

Abstract
Recent results call for the reexamination of the phenotype of wound macrophages and their role in tissue repair. These results include the characterization of distinct circulating monocyte populations with temporally restricted capacities to migrate into wounds and the observation that the phenotype of macrophages isolated from murine wounds partially reflects those of their precursor monocytes, changes with time, and does not conform to current macrophage classifications. Moreover, findings in genetically modified mice lacking macrophages have confirmed that these cells are essential to normal wound healing because their depletion results in retarded and abnormal repair. This mini-review focuses on current knowledge of the phenotype of wound macrophages, their origin and fate, and the specific macrophage functions that underlie their reparative role in injured tissues, including the regulation of the cellular infiltration of the wound and the production of transforming growth factor-β and vascular endothelial growth factor.
The resolution of injury through replacement by scar has been preserved in evolution and applies to virtually all soft tissues in mammals. A healed wound is the result of the activities of cells that constitute what can be conceptually described as a wound organ. This transient neoorgan is assembled at the time and site of injury and disassembled once repair is complete, leaving in place a scar as evidence of the repair process. The parenchyma of the wound organ is composed of a temporally changing assembly of inflammatory cells, neovessels, fibroblasts and myofibroblasts, regenerating nerves, and other cells specific to the site of the injury (eg, keratinocytes, osteocytes, and hepatocytes).
Two recent developments led to this review of wound macrophages. First, studies in both humans and rodents have demonstrated the existence of diverse subpopulations of circulating monocytes with, at least in mice, distinct and time-restricted abilities to migrate into wounds and other sites of tissue injury (Fig. 1). These immediate precursors of wound macrophages are required for repair by scarring in organs as diverse as skin, heart, liver, and kidney. Second, the concept that macrophages involved in tissue repair express the alternatively activated phenotype first proposed by Gordon1 has been retained in the literature.2 Results from studies of macrophages isolated from wounds, however, contest this paradigm by demonstrating that they are not alternatively activated. Wound macrophages exhibit phenotypes that partially reflect those of their monocyte precursors, change with time, and fail to be adequately described by macrophage classifications based on activation profiles or functional capacity (Fig. 2).3
Figure 1.
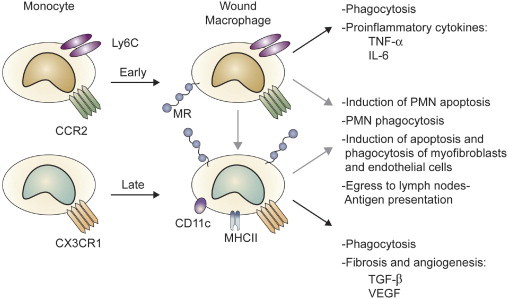
Blood monocyte and wound macrophage phenotypes in the mouse. Macrophages in wounds originate mainly from circulating monocytes. The figure depicts the two subpopulations of murine blood monocytes that are distinguished by their expression of Ly-6C and chemokine receptors CCR2 and CX3CR1. Ly-6ChighCCR2high monocytes are recruited into wounds early after injury and are known to produce proinflammatory cytokines and to clear wound debris by phagocytosis. Ly-6ClowCX3CR1high monocytes subsequently infiltrate wounds and express TGF-β and VEGF. There is conflicting evidence in the literature as to whether Ly-6ChighCCR2high cells can become Ly-6ClowCX3CR1high in the wound. Functions specifically associated with early or late wound macrophages are linked by a black arrow. Other wound macrophage functions not yet clearly assigned to either macrophage population are indicated with a gray arrow. MHCII, major histocompatibility complex type 2; MR, mannose receptor-1; PMN, polymorphonuclear leukocytes.
Figure 2.
