

Abstract
Early growth response 1 (EGR-1) contributes to the development of chronic obstructive pulmonary disease in the lungs of smokers by mediating pulmonary inflammatory responses, but the direct downstream genes of EGR-1 that regulate this process remain unknown. We show that a new EGR-1 target gene, geranylgeranyl diphosphate synthase (GGPPS), which controls protein prenylation, can regulate the proinflammatory function of EGR-1 by activating MAPK signaling. When C57BL/6 mice were exposed to cigarette smoke, EGR-1 and GGPPS levels increased in their lungs, and the inflammatory responses were augmented, whereas these effects could be reversed by the down-regulation of EGR-1 transcription activity. The accumulation of EGR-1 and GGPPS was induced by MAPK/ERK pathway activation when Beas-2B human bronchial epithelial cells were exposed to cigarette smoke extract (CSE). Further examination showed that EGR-1 in turn regulated Erk1/2 activity because inhibition of EGR-1 transcription activity decreased CSE-induced Erk1/2 phosphorylation. Furthermore, EGR-1–promoted Erk1/2 activation was dependent on GGPPS transcription. Knockdown of GGPPS expression with small-interfering RNA abolished the EGR-1–activated Erk1/2 activity. Both EGR-1 transcription inhibition and GGPPS expression knockdown decreased the inflammatory response induced by CSE in Beas-2B cells. Our results reveal a new EGR-1/GGPPS/MAPK signaling pathway that controls cigarette smoke–induced pulmonary inflammation, and this may shed light on our understanding of the mechanism of cigarette smoke–related pulmonary diseases such as chronic obstructive pulmonary disease.
Although cigarette smoke is a major risk factor for chronic obstructive pulmonary disease (COPD), genetic susceptibility may also be crucial to the progression of the disease because only 15% to 20% of heavy smokers develop clinically significant airway obstruction.1 However, the genes that link cigarette smoking and COPD susceptibility remain poorly understood. A report from Chan et al2 demonstrated that the early growth response 1 (EGR-1) polymorphisms are significantly associated with plasma total IgE and atopy in patients with asthma. Our previous report also showed that EGR-1 is a genetic susceptibility factor that can control the production of matrix metalloproteinase (MMP-2 and MT1-MMP) to overwhelm the antiprotease systems and lead to destruction of alveolar septal architecture.3,4 In addition, EGR-1 can also stimulate heat shock protein 70 elevation in an Erk1/2-dependent manner, which may regulate the cigarette smoke–induced inflammatory processes.5
The transcription factor EGR-1 is the product of the EGR-1 gene, which belongs to the immediate-early gene family of proteins and is often rapidly and transiently activated by a variety of stress signals, such as osmotic pressure variation, heat shock, hypoxia, DNA-damaging agents, radiation, injury, and stretch.6–13 As an important zinc finger transcription factor, the function of EGR-1 is dependent on the expression of its target genes. EGR-1 can recognize highly conserved consensus GC-rich nucleotide sequences (GCGG/TGGGCG)13–16 and directly activate the transcription of many genes, including immune effector genes, such as cytokine interleukin (IL) 217 and proinflammation immune mediator tumor necrosis factor α (TNF-α),18 and cell surface molecules, such as IL-2 receptor,19 Fas/CD95,20 and intracellular adhesion molecule 1.21 Through the regulation of its target genes, EGR-1 plays important roles in various cellular programs, including cell proliferation, differentiation, and apoptosis.13 Although several downstream genes may be related to the pulmonary inflammatory response,4,5,22,23 a detailed mechanism for the function of EGR-1 in cigarette smoke–related pulmonary diseases still remains largely unknown.
We have reported that human primary lung fibroblasts can synthesize MMP and chemokines in an EGR-1–dependent manner when exposed to cigarette smoke extract (CSE) stress.3–5 To further understand the pathological functions of EGR-1 in pulmonary diseases during cigarette smoke–induced stress, we used chromatin immunoprecipitation methods to identify a new target gene, geranylgeranyl diphosphate synthase (GGPPS). GGPPS is a branch-point enzyme in the mevalonate pathway that catalyzes the synthesis of geranylgeranyl diphosphate24,25 from farnesyl diphosphate,26 which is used for the geranylgeranylation of proteins such as Ras and Ras-related small GTP-binding proteins with a CaaX motif at the C-terminus. Ras prenylation enhances its membrane association and facilitates the activation of Ras/Raf/MAPK signaling.27,28
In this report, we find that GGPPS is a novel target gene of EGR-1 that can mediate cigarette smoke–related pulmonary diseases. Furthermore, we also propose a mechanism by which EGR-1/GGPPS can augment Erk1/2 MAPK activation under constant, long-term cigarette smoke stimulation to promote the chronic inflammation that causes airway abnormalities, emphysema, and consequently, COPD in susceptible smokers.29–32 MAPK, including Erk1/2, p38, and JNK, is able to induce EGR-1 expression, depending on the stimulus and cell type.33–35 We present evidence that not only can EGR-1 stimulate the expression of GGPPS in an Erk1/2 MAPK–dependent manner, but also that EGR-1/GGPPS can reactivate Erk1/2 MAPK by promoting the transcription of GGPPS. This regulation might be responsible for the expression of TNF-α, IL-8, and MMP-2 during CSE exposure, which could promote cigarette smoke–related pathological processes in the lung.
Materials and Methods
Preparation and Treatment of CSE
Nonfiltered research reference cigarettes (1R3F cigarettes) were purchased from the University of Kentucky in Lexington. CSE was prepared at a concentration of 1 cigarette per 5 ml of serum-free Dulbecco's modified Eagle's medium (Gibco/BRL, Gaithersburg, MD) as previously described36 with modifications. This medium was defined as 100% CSE and was used after adjusting the pH to 7.4 and filtering through a 0.22-μm filter. Cells were grown to 90% confluence in 10-cm cell culture plates and rendered quiescent in a medium containing 0.5% fetal bovine serum before CSE treatment. During the CSE treatment, the cells were always cultured in the presence of CSE until the cells were lysed. For the experiment using the MEK inhibitor U0126, cells were preincubated for 2 hours with U0126. The concentration used was 10 μmol/L, which could inhibit MAPK without cytotoxicity. The inhibitor was dissolved in dimethyl sulfoxide (DMSO) as recommended by the manufacturer. The vehicle contained the same concentration of DMSO that was used in the treatment group, and the maximum DMSO content did not exceed 0.1% of medium.
Cell Culture
The Beas-2B human bronchial epithelial cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (vol/vol) fetal bovine serum and penicillin (100 IU/ml)/streptomycin (100 ng/ml) at 37°C in a water-saturated atmosphere with 5% carbon dioxide.
