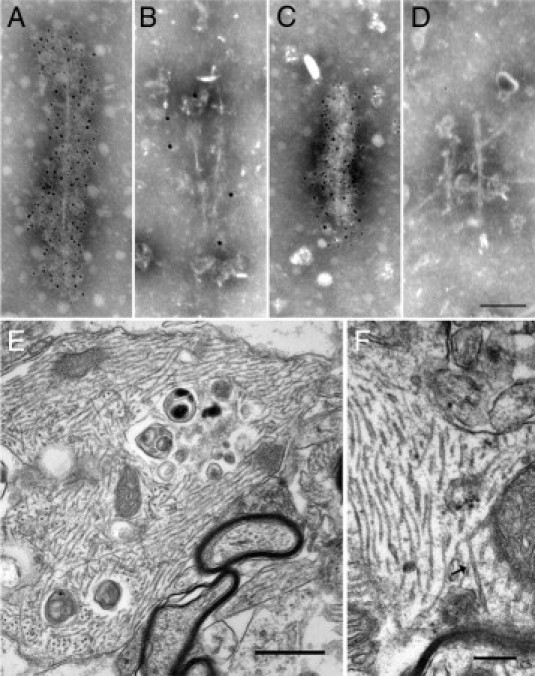
Electron microscopy of PHF-tau in Tg30 and Tg30xtau−/− mice. A–D: Immunolabeling in electron microscopy of abnormal filaments present in the sarkosyl-insoluble fraction of Tg30 (A and B) and Tg30xtau−/− (C and D) mice. A, C: Anti-human tau antibody (BR21). A strong labeling was observed both in Tg30 and in Tg30xtau−/− mice. B, D: Anti-mouse tau antibody (mTau5). A weak labeling was observed in filaments from Tg30 mice but was absent in Tg30xtau−/− mice. Scale bar = 50 nm (A–D). E, F: Ultrastructural aspects of abnormal filaments in Tg30xtau−/− mice (subiculum). E: A dilated neurite contains bundles of 19-nm straight filaments, admixed with degraded organelles and lysosomal vacuoles. Scale bar = 0.5 μm. F: Under higher magnification, straight filaments and a PHF-like filament (arrow) can be identified. Scale bar = 0.25 μm.
Abnormal Filaments Are Mainly Straight Filaments in Tg30 and Tg30xtau−/− Mice
The ultrastructural features of intraneuronal fibrillary inclusions in the hippocampus and in the anterior horn of the spinal cord were analyzed in Tg30 and in Tg30xtau−/− mice. As previously reported in Tg30 mice,24 hippocampal neurofibrillary inclusions inTg30xtau−/− mice were made mainly of bundles of straight filaments, 19 nm wide (Figure 9E), with occasional twisted filaments 25 nm wide, with regular constrictions, looking like the paired helical filaments observed in AD and other tauopathies (Figure 9F).
Proteolytic Tau Fragments and LC3 Levels
Because proteolytic cleavage of tau has been suggested to favor tau aggregation, we next compared by Western blotting the profile of soluble and insoluble tau species in Tg30 and Tg30xtau−/− mice, using tau antibodies to the extreme N-terminus (TP007) or C-terminus (TP70) of tau, and to tau cleaved at Asp421 by caspase 3. The labeling pattern of soluble tau and sarkosyl-insoluble tau by TP007 and TP70 was similar in Tg30 and Tg30xtau−/− mice (data not shown). Soluble and sarkosyl-insoluble-tau were not labeled by the anti-tau Asp421. By immunocytochemistry, NFTs in Tg30 and Tg30xtau−/− mice were immunoreactive with both TP007 and TP70 tau antibodies (data not shown), and a few NFTs were positive with the anti-tau Asp421 (data not shown).
The level of LC3, an autophagy marker, was not significantly different between Tg30 and Tg30xtau−/− (data not shown). The level of β-catenin, a target protein for the ubiquitin-proteasome pathway, was comparable in Tg30 and Tg30xtau−/− (data not shown).
Increased Number of Neuronal Pericaryal Inclusions in the Spinal Cord of Tg30xtau−/− Mice
In addition to NFTs composed of abnormal filaments and abundant in the forebrain, Tg30 mice developed abundant pericaryal rounded inclusions in the spinal cord (Figure 10, A and B), made of abnormal filaments admixed with abundant neurofilaments24 (Figure 10, E and F). Similar pericaryal inclusions were identified in the spinal cord of Tg30xtau−/− mice. The density of these inclusions was significantly higher in Tg30xtau−/− mice, compared with Tg30 mice (Figure 10, C).
Figure 10.