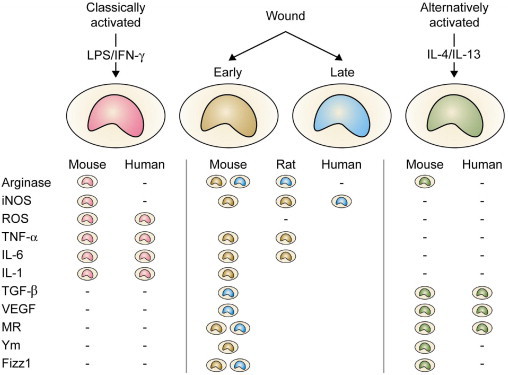
Wound macrophage phenotypes. The figure depicts a simplified view of the polarization of macrophages toward classic and alternative activation phenotypes to illustrate the concept that wound macrophages share traits with both phenotypes. As discussed in the text, the phenotype of wound macrophages changes as the wound heals. This temporal variability is represented in the table by color-coded symbols indicating selected phenotypic traits of early versus late wound macrophages. Dash marks signify the phenotype marker is not expressed. Table cells are empty when there are no data to confirm or deny expression. Inducible nitric oxide synthase (iNOS) is expressed by macrophages in sterile rat wounds, in infected murine wounds, and in chronic and infected human wounds. Reactive oxygen species (ROS) are produced through the NADPH-dependent respiratory burst oxidase. Heparin-binding proteins (Ym1 and Ym2) are induced in alternatively activated murine macrophages during helminthic infections. Fizz1 (found in inflammatory zone 1) is also known as hypoxia-induced mitogenic factor or Relm-α. LPS, lipopolysaccharide; MR, mannose receptor-1.
This review addresses the origin and fate of wound macrophages, their regulatory interactions with other cellular populations in the wound, and the roles of macrophage-derived transforming growth factor-β (TGF-β) and vascular endothelial growth factor (VEGF) in the regulation of wound fibrosis and neovascularization, respectively.
Origin of Wound Macrophages
Wound macrophages, which derive mostly from circulating monocytes, are recruited to the site of injury. Monocytes rapidly acquire certain macrophage phenotypic traits on arrival at the wound (ie, mannose receptor-13), while retaining others present in circulating cells (ie, Ly-6C and dectin-13). Recent work has revealed the existence of at least two distinct monocyte subpopulations in mice.4 Inflammatory monocytes, characterized as Ly-6ChighCCR2highCX3CR1low, migrate into sites of inflammation, including wounds, during the early phase of the response to injury.3,5 A second population of murine monocytes, defined as Ly-6ClowCCR2lowCX3CR1high and originally thought to be the precursors of tissue-resident macrophages,6 egress the circulation into wounds and other sites of inflammation after the Ly-6ChighCCR2high CX3CR1low cells. CX3CR1 is expressed in murine skin wounds by infiltrating macrophages, myofibroblasts, and endothelial cells, and its ligand CX3CL1 (fractalkine) is found in wound macrophages and endothelial cells. Cutaneous wound healing is retarded in CX3CR1 knockout animals, where wounds exhibit reduced numbers of macrophages and myofibroblasts and decreased levels of TGF-β1 and VEGF.7
The roles of CCR2 and CX3CR1 in the recruitment of monocytes into sites of inflammation in the mouse are not restricted to skin wounds. A study of experimental myocardial infarction in mice confirmed that both populations of monocytes migrate into the area of ischemic injury and, more importantly, that they do so in a temporally segregated fashion, with Ly-6ChighCCR2highCX3CR1low monocytes predominating during the first 3 days after coronary artery ligation and Ly-6ClowCCR2lowCX3CR1high cells increasing thereafter. Ly-6Chigh cells produce proinflammatory cytokines and require the engagement of CCR2 for recruitment into the ischemic lesion. The Ly-6ClowCCR2lowCX3CR1high macrophages release VEGF and are needed for collagen accumulation in the infarct.5 The sequential infiltration by Ly-6Chigh and Ly-6Clow monocytes/macrophages was also found in the authors' laboratory in murine soft tissue wounds, along with evidence that wound macrophages isolated 1 day after wounding predominantly produce proinflammatory cytokines (tumor necrosis factor-α [TNF-α] and interleukin-6 [IL-6]), whereas those harvested from 7-day-old wounds release more TGF-β.3
Although Ly-6ClowCCR2lowCX3CR1high monocytes/macrophages are essential for the fibrotic response to infarction, elimination of the initial infiltrate of Ly-6ChighCCR2highCx3CR1low cells also results in decreased collagen deposition.5 It appears that Ly-6ChighCCR2high CX3CR1low cells set the stage for the repair of the injury by scarring. Supporting this view, the inactivation of CCR2 or CX3CR1 by either genetic or pharmacological means suppresses macrophage infiltration and the development of hepatic, renal, and pulmonary fibrosis in a variety of murine models.8–11
The expression of Ly-6C provides a useful marker to distinguish different populations of murine monocytes and macrophages. Its function in these cells remains unknown. Ly-6C belongs to a family of cell surface phosphatidylinositol-anchored glycoproteins expressed in varying degrees in leukocytes and some endothelial cells.12 No in vivo ligands for Ly-6 family members have been identified. Because these molecules lack transmembrane or intracellular domains, a variety of transduction mechanisms for Ly-6 have been proposed, including binding to intracellular Src tyrosine kinases through cytoskeletal proteins in cells of monocytic lineage.12,13 Antibody cross-linking of Ly-6C in CD8+ lymphocytes results in integrin activation and enhances adherence to endothelia.13 A similar function in monocytes could regulate their transvascular migration into wounds, but this possibility has not been reported.