Plasmids, siRNA, and Adenovirus Construction
Human EGR-1 and dnEGR-1 (dominant negative EGR-1) cDNAs were a gift from Professor J. M. Baraban. To generate adenoviruses overexpressing EGR-1, dnEGR-1, and GGPPS, the sequences of EGR-1 and dnEGR-1 were cloned from their original vectors into the pAdTrack-CMV vector. Human GGPPS cDNA was cloned by reverse transcription PCR (RT-PCR) from the total RNA of HEK293 cells into the pAdTrack-CMV vector. These pAdTrack-CMV vectors carrying EGR-1, dnEGR-1, and GGPPS were recombined into an adenovirus backbone with the AdEasy System according to the manufacturer's protocol. The adenovirus vector has a Green fluorescent protein (GFP) cassette as an infection marker. To generate an adenovirus expressing human GGPP small-interfering RNA (siRNA), a unique 19-nucleotide sequence targeting human GGPPS (5′-GTCCCACTGAAGAAGAATA-3′) and the scrambled sequence (5′-TTCTCCGAACGTGTCACGT-3′) were inserted into the pShuttle-H1 vector and subjected to adenovirus recombination with the AdEasy System according to the manufacturer's protocol. The siRNAs of EGR-1 were purchased from Invitrogen Corp (Carlsbad, CA) and designed to target the following cDNA sequence of EGR-1: 5′-TCTCCCAGGACAATTGAAATTTGCT-3′. The scrambled sequence was used as control: 5′-CCTACGCCACCAATTTCGT-3′.
Western Blotting
Beas-2B whole-cell lysates were prepared according to previously reported standard protocols.3 For Western blotting, equal amounts of protein for each group were resolved on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto polyvinylidene fluoride membrane (Bio-Rad, Hercules, CA). The membranes were then incubated with the appropriate primary antibody as indicated. Bound antibody was visualized using alkaline phosphatase–conjugated or horseradish peroxidase–labeled secondary antibodies.
Real-Time RT-PCR
Total RNA from lung tissues and treated cells was extracted using TRIzol reagent (Invitrogen) and reversely transcribed with the ReverTra Ace-α kit (Toyobo Co Ltd, Osaka, Japan). The resulting cDNA was used for real-time RT-PCR performed on an ABI-7300 (Applied Biosystems. Carlsbad, CA). SYBR Green master mix was purchased from Toyobo. All quantitations were performed in triplicate and normalized to an endogenous 18-strand RNA control. The sequences of primer probes are presented in Table 1.
Table 1.
Primers Used in Real-Time PCR Analysis
| Genes | Accession no. | Forward/reverse primer |
|---|---|---|
| Human | ||
| EGR-1 | NM_001964.2 | 5′-CAGCAGTCCCATTTACTCAG-3′ |
| 5′-GACTGGTAGCTGGTATTG-3′ | ||
| GGPPS | NM_004837.3 | 5′-CCAGGTAAACAAGTGAGAACCAA-3′ |
| 5′-CGTCGGAGTTTTGAGTTGTCT-3′ | ||
| IL-8 | NM_000584.2 | 5′-ATGACTTCCAAGCTGGCCGTGGCT-3′ |
| 5′-TCTCAGCCCTCTTCAAAAACTTCTC-3′ | ||
| TNF-α | NM_000594.2 | 5′-CCCAGGCAGTCAGATCATCTTC-3′ |
| 5′-AGCTGCCCCTCAGCTTGA-3′ | ||
| MMP-2 | NM_001127891.1 | 5′-CCTGTTTGTGCTGAAGGACA-3′ |
| 5′-GTACTTGCCATCCTTCTCAA-3′ | ||
| 18s | NR_003286.2 | 5′-GTCTGTGATGCCCTTAGATG-3′ |
| 5′-AGCTTATGACCCGCACTTAC-3′ | ||
| Mouse | ||
| TNF-α | NM_013693.2 | 5′-GTGGAACTGGCAGAAGAGGC-3′ |
| 5′-AGACAGAAGAGCGTGGTGGC-3′ | ||
| 18s | NR_003286.2 | 5′-GTCTGTGATGCCCTTAGATG-3′ |
| 5′-AGCTTATGACCCGCACTTAC-3′ |
EGR-1, early growth response 1; GGPPS, geranylgeranyl diphosphate synthase; IL-8, interleukin 8; MMP-2, matrix metalloproteinase 2; TNF-α, tumor necrosis factor α; 18s rRNA, 18 svedberg units rRNA.
Animal Experiments
C57BL/6 mice were purchased from the Model Animal Research Center of the Nanjing University Animal Center. Experiments were conducted in accordance with the China Guidelines for the Care and Use of Laboratory Animals. The animals were maintained in individual ventilated cages (Suzhou Fengshi Laboratory Animal Equipment Co Ltd, Huangqiao Town City, Suzhou, China) under specific pathogen-free conditions. The 8-week-old mice were exposed to smoke in a whole-body manner using a method previously published by Witschi et al.37 Cigarette smoke exposure was performed (45 minutes per day and 6 days per week for up to 27 weeks) by burning 1R3F reference cigarettes (1.16 mg of nicotine per cigarette; purchased from the Tobacco Research Institute, University of Kentucky, Lexington) through a smoking machine (model TE-10; Teague Enterprises, Woodland, CA). Each smoldering cigarette was puffed for 2 seconds once every minute for a total of 8 puffs, at a flow rate of 1.05 L/min, to provide a standard puff of 35 cm3. The smoke machine was adjusted to produce a mixture of sidestream smoke (89%) and mainstream smoke (11%) by burning 5 cigarettes at one time. The chamber atmosphere was monitored for total suspended particulates and carbon monoxide, with concentrations of 90 mg/m3 and 350 ppm, respectively. The control mice were exposed to filtered room air. Each experimental group contained at least 5 mice.
To inhibit the transcriptional activity of EGR-1 in the lung tissues of the mice, intranasal administration of dnEGR-1 adenovirus (a dominant-negative EGR-1 that only expresses the zinc finger domain that binds to the GGPPS promoter but that does not have any transcriptional activity)38 [5 × 105 plaque-forming units (PFU)], control adenovirus (5 × 105 PFU) (Ade group), or PBS (mock group) was performed 2 weeks before cigarette smoke stimulation. To maintain the efficiency of the adenoviruses, intranasal administration was performed every 2 weeks during the entire cigarette smoke stimulation process.
Bronchoalveolar Lavage
After the thorax was opened and the trachea exposed, the lung tissues were lavaged with PBS. A total of 0.8 ml of PBS was injected into and retrieved from the trachea twice, and this process was repeated 3 times for each mouse. The fluid was centrifuged at 1500 × g for 10 minutes. The supernatant was stored at −70°C and used for the determination of cytokine concentration by enzyme-linked immunosorbent assay. The cells in the bronchoalveolar lavage were resuspended with 100 μL of PBS. The neutrophils were counted based on morphologic criteria; at least 10 high-powered fields were counted per slide.
