

Abstract
Superior insights into molecular mechanisms of liver failure, which are not fully understood, will help strategies for inducing liver regeneration. We examined hepatotoxic mechanisms in mice homozygous for the severe combined immune deficiency mutation in the protein kinase, DNA-activated, catalytic polypeptide. Mice were treated with rifampicin, phenytoin, and monocrotaline. The ensuing acute liver failure was characterized by serological, histological, and mRNA studies. Subsequently, we studied whether transplantation of hepatocytes could rescue animals with liver failure. We found extensive liver damage in these animals, with mortality over several days. The expression of multiple hepatic genes was rapidly altered, including those representing pathways in oxidative/metabolic stress, inflammation, DNA damage-repair, and ataxia telangiectasia mutant (Atm) signaling pathways. This led to liver cell growth arrest involving cyclin-dependent kinase inhibitor 1A. Transplantation of hepatocytes with microcarriers in the peritoneal cavity efficiently rescued animals with liver failure. Molecular abnormalities rapidly reversed, including in hepatic Atm and downstream signaling pathways; and residual hepatocytes overcame cyclin-dependent kinase inhibitor 1A–induced cell growth arrest. Reseeding of the liver with transplanted hepatocytes was not required for rescue because native hepatocytes overcame cell growth-arrest to regenerate the liver. This likely resulted from paracrine signaling from hepatocytes in the peritoneal cavity. We concluded that Atm signaling played critical roles in the pathological features of liver failure. These studies should help redirect examination of pathophysiologic and therapeutic mechanisms in liver failure.
Drug-induced liver injury (DILI) often results in acute liver failure (ALF).1 Acetaminophen (APAP) is the leading offender in causing ALF; however, despite extensive work, mechanisms of DILI by APAP are incompletely understood.2–5 The identification of molecular pathways initiating or amplifying DILI will be highly significant for drug development and for preventing and/or treating ALF. After exposure to hepatotoxic drugs, perturbations in cell stress and toxicity pathways assume prominence. However, more information is required regarding intracellular signaling pathways that impart susceptibility to DILI. Similarly, more information is required regarding failure of liver regeneration in ALF. Nevertheless, establishing these mechanisms has been difficult in the available animal models of ALF. For instance, after exposure to toxic drugs or chemicals, some animals may exhibit rapid and irreversible mortality (eg, within 24–30 hours after APAP), whereas other animals may recover spontaneously. Therefore, animal models more faithfully reproducing the human condition, in which mortality after onset of ALF occurs over much longer periods,1 will be invaluable.
Recently, studies of DILI in healthy C57BL/6 mice showed that the antitubercular drug, rifampicin (Rif), the anticonvulsant agent, phenytoin (Phen), and the plant-derived pyrrolizidine alkaloid, monocrotaline (MCT), produced liver damage synergistically.6 However, when we sought to reproduce this Rif-Phen-MCT–induced liver injury in mice with severe combined immune deficiency (SCID) mutation in the protein kinase, DNA-activated, catalytic polypeptide (Prkdcscid/J), we instead observed ALF, with mortality over several days. Characterization of the nature of this liver injury and analysis of underlying molecular mechanisms demonstrated that strain-specific regulation of gene expression accounted for differences in outcomes of Rif-Phen-MCT–induced hepatotoxicity in C57BL/6 mice versus these NOD/SCID (C3H/He) mice. Studies of cell stress and toxicity pathways identified changes in ataxia telangiectasia mutant (Atm) signaling in ALF. Depletion of Atm after Rif-Phen-MCT led to alterations in downstream signaling, including DNA damage/repair pathways, which culminated in cyclin-dependent kinase inhibitor 1A (p21)–mediated liver cell growth arrest and accounted for impairment in liver regeneration. Previously, these intracellular cell signaling and hepatocellular growth-arrest mechanisms had not been identified in DILI.2–5
Recapitulation of multiple aspects of ALF in humans, including mortality several days after DILI, permitted us to examine therapeutic mechanisms. This was important because mortality in people with ALF remains extremely high (ie, 50%-70%). Orthotopic liver transplantation (OLT) constitutes the only definitive treatment for ALF. However, severe shortages of donor livers and technical complexities restrict OLT. Therefore, alternatives, such as cell therapy, are of much interest.7 Conceptually, if cell therapy would prolong life through provision of hepatic support, people with ALF could be bridged to OLT. On the other hand, if reseeding of the liver with healthy transplanted cells would promote liver regeneration, OLT could be avoided altogether. Despite multiple cell transplantation studies in various models of ALF, these key mechanisms had not been effectively addressed.8
Previous studies8,9 established that transplantation of hepatocytes into hepatic sinusoids was most effective for survival and function of transplanted cells in the liver. However, because the capacity of the hepatic vascular bed for transplanted cells is small,10 this posed questions about the feasibility of significant hepatic support by this method. Cell transplantation in liver sinusoids may even be undesirable in ALF because occlusion of blood flow by transplanted cells might worsen hepatic injury.11–14 Moreover, transplanted hepatocytes integrate in liver parenchyma over several days and take far longer to proliferate,6,8 casting doubt on the importance of reseeding of the liver in ALF. By contrast, spacious extrahepatic sites (eg, the peritoneal cavity) may accommodate many transplanted cells.9,15,16 With appropriate scaffolds, transplanted hepatocytes may engraft in the peritoneal cavity and express metabolic or synthetic functions. Because NOD/SCID mice with Rif-Phen-MCT–induced ALF tolerated cell transplantation in the liver and peritoneal cavity, we were able to study cell therapy mechanisms in ALF.
Materials and Methods
Drugs, chemicals, and reagents were obtained from Sigma Chemical Co, St Louis, MO.
Animals
The Animal Care and Use Committee of Albert Einstein College of Medicine, Bronx, NY, approved protocols according to the National Research Council's Guide for the Care and Use of Laboratory Animals.17 Male NOD.CB17-Prkdcscid/J (NOD/SCID) mice, aged 6 to 7 weeks; B6.129S7-Gt(ROSA)26Sor/J (Rosa26) mice, and C57BL/6-TgCAG-EGFP/1Osb/J [green fluorescent protein (GFP)] mice were obtained from Jackson Laboratories, Bar Harbor, ME. The NOD.CB17-Prkdcscid/J mice were in a C3H/HeJ background. Fischer 344 (F344) rats and C57BL/6 mice were obtained from the National Cancer Institute–Frederick Animal Production Program, Bethesda, MD. To induce ALF, NOD/SCID mice were treated for 3 days by i.p. injections (in normal saline) of Rif (75 mg/kg) and Phen (30 mg/kg), followed on day 4 by MCT (100–150 mg/kg), as previously described.6
Cell Isolation and Transplantation
Hepatocytes were isolated from healthy F344 rats, C57BL/6 mice, GFP mice, or Rosa26 mice by collagenase perfusion of the liver, as previously described.6,8 Cell viability was tested by trypan blue dye exclusion and was 80% or greater. For cell transplantation studies, NOD/SCID mice treated with Rif-Phen-MCT were given 5 × 106 to 50 × 106 cells i.p., along with 1-ml microcarriers (Cytodex 3; Amersham Biosciences Corp, Piscataway, NJ) in a total volume of 1.2 to1.5 ml. In early studies, hepatocytes from C57BL/6 mice were transplanted into NOD/SCID mice with induction of ALF. These animals were studied for reversal of mortality over 2 weeks versus untreated mice with ALF. To determine whether, after i.p. transplantation, donor cells were distributed to various organs, we transplanted GFP transgenic mouse hepatocytes into NOD/SCID mice with ALF, followed by analysis of the presence of transplanted cells in the peritoneal cavity, liver, spleen, and lungs after 24 hours, 48 hours, and 7 days by tissue staining for GFP. To demonstrate the benefits of cell therapy in ALF, we performed studies in NOD/SCID mice with ALF, followed by transplantation of C57BL/6 mouse or F344 rat hepatocytes, ranging from 5 × 106 to 50 × 106 cells. Animal outcomes were analyzed over time, including after early (1–3 days) and later (4 days to 12 weeks) times. In some mice, 2 × 106 Rosa26 cells were transplanted simultaneously via the spleen for deposition into the liver, as previously described.13 Cells were transplanted within 2 hours after isolation. Multiple groups of animals were established, as indicated in individual studies, including control mice treated with vehicle and microcarriers alone. All analyses were with groups of three to six mice or more. Untreated control mice were included as necessary.
Quantitative Real-Time PCR
RNA was extracted from three animals per experimental condition. Total RNA was extracted by Trizol solution (Invitrogen Corp, Carlsbad, CA), cleaned with a kit (RNeasy Kit), and treated with DNase (Qiagen Corp, Valencia, CA). cDNA was prepared from 1 μg of total RNA with a kit (First Strand cDNA Synthesis Kit; C-01, SA Biosciences, Frederick, MD). The expression of 84 genes was studied by RT2 (Real-Time SYBR Green PCR Array; Mouse Stress and Toxicity Pathway Finder, SA Biosciences, Frederick, MD) in an instrument (ABI 7000; Applied Biosystems, Foster City, CA). Amplifications were in 25 μL, with denaturation for 10 minutes at 95°C, 40 cycles for 15 seconds at 95°C, and annealing for 60 seconds at 60°C. Gene expression was normalized to β-actin in each sample before comparisons. Threshold cycle values were determined with computer software (ABI Prism 7000 SDS software). Fold changes in gene expression were analyzed as follows: 2^(−ΔΔThreshold Cycle). Genes of interest were selected based on a 2-fold change or greater, either upward or downward, with P < 0.05 in group comparisons. Curated gene pathways of interest were mapped by software (PathwayStudio5.0; Ariadne Genomics, Rockville, MD).
Cytochrome P450 3a4 Expression
Tissue samples were dounce homogenized in 0.25-mol/L sucrose, 10-mmol/L Tris hydrochloride (pH 7.5), and protease inhibitor cocktail (Calbiochem 539134; EMD Biosciences Inc, San Diego, CA). Lysates were centrifuged at 2000 × g for 25 minutes and then at 100,000 × g for 1 hour at 4°C. Pellets were sonicated, and 75 μg of total proteins was separated in 10% SDS–polyacrylamide gel electrophoresis. After transfer, nitrocellulose membranes were incubated for 1 hour with anti–cytochrome P450 (CYP) 3A4 (1:4000, A4100; Xenotech, Lenexa, KA), followed by peroxidase-conjugated anti–rabbit IgG (1:5000, 321804; Amersham Biosciences, Piscataway, NJ) for 1 hour for enzymatic chemiluminescence. Blots were stained with ponceau red to verify equivalent protein loading.