Although strong evidence supports the roles of CCR2/CCL2 and CX3CR1/CX3CL1 in the sequential recruitment of distinct macrophage precursors into wounds and other inflammatory lesions,7,14 it appears that a more complex signal network is required for the normal migration of monocytes into wounds. In this regard, an antiserum against CCL3 (macrophage inflammatory protein-1α, a chemokine that signals through chemokine receptors CCR1, 3, 5, and 9) decreased macrophage accumulation in wounds.15 In addition, a variety of genetic manipulations, including deletions of TNF-α receptor p55, β-1,4 galactosyltransferase, P/E selectin, or intercellular adhesion molecule-1, result in reduced recruitment of macrophages to wounds and, with the exception of the TNF-α receptor p55 knockouts, in impaired wound healing.16
The relative roles of monocyte-derived and resident tissue macrophages (such as osteoclasts, pulmonary alveolar macrophages, microglia, and tissue histiocytes) in the repair of specific organs are not completely defined. However, there is evidence of cooperation between blood-derived and tissue macrophages in wound healing. For example, resident Kupffer cells participate in the fibrotic response to a variety of hepatic insults by secreting cytokines and growth factors and by activating hepatic stellate cells, but the extent of fibrosis is markedly reduced in CCR2 knockouts unable to recruit blood monocytes into the injured liver.8
Distinct populations of monocytes have also been described in humans, where they are distinguished by the expression of CD16.4 CD16+ cells, which normally constitute 5% to 15% of circulating monocytes, express high levels of CX3CR1 and may be the ortholog of Ly-6ClowCX3CR1high murine monocytes. The normally more abundant CD16− monocytes are CX3CR1low and resemble Ly-6Chigh murine monocytes in their expression of CCR2. A third population of human monocytes expressing both CD14 and CD16 has been shown to comprise at least 2 subpopulations with distinct capacities to release TNF-α and IL-1 in response to lipopolysaccharide. The function of these CD14+CD16+ cells remains unclear, but their frequency in the circulation increases in septic diseases.4 Whether human monocytes parallel their murine counterparts in their capacity to migrate into wounds in a temporally restricted manner and have differential secretory and functional profiles at a site of injury remains to be investigated.