Immunohistochemical Analysis
Mice were sacrificed at indicated times 4 hours after cigarette smoke exposure, and lung tissue was collected for immunostaining. Lung tissues were fixed, paraffin embedded, and cut 6 to 7 μm thick. The sections were deparaffinized, stained with hematoxylin-eosin using standard protocols, and then immunostained using antihuman EGR-1 antibody and antihuman GGPPS antibody. Next, the sections were incubated with peroxidase-labeled secondary antibody and stained with diaminobenzidine solution (0.05% diaminobenzidine, 50 mmol/L Tris hydrochloride, pH 7.4, freshly prepared 0.01% hydrogen peroxide).
Results
Cigarette Smoke Exposure Caused EGR-1 and GGPPS Elevation in Mouse Lung Tissue
We monitored the expression of EGR-1 and GGPPS in the lungs of mice when they were exposed to cigarette smoke. EGR-1 was found to increase over time with cigarette smoke exposure (Figure 1). With a cigarette smoke challenge every day for 5 weeks, EGR-1 was found mainly in the epithelial cells and stromal cells of the airway epithelium, whereas it was found mainly in the airway epithelium and alveolar epithelial cells after 15 weeks of exposure. After 27 weeks of exposure, EGR-1 was found in the airway epithelium, alveolar epithelial cells, and invasive inflammatory cells. Many invasive inflammatory cells were found in the airway and alveolar epithelia after 27 weeks of exposure. GGPPS, a new target gene of EGR-1 we recently indentified (N. Shen, unpublished data), was also found to increase in the lung tissues of mice exposed to cigarette smoke challenge for 27 weeks. The increase in GGPPS was mainly observed in the airway epithelium, alveolar epithelial cells, and invasive inflammatory cells, similar to EGR-1 (Figure 1).
Figure 1.
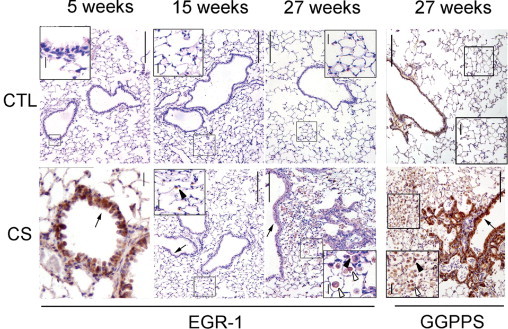
Cigarette smoke exposure increased early growth response 1 (EGR-1) and geranylgeranyl diphosphate synthase (GGPPS) expression in normal mouse lung tissues. Immunostaining of EGR-1 and GGPPS in lung tissue of normal mice exposed to cigarette smoke. EGR-1 levels increased in the cigarette smoking group (CS) compared with the control group (CTL) in the airway epithelium (arrow) after 5 weeks of challenge, alveolar epithelial cells (arrowhead) after 15 weeks, and airway epithelium (arrow) and invasive inflammatory (hollow arrowhead) cells after 27 weeks of exposure. GGPPS was also found in the airway epithelium (arrow), alveolar epithelial cells (arrowhead), and invasive inflammatory cells (hollow arrowhead) as EGR-1 did after 27 weeks of exposure. Scale bars: 200 μm, insets: 12.5 μm.
Inhibition of EGR-1 Transcription Function Decreased Cigarette Smoke–Induced Inflammatory Response
We measured the cytokines and neutrophils in lung tissue and lavage after cigarette smoke exposure. The mRNA and protein levels of the cytokine TNF-α were significantly increased by smoke exposure in lung tissues and in the lavage, respectively (Figure 2, A and B). The number of neutrophils was also elevated (Figure 2C). If dnEGR-1 adenovirus was administrated during cigarette smoke exposure (the efficiency of overexpression of dnEGR-1 in lungs is shown in Figure 2D), the TNF-α mRNA level in the lung tissues was significantly reduced (Figure 2A). The protein level of TNF-α and the number of neutrophils also decreased in the lavage, although the difference was not significant (Figure 2, B and C). The results suggest that EGR-1 might mediate cigarette smoke–induced pulmonary inflammation.
Figure 2.
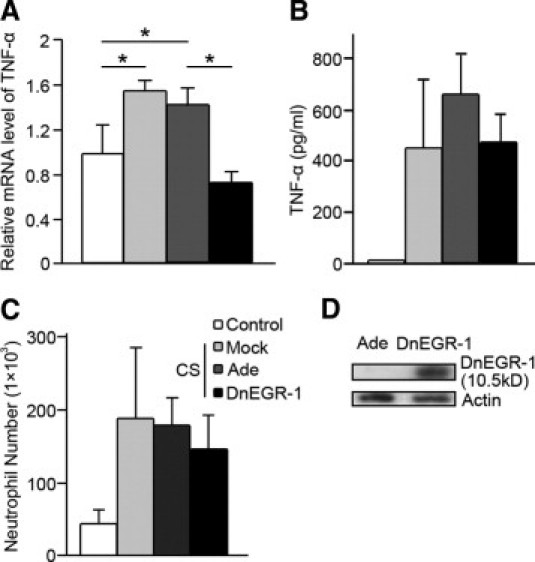
Inhibition of early growth response 1 (EGR-1) transcriptional activity decreased the cigarette smoke–induced inflammatory response. mRNA levels of tumor necrosis factor α (TNF-α) in mouse lung (A), TNF-α protein in the lavage (B), and neutrophil number (C) in the lavage. The mice were infected with dominant negative EGR-1 (dnEGR-1 group) adenovirus, control adenovirus (Ade group), or PBS (mock group) through nasal incubation. The TNF-α mRNA level (A) in lung tissue, as detected by quantitative PCR, decreased significantly in the dnEGR-1 group. The protein level of TNF-α (B) in lavage, as detected by enzyme-linked immunosorbent assay, was also decreased in the dnEGR-1 group. The number of neutrophils (C) from the bronchoalveolar lavage was decreased in the dnEGR-1 group. However, there was no significant difference in the TNF-α protein level and the number of neutrophils between the Ade control group and the dnEGR-1 group. D: The efficiency of dnEGR-1 adenovirus in mouse lung tissues was detected using Western blot. The protein was extracted from the lung tissues of the mice, which had been infected with the control adenovirus (Ade) or the dnEGR-1 adenovirus (dnEGR-1) for 27 weeks (intranasal administration every 2 weeks) and subjected to Western blot. The overexpressed dnEGR-1 is a zinc finger protein that can bind the promoters of EGR-1 target genes but has no transcriptional activity. It is a 10.5-kDa protein, which can be detected with the EGR-1 antibody (Santa Cruz, Biotechnology). *P < 0.05.
The Erk1/2 Pathway Is Involved in EGR-1 Expression and Could Be Regulated by Elevated EGR-1
It has been reported that EGR-1 is regulated by various MAPK pathways, in particular the Erk1/2 pathway.33,39–43 Furthermore, recent studies have shown that CSE exerts its biological effects via the MAPK signaling pathway.42,43 We also found that CSE was able to significantly increase Erk1/2 phosphorylation within 10 to 30 minutes after exposure in Beas-2B cells (Figure 3A). When cells were treated with U0126, an inhibitor of MEK (a specific kinase that activates Erk1/2 MAPK), the CSE-induced EGR-1 expression was significantly decreased (Figure 3B). Meanwhile, we found that Erk1/2-activated EGR-1 was able to promote Erk1/2 reactivation. As shown in Figure 3C, overexpression of exogenous EGR-1 with adenovirus could mimic CSE stress to elevate Erk1/2 activation (Figure 3C). At the same time, inhibiting EGR-1 transcriptional activity with the dnEGR-1 adenovirus could block the phosphorylation of Erk1/2 after CSE stimulation for 4 hours (Figure 3D). These results suggest that the reactivation of Erk1/2 is regulated by CSE-induced EGR-1.