Histological Studies
Liver samples were fixed in 10% formalin, and sections were stained with hematoxylin-eosin. Tissues were frozen in methylbutane to −80°C for 5-μm-thick cryosections. Bacterial β-galactosidase (LacZ), glycogen, and glucose-6-phosphatase were stained histochemically, as previously described.6,18 Immunostainings were for p21 (1:50, sc-397; Santa Cruz Biotechnology Inc, Santa Cruz, CA), proliferation-related Ki-67 antigen (Ki-67) (1:1000, VP-K451; Vector Labs Inc, Burlingame, CA), heme oxygenase 1 (1:50, AB128; Chemicon International Inc, Temecula, CA), glutathione-S-transferase 1 (1:200, primary antibody in rabbit from Dr I. Listowski, Albert Einstein College of Medicine), ATM (1:150, AB370; Chemicon International Inc), oxidative DNA adducts on guanine residues (4359-MC-100; Trevigen Inc, Gaithersburg, MD), and CD11b (1:100 phycoerythrin-conjugated antibody, 553310; BD Pharmingen, San Diego, CA). Peroxidase-conjugated goat anti-mouse or anti-rabbit Igs (1:500 to 1:600 of No. 3682 and AO545, respectively; Sigma Chemical Co) were used with color development by diaminobenzidine (K3465; Dako Corp, Carpinteria, CA). In negative controls, primary antibodies were omitted. For Ki-67, the positive control was liver 40 hours after two-thirds partial hepatectomy. Apoptosis was shown with a kit (ApopTag Peroxidase In Situ Kit; Chemicon International Inc). For GFP staining, tissues were equilibrated in 30% sucrose and then frozen. Cryostat sections were fixed with 4% paraformaldehyde and blocked in 5% goat serum for incubation with rabbit anti-GFP (1:300; Molecular Probes, Eugene, OR) and then incubated with conjugated goat anti-rabbit IgG (Alexa Fluor 488; Molecular Probes). Morphometric quantitation was in multiple sections per animal (n = ≥ 3 per condition). Typically, 25 tissue sections were scored.
Outcomes in Animals with ALF
Mice were observed daily for grading of encephalopathy and for mortality. Encephalopathy was graded as follows: 0, normal behavior of animals; 1, decreased motor activity and lethargy; 2, sedated appearance and immobility or reticence to move in cages; and 3, comatose condition. The duration of survival was monitored for at least 2 weeks. Animal studies extended up to 12 weeks, with continued observation and blood and tissue sampling from some mice after 4, 8, and 12 weeks. Some animals were sacrificed for interval studies (n = 3–6), as described later.
Serological Assays
Serum samples were stored at −20°C for alanine aminotransferase (ALT), alkaline phosphatase, and total bilirubin measurements in automated clinical systems. Prothrombin time was measured by thromboplastin (Plastinex, 101158; Bio/Data Corp, Horsham, PA) with blood in 0.11-mol/L sodium citrate. Plasma was diluted in water (7:3, v/v). To 0.1-ml plasma, 0.2-ml thromboplastin reconstituted in water and preincubated with plasma at 37°C for 5 minutes was added and time to clotting was measured.
Statistical Analyses
Data are shown as mean ± SD. Differences were analyzed by t-test, χ2 test, analysis of variance with Holm-Sidak pairwise comparisons, or log-rank tests in survival curves by computer software (SigmaStat 3; Systat Software Inc, Point Richmond, CA). P < 0.05 was considered significant.
Results
In the first set of studies, we characterized ALF in NOD/SCID mice. This was followed by cell transplantation studies in the next part to demonstrate mechanisms in the rescue of animals with ALF.
Characterization of ALF
We observed dose-dependent DILI in mice given fixed amounts of Rif (75 mg/kg) and Phen (30 mg/kg), followed by 100-, 125-, or 150-mg/kg MCT. The NOD/SCID mice tolerated Rif, Phen, and 100-mg/kg MCT without overt hepatotoxicity or liver necrosis. However, severe liver injury was evident after Rif plus Phen, followed by either 125- or 150-mg/kg MCT. Serum ALT and total bilirubin levels in mice treated with Rif, Phen, and 125-mg/kg MCT (n = 30) or Rif, Phen, and 150-mg/kg MCT (n = 30) showed significant changes compared with vehicle-treated mice (n = 20) (Figure 1, A and B). Serum ALT and total serum bilirubin levels were 58 ± 3 U/L and 0.3 ± 0.1 mg/dL, respectively, in control mice. By contrast, in mice 1, 2, 3, and 6 to 8 days after Rif, Phen, and 150-mg/kg MCT, serum ALT levels were 2350 ± 180, 5500 ± 670, 6380 ± 689, and 4689 ± 300 U/L, respectively; and serum bilirubin levels were 1.1 ± 0.5, 0.8 ± 0.4, 3.0 ± 1.0, and 2.5 ± 0.7 mg/dL, respectively (P < 0.05, analysis of variance with Holm-Sidak tests). Prothrombin time was prolonged by 2- to 4-fold above normal in drug-treated mice, reflecting severe liver injury. Virtually all mice exhibited encephalopathy 6 days after drugs, which was worse in mice given 150-mg/kg MCT (Figure 1C). Most mice were in grade 3 to 4 encephalopathy before death. As expected, vehicle-treated control mice remained alive and well, whereas extensive mortality was observed in drug-treated mice (Figure 1D). Analysis of mortality showed that after Rif, Phen, and 150-mg/kg MCT, 50% died in 5.5 days and 100% died in 7 days (P < 0.05, log-rank tests versus controls). The duration of survival was slightly longer after Rif, Phen, and 125-mg/kg MCT, with 40% dying by 5.5 days, 67% dying by 7 days, and 87% dying by 14 days (P < 0.05 versus controls and P > 0.05 versus 150-mg/kg MCT). In view of slightly longer survival in mice treated with 125-mg/kg MCT, we performed further studies of animals under this condition. Liver morphological features in control mice were normal (Figure 1E), although drug-treated mice showed extensive liver necrosis; 1 day after Rif, Phen, and 125-mg/kg MCT, 30% to 40% of the liver parenchyma was destroyed (Figure 1F). After 4 days, 50% to 70% of the liver parenchyma was lost (Figure 1G). This parenchymal loss continued, with 50% to 70% hepatic necrosis before death, typically after 6 to 9 days (Figure 1H).
Figure 1.
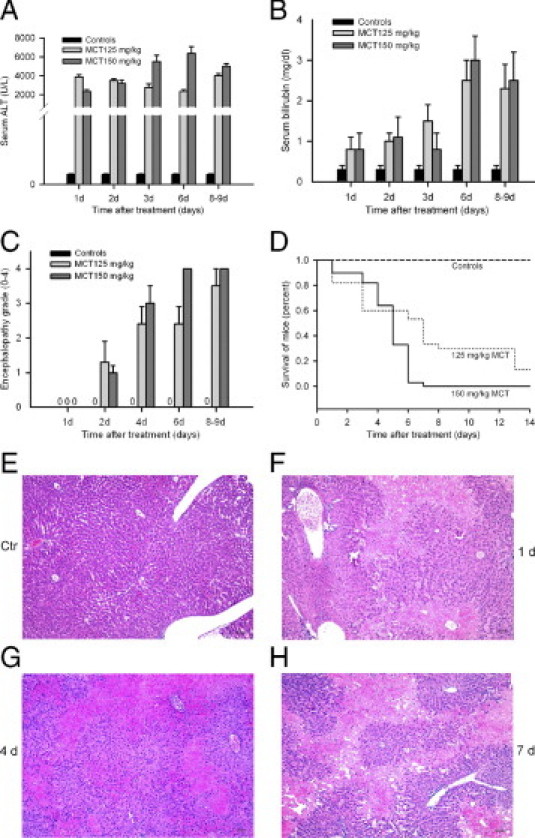
Hepatic injury in NOD/SCID mice. Vehicle-treated control mice and mice treated with Rif and Phen for 3 days, followed by either 125- or 150-mg/kg MCT, are shown. A: Serum ALT levels. B: Serum bilirubin levels. C: Grade of encephalopathy. D: Survival curves in animals. E through H: Normal liver histological features in a vehicle-treated control mouse (E), whereas mice 1 day (F), 4 days (G), and 7 days (H) after Rif, Phen, and 125-mg/kg MCT showed loss of liver parenchyma (pink areas without blue nuclei). Original magnification, ×100. Scale bar = 100 μm (hematoxylin-eosin stain).
The course of morphological changes agreed with abnormalities in liver test results, including prolongation of prothrombin time, encephalopathy, and mortality. We did not observe proliferation of bile duct cells or appearance of other liver cell subpopulations (eg, oval cells), indicating that liver injury did not activate putative hepatic stem cells. The liver was not grossly infiltrated with inflammatory cells (eg, neutrophils, macrophages, eosinophils, or basophils), which represented the innate immune system. The NOD/SCID mice lacked T and B lymphocytes.
To determine the effects of Rif-Phen-MCT on liver cell turnover, we studied apoptosis and cell proliferation in multiple liver sections (Figure 2). In healthy control mice, only 0 to 1 apoptotic cell per liver section was found (Figure 2A). Apoptosis increased slightly in drug-treated mice, with 5 to 8 cells per section (Figure 2, C, E, and G). This mostly nonapoptotic injury was in agreement with extensive tissue necrosis. In healthy livers, we observed <0.01% cells with Ki-67, indicating little cell turnover (Figure 2B). By contrast, Ki-67 expression increased extensively after partial hepatectomy, as expected (Figure 2D). In drug-treated mice, hepatocytes in periportal areas expressed Ki-67 (Figure 2, F and H). Morphometry showed Ki-67 expression in 4% ± 3% of hepatocytes in mice 1 to 3 days after ALF and in 7% ± 2% of hepatocytes in mice 4 to 8 days after ALF (P < 0.05). This extent of hepatocellular proliferation was insufficient for restoring the liver and rescuing animals from ALF in most cases.
Figure 2.