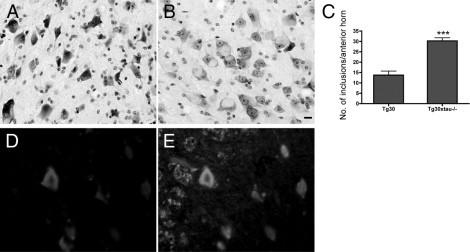
Neurofilament-positive neuronal inclusions in spinal cord were more frequent in Tg30xtau−/− mice than in Tg30 mice. A, B: Nissl staining for hyaline inclusions in the anterior horn of the lumbar spinal cord in Tg30 (A) and Tg30xtau−/− (B) mice. C: The density of inclusions in the anterior horn was significantly increased in Tg30xtau−/− mice. ***P < 0.001 by Student's t-test (n = 5 mice for each genotype). D–E: Double immunolabeling of the anterior horn of Tg30xtau−/− mouse with the PHF-1 tau antibody (D) and a neurofilament antibody (E). Most of the neuronal inclusions were positive for both phosphotau and neurofilament. Scale bar = 20 μm.
Neuronal Cell Numbers Are Similar in Tg30 and Tg30xtau−/− in Hippocampal CA1–CA3 Sectors and in Spinal Cord
To assess potential neuronal cell death in the hippocampus, we performed a stereological cell count of the total number of pyramidal neurones in hippocampal sectors CA1–CA4 in 12-month-old animals of each genotype. The number of neurons was not statistically different among genotypes: wild-type mice, (440,900 ± 19,240 (mean ± SEM, n = 4); tau−/− mice, 418,500 ± 32,150 (mean ± SEM, n = 5); Tg30 mice, 387,750 ± 22,400 (mean ± SEM, n = 5); and Tg30xtau−/− mice, 443,900 ± 50,760 (mean ± SEM, n = 3).
A stereological estimation of neuronal density was also performed in the left anterior horn of the cervical spinal cord. These neuronal densities (estimated as the neuron number relative to the volume of spinal gray matter), although tending to be lower in Tg30tau and in Tg30txtau−/− mice, were not statistically different among different genotypes (one-way analysis of variance, P = 0.31).
Estimations of the volumes of the brain, cortex, hippocampal formation, and cervical spinal cord segment (used for cell counting) with the Cavalieri method were also made in 12-month-old animals of each genotype. These volumes were not statistically different among the different genotypes (data not shown).
Axonopathy in the Sciatic Nerve
Tg30tau mice show a loss of axons and a reduction of their mean cross-sectional area in the sciatic nerve. We confirmed the presence of this axonopathy in Tg30 mice and observed it also in Tg30xtau−/− mice; however, the mean axonal cross-sectional area and the mean number of axons in the sciatic nerve did not differ between Tg30 and Tg30xtau−/− mice (see Supplemental Figure S1 at http:ajp.amjpathol.org).
Discussion
Absence of Endogenous Murine Tau Decreases Survival and Enhances Motor Deficits of Mice Expressing a Mutant Tau
We have characterized in the present study a mouse model in which a human mutant tau protein is expressed in absence of endogenous mouse tau proteins. These Tg30xtau−/− mice had a reduced survival with an accelerated mortality during the first 3 months of age, before they developed significant motor deficits, suggesting that this phenotype was due to a gain of toxic function of the human mutant tau in absence of wild-type mouse tau, because tau−/− mice did not show reduced survival. Tg30xtau−/− mice also showed an earlier and a more severe motor phenotype than Tg30 mice, which also might have contributed to the reduced survival after 12 months. Tg30xtau−/− mice had an increased number of pericaryal neuronal inclusions and of sarkosyl-insoluble tau in the spinal cord, compared with Tg30 mice, suggesting that these lesions were responsible for the more severe motor phenotype. Thus, even though Tg30 mice have a lower survival than wild-type mice and develop a motor deficit, the presence of mouse tau in Tg30 mice to some extent slows down this phenotype, suggesting that the absence of mouse wild-type tau rather revealed the toxicity of human tau, which was expressed at similar level in Tg30 and Tg30xtau−/− mice.
Relationship among Sarkosyl-Insoluble Tau, Tangles Load, and Neuronal Death
Sarkosyl-insoluble tau increased in the whole brain and the spinal cord of Tg30xtau−/− mice, but tangles load was increased only in the hippocampus, not in the cortex or spinal cord. Sarkosyl-insoluble fractionation allows isolating a relatively soluble population of PHF-tau without disrupting large structures such as NFTs.46 Thus, sarkosyl-insoluble tau can to some extent be uncoupled from tangles load in this murine model, as observed in human brain studies.47,48 Also, we did not observe significantly increased cell death in the hippocampus or in spinal cord of Tg30xtau−/− mice. Altogether, these findings suggest that sarkosyl-insoluble tau was more correlated than tangles with decreased survival and increased motor deficits. Neither type of tau accumulations was associated with significant neuronal loss in this model, but intermediate tau species (eg, oligomeric species) might have been responsible for neuronal dysfunction, leading to observed decreased survival and motor problems.