Figure 3.
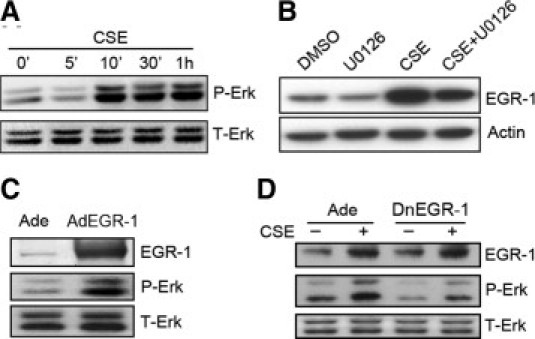
The Erk1/2 pathway is involved in early growth response 1 (EGR-1) expression and is regulated by elevated EGR-1. A: Beas-2B cells were exposed to cigarette smoke extract (CSE), and the samples were collected at the indicated times. Erk1/2 was phosphorylated within 10 minutes and lasted for at least 1 hour. B: Beas-2B cells were pretreated with the Erk1/2 activation inhibitor, U0126, for 2 hours before CSE exposure for another 1 hour and subjected to Western blot. The accumulation of EGR-1 was decreased by U0126. C: Beas-2B cells were infected with exogenous EGR-1 adenovirus or control adenovirus for 48 hours and subjected to Western blot. Overexpression of exogenous EGR-1 largely increased the phosphorylated Erk1/2. D: Beas-2B cells were infected with dominant-negative EGR-1 (dnEGR-1) adenovirus or control adenovirus for 48 hours. The protein samples were collected and subjected to Western blot after CSE exposure for 4 hours. The increase in Erk1/2 activity under CSE treatment was blocked by dnEGR-1 overexpression.
The Promotion of GGPPS Transcription by EGR-1 Is Critical to MAPK Erk1/2 Reactivation
When cells were treated with U0126, the level of GGPPS mRNA transcription significantly decreased (Figure 4A). This result implies that the early phase of Erk1/2 activation is also critical for GGPPS expression. As shown in Figure 4B, exogenous expression of GGPPS with adenovirus increased the phosphorylation of Erk1/2, like CSE and EGR-1 overexpression did. When we introduced GGPPS siRNA into cells (the efficiency of GGPPS siRNA is shown in Figure 4C), knockdown of GGPPS blocked EGR-1 overexpression–induced Erk1/2 activation (Figure 4D). These results suggest that Erk1/2 activity is regulated through the function of EGR-1 in promoting GGPPS expression.
Figure 4.
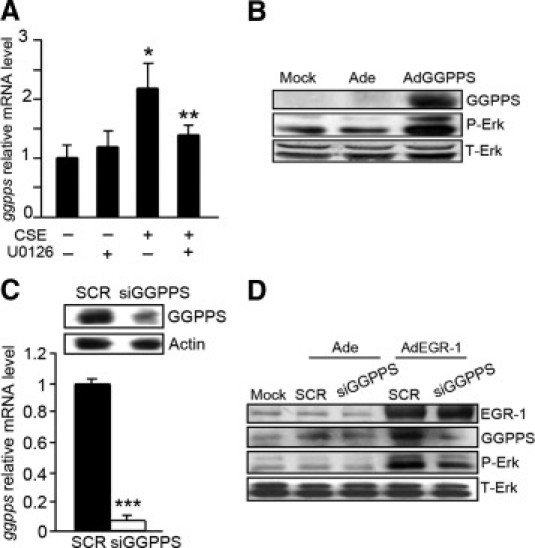
The promotion of geranylgeranyl diphosphate synthase (GGPPS) transcription by early growth response 1 (EGR-1) is critical to MAPK Erk1/2 reactivation. A: The mRNA level of GGPPS in Beas-2B cells. The cells were pretreated with or without U0126 for 2 hours before cigarette smoke extract (CSE) exposure. Sampling was performed after stimulation of cells with or without CSE for another 4 hours. The elevation of GGPPS mRNA by CSE is down-regulated by U0126. B: Beas-2B cells were infected with GGPPS adenovirus (AdGGPPS) or control adenovirus (Ade) for 48 hours. The cell lysates were collected and subjected to Western blot. The phosphorylation of Erk1/2 is enhanced by overexpression of GGPPS. C: Beas-2B cells were infected with GGPPS small-interfering RNA (siRNA) adenovirus (siGGPPS) or the scrambled siRNA adenovirus (SCR) and subjected to Western blot and quantitative PCR. D: Beas-2B cells were coinfected with EGR-1 adenovirus and with or without GGPPS siRNA adenovirus. GGPPS and Erk1/2 activation increases after EGR-1 overexpression, whereas it is blocked by GGPPS knockdown with siRNA. *P < .05 compared with the control group without any treatment, **P < 0.05 compared with CSE treatment only, ***P < .01 compared with the scrambled group.
The EGR-1/GGPPS/Erk1/2 Pathway Is Responsible for the Production of Proinflammatory Cytokines and an MMP
To determine the biological function of the EGR-1/GGPPS/Erk1/2 pathway, we checked the expression level of proinflammatory cytokines and an MMP, which are involved in EGR-1–mediated pulmonary disease development3–5 in Beas-2B cells after smoke treatment. We showed that CSE stress challenge stimulated the Beas-2B cells to synthesize the proinflammatory cytokines IL-8 (Figure 5A) and TNF-α (Figure 5B) (which promote inflammatory responses) and to express MMP-2 (Figure 5C) (which might be related to alveolar destruction during pulmonary fibrosis). When the transcriptional activity of EGR-1 was inhibited with dnEGR-1 and the expression of GGPPS was knocked down with siRNA, the synthesis of IL-8, TNF-α, and MMP-2 was significantly decreased. The results suggest that the EGR-1/GGPPS/Erk1/2 pathway might mediate the progression of cigarette smoke–related pulmonary disease.
Figure 5.