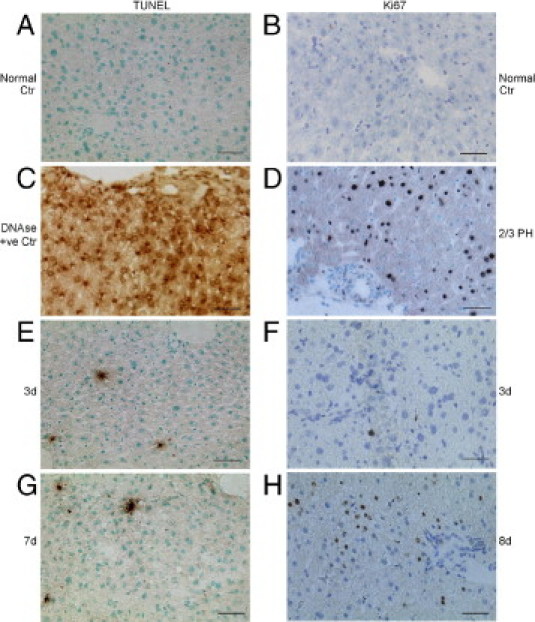
Hepatic apoptosis and proliferation after drug-induced damage in NOD/SCID mice. The terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assays (A, C, E, and G) and Ki-67 immunostaining (B, D, F, and H) are shown. A and B: Normal liver with absence of TUNEL or Ki-67 expression. C: The DNase-treated liver as a positive control for TUNEL. D: Mouse liver 40 hours after partial hepatectomy showing widespread Ki-67 expression in hepatocytes. E and F: Infrequent TUNEL-positive cells (E) and hepatocytes with Ki-67 (F) 3 days after Rif, Phen, and 125-mg/kg MCT. G: Infrequent TUNEL-positive cells 7 days after Rif, Phen, and MCT. H: Ki-67 expression in periportal hepatocytes 8 days after Rif, Phen, and MCT. Original magnification, ×200. Scale bar = 100 μm (left, methylgreen counterstain; right, toluidine blue counterstain).
Liver Gene Expression in Healthy NOD/SCID and Healthy C57BL/6 Mice
To examine the basis for differences in DILI in NOD/SCID and C57BL/6 mice, we analyzed genes in stress and toxicity pathways. Of 84 such genes, expression of 15 (18%) was 2-fold or more higher and expression of 47 (56%) was 2-fold or more lower in NOD/SCID mice versus C57BL/6 mice (Table1).
Table 1.
Differences in Baseline Expression Levels of Genes in Healthy NOD/SCID Mice Versus C57BL/6 Mice
| Gene description | Gene symbol | Expression level for NOD/SCID/C57/BL6 |
|---|---|---|
| Oxidative or metabolic stress | ||
| Up-regulated | ||
| Cytochrome P450, family 2, subfamily a, polypeptide 5 | Cyp2a5 | 741.00 |
| Cytochrome P450, family 2, subfamily b, polypeptide 10 | Cyp2b10 | 205.00 |
| Cytochrome P450, family 2, subfamily b, polypeptide 9 | Cyp2b9 | 46.00 |
| Cytochrome P450, family 2, subfamily c, polypeptide 29 | Cyp2c29 | 39.00 |
| Flavin-containing monooxygenase 4 | Fmo4 | 2.30 |
| Glutathione-S-transferase, μ3 | Gstm3 | 3.38 |
| Superoxide dismutase 2, mitochondrial | Sod2 | 177.00 |
| Down-regulated | ||
| Cytochrome P450, family 1, subfamily a, polypeptide 1 | Cyp1a1 | 0.14 |
| Cytochrome P450, family 1, subfamily b, polypeptide 1 | Cyp1b1 | 0.02 |
| Cytochrome P450, family 3, subfamily a, polypeptide 11 | Cyp3a11 | 0.15 |
| Cytochrome P450, family 4, subfamily a, polypeptide 10 | Cyp4a10 | 0.20 |
| Cytochrome P450, family 4, subfamily a, polypeptide 14 | Cyp4a14 | 0.01 |
| Flavin-containing monooxygenase 1 | Fmo1 | 0.21 |
| Flavin-containing monooxygenase 5 | Fmo5 | 0.33 |
| Glutathione peroxidase 1 | Gpx1 | 0.12 |
| Glutathione reductase | Gsr | 0.29 |
| Glutathione-S-transferase, 1 | Gstm1 | 0.20 |
| Heme oxygenase (decycling) 2 | Hmox2 | 0.16 |
| Superoxide dismutase 1, soluble | Sod1 | 0.00 |
| Heat shock | ||
| Up-regulated | ||
| Heat shock protein 41 | Hspa4 | 7.90 |
| Down-regulated | ||
| DnaJ (Hsp40) homolog, subfamily A, member 1 | Dnaja1 | 0.10 |
| Hsp 1 | Hspa1b | 0.00 |
| Hsp 1–like | Hspa1l | 0.09 |
| Hsp 5 | Hspa5 | 0.16 |
| Hsp 8 | Hspa8 | 0.30 |
| Hsp β-1 | Hspb1 | 0.03 |
| Hsp 1 (chaperonin) | Hspd1 | 0.19 |
| Hsp 1 (chaperonin 10) | Hspe1 | 0.35 |
| Heat shock transcription factor 1 | Hsf1 | 0.23 |
| Proliferation and carcinogenesis | ||
| Up-regulated | ||
| Cyclin C | Ccnc | 16.33 |
| Cyclin D1 | Ccnd1 | 20.25 |
| Down-regulated | ||
| Colony-stimulating factor 2 (granulocyte-macrophage) | Csf2 | 0.06 |
| E2F transcription factor 1 | E2f1 | 0.04 |
| Early growth response 1 | Egr1 | 0.50 |
| Insulinlike growth factor binding protein 6 | Igfbp6 | 0.33 |
| Growth arrest and senescence | ||
| Down-regulated | ||
| Cyclin-dependent kinase inhibitor 1A (p21) | Cdkn1a | 0.01 |
| Transformed mouse 3T3 cell double minute 2 | Mdm2 | 0.09 |
| Tumor protein p53 | Trp53 | 0.46 |
| Inflammation | ||
| Up-regulated | ||
| Interleukin 1β | Il1b | 2.19 |
| Down-regulated | ||
| Chemokine (C-C motif) ligand 3 | Ccl3 | 0.10 |
| Chemokine (C-C motif) ligand 4 | Ccl4 | 0.05 |
| Chemokine (C-C motif) ligand 21 | Ccl21b | 0.07 |
| Interleukin 6 | Il6 | 0.12 |
| Nuclear factor of κ light polypeptide gene enhancer in B-cell inhibitor, α | Nfkbia | 0.34 |
| Nitric oxide synthase 2, inducible, macrophage | Nos2 | 0.07 |
| Serine (or cysteine) peptidase inhibitor, clade E, member 1 | Serpine1 | 0.49 |
| DNA damage and repair | ||
| Up-regulated | ||
| RAD50 homolog (Saccharomyces cerevisiae) | Rad50 | 3.54 |
| Down-regulated | ||
| Ataxia telangiectasia mutated homolog (human) | Atm | 0.50 |
| Cyclin G1 | Ccng1 | 0.50 |
| DNA damage–inducible transcript 3 | Ddit3 | 0.29 |
| Excision repair cross complementing rodent repair deficiency, complementation group 1 | Ercc1 | 0.22 |
| RAD23 homolog A (S cerevisiae) | Rad23a | 0.41 |
| X-ray repair complementing defective repair in Chinese hamster cells 1 | Xrcc1 | 0.20 |
| X-ray repair complementing defective repair in Chinese hamster cells 4 | Xrcc4 | 0.42 |
| Apoptosis signaling | ||
| Up-regulated | ||
| Bcl2–like-1 | Bcl2l1 | 4.20 |
| Caspase 8 | Casp8 | 19.65 |
| Tumor necrosis factor receptor superfamily, member 1a | Tnfrsf1a | 7.00 |
| Down-regulated | ||
| Annexin A5 | Anxa5 | 0.16 |
| Bcl2-associated X protein | Bax | 0.10 |
| Caspase 1 | Casp1 | 0.16 |
| Lymphotoxin α | Lta | 0.08 |
| TNFRSF1A associated via death domain | Tradd | 0.13 |
NOD, natural-onset diabetes; SCID, severe combined immunodeficiency; Hsp, heat shock protein.
Several Cyp genes were expressed more in NOD/SCID mice compared with C57BL/6 mice (Cyp2a5, Cyp2b10, Cyp2b9, and Cyp2c29), whereas other Cyp genes were expressed at lower levels in NOD/SCID mice (Cyp1a1, Cyp1b1, Cyp3a11, Cyp4a10, and Cyp4a14), which reflected differences in metabolic activity or stress and toxicity conditions (Table 1). Similarly, genes involved in oxidative stress were either expressed more (eg, superoxide dismutase 2) or less (eg, superoxide dismutase 1) in NOD/SCID mice. Several heat shock protein (Hsp) genes were expressed at lower levels, although Hspa4 was expressed at higher levels in NOD/SCID versus C57BL/6 mice, suggesting less hepatic preconditioning by environmental stresses (eg, lack of microbial exposures under pathogen-free conditions). This seemed consistent with lower expression of inflammation-associated genes in NOD/SCID mice (eg, Il6, nitric oxide synthase, and C-C motif chemokines 3 and 4). By contrast, interleukin-1b was expressed at higher levels in NOD/SCID versus C57BL/6 mice, possibly secondary to T- and B-cell deficiency. Differences in apoptosis genes, some expressed at higher and others at lower levels in NOD/SCID mice, were of unclear significance, given that hepatic apoptosis was absent (Figure 2A).
If NOD/SCID mice were undergoing genotoxic liver injury, expression of relevant genes should have been altered. However, DNA damage/repair genes were expressed 2- to 5-fold less in NOD/SCID mice, including Atm, cyclin G, Chek 2, CHK2 checkpoint homolog 1, DNA damage–inducible transcript 3 (Ddit3), excision repair cross-complementing rodent repair deficiency, complementation group 1, RAD23 homolog A (Saccharomyces cerevisiae), X-ray repair complementing defective repair in Chinese hamster cells 1 (Xrcc1), and Xrcc4 genes (Table 1). This agreed with lower basal hepatic genotoxicity in NOD/SCID mice compared with C57BL/6 mice. The significance of Atm expression became apparent in further studies (explained later). Among this group of DNA damage/repair genes, only RAD50 homolog (S cerevisiae), which repairs double-strand DNA breaks, was expressed at 3.5-fold higher levels in NOD/SCID mice. Cell growth-arrest or senescence genes [eg, Cdkn1a (p21), transformed mouse 3T3 cell double minute 2 (Mdm2), and transformation-related protein 53] were 2- to 100-fold lower in NOD/SCID mice, which was in agreement with less cell damage. Because cyclin C and cyclin D1 levels were 16- and 20-fold higher, respectively, we considered hepatocytes were in G0/G1 (ie, their expected state) in NOD/SCID mice. This agreed with absence of hepatic Ki-67 expression (Figure 2B) in NOD/SCID mice because Ki-67 characterizes cells in late G1, S, or G2/M.
Despite differences in stress and toxicity pathways, including several Cyp genes, after Rif and Phen for 3 days in NOD/SCID mice, Cyp3a4 protein was induced (Figure 3), which metabolizes MCT to toxic intermediates.6 This was similar to Cyp3a4 induction in C57BL/6J mice.
Figure 3.