Murine Tau Contributes to a Minor Proportion of Aggregated Tau
Sarkosyl-insoluble tau was composed predominantly of human mutant tau in Tg30 mice, and was composed solely of human mutant tau in Tg30xtau−/− mice. We also observed that PHFs present in this fraction were more strongly labeled by the anti-human tau antibody than by the anti-murine tau antibody, in agreement with a previous study showing that anti-murine tau antibody labeled only a small number of PHF-tau purified from P301S transgenic mouse brain, whereas an anti-human tau antibody labeled most of PHF-tau.49 A study of P301L mice also indicated that sarkosyl-insoluble tau contained predominantly human tau.50 Furthermore, injection of brain homogenate of P301L transgenic mouse, which contains PHF-tau, induced PHF-tau in mice expressing human non-mutant tau but not in wild-type mice.51 These studies support the idea that murine tau does not have the same propensity for aggregation as human tau in vivo.
Nonetheless, some PHFs were labeled by both human and murine tau antibodies in our study, indicating that mouse tau, even at low levels, was coaggregated with human mutant tau in the same filament in Tg30 mice. Several mouse tau isoforms were probably recruited in PHFs in Tg30 mice, because NFTs labeled with the anti-insert 3 tau antibody (BR10) and the sarkosyl-insoluble tau contained a 110-kDa tau species (absent in Tg30xtau−/− mice), consistent with mouse big tau isoform.
Murine tau can nevertheless assemble in PHFs in vitro,52,53 and NFT-like tau aggregates of endogenous murine tau have been reported in mice overexpressing the p25 activator of Cdk554 or lacking the prolyl-isomerase Pin1.55 The coaggregation of murine tau with a truncated human mutant tau and the persistence of NFTs made of mouse tau was also observed in an inducible mouse model.56 Mice overexpressing wild-type murine tau also accumulated sarkosyl-insoluble tau, but not NFTs.57
Absence of Endogenous Murine Tau Increases Aggregation of Human Mutant Tau
In transgenic mice overexpressing human wild-type tau, previous studies indicated that absence of murine tau either favored17 or had no effect19 on the aggregation of this wild-type human tau. In the present study, the aggregation of human mutant tau was accelerated in the absence of murine tau, as shown by increased proportions of sarkosyl-insoluble tau in the brain and spinal cord, and increased NFT density in the hippocampus in Tg30xtau−/− mice. This increased tau aggregation in Tg30xtau−/− was not due to increased tau expression. because the total level of tau was lower in Tg30xtau−/− than in Tg30 mice and the level of total human tau was similar in the two lines.
The degree of phosphorylation of sarkosyl-insoluble tau was increased on several epitopes in Tg30xtau−/− mice, suggesting that increased tau aggregation in these mice might have resulted from increased tau phosphorylation. This increased phosphorylation might be related to the increased levels of the active form of GSK-3 in Tg30xtau−/− mice. The phosphorylation level of soluble tau was generally decreased in Tg30xtau−/− mice, possibly indicating that the pool of phosphorylated tau in Tg30xtau−/− mice was more rapidly integrated into sarkosyl-insoluble tau. Increased levels of active GSK-3β have been observed in AD frontal cortex,58 and an immunoreactivity for active GSK-3 has been detected in tangles in AD58,59 and in tau transgenic animals.24,60 The Fyn tyrosine kinase can activate GSK-3β,61 and because tau regulates intracellular localization of Fyn,62 it seems possible that absence of murine tau disturbs Fyn subcellular localization, leading to increased phosphorylation of Y216 GSK-3β.
Our data also suggest that the increased tau aggregation in Tg30xtau−/− mice was related to the absence of an inhibitory effect (or slowness) due to the absence of wild-type mouse tau. The mechanisms by which murine tau could interfere with human mutant tau aggregation remain unclear, but we here propose several potential mechanisms by which the coaggregation of murine tau with human mutant tau might affect the efficiency of PHF formation.
In a previous study of mice expressing human 6 tau isoforms in the absence of murine tau,17 an imbalance of 3R/4R tau isoform composition was identified as a potential mechanism for increased aggregation of wild-type human tau proteins. It has also been recently observed in vitro that 3R tau isoforms inhibit 4R tau assembly.63 In the Tg30 and Tg30xtau−/− models, however, only 4R tau isoforms are expressed. Thus, this imbalance of 3R/4R tau cannot explain the acceleration of PHF formation in the absence of murine tau in our Tg30xtau−/− model. In addition, wild-type adult mice do not express significant levels of 3R tau isoforms and do not develop NFTs.
An imbalance in the ratios of tau isoforms with or without N-terminal inserts might nevertheless play a role in the acceleration of PHF formation in Tg30xtau−/− mice, because these mice express only the 1N4R tau isoform, whereas Tg30 express 0N4R, 1N4R, and 2N4R tau isoforms.