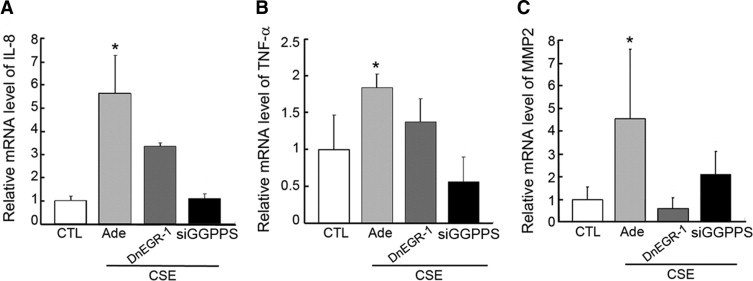
The EGR-1/GGPPS/Erk1/2 pathway is responsible for the production of proinflammatory cytokines and a matrix metalloproteinase (MMP). The expression level of interleukin (IL)-8 (A), tumor necrosis factor α (TNF-α) (B), and MMP-2 (C) in Beas-2B cells. Cells were infected with adenovirus (Ade) carrying either dominant-negative early growth response 1 (dnEGR-1) or geranylgeranyl diphosphate synthase (GGPPS) small-interfering RNA (siRNA) and then exposed to cigarette smoke extract (CSE) for 6 hours. Total RNA was extracted and subjected to quantitative PCR. CSE could stimulate Beas-2B cells to produce proinflammatory cytokines IL-8 and TNF-α, as well as the MMP-2. However, the role of CSE was reversed by overexpression of dnEGR-1 or GGPPS siRNA. *P < 0.05 compared with other shown groups.
Discussion
Although more and more people are developing COPD, the cause of this condition is complex and still incompletely understood. One of the key factors is the genetic susceptibility. A series of studies in diverse populations have shown a large genetic contribution to the variability in pulmonary function and COPD.44,45 Polymorphisms in multiple genes have been reported to be associated with COPD, such as microsomal epoxide hydrolase,46 vitamin D–binding protein,47 TNF-α,48 SERPINE2,49 and α-nicotinic acetylcholine receptor, such as neuronal acetylcholine receptor subunit alpha-3/5 (CHRNA 3/5).50,51 Notably, Chan et al2 reported that EGR-1 polymorphisms are significantly associated with plasma total IgE and atopy in asthmatic patients. Thus, EGR-1 may be a genetic susceptibility factor for COPD.
After long-term exposure to cigarette smoke, strains of mice with different susceptibilities for the development of smoke-induced emphysema have varied levels of EGR-1. EGR-1 is markedly increased in the lungs of the emphysema-susceptible AKR/J mice but only minimally increased in the resistant NZWLac/J mice.22 In the moderately susceptible C57BL/6 strain we used, both EGR-1 and its downstream target gene, GGPPS, increased in lungs after long-term exposure to cigarette smoke. Meanwhile, the inflammatory responses were augmented, which could be reversed by down-regulation of EGR-1 transcriptional activity. We also found that GGPPS is a crucial mediator of the proinflammatory function of EGR-1 because it may reactivate Erk1/2 through enhancing Ras prenylation (N. Shen, unpublished data). Thus, we provide the first evidence, to our knowledge, that EGR-1 can activate MAPK Erk1/2 in a positive feedback manner through promoting GGPPS transcription in lung epithelial cells when they are exposed to long-term constant cigarette smoke stimulation (Figure 6). This pathway can regulate the production of proinflammatory cytokines, such as IL-8 and TNF-α, as well as MMP-2, and all of these factors can promote chronic inflammation that causes airway abnormalities, emphysema, and consequently, COPD in susceptible smokers.
Figure 6.
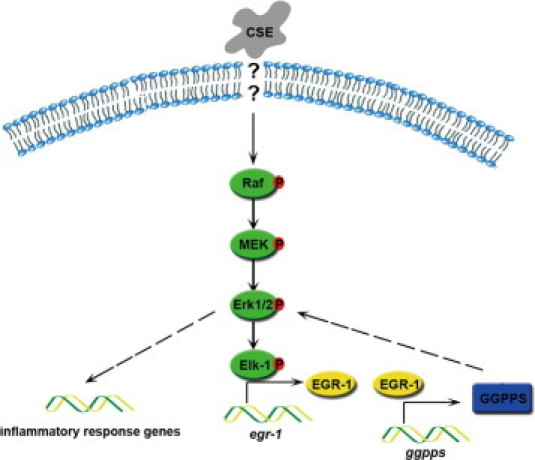
Model of the EGR-1/GGPPS/Erk1/2 pathway in the promotion of the inflammatory response during cigarette smoke exposure. Cigarette smoke extract (CSE) exposure leads to early growth response 1 (EGR-1) elevation by activating the Erk1/2 MAPK pathway. The up-regulated EGR-1 can activate transcription of its target gene geranylgeranyl diphosphate synthase (GGPPS), which is responsible for the reactivation of the Erk1/2 MAPK pathway. This positive feedback pathway can constantly activate Erk1/2, which is crucial for the inflammatory response.
Cigarette smoke–induced chronic inflammation has also long been viewed as central to the pathogenesis of COPD. The inhaled substances, such as cigarette smoke, cause an inflammatory response in the lung, which results from the oxidant–antioxidant imbalance and protease–antiprotease imbalance induced by oxidants or mutagenic substances in cigarette smoke.52 The balance of the protease–antiprotease system is regulated by EGR-1 in that it can directly activate the transcription of MMPs in lung fibroblasts, which may affect extracellular matrix turnover in the development of COPD-associated emphysema.3,4,53 Our report also suggests that EGR-1 can directly up-regulate heme oxygenase 1 transcription when C57BL/6 mice are exposed for long periods to cigarette smoke, to protect the lung from oxidant damage.54 Furthermore, the inflammatory response resulting from the oxidant–antioxidant imbalance and protease–antiprotease imbalance is also regulated by EGR-1. Cytokines such as IL-1β and TNF-α in pulmonary epithelial cells and IL-8 in human fibroblasts can be up-regulated by CSE via EGR-1 activation.5,22 Interestingly, an analysis of the promoters of these cytokines revealed no direct EGR-1 response elements, which suggests that the transcription of these cytokines requires indirect transcriptional regulation that is induced by EGR-1 through the activation of an additional signaling pathway.
GGPPS is a new target gene of EGR-1 that was identified and confirmed through chromatin immunoprecipitation and electrophoretic mobility shift assay methods (N. Shen, unpublished data). GGPPS is an enzyme that catalyzes its substrate for protein prenylation. Most proteins of the Ras-like GTPase superfamily need to be posttranslationally modified by prenyl groups to associate with cellular membranes and to activate their downstream effectors. The mouse lung chocolate mutation has been attributed to a G146T transversion in the conserved GTP/GDP-interacting domain of Rab38 (Rab38cht). The molecular basis for the mutant phenotype is defective membrane-binding activity of Rab38cht due to a failure in its prenyl modification but is not due to the loss of its GTP-binding activity.55 In addition, statin, which is an inhibitor of HMG-CoA reductase (a key upstream component of the mevalonate pathway), can inhibit CSE-mediated MMP-9 induction by blocking prenylation of Ras in MAPK pathways.56 The inhibition of geranylgeranyltransferase (GGTase) by geranylgeranyltransferase inhibitor (GGTI) is also effective in inhibiting the infiltration of inflammatory cells into airways in mouse experimental asthma models.57 Thus, prenylation of Ras can augment inflammatory responses in lung pathological processes. We found that EGR-1 could enhance Ras prenylation and membrane association in a GGPPS-dependent manner, and then it augments Erk1/2 activation in lung epithelial cells (N. Shen, unpublished data). Because EGR-1 activation is also dependent on Erk1/2, the enhancement of GGPPS transcription by EGR-1 can reactivate Erk1/2 in a positive feedback manner, which might affect cigarette smoke–related lung pathological processes.