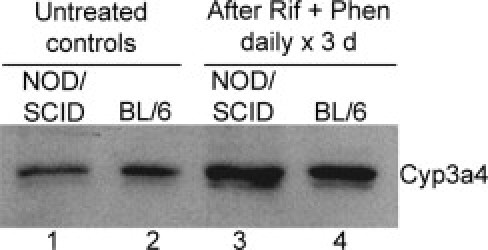
Induction of hepatic Cyp3a4 by Rif and Phen for 3 days in mice. Western blot showing Cyp3a4 in NOD/SCID and C57BL/6 mice: lanes 1 and 2, drug-untreated controls; and lanes 3 and 4, after drug treatments.
Taken together, these findings indicated that, although the baseline pattern of hepatic gene expression in NOD/SCID mice differed from C57BL/6 mice, we did not find evidence for greater ongoing hepatic DNA damage or repair in the former.
Molecular Changes During Rif-Phen-MCT–Induced Hepatic Injury in NOD/SCID Mice
To determine hepatic stress and toxicity pathway genes in NOD/SCID mice after drugs, we used quantitative real-time RT-PCR gene arrays, with healthy NOD/SCID mice as controls. Analysis of tissues from NOD/SCID mice 1, 3, and 7 days after Rif, Phen, and 125-mg/kg MCT showed that, of 84 total genes, 28 to 37 (33%–44%) were expressed at 2-fold higher or lower levels (or more), compared with baseline levels in control mice (Table 2).
Table 2.
Gene Expression Levels in NOD/SCID Mice Treated by Rif-Phen-MCT Versus Healthy NOD/SCID Mice
| Gene description | Gene symbol | NOD/SCID baseline | Time after drugs, days |
||
|---|---|---|---|---|---|
| 1 | 3 | 7 | |||
| Oxidative or metabolic stress | |||||
| Up-regulated | |||||
| Cytochrome P450, family 2, subfamily b, polypeptide 10 | Cyp2b10 | 1.0 | 1.2⁎ | 5.0 | 1.1⁎ |
| Cytochrome P450, family 3, subfamily a, polypeptide 11 | Cyp3a11 | 1.0 | 2.8 | 1.8⁎ | 3.4 |
| Glutathione peroxidase 2 | Gpx2 | 1.0 | 4.2 | 2.0 | 4.1 |
| Glutathione-S-transferase, μ3 | Gstm3 | 1.0 | 15.7 | 3.7 | 16.0 |
| Heme oxygenase (decycling) 1 | Hmox1 | 1.0 | 0.5⁎ | 5.2 | 0.5⁎ |
| Down-regulated | |||||
| Cytochrome P450, family 1, subfamily b, polypeptide 1 | Cyp1b1 | 1.0 | 0.3 | 0.7⁎ | 0.3 |
| Cytochrome P450, family 2, subfamily a, polypeptide 5 | Cyp2a5 | 1.0 | 0.3 | 0.1 | 0.3 |
| Cytochrome P450, family 2, subfamily b, polypeptide 9 | Cyp2b9 | 1.0 | 0.0 | 0.0 | 0.0 |
| Cytochrome P450, family 4, subfamily a, polypeptide 10 | Cyp4a10 | 1.0 | 0.1 | 0.1 | 0.1 |
| Cytochrome P450, family 4, subfamily a, polypeptide 14 | Cyp4a14 | 1.0 | 0.1 | 0.0 | 0.1 |
| Cytochrome P450, family 7, subfamily a, polypeptide 1 | Cyp7a1 | 1.0 | 0.1 | 0.0 | 0.1 |
| Flavin-containing monooxygenase 1 | Fmo1 | 1.0 | 0.8⁎ | 0.2 | 0.8⁎ |
| Flavin-containing monooxygenase 4 | Fmo4 | 1.0 | 0.4 | 0.2 | 0.4 |
| Heat shock | |||||
| Up-regulated | |||||
| Hsp 1 | Hspb1 | 1.0 | 3.5 | 27.4 | 3.6 |
| Hsp 1–like | Hspa1l | 1.0 | 2.0 | 0.4 | 2.4 |
| Hsp 8 | Hspa8 | 1.0 | 0.6⁎ | 2.4 | 0.0 |
| Down-regulated | |||||
| DnaJ (Hsp40) homolog, subfamily A, member 1 | Dnaja1 | 1.0 | 0.3 | 0.4 | 0.3 |
| Hsp 1 (chaperonin) | Hspd1 | 1.0 | 0.0 | 0.5 | 0.3 |
| Hsp 1 (chaperonin 10) | Hspe1 | 1.0 | 0.4 | 0.7 | 0.5 |
| Proliferation and carcinogenesis | |||||
| Up-regulated | |||||
| E2F transcription factor 1 | E2f1 | 1.0 | 2.4 | 0.7⁎ | 0.5⁎ |
| Down-regulated | |||||
| Cyclin D1 | Ccnd1 | 1.0 | 0.6⁎ | 0.1 | 0.5⁎ |
| Early growth response 1 | Egr1 | 1.0 | 0.3 | 9.9 | 0.2 |
| Growth arrest and senescence | |||||
| Up-regulated | |||||
| Cyclin-dependent kinase inhibitor 1A (p21) | Cdkn1a | 1.0 | 277.0 | 434.0 | 277.0 |
| DNA damage–inducible transcript 3 | Ddit3 | 1.0 | 1.2⁎ | 11.5 | 1.3⁎ |
| Growth arrest and DNA damage–inducible 45α | Gadd45a | 1.0 | 0.8⁎ | 74.0 | 1.0⁎ |
| Transformed mouse 3T3 cell double minute 2 | Mdm2 | 1.0 | 9.3 | 4.4 | 6.2 |
| Inflammation | |||||
| Up-regulated | |||||
| Chemokine (C-X-C motif) ligand 10 | Cxcl10 | 1.0 | 0.7⁎ | 3.1 | 0.9⁎ |
| Interleukin 6 | Il6 | 1.0 | 0.8⁎ | 4.5 | 0.6⁎ |
| Nitric oxide synthase 2, inducible, macrophage | Nos2 | 1.0 | 1.0⁎ | 5.7 | 0.8⁎ |
| Serine (or cysteine) peptidase inhibitor, clade E, member 1 | Serpine1 | 1.0 | 5.4 | 42.2 | 11.1 |
| Down-regulated | |||||
| Interleukin 1β | Il1b | 1.0 | 0.0 | 0.6⁎ | 0.1 |
| Interleukin 18 | Il18 | 1.0 | 0.4 | 0.5⁎ | 0.4 |
| DNA damage and repair | |||||
| Up-regulated | |||||
| Cyclin G1 | Ccng1 | 1.0 | 8.8 | 4.3 | 8.4 |
| Uracil DNA glycosylase | Ung | 1.0 | 1.3⁎ | 2.5 | 1.4⁎ |
| Down-regulated | |||||
| Ataxia telangiectasia mutated homolog (human) | Atm | 1.0 | 0.4 | 0.1 | 0.6⁎ |
| CHK2 checkpoint homolog (Schizosaccharomyces pombe) | Chek2 | 1.0 | 1.5⁎ | 0.2 | 1.7⁎ |
| RAD50 homolog (Saccharomyces cerevisiae) | Rad50 | 1.0 | 0.6⁎ | 0.1 | 0.7⁎ |
| X-ray repair complementing defective repair in Chinese hamster cells 4 | Xrcc4 | 1.0 | 0.5 | 0.2 | 0.5 |
| Apoptosis signaling | |||||
| Up-regulated | |||||
| Annexin A5 | Anxa5 | 1.0 | 2.3 | 1.3⁎ | 2.8 |
| Bcl2-associated X protein | Bax | 1.0 | 3.1 | 0.6⁎ | 3.7 |
| Bcl2–like-1 | Bcl2l1 | 1.0 | 0.3 | 2.1 | 0.7⁎ |
| Down-regulated | |||||
| Fas ligand (TNF superfamily, member 6) | Fasl | 1.0 | 0.1 | 0.2 | 0.1 |
| TNF (ligand) superfamily, member 10 | Tnfsf10 | 1.0 | 0.2 | 0.1 | 0.3 |
| TNFRSF1A-associated via death domain | Tradd | 1.0 | 0.5⁎ | 0.2 | 0.5⁎ |
NOD, natural-onset diabetes; SCID, severe combined immunodeficiency; Rif, rifampicin; Phen, phenytoin; MCT, monocrotaline; NS, not significant; Hsp, heat shock protein; TNF, tumor necrosis factor.
Statistically not significant
After DILI, significant changes occurred in stress and toxicity pathways. For instance, Cyp2b10 expression increased by 5-fold at 3 day and Cyp3a11 expression increased by 3-fold at 1 day. Expression of Cyp2a5, Cyp2b9, and other Cyp genes (1b1, 4a10, 4a14, and 7a1) declined by 3- to 10-fold or more. Undetectable expression of Cyp2b9, 4a14, and 7a1 was in agreement with perivenous necrosis after Rif-Phen-MCT because these genes are normally expressed there. Among metabolic/oxidative stress genes, glutathione peroxidase 2, glutathione-S-transferase 3, and heme oxygenase 1 were expressed 2- to 16-fold more, whereas expression of the flavin monooxygenases (ie, 1 and 4) declined by 2.5- to 5-fold. Some Hsp genes were expressed more, by 2- to 27-fold (eg, Hspb1, Hspa1l, and Hspa8); and other Hsp genes decreased, typically by 2- to 3-fold (Dnaja1, Hspd1, and Hspe1). These findings indicated that Rif-Phen-MCT caused extensive hepatic oxidative and metabolic stress, although regulation or counterregulation of individual genes was clearly complex.
We observed 3- to 42-fold greater expression of inflammation-associated genes, including C-X-C motif ligand 10, Il6, nitric oxide synthase 2, and serine (or cysteine) peptidase inhibitor, clade E, member 1 (Table 2). These genes were expressed at lower levels compared with NOD/SCID mice versus C57BL/6 mice under basal conditions. By contrast, expression of interleukin-1b and interleukin-18 declined after drugs. The apoptosis genes (ie, annexin A5, BclII-associated X protein, and BclII-like-1) were expressed 2- to 4-fold more, whereas Fas ligand, tumor necrosis factor (ligand) superfamily member 10, and tumor necrosis factor receptor type 1–associated death domain protein were expressed 3- to 10-fold less, in the background of limited terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling positivity in liver tissues.