The N-terminal projection domain has only 75% homology between human and murine, whereas the C-terminal microtubule-binding domain is almost perfectly identical.52 Thus, in view of these sequence differences, the N-terminal domain of murine tau might interfere with human mutant tau aggregation. First, N-terminal domain of murine tau might be less phosphorylated than human tau. At the N-terminal 150 amino acids of the longest isoforms (alignment shown in reference 52), human tau includes 10 residues that can be potentially phosphorylated (Ser, Thr, or Tyr) and are not found in murine tau (Thr17, Tyr18, Thr39, Thr50, Thr52, Ser56, Thr101, Ser113, Ser131, Ser137), whereas the murine tau contains only one Thr residue (Thr12) that is not included in human tau. Importantly, phosphorylation of Thr39, Thr52, and Ser56 could be induced by casein kinase II,64,65 which is dysregulated in AD brains.66 Tyr18 is phosphorylated by Fyn, and phospho-Tyr18 colocalizes with tangles in AD.67 Thr50 can be phosphorylated by SAPK4/p38. Phosphorylation on Thr50 is likely to promote microtubule-binding ability, and this phosphorylation is found in PHF-tau from AD brain.68 Furthermore, Tyr17, Thr50, Thr101, Ser113, and Ser131 could be phosphorylated by casein kinase 1ε in vitro, and phosphorylation of Ser113 was notably detected in PHF-tau from AD brain65 Thus, most of these Thr, Ser, and Tyr residues found in human tau but not found in murine tau can be phosphorylated and could be critical for accelerated PHF formation from human tau.
Second, in considering N-terminal heterogeneity, tau consists of both acidic and basic regions, and the extreme amino terminal projection domain contains many acidic residues. Despite their heterogeneity at the amino terminus, the isoelectric points (pI) of the acidic region of human and murine tau are not greatly different: the pI of the acidic region of human tau (amino acids 1–120) is 3.92, but that of murine tau (amino acids 1–109) is 3.90; however, because of more numerous phosphorylation sites, the pI of the acidic region could be even more negatively changed in humans than in mice. This would increase the interaction between acidic and basic regions of tau more easily in human than in mouse tau, possibly favoring a pathological conformation and dimerization/oligomerization of tau.69
Third, heterogeneity at N-terminus might influence the structure of tau. Whereas tau is originally an extremely soluble protein without a rigid structure,70 tau undergoes pathological conformation change as it forms PHF-tau, in which the N-terminal domain is adjacent to the microtubule binding domain35,71 (reviewed by Binder et al.).72 This conformation change, which is one of the early events of tau pathology,73 can be recognized by specific antibodies such as MC1, Alz50, and TG-335 (reviewed by Binder et al).72 Because the N-terminal sequence is distinctly different in murine and human tau, it is possible that murine tau is less likely to adopt this early pathological conformation change.
Last, N-terminal heterogeneity might be also a key factor for tau truncation. Truncation at both the amino and carboxyl termini takes place in the process of NFT formation.72 Because of the difference in its N-terminus, murine tau may possibly not undergo truncation in the same manner, and this may affect the evolution of tau aggregation into NFTs. However, we could not find a marked difference between Tg30 and Tg30xtau−/− in the pattern of tau fragments when tested with antibodies to the extreme N- and C-terminus. Also, tau in the sarkosyl-insoluble fraction was not immunoreactive with the antibody to tau cleaved at aspartic residue 421, a caspase-mediated truncation of tau,74,75 in either Tg30 or Tg30xtau−/− mice. It thus seems unlikely that tau truncation at D421 contributed to increased tau aggregation in Tg30xtau−/− mice (in agreement with a previous observation in P301S tau transgenic mice76).
An alternative explanation for the increase in tau aggregation in Tg30xtau−/− mice is that absence of murine tau would impede the removal of aggregated human tau. For example, acute reduction of tau in a cellular model has been reported to impair autophagy in cells deficient for the NPC1 gene (Niemann-Pick disease, type C1).77 However, we could not find a change in the levels of the autophagosomal markers LC3 in Tg30xtau−/− mice. The levels of β-catenin, a substrate of the proteasome, likewise did not differ between Tg30 and Tg30xtau−/− mice in the brain, suggesting that the activity of the ubiquitin-proteasomal system was not markedly different in Tg30 and Tg30xtau−/− mice.
Human tau contains three lysine residues absent in murine tau (Lys24, Lys131, and Lys257) that could potentially be ubiquitinated. However, a liquid chromatography-tandem mass spectrometry study has only identified three ubiquitination sites at Lys-254, Lys-311 and Lys-353 (common to mouse and human tau) from the human soluble PHF-tau affinity purified with MC1 antibody.78
Thus, our results suggest that increased aggregation of human mutant tau in absence of murine tau did not result from increased human tau expression or decreased tau clearance, but rather from the absence of interference of wild-type murine tau. Tg30xtau−/− mice thus represent a better model of tauopathies for study of the mechanisms of toxicity of human mutant tau, as exemplified here by the development of a more severe phenotype in Tg30xtau−/− mice than in Tg30 mice.