EGR-1 has been termed an immediate-early response protein based on the brisk kinetics of its induction within minutes of a stimulus and on its rapid decay, often within hours.14 This kind of immediate-early response protein is usually thought to activate the expression of various target genes in response to stress. Many stress signals, such as osmotic pressure variation, heat shock, hypoxia, DNA-damaging agents, radiation, injury, and stretch, can stimulate EGR-1 expression. EGR-1 can also directly activate the transcription of many genes that are usually crucial for the proper functioning in a cell system. The mechanism that EGR-1 promoted GGPPS transcription is not specific to the cigarette smoke stress and may also be involved in other stresses, such as hyperinsulinism exposure to adipocytes that enhances insulin resistance development (N. Shen, unpublished data).
As an immediate-early gene, EGR-1 has been observed in cells responding not only to acute injury or environmental manipulation but also to prolonged long-term insults, such as cigarette smoke.4,5 We provide a molecular mechanism to explain the role of EGR-1 in chronic diseases such as emphysema.58 The short-term increase of EGR-1 in response to CSE can reactivate Erk1/2 by promoting GGPPS transcription in such a way that EGR-1 can reactivate the Erk1/2 as long as CSE or cigarette smoke stimulation occurs. It is well known that the activated Erk1/2 can promote chronic inflammation. For example, the release of IL-8 in human bronchial epithelial cells by acetylcholine is dependent on NF-κB/Erk.59 In addition, activated Erk1/2 is also involved in the formation of CSE-induced death-inducing signaling complex in human lung fibroblasts60 and results in sustained lung damage. Thus, we proposed a possible mechanism by which the long-term cigarette smoke exposure leads to severe inflammatory responses and EGR-1 feedback activation involving GGPPS-promoted Ras prenylation that leads to reactivation of Erk1/2 and thus enhances the imbalance of oxidant–antioxidant and/or protease–antiprotease that will damage lung structures.
In conclusion, our research reveals a new molecular mechanism by which EGR-1 orchestrates pulmonary cellular inflammatory responses to long-term exposure to cigarette smoke. EGR-1 is induced by short-term cigarette smoke exposure and leads to the increased secretion of proinflammatory cytokines by pulmonary epithelial cells. This process is often transient and reversible. However, when the cells are exposed to long-term constant cigarette smoke stimulation, the induced EGR-1 can intermittently activate MAPK Erk1/2 in a positive feedback manner by promoting GGPPS transcription. Continued activation of Erk1/2 can promote chronic inflammation through the production of the proinflammatory cytokines, IL-8, and TNF-α, as well as MMP-2, which might be involved in pulmonary abnormalities in susceptible smokers.
Footnotes
Supported by the National Basic Research Program of China (2009CB918703) Key Grant of Jiangsu Natural Science Foundation (06KJA31024 to C-J.L.); and by the Natural Science Foundation of Jiangsu Province of China (BK2007598 and 07KJB180053 to B.X.).
References
- 1.Mayer A.S., Newman L.S. Genetic and environmental modulation of chronic obstructive pulmonary disease. Respir Physiol. 2001;128:3–11. doi: 10.1016/s0034-5687(01)00258-4. [DOI] [PubMed] [Google Scholar]
- 2.Chan I.H.S., Tang N.L.S., Leung T.F., Huang W., Lam Y.Y.O., Wong G.W.K., Chan J.C.N., Chan M.H.M., Wong C.K., Zhang Y.P., Lam C.W.K. Association of early growth response-1 gene polymorphisms with total IgE and atopy in asthmatic children. Pediatr Allergy Immunol. 2009;20:142–150. doi: 10.1111/j.1399-3038.2008.00757.x. [DOI] [PubMed] [Google Scholar]
- 3.Ning W., Li C.J., Kaminski N., Feghali-Bostwick C.A., Alber S.M., Di Y.P., Otterbein S.L., Song R., Hayashi S., Zhou Z. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2004;101:14895–14900. doi: 10.1073/pnas.0401168101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ning W., Dong Y., Sun J., Li C., Matthay M.A., Feghali-Bostwick C.A., Choi A.M. Cigarette smoke stimulates matrix metalloproteinase-2 activity via EGR-1 in human lung fibroblasts. Am J Respir Cell Mol Biol. 2007;36:480–490. doi: 10.1165/rcmb.2006-0106OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C.J., Ning W., Matthay M.A., Feghali-Bostwick C.A., Choi A.M.K. MAPK pathway mediates EGR-1-HSP70-dependent cigarette smoke-induced chemokine production. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1297. doi: 10.1152/ajplung.00194.2006. [DOI] [PubMed] [Google Scholar]
- 6.Yan S.F., Lu J., Zou Y.S., Soh-Won J., Cohen D.M., Buttrick P.M., Cooper D.R., Steinberg S.F., Mackman N., Pinsky D.J., Stern D.M. Hypoxia-associated induction of early growth response-1 gene expression. J Biol Chem. 1999;274:15030–15040. doi: 10.1074/jbc.274.21.15030. [DOI] [PubMed] [Google Scholar]
- 7.Rolli M., Kotlyarov A., Sakamoto K.M., Gaestel M., Neininger A. Stress-induced stimulation of early growth response gene-1 by p38/stress-activated protein kinase 2 is mediated by a cAMP-responsive promoter element in a MAPKAP kinase 2-independent manner. J Biol Chem. 1999;274:19559–19564. doi: 10.1074/jbc.274.28.19559. [DOI] [PubMed] [Google Scholar]
- 8.O'Donovan K.J., Tourtellotte W.G., Millbrandt J., Baraban J.M. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 1999;22:167–173. doi: 10.1016/s0166-2236(98)01343-5. [DOI] [PubMed] [Google Scholar]
- 9.Liu C., Rangnekar V.M., Adamson E., Mercola D. Suppression of growth and transformation and induction of apoptosis by EGR-1. Cancer Gene Ther. 1998;5:3–28. [PubMed] [Google Scholar]
- 10.Lim C.P., Jain N., Cao X. Stress-induced immediate-early gene, egr-1, involves activation of p 38/JNK 1. Oncogene. 1998;16:2915–2926. doi: 10.1038/sj.onc.1201834. [DOI] [PubMed] [Google Scholar]
- 11.Huang R.P., Adamson E.D. A biological role for Egr-1 in cell survival following ultra-violet irradiation. Oncogene. 1995;10:467–475. [PubMed] [Google Scholar]