Expression of DNA damage/repair, proliferation, and growth-arrest genes was most remarkable (Table 2). Atm expression decreased by 2- to 10-fold (Table 1). Expression of DNA damage/repair genes [ie, Chek2, RAD50 homolog (S cerevisiae), and Xrcc4] declined by 5- to 10-fold. The consequence of these changes should have been inefficient DNA repair, which was verified by analysis of oxidative DNA adducts (explained later). If DNA damage was significant, cell cycling or cell growth should have been perturbed, which was shown by increased p21 expression (277- to 434-fold), along with cell growth-arrest genes [ie, Ddit3 (11-fold), growth arrest and DNA damage–inducible 45α (74-fold), and Mdm2 (4- to 9-fold)]. The expression of cell cycle proliferation-associated genes (eg, cyclin D1 and Egr1) altered by 3- to 10-fold, including no change, decline, or increase in expression; however, E2f1 expression increased by 2.4-fold after 1 day. This reflected changes in DNA synthesis, as shown by Ki-67 staining at early or later times after drugs (previously described).
We verified the relevance of mRNA level changes by tissue staining. This confirmed hepatic oxidative/metabolic stress because glutathione-S-transferase expression increased (Figure 4, A and B), and extensive oxidative DNA adducts on guanosine residues (Figure 4, C and D) decreased Atm expression (Figure 4, E and F) and increased p21 expression (Figure 4, G and H). These changes were obvious 1 day after Rif-Phen-MCT and persisted subsequently until death of animals over several days.
Figure 4.
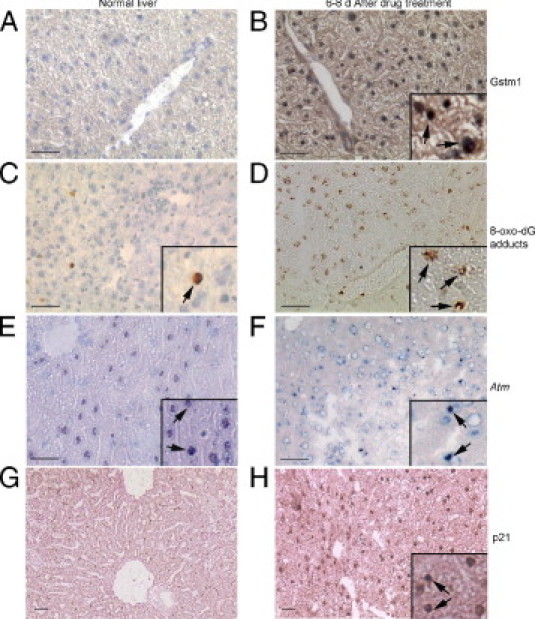
Verification of gene expression changes by tissue staining with diaminobenzidine-based methods in Rif-Phen-MCT–treated mice. A and B: Glutathione-S-transferase 1 (Gstm1) in normal liver, which was lower (A) than in drug-treated mice (B, inset, cytoplasmic plus nuclear Gstm1). C and D: The oxidative DNA adducts on guanine residues in occasional cells in normal liver (C) and in far more cells in drug-treated mice (D, insets, magnified views of cells with adducts, arrows). E and F:Atm in normal liver (E) and in drug-treated mice (F), where fewer hepatocytes expressed Atm and where Atm content seemed lower in cells (arrows, insets). G and H: No p21 expression in normal liver (G) and widespread p21 expression in cell nuclei from drug-treated mice (H, arrows in inset). Original magnification, ×200. Scale bar = 100 μm. A through C, E, and F: Toluidine blue counterstain. G and H: Eosin counterstain.
Mapping of Atm Signaling Pathways
We considered that lower Atm expression associated with perturbations in Atm targets intimately involved in DNA damage/repair and cell growth arrest indicated roles for Atm signaling. Therefore, we mapped genes in curated Atm signaling pathways. This showed perturbations in key limbs of Atm signaling in NOD/SCID mice after Rif-Phen-MCT–induced liver injury versus control NOD/SCID mice (Figure 5, A and B). In drug-treated mice, markedly increased expression of p21 and other cell growth-arrest genes suggested that hepatic DNA damage activated cell cycle checkpoint controls, which was consistent with restriction of hepatocytes in G1, S, or G2/M, according to Ki-67 staining (Figure 2H).
Figure 5.
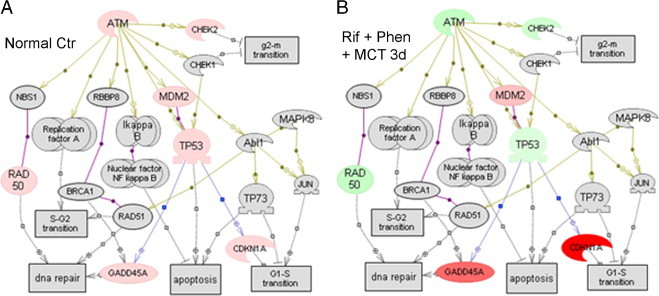
Regulation of Atm pathway genes after Rif-Phen-MCT. A: Expression of genes in normal liver from the array of 84 genes studied (Table 2 describes gene symbols). Genes in gray boxes were absent from the array studied. Genes in pink boxes were expressed. B: Down-regulated genes (green boxes) and up-regulated genes (red boxes) compared with normal liver 3 days after Rif-Phen-MCT. These changes in gene expression were similar 1 or 7 days after drug treatments. In particular, Atm expression declined with evidence for impaired protection against DNA damage, leading to activation of p21 and growth-arrest of damaged cells. Specific pathways in differentially expressed gene lists were mapped (PathwayStudio 5.0; Aridane Genomics, Rockville, MD).
Taking these findings together, we concluded that gene expression under basal and liver injury conditions in NOD/SCID mice was informative regarding the role of Atm signaling in cell growth-arrest in ALF. We then examined cell therapy mechanisms in this mouse model of ALF.
Role of Cell Therapy in Rescuing Animals with ALF and Reversing Molecular Abnormalities
We performed studies to determine whether suitable liver support would permit hepatic repair and regrowth with or without reseeding of the damaged liver with healthy transplanted cells. Because hepatocytes can be transplanted in large numbers in the peritoneal cavity, readily equaling or exceeding those in the liver, we first examined the benefits of graded hepatic support from transplanted cells.
We transplanted 30 × 106 hepatocytes from healthy C57BL/6 donor mice i.p. into some NOD/SCID mice (n = 6) and simultaneously treated mice with microcarriers alone (n = 6) 2 days after Rif-Phen-MCT administration. Mortality in hepatocyte-treated animals declined significantly over 2 weeks compared with control animals (17% versus 83%; P < 0.05).
To verify that transplanted hepatocytes remained in the peritoneal cavity without migrating elsewhere, particularly to the liver, which could have incriminated alternative mechanisms of liver regeneration, we established further groups of NOD/SCID mice 2 days after Rif-Phen-MCT. To localize donor cells, we transplanted i.p. 5 × 106 reporter hepatocytes from GFP donor mice, along with microcarriers. We transplanted GFP donor cells to permit multicolor tissue staining if that were required for characterizing GFP+ cells in organs. Tissue analysis from control NOD/SCID mice, donor transgenic mice, and cell recipients 24 or 48 hours after transplantation (n = 3 each) showed GFP+ transplanted hepatocytes in the peritoneal cavity (Figure 6, A and B). The GFP expression in donor cells was verified in the liver, spleen, and lungs (Figure 6, C, E, and G). However, we did not find GFP+ cells in the liver, spleen, or lungs 1, 2, or 7 days after i.p. cell transplantation (Figure 6, D, F, and H), which excluded migration of intact hepatocytes or redistribution of their fragments by other cells (eg, macrophages).
Figure 6.
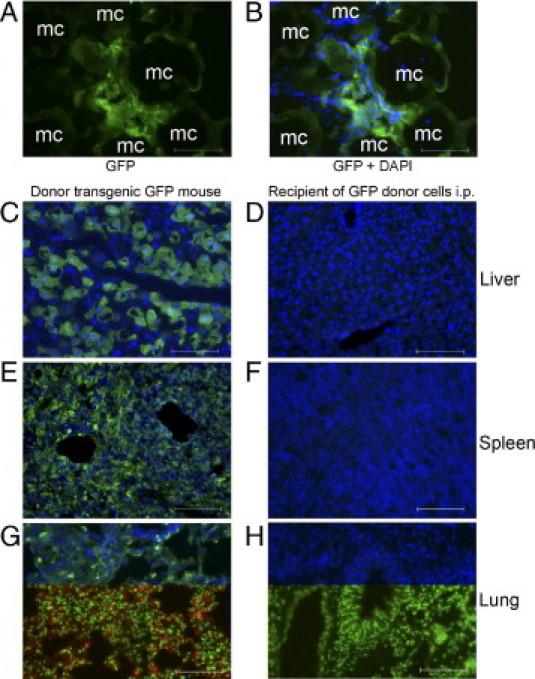
Transplanted cell distributions in liver, spleen, and lungs 24 hours after hepatocyte transplantation with microcarriers in the peritoneal cavity. A and B: Transplanted hepatocytes adjacent to microcarriers (mc) in NOD/SCID mice with GFP immunostaining (green) and merge of GFP plus 4′,6-diamidino-2-phenylindole staining of cell nuclei (blue) (B). C, E, and G: Tissues from donor GFP transgenic mice demonstrating widespread distribution of GFP+ cells in the liver, spleen, and lungs. D, F, and H: Absence of GFP+ cells in the liver, spleen, or lungs. Transplanted cells were also not found in the liver, spleen, and lungs of mice 48 hours or 7 days after transplantation. Original magnification, ×200. Scale bar = 100 μm.
To determine whether transplantation of xenogeneic cells would have produced similar benefits, which should be of great value for characterizing human cell populations, we next used healthy donor F344 rat hepatocytes. This was also experimentally convenient because only a few hepatocytes may be isolated from mice (typically 30 × 106 to 40 × 106 viable cells per mouse). We administered Rif, Phen, and 125-mg/kg MCT, followed 2 days later by i.p. injection of microcarriers alone (n = 20) or 5 × 106, 10 × 106, and 50 × 106 rat hepatocytes with microcarriers (n = 20). This represented transplantation of 0%, 5%, 10%, and 50%, respectively, of the hepatocyte mass in the mouse liver.
All mice with ALF given microcarriers alone died in 2 weeks, whereas mice treated with rat hepatocytes survived, including after transplantation of only 5 × 106 hepatocytes (P < 0.001, log-rank test) (Figure 7A). Therefore, extrahepatic liver support was sufficient for rescuing animals with ALF. We found that mice were healthy 1 day after cell therapy, whereas control mice treated by microcarriers alone worsened with encephalopathy, indicating serious hepatic insufficiency (Figure 7B). In animals with cell therapy, serum bilirubin levels decreased 1, 2, 3, 6, and 8 days after Rif-Phen-MCT to 0.9 ± 0.2, 1.0 ± 0.2, 0.8 ± 0.3, 0.7 ± 0.5, and 0.8 ± 0.5 mg/dL, respectively (Figure 7C). In control mice given microcarriers alone, the prothrombin time was 2.8 ± 0.4-fold and 2.9 ± 0.6-fold above normal after 6 and 8 days, respectively (Figure 7D). By contrast, prothrombin time after cell therapy improved and was only 1.4 ± 0.1-fold and 1.3 ± 0.3-fold above normal (P < 0.05). Therefore, hepatic support from transplanted hepatocytes in the peritoneal cavity improved critical parameters of liver injury in ALF. Transplanted hepatocytes were identified and were morphologically intact in the peritoneal cavity (Figure 7E). These cells contained glycogen, and glucose-6-phosphatase, which are critical for glucose metabolism (Figure 7F).