Acknowledgments
We thank Dr. Luc Buée (INSERM, U837) for providing the Tg30 line, Dr. Marc Mercken (Johnson & Johnson, Beerse, Belgium) for providing the mTau5 antibody, and Dr. Peter Davies (Albert Einstein College of Medicine, New York, NY) for providing the CP13, PHF-1, and MC1 antibodies.
Footnotes
Supported by the Interuniversity Attraction Poles program (P6/43) of the Belgian Federal Science Policy Office, by the Diane program (Wallon region) (816856), by grants from the Fund for Medical Scientific Research (Fonds de la Recherche Scientifique Médicale) (3.4504.10), and the Foundation for Alzheimer's Disease Research (Fondation pour la Recherche sur la maladie d'Alzheimer/Stichting voor Alzheimer Onderzoek) (08604).
Supplemental material for this article can be found on http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.10.034.
Supplementary data
Supplemental Figure s1.
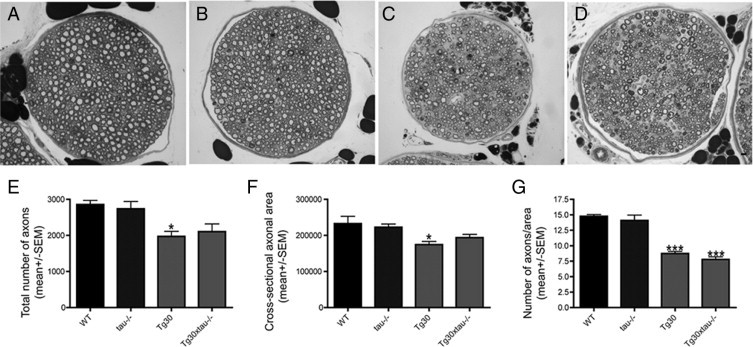
Semi-thin cross-section of the sciatic nerve in wild-type (A), tau−/− (B), Tg30 (C), and Tg30xtau−/− mice (D). The total number of axons (E), mean cross-sectional area (F), and density of axons (G) were similar in wild-type and tau−/− mice, but reduced in Tg30 and Tg30xtau−/− mice. *P <0.05 and *** P <0.001 by one-way ANOVA with Bonferroni post hoc tests (n = 3 for wild-type; n = 4 for tau−/−; n = 6 for Tg30 and Tg30xtau−/−). Scale bar, 50 μm (A–D).
References
- 1.Selkoe D.J. The origins of Alzheimer disease—A is for amyloid. JAMA. 2000;283:1615–1617. doi: 10.1001/jama.283.12.1615. [DOI] [PubMed] [Google Scholar]
- 2.Ballatore C., Lee V.M.Y., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 3.van Swieten J., Spillantini M.G. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73. doi: 10.1111/j.1750-3639.2007.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denk F., Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging. 2009;30:1–13. doi: 10.1016/j.neurobiolaging.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis J., Dickson D.W., Lin W.L., Chisholm L., Corral A., Jones G., Yen S.H., Sahara N., Skipper L., Yager D., Eckman C., Hardy J., Hutton M., McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 6.Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y., LaFerla F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 7.Pérez M., Ribe E., Rubio A., Lim F., Morán M.A., Ramos P.G., Ferrer I., Isla M.T., Avila J. Characterization of a double (amyloid precursor protein-tau) transgenic: tau phosphorylation and aggregation. Neuroscience. 2005;130:339–347. doi: 10.1016/j.neuroscience.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 8.Terwel D., Muyllaert D., Dewachter I., Borghgraef P., Croes S., Devijver H., Van Leuven F. Amyloid activates GSK-3 beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008;172:786–798. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Götz J., Ittner L.M. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- 10.Brion J.P., Tremp G., Octave J.N. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer disease. Am J Pathol. 1999;154:255–270. doi: 10.1016/S0002-9440(10)65272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Götz J., Probst A., Spillantini M.G., Schäfer T., Jakes R., Bürki K., Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higuchi M., Ishihara T., Zhang B., Hong M., Andreadis A., Trojanowski J.Q., Lee V.M.Y. Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron. 2002;35:433–446. doi: 10.1016/s0896-6273(02)00789-4. [DOI] [PubMed] [Google Scholar]
- 13.Ishihara T., Hong M., Zhang B., Nakagawa Y., Lee M.K., Trojanowski J.Q., Lee V.M.Y. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 14.Ishihara T., Zhang B., Higuchi M., Yoshiyama Y., Trojanowski J.Q., Lee V.M.Y. Age-dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am J Pathol. 2001;158:555–562. doi: 10.1016/S0002-9440(10)63997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Probst A., Götz J., Wiederhold K.H., Tolnay M., Mistl C., Jaton A.L., Hong M., Ishihara T., Lee V.M., Trojanowski J.Q., Jakes R., Crowther R.A., Spillantini M.G., Bürki K., Goedert M. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 16.Duff K., Knight H., Refolo L.M., Sanders S., Yu X., Picciano M., Malester B., Hutton M., Adamson J., Goedert M., Burki K., Davies P. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- 17.Andorfer C., Kress Y., Espinoza M., De Silva R., Tucker K.L., Barde Y.A., Duff K., Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 18.Andorfer C., Acker C.M., Kress Y., Hof P.R., Duff K., Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terwel D., Lasrado R., Snauwaert J., Vandeweert E., Van Haesendonck C., Borghgraef P., Van Leuven F. Changed conformation of mutant tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of tau-4R/2N transgenic mice. J Biol Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- 20.Schindowski K., Bretteville A., Leroy K., Begard S., Brion J.P., Hamdane M., Buée L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leroy K., Bretteville A., Schindowski K., Gilissen E., Authelet M., De Decker R., Yilmaz Z., Buee L., Brion J.P. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am J Pathol. 2007;171:976–992. doi: 10.2353/ajpath.2007.070345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tucker K.L., Meyer M., Barde Y.A. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 23.Boutajangout A., Authelet M., Blanchard V., Touchet N., Tremp G., Pradier L., Brion J.P. Cytoskeletal abnormalities in mice transgenic for human tau and familial Alzheimer's disease mutants of APP and presenilin-1. Neurobiology of disease. 2004;15:47–60. doi: 10.1016/j.nbd.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 24.Leroy K., Bretteville A., Schindowski K., Gilissen E., Authelet M., De Decker R., Yilmaz Z., Buée L., Brion J.P. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am J Pathol. 2007;171:976–992. doi: 10.2353/ajpath.2007.070345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leroy K., Ando K., Heraud C., Yilmaz Z., Authelet M., Boeynaems J.M., Buee L., De Decker R., Brion J.P. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J Alzheimers Dis. 2010;19:705–719. doi: 10.3233/JAD-2010-1276. [DOI] [PubMed] [Google Scholar]
- 26.Holcomb L.A., Gordon M.N., Jantzen P., Hsiao K., Duff K., Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 27.Brion J.P., Hanger D.P., Bruce M.T., Couck A.M., Flament-Durand J., Anderton B.H. Tau in Alzheimer neurofibrillary tangles: N- and C-terminal regions are differentially associated with paired helical filaments and the location of a putative abnormal phosphorylation site. Biochem J. 1991;273:127–133. doi: 10.1042/bj2730127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanger D.P., Gibb G.M., de Silva R., Boutajangout A., Brion J.P., Revesz T., Lees A.J., Anderton B.H. The complex relationship between soluble and insoluble tau in tauopathies revealed by efficient dephosphorylation and specific antibodies. FEBS Lett. 2002;531:538–542. doi: 10.1016/s0014-5793(02)03611-6. [DOI] [PubMed] [Google Scholar]
- 29.Sennvik K., Boekhoorn K., Lasrado R., Terwel D., Verhaeghe S., Korr H., Schmitz C., Tomiyama T., Mori H., Krugers H., Joels M., Ramakers G.J.A., Lucassen P.J., Van Leuven F. Tau-4R suppresses proliferation and promotes neuronal differentiation in the hippocampus of tau knockin/knockout mice. FASEB J. 2007;21:2149–2161. doi: 10.1096/fj.06-7735com. [DOI] [PubMed] [Google Scholar]
- 30.Davis D.R., Brion J.P., Couck A.M., Gallo J.M., Hanger D.P., Ladhani K., Lewis C., Miller C.C.J., Rupniak T., Smith C., Anderton B.H. The phosphorylation state of the microtubule-associated protein tau as affected by glutamate, colchicine and beta-amyloid in primary rat cortical neuronal cultures. Biochem J. 1995;309:941–949. doi: 10.1042/bj3090941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brion J.P., Couck A.M., Robertson J., Loviny T.L.F., Anderton B.H. Neurofilament monoclonal antibodies RT97 and 8D8 recognize different modified epitopes in PHF-tau in Alzheimer's disease. J Neurochem. 1993;60:1372–1382. doi: 10.1111/j.1471-4159.1993.tb03298.x. [DOI] [PubMed] [Google Scholar]
- 32.Goedert M., Jakes R., Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–170. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- 33.Goedert M., Jakes R., Crowther R.A., Cohen P., Vanmechelen E., Vandermeeren M., Cras P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J. 1994;301:871–877. doi: 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otvos L., Jr, Feiner L., Lang E., Szendrei G.I., Goedert M., Lee V.M. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 35.Jicha G.A., Bowser R., Kazam I.G., Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 36.Planel E., Krishnamurthy P., Miyasaka T., Liu L., Herman M., Kumar A., Bretteville A., Figueroa H.Y., Yu W.H., Whittington R.A., Davies P., Takashima A., Nixon R.A., Duff K.E. Anesthesia-induced hyperphosphorylation detaches 3-repeat tau from microtubules without affecting their stability in vivo. J Neurosci. 2008;28:12798–12807. doi: 10.1523/JNEUROSCI.4101-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brion J.P., Hanger D.P., Couck A.M., Anderton B.H. A68 proteins in Alzheimer's disease are composed of several tau isoforms in a phosphorylated state which affects their electrophoretic mobilities. Biochem J. 1991;279:831–836. doi: 10.1042/bj2790831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.West M.J., Slomianka L., Gundersen H.J. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 39.Williams R.W., Rakic P. Three-dimensional counting: an accurate and direct method to estimate numbers of cells in sectioned material [Erratum appeared in J Comp Neurol 1989, 281:335] J Comp Neurol. 1988;278:344–352. doi: 10.1002/cne.902780305. [DOI] [PubMed] [Google Scholar]