- 12.Datta R., Taneja N., Sukhatme V.P., Qureshi S.A., Weichselbaum R., Kufe D.W. Reactive oxygen intermediates target CC(A/T)6GG sequences to mediate activation of the early growth response 1 transcription factor gene by ionizing radiation. Proc Natl Acad Sci U S A. 1993;90:2419–2422. doi: 10.1073/pnas.90.6.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chavrier P., Zerial M., Lemaire P., Almendral J., Bravo R., Charnay P. A gene encoding a protein with zinc fingers is activated during G0/G1 transition in cultured cells. EMBO J. 1988;7:29–35. doi: 10.1002/j.1460-2075.1988.tb02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan S.F., Pinsky D.J., Mackman N., Stern D.M. Egr-1: is it always immediate and early. J Clin Invest. 2000;105:553. doi: 10.1172/JCI9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gashler A., Sukhatme V.P. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 16.Cao X.M., Guy G.R., Sukhatme V.P., Tan Y.H. Regulation of the Egr-1 gene by tumor necrosis factor and interferons in primary human fibroblasts. J Biol Chem. 1992;267:1345–1349. [PubMed] [Google Scholar]
- 17.Skerka C., Decker E.L., Zipfel P.F. A regulatory element in the human interleukin 2 gene promoter is a binding site for the zinc finger proteins Sp1 and EGR-1. J Biol Chem. 1995;270:22500–22506. doi: 10.1074/jbc.270.38.22500. [DOI] [PubMed] [Google Scholar]
- 18.Kramer B., Meichle A., Hensel G., Charnay P., Kronke M. Characterization of an Krox-24/Egr-1-responsive element in the human tumor necrosis factor promoter. Biochim Biophys Acta. 1994;1219:413–421. doi: 10.1016/0167-4781(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 19.Lin J.X., Leonard W.J. The immediate-early gene product Egr-1 regulates the human interleukin-2 receptor beta-chain promoter through noncanonical Egr and Sp1 binding sites. Mol Cell Biol. 1997;17:3714–3722. doi: 10.1128/mcb.17.7.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinkel A., Aicher W.K., Haas C., Zipfel P.F., Peter H.H., Eibel H. Transcription factor Egr-1 activity down-regulates Fas and CD23 expression in B cells. J Immunol. 1997;159:2678–2684. [PubMed] [Google Scholar]
- 21.Maltzman J.S., Carmen J.A., Monroe J.G. Transcriptional regulation of the Icam-1 gene in antigen receptor- and phorbol ester-stimulated B lymphocytes: role for transcription factor EGR1. J Exp Med. 1996;183:1747–1759. doi: 10.1084/jem.183.4.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynolds P.R., Cosio M.G., Hoidal J.R. Cigarette smoke-induced Egr-1 upregulates proinflammatory cytokines in pulmonary epithelial cells. Am J Respir Cell Mol Biol. 2006;35:314–319. doi: 10.1165/rcmb.2005-0428OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Z.-H., Kim H.P., Sciurba F.C., Lee S.-J., Feghali-Bostwick C., Stolz D.B., Dhir R., Landreneau R.J., Schuchert M.J., Yousem S.A., Nakahira K., Pilewski J.M., Lee J.S., Zhang Y., Ryter S.W., Choi A.M.K. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE. 2008;3:e3316. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rilling H.C., Breunger E., Epstein W.W., Crain P.F. Prenylated proteins: the structure of the isoprenoid group. Science. 1990;247:318–320. doi: 10.1126/science.2296720. [DOI] [PubMed] [Google Scholar]
- 25.Farnsworth C.C., Gelb M.H., Glomset J.A. Identification of geranylgeranyl-modified proteins in HeLa cells. Science. 1990;247:320–322. doi: 10.1126/science.2296721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farnsworth C.C., Wolda S.L., Gelb M.H., Glomset J.A. Human lamin B contains a farnesylated cysteine residue. J Biol Chem. 1989;264:20422–20429. [PMC free article] [PubMed] [Google Scholar]
- 27.Plowman S.J., Hancock J.F. Ras signaling from plasma membrane and endomembrane microdomains. Biochim Biophys Acta. 2005;1746:274–283. doi: 10.1016/j.bbamcr.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Wright L.P., Philips M.R. Thematic review series: lipid Posttranslational modifications: CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47:883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Black L.F., Kueppers F. alpha1-Antitrypsin deficiency in nonsmokers. Am Rev Respir Dis. 1978;117:421–428. doi: 10.1164/arrd.1978.117.3.421. [DOI] [PubMed] [Google Scholar]
- 30.Gadek J.E., Fells G.A., Crystal R.G. Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humans. Science. 1979;206:1315–1316. doi: 10.1126/science.316188. [DOI] [PubMed] [Google Scholar]
- 31.Hunninghake G.W., Crystal R.G. Cigarette smoking and lung destruction: Accumulation of neutrophils in the lungs of cigarette smokers. Am Rev Respir Dis. 1983;128:833–838. doi: 10.1164/arrd.1983.128.5.833. [DOI] [PubMed] [Google Scholar]
- 32.Janus E.D., Phillips N.T., Carrell R.W. Smoking, lung function, and alpha 1-antitrypsin deficiency. Lancet. 1985;1:152–154. doi: 10.1016/s0140-6736(85)91916-6. [DOI] [PubMed] [Google Scholar]
- 33.Baron V., Duss S., Rhim J., Mercola D. Antisense to the early growth response-1 gene (Egr-1) inhibits prostate tumor development in TRAMP mice. Ann N Y Acad Sci. 2003;1002:197–216. doi: 10.1196/annals.1281.024. [DOI] [PubMed] [Google Scholar]
- 34.Yu H.-W., Liu Q.-F., Liu G.-N. Positive regulation of the Egr-1/osteopontin positive feedback loop in rat vascular smooth muscle cells by TGF-[beta], ERK, JNK, and p38 MAPK signaling. Biochem Biophys Res Commun. 2010;396:451–456. doi: 10.1016/j.bbrc.2010.04.115. [DOI] [PubMed] [Google Scholar]
- 35.Allen K., Kim N.D., Moon J.-O.K., Copple B.L. Upregulation of early growth response factor-1 by bile acids requires mitogen-activated protein kinase signaling. Toxicol Appl Pharmacol. 2010;243:63–67. doi: 10.1016/j.taap.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vayssier-Taussat M., Camilli T., Aron Y., Meplan C., Hainaut P., Polla B.S., Weksler B. Effects of tobacco smoke and benzo[a]pyrene on human endothelial cell and monocyte stress responses. Am J Physiol Heart Circ Physiol. 2001;280:H1293-–H1300. doi: 10.1152/ajpheart.2001.280.3.H1293. [DOI] [PubMed] [Google Scholar]
- 37.Witschi H., Uyeminami D., Moran D., Espiritu I. Chemoprevention of tobacco-smoke lung carcinogenesis in mice after cessation of smoke exposure. Carcinogenesis. 2000;21:977–982. doi: 10.1093/carcin/21.5.977. [DOI] [PubMed] [Google Scholar]