Figure 7.
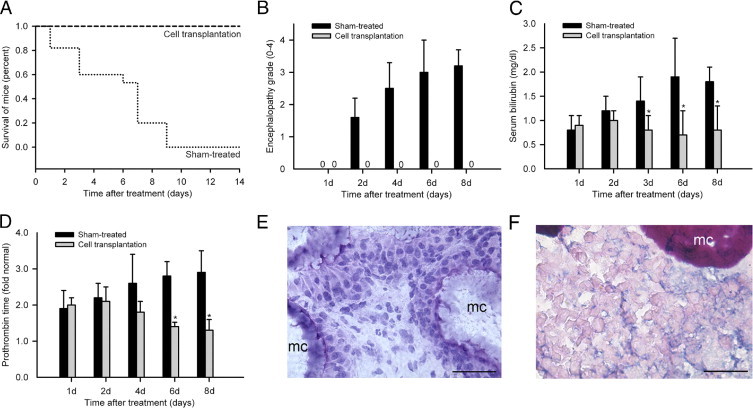
Outcomes in animals with ALF after cell therapy. A: Survival curves in mice with ALF after sham treatment with microcarriers alone and reversal of mortality after i.p. transplantation of 1 × 106 F344 rat hepatocytes (*P < 0.01, log-rank test). B: Encephalopathy in sham-treated mice and its absence in mice treated by transplantation of F344 rat hepatocytes. C: Decreases in serum bilirubin levels in treated mice. D: Improvements in prothrombin times after cell therapy. All data were from three to five mice per time point. *P < 0.01. E: Healthy transplanted hepatocytes and microcarriers (mc) in conglomerates recovered after 14 days from the peritoneal cavity (hematoxylin stain). F: Histochemical staining showing glycogen in transplanted hepatocytes (pink) adjacent to mc in the peritoneal cavity. Original magnification, ×200. Scale bar = 100 μm.
To confirm that improved survival in animals was due to intact cells, we established another group of NOD/SCID mice with Rif-Phen-MCT–induced ALF (n = 10), in which medium from primary rat hepatocytes cultured in serum-free conditions for 2 days was injected i.p. once daily. This did not alter mortality after ALF, indicating that transplantation of hepatocytes was necessary for rescuing animals with ALF.
Analysis of livers by quantitative real-time RT-PCR arrays in mice 7 days after hepatocyte transplantation in the peritoneal cavity showed that only two genes were differentially expressed (ie, Bcl2l1-associated X protein and p21) compared with previous differential expression of 28 genes. Moreover, these two genes were expressed at lower levels compared with mice without cell therapy [ie, 2-fold (Bcl2l1-associated X protein) and 10-fold (p21) lower]. This indicated substantial correction of molecular abnormalities in ALF after cell therapy.
To verify that hepatic support by transplanted cells in the peritoneal cavity promoted liver regeneration in ALF, we examined cell cycling by Ki-67 staining. Subsequent to cell transplantation, native hepatocytes were proliferating 3 days, and 6 to 8 days, after the onset of ALF (Figure 8A). The proliferation indices were 5% ± 2% and 11% ± 4%, respectively. This proliferation activity was greater than in animals without cell transplantation at corresponding times, although the differences did not reach statistical significance. Ki-67 was also expressed in bile duct cells (Figure 8B). However, ductular or juxtaductular cells did not expand to suggest recruitment of stem cells during hepatic recovery. Also, hepatic expression of Atm returned to normal and p21 was no longer detected by immunostaining (Figure 8, C and D). Liver morphological features were normal after cell therapy for up to 12 weeks.
Figure 8.
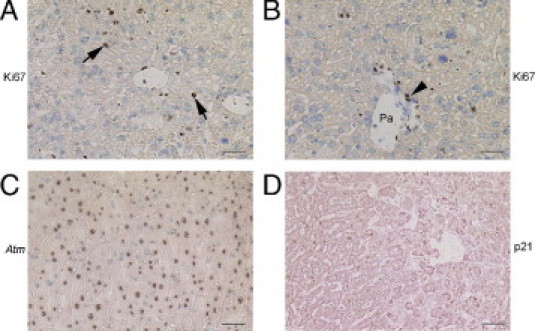
Changes during hepatic regeneration after cell therapy in mice with ALF. A and B: Ki-67 staining 7 days after cell transplantation in the peritoneal cavity alone, indicating proliferation in native hepatocytes (arrows, dark nuclei) and bile duct cells (arrowhead, dark nuclei). C: Hepatic Atm expression returned to normal at this point. D: Absence of p21 immunostaining in the liver 7 days after transplantation of cells in the peritoneal cavity alone. Original magnification, ×200. Scale bar = 100 μm. Toluidine blue (A–C) and eosin (D) counterstains were used. Pa, Portal area.
Finally, to address whether reseeding of the damaged liver with healthy cells would have provided advantages for liver regeneration in ALF, we performed studies with NOD/SCID mice given Rif, Phen, and 125-mg/kg MCT (n = 30). To localize transplanted cells in the liver, these studies were performed with Rosa26 donor hepatocytes expressing the LacZ gene, which was shown in previous studies to not activate deleterious immune responses in NOD/SCID mice.18 By contrast, GFP expression in donor transplanted cells activates host immune responses, resulting in clearance of transplanted cells after 7 days, even in NOD/SCID mice. We induced ALF by Rif-Phen-MCT; after 2 days, 2 × 106 Rosa26 donor hepatocytes were transplanted into the liver via the spleen and 5 × 106 F344 rat hepatocytes were simultaneously transplanted into the peritoneal cavity. Once again, all mice treated by cell transplantation survived beyond 14 days, which permitted examination of the time course of transplanted cell engraftment and proliferation.
We observed transplanted LacZ+ hepatocytes in the liver 1 and 3 days, and 1, 2, 4, and 12 weeks, after cell transplantation (n = 3 each). All donor hepatocytes expressed LacZ, and cell recipients were negative for LacZ (Figure 9, A and B). The LacZ+ transplanted hepatocytes were in liver sinusoids 1 day after cell transplantation, as was expected, and were integrated in the liver parenchyma at later times (ie, after 7 or 14 days) (Figure 9C). Transplanted cells survived in the liver throughout the studies, for up to 12 weeks (Figure 9D). Morphometric analysis of the number of transplanted LacZ+ cells in the liver showed absence of transplanted cell proliferation because only 2 to 4 transplanted cells were observed in the liver parenchyma adjacent to individual portal areas under all conditions, including after 3 and 7 days and after 1, 2, 4, and 12 weeks. No instances of transplanted cells in clusters were found. This confirmed that liver regenerated by proliferation of native hepatocytes alone after prolongation of survival through hepatic support from transplanted cells in the peritoneal cavity.
Figure 9.
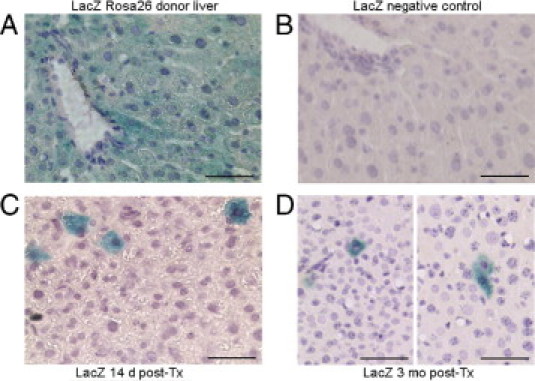
Transplanted hepatocytes in the liver of mice with ALF. A: LacZ staining (cytoplasmic blue) in Rosa26 donor liver. B: Absence of LacZ staining in recipient NOD/SCID mouse liver. C and D: Representative examples of LacZ+ transplanted hepatocytes (blue) integrated in the mouse liver 14 days (C) or 3 months (D) after intrasplenic transplantation. Transplanted hepatocytes are arranged individually, agreeing with absence of their proliferation over time and indicating liver recovery occurred by proliferation of native hepatocytes. Original magnification, ×200. Scale bar = 100 μm. A hematoxylin counterstain was used.
Discussion
These studies provide several important insights into the mechanisms of DILI. First, we identified that Atm signaling pathways were dysregulated in drug-induced ALF, which, to our knowledge, was not previously known; this offers opportunities for further studies. Second, liver injury induced by Rif-Phen-MCT in NOD/SCID mice reproduced key aspects of ALF in humans, including extensive hepatic necrosis, abnormal liver test results, encephalopathy, coagulopathy, and mortality over several days. Third, this model of ALF provided critical insights into cell therapy mechanisms, particularly by establishing that reseeding the liver with healthy cells was unnecessary. By contrast, metabolic support and perhaps release of paracrine signals from transplanted hepatocytes in the peritoneal cavity permitted native hepatocytes to regenerate the liver in ALF. Fourth, this ALF model in xenotolerant NOD/SCID mice will permit translational studies with candidate human cells, including cells generated from stem cells. This will be of extensive significance for cell therapy in people with ALF.
In the first part, our studies of DILI in NOD/SCID mice reproduced several aspects of APAP-induced changes in oxidative/metabolic stress pathways, including glutathione, heme oxygenase, monooxygenase or Cyp genes, Hsp genes, cytokines, and other inflammatory mechanisms.19 This agreed with broad similarities with other drug toxicities. Characterization of liver injury in NOD/SCID mice showed typical manifestations of ALF because of extensive hepatic necrosis, including abnormal liver test results, coagulopathy, and encephalopathy. Moreover, evidence of hepatic DNA synthesis, albeit insufficient for completing liver regeneration, was similar to that in people with ALF.20
In the NOD/SCID setting, deficiencies in DNA repair are driven by mutant catalytic subunit of DNA-dependent protein kinase (Prkdc), which is applicable to T and B lymphocytes with a propensity for lymphoma and thymoma.21,22 By contrast, Prkdc mutation is of no consequence for liver cells. We confirmed this by normal liver test results and hepatic morphological features in healthy control NOD/SCID mice, including absence of apoptosis or of increased Ki-67 expression. Also, studies of liver from healthy NOD/SCID mice by quantitative real-time PCR arrays showed that expression levels of DNA damage-repair and growth-arrest genes were actually lower than in healthy C57BL/6 mice. If lymphocyte deficiency were to impart greater susceptibility for liver injury, this should have been apparent in immunodeficient people. However, in individuals with low levels of lymphocytes, susceptibility to DILI neither increased nor decreased,23 excluding roles of immunocytes in altering drug hepatotoxicity. The basis of altered gene expression and susceptibility to DILI in NOD/SCID mice (in a C3H/HeJ background) versus C57BL/6 mice likely emanated from strain-dependent genetic and epigenetic regulation of gene expression,24 which essentially reflects individual-specific differences in people.