- 40.Schmitz C., Hof P.R. Recommendations for straightforward and rigorous methods of counting neurons based on a computer simulation approach. J Chem Neuroanat. 2000;20:93–114. doi: 10.1016/s0891-0618(00)00066-1. [DOI] [PubMed] [Google Scholar]
- 41.Schmitz C., Hof P.R. Design-based stereology in neuroscience. Neuroscience. 2005;130:813–831. doi: 10.1016/j.neuroscience.2004.08.050. [DOI] [PubMed] [Google Scholar]
- 42.Slomianka L., West M.J. Estimators of the precision of stereological estimates: an example based on the CA1 pyramidal cell layer of rats. Neuroscience. 2005;136:757–767. doi: 10.1016/j.neuroscience.2005.06.086. [DOI] [PubMed] [Google Scholar]
- 43.Cavalieri B. Geometria Indivisibilibus Continuorum [Geometry of Indivisible Continua]. Latin. Bononia [Bologna]: Typis Clementis Ferronij, 1635: Translated as Geometria Degli Indivisibili [Geometry of Indivisibles]. Italian. In: Lombardo-Radice L., editor. Unione Tipografico-Editrice Torinese; Turin, Italy: 1966. [Google Scholar]
- 44.Paxinos G., Franklin K.B.J. 2nd ed. Academic Press; London: 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- 45.Goñi-Oliver P., Lucas J.J., Avila J., Hernández F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- 46.Greenberg S.G., Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sahara N., Maeda S., Yoshiike Y., Mizoroki T., Yamashita S., Murayama M., Park J.M., Saito Y., Murayama S., Takashima A. Molecular chaperone-mediated tau protein metabolism counteracts the formation of granular tau oligomers in human brain. J Neurosci Res. 2007;85:3098–3108. doi: 10.1002/jnr.21417. [DOI] [PubMed] [Google Scholar]
- 48.Katsuno T., Morishima-Kawashima M., Saito Y., Yamanouchi H., Ishiura S., Murayama S., Ihara Y. Independent accumulations of tau and amyloid beta-protein in the human entorhinal cortex. Neurology. 2005;64:687–692. doi: 10.1212/01.WNL.0000151958.79884.86. [DOI] [PubMed] [Google Scholar]
- 49.Allen B., Ingram E., Takao M., Smith M.J., Jakes R., Virdee K., Yoshida H., Holzer M., Craxton M., Emson P.C., Atzori C., Migheli A., Crowther R.A., Ghetti B., Spillantini M.G., Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sahara N., Lewis J., DeTure M., McGowan E., Dickson D.W., Hutton M., Yen S.H. Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility. J Neurochem. 2002;83:1498–1508. doi: 10.1046/j.1471-4159.2002.01241.x. [DOI] [PubMed] [Google Scholar]
- 51.Clavaguera F., Bolmont T., Crowther R.A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A.K., Beibel M., Staufenbiel M., Jucker M., Goedert M., Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kampers T., Pangalos M., Geerts H., Wiech H., Mandelkow E. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer's disease. FEBS Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- 53.Chohan M.O., Haque N., Alonso A., El-Akkad E., Grundke-Iqbal I., Grover A., Iqbal K. Hyperphosphorylation-induced self assembly of murine tau: a comparison with human tau. J Neural Transm. 2005;112:1035–1047. doi: 10.1007/s00702-004-0241-9. [DOI] [PubMed] [Google Scholar]
- 54.Cruz J.C., Tseng H.C., Goldman J.A., Shih H., Tsai L.H. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 55.Liou Y.C., Sun A., Ryo A., Zhou X.Z., Yu Z.X., Huang H.K., Uchida T., Bronson R., Bing G., Li X., Hunter T., Lu K.P. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 56.Mocanu M.M., Nissen A., Eckermann K., Khlistunova I., Biernat J., Drexler D., Petrova O., Schönig K., Bujard H., Mandelkow E., Zhou L., Rune G., Mandelkow E.M. The potential for beta-structure in the repeat domain of Tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adams S.J., Crook R.J., DeTure M., Randle S.J., Innes A.E., Yu X.Z., Lin W.L., Dugger B.N., McBride M., Hutton M., Dickson D.W., McGowan E. Overexpression of wild-type murine tau results in progressive tauopathy and neurodegeneration. Am J Pathol. 