- 38.Levkovitz Y., Baraban J.M. A dominant negative inhibitor of the Egr family of transcription regulatory factors suppresses cerebellar granule cell apoptosis by blocking c-Jun activation. J Neurosci. 2001;21:5893–5901. doi: 10.1523/JNEUROSCI.21-16-05893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mishra S., Fujita T., Lama V.N., Nam D., Liao H., Okada M., Minamoto K., Yoshikawa Y., Harada H., Pinsky D.J. Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci U S A. 2006;103:5191–5196. doi: 10.1073/pnas.0600241103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease. Cell Biochem Biophys. 2005;43:167–188. doi: 10.1385/CBB:43:1:167. [DOI] [PubMed] [Google Scholar]
- 41.McNeil L.K., Starr T.K., Hogquist K.A. A requirement for sustained ERK signaling during thymocyte positive selection in vivo. Proc Natl Acad Sci U S A. 2005;102:13574–13579. doi: 10.1073/pnas.0505110102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy L.O., MacKeigan J.P., Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol. 2004;24:144–153. doi: 10.1128/MCB.24.1.144-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mercer B.A., Kolesnikova N., Sonett J., D'Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem. 2004;279:17690–17696. doi: 10.1074/jbc.M313842200. [DOI] [PubMed] [Google Scholar]
- 44.McCloskey S.C., Patel B.D., Hinchliffe S.J., Reid E.D., Wareham N.J., Lomas D.A. Siblings of patients with severe chronic obstructive pulmonary disease have a significant risk of airflow obstruction. Am J Respir Crit Care Med. 2001;164:1419–1424. doi: 10.1164/ajrccm.164.8.2105002. [DOI] [PubMed] [Google Scholar]
- 45.Molfino N.A. Genetics of COPD. Chest. 2004;125:1929–1940. doi: 10.1378/chest.125.5.1929. [DOI] [PubMed] [Google Scholar]
- 46.Christopher A.D.S., David J.H. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet. 1997;350:630–633. doi: 10.1016/S0140-6736(96)08061-0. [DOI] [PubMed] [Google Scholar]
- 47.Schellenberg D., Pare P.D., Weir T.D., Spinelli J.J., Walker B.A.M., Sandford A.J. Vitamin D binding protein variants and the risk of COPD. Am J Respir Crit Care Med. 1998;157:957–961. doi: 10.1164/ajrccm.157.3.9706106. [DOI] [PubMed] [Google Scholar]
- 48.Huang S.-L., Su C.-H., Chang S.-C. Tumor necrosis factor-alpha gene polymorphism in chronic bronchitis. Am J Respir Crit Care Med. 1997;156:1436–1439. doi: 10.1164/ajrccm.156.5.9609138. [DOI] [PubMed] [Google Scholar]
- 49.Zhu G., Warren L., Aponte J., Gulsvik A., Anderson W.H., Lomas D.A., Silverman E.K., Pillai S.G., International COPD Genetics Network (ICGN) Investigators The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am J Respir Crit Care Med. 2007;176:167–173. doi: 10.1164/rccm.200611-1723OC. [DOI] [PubMed] [Google Scholar]
- 50.Spitz M.R., Amos C.I., Dong Q., Lin J., Wu X. The CHRNA5-A3 region on chromosome 15q24-25.1 is a risk factor both for nicotine dependence and for lung cancer. J Natl Cancer Inst. 2008;100:1552–1556. doi: 10.1093/jnci/djn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pillai S.G., Ge D., Zhu G., Kong X., Shianna K.V., Need A.C., Feng S., Hersh C.P., Bakke P., Gulsvik A., Ruppert A., Lødrup Carlsen K.C., Roses A., Anderson W., Rennard S.I., Lomas D.A., Silverman E.K., Goldstein D.B. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacNee W. Oxidative stress and lung inflammation in airways disease. Eur J Pharmacol. 2001;429:195–207. doi: 10.1016/s0014-2999(01)01320-6. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Y.K., Liu X., Ertl R.F., Kohyama T., Wen F.Q., Wang H., Spurzem J.R., Romberger D.J., Rennard S.I. Retinoic acid attenuates cytokine-driven fibroblast degradation of extracellular matrix in three-dimensional culture. Am J Respir Cell Mol Biol. 2001;25:620–627. doi: 10.1165/ajrcmb.25.5.4495. [DOI] [PubMed] [Google Scholar]
- 54.Chen H., Wang L., Gong T., Yu Y., Zhu C., Li F., Wang L., Li C. EGR-1 regulates Ho-1 expression induced by cigarette smoke. Biochem Biophys Res Commun. 2010;396:388–393. doi: 10.1016/j.bbrc.2010.04.102. [DOI] [PubMed] [Google Scholar]
- 55.Osanai K., Oikawa R., Higuchi J., Kobayashi M., Tsuchihara K., Iguchi M., Jongsu H., Toga H., Voelker D.R. A mutation in Rab38 small GTPase causes abnormal lung surfactant homeostasis and aberrant alveolar structure in mice. Am J Pathol. 2008;173:1265–1274. doi: 10.2353/ajpath.2008.080056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim S.E., Thuy Than T.T., Lee J.H., Ro J.Y., Bae Y.A., Kong Y., Ahn J.Y., Lee D.S., Oh Y.M., Lee S.D., Lee Y.S. Simvastatin inhibits induction of matrix metalloproteinase-9 in rat alveolar macrophages exposed to cigarette smoke extract. Exp Mol Med. 2009;41:277–287. doi: 10.3858/emm.2009.41.4.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiba Y., Sato S., Misawa M. GGTI-2133, an inhibitor of geranylgeranyltransferase, inhibits infiltration of inflammatory cells into airways in mouse experimental asthma. Int J Immunopathol Pharmacol. 2009;22:929–935. doi: 10.1177/039463200902200408. [DOI] [PubMed] [Google Scholar]
- 58.Zhang W., Yan S.D., Zhu A., Zou Y.S., Williams M., Godman G.C., Thomashow B.M., Ginsburg M.E., Stern D.M., Yan S.-F. Expression of Egr-1 in late stage emphysema. Am J Pathol. 2000;157:1311–1320. doi: 10.1016/S0002-9440(10)64646-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Profita M., Bonanno A., Siena L., Ferraro M., Montalbano A.M., Pompeo F., Riccobono L., Pieper M.P., Gjomarkaj M. Acetylcholine mediates the release of IL-8 in human bronchial epithelial cells by a NFkB/ERK-dependent mechanism. Eur J Pharmacol. 2008;582:145–153. doi: 10.1016/j.ejphar.2007.12.029. [DOI] [PubMed] [Google Scholar]
- 60.Park J.W., Yoon J.Y., Kim Y.J., Kyung S.Y., Lee S.P., Jeong S.H., Moon C. Extracellular signal-regulated kinase (ERK) inhibition attenuates cigarette smoke extract (CSE) induced-death inducing signaling complex (DISC) formation in human lung fibroblasts (MRC-5) cells. J Toxicol Sci. 2010;35:33–39. doi: 10.2131/jts.35.33. [DOI] [PubMed] [Google Scholar]