Although induction of Cyp3a4 by Rif and other drugs, including Phen and APAP, is thought to worsen hepatotoxicity,6,25 we found that Rif and Phen induced Cyp3a4 in both C57BL/6 and NOD/SCID mice, suggesting drug metabolic differences did not account for outcomes of DILI in these animal strains. Examination of alternative or additional mechanisms led us to Atm pathways. The NOD/SCID mice expressed Atm at lower levels compared with the C57BL/6 mice. After Rif-Phen-MCT in NOD/SCID mice, Atm expression declined further, and greater perturbations in Atm signaling were observed in NOD/SCID mice with ALF. Examination of APAP-induced liver gene expression in an experimental study in rats,19 in which gene expression was determined with limited probe sets 24 hours after APAP, showed 3-fold alterations in oxidative/metabolic stress genes (eg, heme oxygenase 1, Cyp7a1, flavin monooxygenase 1, Hspb1, Hspa8, and DnaJ) and in cell proliferation, growth arrest, and senescence genes (eg, Egr1, Ddit3, growth arrest and DNA damage–inducible 45α, and Mdm2), which was similar to our results herein. These types of data should strengthen the role of Atm signaling in other forms of DILI; also, it should be appropriate to consider how regulation of Atm expression may have altered the susceptibility to drug-induced ALF.
Previously, Atm−/− mice were shown to have impairment in liver regeneration, including mortality in some cases after partial hepatectomy.26 Similarly, Atm deficiency amplified genotoxicity induced by Phen.27 Also, inhibition of Atm expression by caffeine, a well-characterized ATM kinase inhibitor,28 significantly increased APAP hepatotoxicity.29 Because expression of Atm is largely governed by promoter regulation,30 and is negatively regulated by displacement of positive transcriptional transactivators with soluble factors (eg, epidermal growth factor or hepatocyte growth factor),31,32 it should be reasonable to consider whether soluble factors will be relevant in DILI and Atm expression and in improvement of Atm expression after cell therapy. For instance, positive regulators of Atm transcription have also been identified (eg, granulocyte colony-stimulating factor),30 which protects liver from injury by unknown mechanisms.33 Because multifold increases in Atm levels are normally observed during protection of cells from DNA damage,34 lowering of Atm levels in Rif-Phen-MCT–treated NOD/SCID mice should have caused serious problems, as represented by multiple perturbations in downstream Atm signaling.
The role of Atm as master regulator of DNA damage through phosphorylation of multiple effectors is well established.35 After DNA damage, Atm, along with other genes (eg, Chek1 and Chek2) phosphorylates p53. In turn, p53 activates p21, leading to arrest of damaged cells in G1/S through corresponding cyclins and cyclin-dependent kinases.36 However, p21 may also be regulated by p53-independent mechanisms.37 We consider it most likely that persistent Atm down-regulation in mice after Rif- Phen-MCT led to inadequate DNA repair, as indicated by down-regulation of RAD50 homolog (S cerevisiae) and Xrcc4 genes. This should have recruited p21-mediated G1/S checkpoint arrest. We found that cyclin G, Chek 2, CHK2 checkpoint homolog 1 was up-regulated, whereas cyclin D1, Egr1, and E2F were up- or down-regulated at various times. Up-regulation of growth arrest and DNA damage–inducible 45α, Ddit3, and Mdm2 provided further evidence for cell growth arrest.38 These mechanisms agreed with our observations of relatively few hepatocytes with Ki-67, which characterizes cells in late G1, S, and G2/M.
Previously, only the Mad transcription factor, which dominantly antagonizes the activity of the c-myelocytomatosis protooncogene, induced ALF through a discrete molecular mechanism, although liver injury was predominantly apoptotic.39 Toxicity through transgenes (eg, pro–drug-activating herpes simplex virus thymidine kinase) produced liver injury, although this damage differed from ALF in people.40 By contrast, ALF induced by Rif-Phen-MCT in NOD/SCID mice was similar to the human condition, including extensive hepatic necrosis, liver test abnormalities, encephalopathy, high mortality rates, and the presence of some hepatocytes with DNA synthesis.20
In the second part of this study, we addressed critical cell therapy mechanisms. The ability to rescue subjects with ALF by transplanting cells into the peritoneal cavity without reseeding of the liver will be of immense clinical significance in view of the simplicity of the former procedure. A few people with ALF have already been treated by transplantation of hepatocytes in the peritoneal cavity,41 although those studies did not incorporate extracellular matrix support, without which hepatocytes are rapidly destroyed. We found that after cell therapy, abnormalities in hepatic gene expression reversed and liver regeneration was then completed by proliferation in previously growth–arrested native hepatocytes. Cell transplantation experiments with reporter hepatocytes in the liver provided unequivocal evidence for this mechanism. This was in agreement with the possibility that cell proliferation may be restored under appropriate circumstances, despite cell cycle checkpoint–mediated arrest, such as by p21.42 We excluded migration of transplanted cells from the peritoneal cavity to the native liver or other organs as potential alternative explanations.
As transplanted cells were organized in the peritoneal cavity with liver gene expression, beneficial effects of cell transplantation in ALF included hepatic support from transplanted hepatocytes. Moreover, hepatocytes secrete multiple cytokines, growth factors, and other molecules,43 which could have emanated from transplanted cells in the peritoneal cavity to serve beneficial roles in the recovery of the damaged liver, although further studies of these mechanisms will be appropriate. Soluble factors (eg, granulocyte colony-stimulating factor) restored Atm levels in cells through transcriptional control mechanisms.31 Injection of cell culture medium did not rescue animals with ALF in our studies. However, this did not exclude the possible role of paracrine factors released by transplanted hepatocytes on other aspects of hepatic recovery.
Finally, this ALF model in NOD/SCID mice will be helpful for defining the therapeutic potential of candidate human stem cell–derived cells because these mice tolerate human xenografts.44 Studies of additional mechanisms regulating liver regeneration in ALF should also be of great value.
Footnotes
Supported in part by the NIH (grants R01 DK071111 and P30-DK41296) and by a Druckenmiller fellowship from the New York Stem Cell Foundation (S.B.).
A guest editor acted as editor-in-chief for this manuscript. No person at Thomas Jefferson University or Albert Einstein College of Medicine was involved in the peer review process or final disposition of this article.
Current address of E.B., Institute of Experimental Medicine, Tbilisi State Medical University, Tbilisi, Republic of Georgia; of R.S., St Luke's Hospital, New York, New York; of Y.M.W., Department of Surgery, National Taiwan University Hospital, Taipei, Taiwan; and of K.C., Long Island Jewish Medical Center, New Hyde Park, New York.
References
- 1.Murray K.F., Hadzic N., Wirth S., Bassett M., Kelly D. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47:395–405. doi: 10.1097/MPG.0b013e3181709464. [DOI] [PubMed] [Google Scholar]
- 2.Jaeschke H. Innate immunity and acetaminophen-induced liver injury: why so many controversies? Hepatology. 2008;48:699–701. doi: 10.1002/hep.22556. [DOI] [PubMed] [Google Scholar]
- 3.Avraham Y., Grigoriadis N.C., Magen I., Poutahidis T., Vorobiav L., Zolotarev O., Ilan Y., Mechoulam R., Berry E.M. Capsaicin affects brain function in a model of hepatic encephalopathy associated with fulminant hepatic failure in mice. Br J Pharmacol. 2009;158:896–906. doi: 10.1111/j.1476-5381.2009.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bémeur C., Vaquero J., Desjardins P., Butterworth R.F. N-acetylcysteine attenuates cerebral complications of non-acetaminophen-induced acute liver failure in mice: antioxidant and anti-inflammatory mechanisms. Metab Brain Dis. 2010;25:241–249. doi: 10.1007/s11011-010-9201-2. [DOI] [PubMed] [Google Scholar]
- 5.Shinohara M., Ybanez M.D., Win S., Than T.A., Jain S., Gaarde W.A., Han D., Kaplowitz N. Silencing glycogen synthase kinase-3beta inhibits acetaminophen hepatotoxicity and attenuates JNK activation and loss of glutamate cysteine ligase and myeloid cell leukemia sequence 1. J Biol Chem. 2010;285:8244–8255. doi: 10.1074/jbc.M109.054999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Y.M., Joseph B., Berishvili E., Kumaran V., Gupta S. Hepatocyte transplantation and drug-induced perturbations in liver cell compartments. Hepatology. 2008;47:279–287. doi: 10.1002/hep.21937. [DOI] [PubMed] [Google Scholar]
- 7.Fisher R.A., Strom S.C. Human hepatocyte transplantation: worldwide results. Transplantation. 2006;82:441–449. doi: 10.1097/01.tp.0000231689.44266.ac. [DOI] [PubMed] [Google Scholar]
- 8.Gupta S., Rajvanshi P., Irani A.N., Palestro C.J., Bhargava K.K. Integration and proliferation of transplanted cells in hepatic parenchyma following D-galactosamine-induced acute injury in F344 rats. J Pathol. 2000;190:203–210. doi: 10.1002/(SICI)1096-9896(200002)190:2<203::AID-PATH521>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Gupta S., Vemuru R.P., Lee C.-D., Yerneni P., Aragona E., Burk R.D. Hepatocytes exhibit superior transgene expression after transplantation into liver and spleen compared with peritoneal cavity or dorsal fat pad: implications for hepatic gene therapy. Human Gene Ther. 1994;5:959–967. doi: 10.1089/hum.1994.5.8-959. [DOI] [PubMed] [Google Scholar]