Fate of Wound Macrophages
Macrophages disappear from wounds during the resolution of the inflammatory phase of repair. Some die at the wound, as evidenced by the appearance of intracellular macrophage molecules in the wound extracellular fluid (most notably macrophage arginase I in rat wounds).17 Others are likely to migrate to draining lymph nodes. Although this possibility has not been reported specifically for wound macrophages, evidence provided by studies of the resolution of cellular infiltrates in other models of inflammation suggests its likelihood.18,19 Whether these emigrating cells are true macrophages or monocyte-derived dendritic cells remains a topic in need of further investigation.20–22
Wound Macrophages and the Alternatively Activated Phenotype
The capacity of macrophages to exhibit a variety of phenotypic profiles in response to different stimuli has been used for their classification into distinct functional categories.23 In its most simplified version, the current paradigm holds that macrophages polarize either to a cytotoxic and microbicidal phenotype after stimulation with interferon-γ and lipopolysaccharide (the classically activated macrophage) or to an alternatively activated phenotype that is associated with parasitic infections and allergic reactions and induced by IL-4/IL-13.1 Because these extreme phenotypes are not expressed in pure form in vivo, recent publications have embraced the view that macrophage activation is best described by a continuous spectrum of phenotypic characteristics.2 The concept that wound macrophages express the alternatively activated phenotype has, however, been retained.2,24 Furthermore, it has been proposed that alternatively activated macrophages be reclassified as wound healing macrophages.2 This recommendation is not supported by studies of macrophages directly isolated from wounds. Findings from this laboratory in sterile murine wounds demonstrated that wound macrophages exhibit a mixed and temporally dependent phenotype with traits of both classically and alternatively activated cells and that neither IL-4 nor IL-13, the proposed determinants of the wound healing macrophage phenotype, are detectable in murine deep tissue wounds.3
The expression of the enzyme arginase by alternatively activated1 or wound healing2 macrophages has been singled out as central to their putative role in tissue repair. Arginases catalyze the degradation of arginine to ornithine and urea. Ornithine can, in turn, be metabolized to proline, an amino acid that is abundant in collagens. The demonstration of extracellular arginase activity in rat wounds led this laboratory to propose that ornithine-derived proline could promote or facilitate collagen synthesis by increasing its availability to fibroblasts and myofibroblasts.17 However, there is no direct evidence that native proline is limiting to collagen synthesis in wounds and, thus, that ornithine-derived proline is essential to repair. In fact, rat wound fibroblasts preferentially incorporate preformed, rather than ornithine-derived, proline into collagen.25 Moreover, native proline suppresses the use of ornithine-derived proline during collagen synthesis by fibroblasts. Providing further evidence against arginase as a necessary regulator of collagen synthesis, a recent report demonstrated a paradoxical increase in fibrosis after infection with Schistosoma mansoni in animals with macrophage-specific deletion of arginase I.26
Most importantly, arginase I is not expressed in human monocytes or macrophages, even after polarization toward classic or alternatively activated phenotypes.27,28 Contrasting the macrophage-restricted expression of arginase I in rodent wounds,17 arginase has been found in neutrophils in acute human wounds and in fibroblasts in chronic wounds and ulcers but not in macrophages.29,30 Arginase I is also expressed in human, but not in rodent, erythrocytes,31 and lysed red blood cells contribute arginase to human wounds. Arginase activity is highest in postmastectomy wound fluids within 24 hours of surgery and correlates with the hemoglobin concentration in the fluids (unpublished observations). The potential role of mammalian red blood cell arginase I in early inflammation and repair has not been identified. Arginase expression by wound macrophages is, thus, species restricted and probably not essential for normal collagen deposition in wounds.
Macrophage-Neutrophil Interactions in Wounds
Macrophages control the cellularity of wounds through their capacity to induce apoptosis and phagocytose a variety of wound cells. Macrophage-targeted cells include neutrophils during the inflammatory phase of repair and fibroblasts and endothelial cells during its resolution.
Tissue injury is followed by the sequential infiltration of the wound by neutrophils and macrophages. The number of neutrophils decreases as the wound matures and macrophages become the predominant inflammatory cell. Neutrophils undergo apoptosis in the wound and are recognized and ingested by macrophages.32,33 In addition, rat wound macrophages actively induce apoptosis in neutrophils.34 The induction of neutrophil apoptosis requires the expression of membrane-bound TNF-α (but not soluble TNF-α), β3-integrins, and CD36 by the effector macrophages. Neither resident nor Propionibacterium acnes–elicited peritoneal macrophages induced apoptosis in neutrophils, indicating this ability to be particular to wound macrophages.34
The phagocytosis of apoptotic neutrophils or other cells has been shown to induce an anti-inflammatory phenotype in macrophages. This phenotype includes the release of TGF-β and prostaglandin E2 and a reduced ability to produce proinflammatory mediators, such as TNF-α, after lipopolysaccharide stimulation.35 Because Ly-6Chigh wound macrophages produce TNF-α and Ly-6Clow murine monocytes are more efficient than Ly-6Chigh cells in the ingestion of apoptotic cells,36 it appears reasonable to propose that Ly-6Chigh cells induce apoptosis in wound neutrophils, whereas Ly-6Clow cells remove postapoptotic debris from the wound space.