2009;175:1598–1609. doi: 10.2353/ajpath.2009.090462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leroy K., Yilmaz Z., Brion J.P. Increased level of active GSK-3beta in Alzheimer's disease and accumulation in argyrophilic grains and in neurons at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. 2007;33:43–55. doi: 10.1111/j.1365-2990.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- 59.Pei J.J., Tanaka T., Tung Y.C., Braak E., Iqbal K., Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- 60.Ishizawa T., Sahara N., Ishiguro K., Kersh J., McGowan E., Lewis J., Hutton M., Dickson D.W., Yen S.H. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am J Pathol. 2003;163:1057–1067. doi: 10.1016/S0002-9440(10)63465-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lesort M., Jope R.S., Johnson G.V. Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J Neurochem. 1999;72:576–584. doi: 10.1046/j.1471-4159.1999.0720576.x. [DOI] [PubMed] [Google Scholar]
- 62.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B.C., Christie M.J., Napier I.A., Eckert A., Staufenbiel M., Hardeman E., Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 63.Adams S.J., DeTure M.A., McBride M., Dickson D.W., Petrucelli L. Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. PLoS One. 2010;5:e10810. doi: 10.1371/journal.pone.0010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greenwood J.A., Scott C.W., Spreen R.C., Caputo C.B., Johnson G.V.W. Casein kinase II preferentially phosphorylates human tau isoforms containing an amino-terminal insert: Identification of threonine 39 as the primary phosphate acceptor. J Biol Chem. 1994;269:4373–4380. [PubMed] [Google Scholar]
- 65.Hanger D.P., Byers H.L., Wray S., Leung K.Y., Saxton M.J., Seereeram A., Reynolds C.H., Ward M.A., Anderton B.H. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J Biol Chem. 2007;282:23645–23654. doi: 10.1074/jbc.M703269200. [DOI] [PubMed] [Google Scholar]
- 66.Saitoh T., Iimoto D. Aberrant protein phosphorylation and cytoarchitecture in Alzheimer's disease. Prog Clin Biol Res. 1989;317:769–780. [PubMed] [Google Scholar]
- 67.Lee G., Thangavel R., Sharma V.M., Litersky J.M., Bhaskar K., Fang S.M., Do L.H., Andreadis A., Van Hoesen G., Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feijoo C., Campbell D.G., Jakes R., Goedert M., Cuenda A. Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38 delta at Thr50 promotes microtubule assembly. J Cell Sci. 2005;118:397–408. doi: 10.1242/jcs.01655. [DOI] [PubMed] [Google Scholar]
- 69.Rosenberg K.J., Ross J.L., Feinstein H.E., Feinstein S.C., Israelachvili J. Complementary dimerization of microtubule-associated tau protein: implications for microtubule bundling and tau-mediated pathogenesis. Proc Natl Acad Sci USA. 2008;105:7445–7450. doi: 10.1073/pnas.0802036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mandelkow E.M., Schweers O., Drewes G., Biernat J., Gustke N., Trinczek B., Mandelkow E. Structure, microtubule interactions, and phosphorylation of tau protein. Ann N Y Acad Sci. 1996;777:96–106. doi: 10.1111/j.1749-6632.1996.tb34407.x. [DOI] [PubMed] [Google Scholar]
- 71.Carmel G., Mager E.M., Binder L.I., Kuret J. The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 72.Binder L.I., Guillozet-Bongaarts A.L., Garcia-Sierra F., Berry R.W. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 73.Weaver C.L., Espinoza M., Kress Y., Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21:719–727. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 74.Gamblin T.C., Chen F., Zambrano A., Abraha A., Lagalwar S., Guillozet A.L., Lu M., Fu Y., Garcia-Sierra F., LaPointe N., Miller R., Berry R.W., Binder L.I., Cryns V.L. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rissman R.A., Poon W.W., Blurton-Jones M., Oddo S., Torp R., Vitek M.P., LaFerla F.M., Rohn T.T., Cotman C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Delobel P., Lavenir I., Fraser G., Ingram E., Holzer M., Ghetti B., Spillantini M.G., Crowther R.A., Goedert M. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol. 2008;172:123–131. doi: 10.2353/ajpath.2008.070627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pacheco C.D., Elrick M.J., Lieberman A.P. Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum Mol Genet. 2009;18:956–965. doi: 10.1093/hmg/ddn423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cripps D., Thomas S.N., Jeng Y., Yang F., Davies P., Yang A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-tau is polyubiquitinated through Lys-48: Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–10838. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]