- 10.Rajvanshi P., Kerr A., Bhargava K.K., Burk R.D., Gupta S. Studies of liver repopulation using the dipeptidyl peptidase IV deficient rat and other rodent recipients: cell size and structure relationships regulate capacity for increased transplanted hepatocyte mass in the liver lobule. Hepatology. 1996;23:482–496. doi: 10.1002/hep.510230313. [DOI] [PubMed] [Google Scholar]
- 11.Gupta S., Rajvanshi P., Malhi H., Sokhi R.P., Slehria S., Vasa S.R.G., Dabeva M., Shafritz D.A., Kerr A. Cell transplantation causes loss of gap junctions and activates GGT expression permanently in host liver. Am J Physiol Gastroint Liver Physiol. 2000;279:G815–G826. doi: 10.1152/ajpgi.2000.279.4.G815. [DOI] [PubMed] [Google Scholar]
- 12.Slehria S., Rajvanshi P., Ito Y., Sokhi R.P., Bhargava K.K., Palestro C.J., McCuskey R.S., Gupta S. Hepatic sinusoidal vasodilators improve transplanted cell engraftment and ameliorate microcirculatory perturbations in the liver. Hepatology. 2002;35:1320–1328. doi: 10.1053/jhep.2002.33201. [DOI] [PubMed] [Google Scholar]
- 13.Joseph B., Malhi H., Bhargava K.K., Palestro C.J., McCuskey R.S., Gupta S. Kupffer cells participate in early clearance of syngeneic hepatocytes transplanted in the rat liver. Gastroenterology. 2002;123:1677–1685. doi: 10.1053/gast.2002.36592. [DOI] [PubMed] [Google Scholar]
- 14.Krohn N., Kapoor S., Enami Y., Follenzi A., Bandi S., Joseph B., Gupta S. Hepatocyte transplantation-induced liver inflammation is driven by cytokines-chemokines associated with neutrophils and Kupffer cells. Gastroenterology. 2009;136:1806–1817. doi: 10.1053/j.gastro.2009.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demetriou A.A., Whiting J.F., Feldman D., Levenson S.M., Chowdhury N.R., Moscioni A.D., Kram M., Chowdhury J.R. Replacement of liver function in rats by transplantation of microcarrier-attached hepatocytes. Science. 1986;233:1190–1192. doi: 10.1126/science.2426782. [DOI] [PubMed] [Google Scholar]
- 16.Kumaran V., Benten D., Follenzi A., Joseph B., Sarkar R., Gupta S. Transplantation of endothelial cells corrects the phenotype in hemophilia A mice. J Thromb Haemost. 2005;3:2022–2031. doi: 10.1111/j.1538-7836.2005.01508.x. [DOI] [PubMed] [Google Scholar]
- 17.Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, National Academy Press; Washinton, D.C.: 1996. Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 18.Gagandeep S., Ott M., Sokhi R., Gupta S. Rapid clearance of syngeneic transplanted hepatocytes following transduction with E-1-deleted adenovirus indicates early host immune responses and offers novel ways for studying viral vector, target cell and host interactions. Gene Ther. 1999;6:729–736. doi: 10.1038/sj.gt.3300871. [DOI] [PubMed] [Google Scholar]
- 19.Morishita K., Mizukawa Y., Kasahara T., Okuyama M., Takashima K., Toritsuka N., Miyagishima T., Nagao T., Urushidani T. Gene expression profile in liver of differing ages of rats after single oral administration of acetaminophen. J Toxicol Sci. 2006;31:491–507. doi: 10.2131/jts.31.491. [DOI] [PubMed] [Google Scholar]
- 20.Quaglia A. Auxiliary transplantation for acute liver failure: histopathological study of native liver regeneration. Liver Transpl. 2008;14:1437–1448. doi: 10.1002/lt.21568. [DOI] [PubMed] [Google Scholar]
- 21.Fulop G.M., Phillips R.A. The scid mutation in mice causes a general defect in DNA repair. Nature. 1990;347:479–482. doi: 10.1038/347479a0. [DOI] [PubMed] [Google Scholar]
- 22.Jhappan C., Morse H.C., 3rd, Fleischmann R.D., Gottesman M.M., Merlino G. DNA-PKcs: a T-cell tumour suppressor encoded at the mouse scid locus. Nat Genet. 1997;17:483–486. doi: 10.1038/ng1297-483. [DOI] [PubMed] [Google Scholar]
- 23.De Lazzari E., León A., Arnaiz J.A., Martinez E., Knobel H., Negredo E., Clotet B., Montaner J., Storfer S., Asenjo M.A., Mallolas J., Miró J.M., Gatell J.M. Hepatotoxicity of nevirapine in virologically suppressed patients according to gender and CD4 cell counts. HIV Med. 2008;9:221–226. doi: 10.1111/j.1468-1293.2008.00552.x. [DOI] [PubMed] [Google Scholar]
- 24.Geisert E.E., Lu L., Freeman-Anderson N.E., Templeton J.P., Nassr M., Wang X., Gu W., Jiao Y., Williams R.W. Gene expression in the mouse eye: an online resource for genetics using 103 strains of mice. Mol Vis. 2009;15:1730–1763. [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng J., Ma X., Krausz K.W., Idle J.R., Gonzalez F.J. Rifampicin-activated human pregnane X receptor and CYP3A4 induction enhance acetaminophen-induced toxicity. Drug Metab Dispos. 2009;37:1611–1621. doi: 10.1124/dmd.109.027565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu S., Shen K.C., Wang Y., Brooks S.C., Wang Y.A. Impaired hepatocyte survival and liver regeneration in Atm-deficient mice. Hum Mol Genet. 2005;14:3019–3025. doi: 10.1093/hmg/ddi333. [DOI] [PubMed] [Google Scholar]
- 27.Bhuller Y., Jeng W., Wells P.G. Variable in vivo embryoprotective role for ataxia-telangiectasia-mutated against constitutive and phenytoin-enhanced oxidative stress in Atm knockout mice. Toxicol Sci. 2006;93:146–155. doi: 10.1093/toxsci/kfl022. [DOI] [PubMed] [Google Scholar]
- 28.Ravi D., Muniyappa H., Das K.C. Caffeine inhibits UV-mediated NF-kappaB activation in A2058 melanoma cells: an ATM-PKCdelta-p38 MAPK-dependent mechanism. Mol Cell Biochem. 2008;308:193–200. doi: 10.1007/s11010-007-9628-x. [DOI] [PubMed] [Google Scholar]
- 29.Sato C., Izumi N. Mechanism of increased hepatotoxicity of acetaminophen by the simultaneous administration of caffeine in the rat. J Pharmacol Exp Ther. 1989;248:1243–1247. [PubMed] [Google Scholar]
- 30.Gueven N., Keating K., Fukao T., Loeffler H., Kondo N., Rodemann H.P., Lavin M.F. Site-directed mutagenesis of the ATM promoter: consequences for response to proliferation and ionizing radiation. Genes Chromosomes Cancer. 2003;38:157–167. doi: 10.1002/gcc.10261. [DOI] [PubMed] [Google Scholar]
- 31.Gueven N., Keating K.E., Chen P., Fukao T., Khanna K.K., Watters D., Rodemann P.H., Lavin M.F. Epidermal growth factor sensitizes cells to ionizing radiation by down-regulating protein mutated in ataxia-telangiectasia. J Biol Chem. 2001;276:8884–8891. doi: 10.1074/jbc.M006190200. [DOI] [PubMed] [Google Scholar]
- 32.Maroni P., Bendinelli P., Matteucci E., Desiderio M.A. GF induces CXCR4 and CXCL12-mediated tumor invasion through Ets1 and NF-kappaB. Carcinogenesis. 2007;28:267–279. doi: 10.1093/carcin/bgl129. [DOI] [PubMed] [Google Scholar]
- 33.Hou X.W., Jiang Y., Wang L.F., Xu H.Y., Lin H.M., He X.Y., He J.J., Zhang S. Protective role of granulocyte colony-stimulating factor against adriamycin induced cardiac, renal and hepatic toxicities. Toxicol Lett. 2009;187:40–44. doi: 10.1016/j.toxlet.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 34.Gatei M., Young D., Cerosaletti K.M., Desai-Mehta A., Spring K., Kozlov S., Lavin M.F., Gatti R.A., Concannon P., Khanna K. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat Genet. 2000;25:115–119. doi: 10.1038/75508. [DOI] [PubMed] [Google Scholar]
- 35.Kanu N., Behrens A. ATMINistrating ATM signalling: regulation of ATM by ATMIN. Cell Cycle. 2008;7:3483–3486. doi: 10.4161/cc.7.22.7044. [DOI] [PubMed] [Google Scholar]
- 36.Kuribayashi K., El-Deiry W.S. Regulation of programmed cell death by the p53 pathway. Adv Exp Med Biol. 2008;615:201–221. doi: 10.1007/978-1-4020-6554-5_10. [DOI] [PubMed] [Google Scholar]
- 37.Li D.Q., Pakala S.B., Reddy S.D., Ohshiro K., Peng S.H., Lian Y., Fu S.W., Kumar R. Revelation of p53-independent function of MTA1 in DNA damage response via modulation of the p21 WAF1-proliferating cell nuclear antigen pathway. J Biol Chem. 2010;285:10044–10052. doi: 10.1074/jbc.M109.079095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J., Chen X. Posttranscriptional regulation of p53 and its targets by RNA-binding proteins. Curr Mol Med. 2008;8:845–849. doi: 10.2174/156652408786733748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gagandeep S., Sokhi R., Slehria S., Gorla G.R., Furgiuele J., DePinho R.A., Gupta S. Hepatocyte transplantation improves survival in mice with liver toxicity induced by hepatic overexpression of Mad1 transcription factor. Mol Ther. 2000;1:358–365. doi: 10.1006/mthe.2000.0051. [DOI] [PubMed] [Google Scholar]
- 40.Braun K.M., Degen J.L., Sandgren E.P. Hepatocyte transplantation in a model of toxin-induced liver disease: variable therapeutic effect during replacement of damaged parenchyma by donor cells. Nat Med. 2000;6:320–326. doi: 10.1038/73179. [DOI] [PubMed] [Google Scholar]
- 41.Habibullah C.M., Syed I.H., Qamar A., Taher-Uz Z. Human fetal hepatocyte transplantation in patients with fulminant hepatic failure. Transplantation. 1994;58:951–952. doi: 10.1097/00007890-199410270-00016. [DOI] [PubMed] [Google Scholar]
- 42.Demidenko Z.N., Zubova S.G., Bukreeva E.I., Pospelov V.A., Pospelova T.V., Blagosklonny M.V. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- 43.Rowell D.L., Eckmann L., Dwinell M.B., Carpenter S.P., Raucy J.L., Yang S.K., Kagnoff M.F. Human hepatocytes express an array of proinflammatory cytokines after agonist stimulation or bacterial invasion. Am J Physiol. 1997;273:G322–G332. doi: 10.1152/ajpgi.1997.273.2.G322. [DOI] [PubMed] [Google Scholar]
- 44.Cho J., Joseph B., Sappal B.S., Giri R.K., Wang R., Ludlow J., Susick R., Gupta S. Analysis of the functional integrity of cryopreserved human liver cells including xenografting in immunodeficient mice to address suitability for clinical applications. Liver Int. 2004;24:361–370. doi: 10.1111/j.1478-3231.2004.0938.x. [DOI] [PubMed] [Google Scholar]