Studies in CD18 knockout mice that model for leukocyte adhesion deficiency syndrome 1, a disorder characterized by recurrent infections and impaired wound healing in humans, have provided further information regarding macrophage/neutrophil interactions in wounds.37 Skin wounds in these animals failed to recruit neutrophils, assembled a normal complement of macrophages, and exhibited severe retardation of closure by contraction. These alterations occurred in the context of increased proinflammatory cytokine accumulation, reduced active TGF-β1 and its type II receptor, and a remarkable scarcity of myofibroblasts. It was proposed from these findings that the lack of apoptotic neutrophils in the wounds of knockout animals deprived macrophages of phagocytic substrate, resulting in reduced production of TGF-β1, a main regulator of myofibroblast differentiation.37
Additional work in mice demonstrated that wound neutrophils release lipid-soluble low-molecular-weight factors that suppress the production of proinflammatory mediators by macrophages.38 The picture that emerges from these studies is that wound neutrophils, by serving as phagocytic targets and by releasing soluble pacifying mediators, actively modulate the wound macrophage phenotype.
Macrophages and the Regulation of Wound Healing
The concept that macrophages are unique among blood-borne components of the wound organ in being essential to normal repair was until recently based on findings in guinea pigs where macrophage depletion via antimacrophage antiserum and glucocorticoids reduced the rate of cutaneous wound healing.39–41 The multiple mechanisms by which adrenal steroids can alter wound healing raised obvious questions as to the macrophage specificity of these early observations. Three recent reports using genetic means to deplete monocytes and macrophages in mice have now provided strong confirmatory evidence for the essential role of these cells in normal repair. The elimination of macrophages via the administration of diphtheria toxin to transgenic mice expressing the diphtheria toxin receptor under the control of the lysozyme M24,41 or the CD11b promoter40 resulted in substantial disruption of the repair process and in reductions of wound healing. The alterations in the process of repair in these animals most prominently include decreases in wound reepithelialization and contraction, which probably result from reductions in myofibroblast differentiation, and in neovascularization and granulation tissue formation associated with diminished VEGF and TGF-β accumulation in the wounds.
The experimental design used by Lucas et al24 using the aforementioned transgenic mice further defined the contribution of macrophages to the healing process. The authors administered diphtheria toxin at different time points in relation to excisional skin wounding to deplete macrophages during the initial inflammatory response to injury (first 4 days after injury), the intermediate phase of granulation tissue formation (4–10 days after injury), or during the consolidation of fibrosis (10–14 days after injury). Depletion during the initial phase resulted in severely altered wound healing with reductions in all components of the healing response, including granulation tissue formation, epithelialization, and scar deposition. Macrophage ablation during the intermediate period was associated with wound hemorrhage, increased apoptosis of neovascular endothelial cells, and collapse of the developing vascular structure that characterizes granulation tissue formation. The authors ascribed these alterations to reductions in TGF-β and VEGF in the macrophage-depleted wounds. Macrophage ablation once the fibrotic phase of repair was ongoing was without effect.
Macrophages synthesize and release a vast array of regulatory molecules relevant to wound healing.42 The intricacy and redundancy of the macrophage synthetic repertoire are in clear contrast with the relative frugality of the endocrine system, where single molecules such as insulin can regulate cellular metabolism, growth, and differentiation. In a comprehensive review of the regulation of wound healing by growth factors and cytokines, Werner and Grose42 listed 18 families of signaling molecules associated with different aspects of repair. Each mediator family comprises multiple individual molecules, each of which may exist as multiple isoforms with broadly overlapping activities. The authors identified 11 different mediators of angiogenesis, eight regulators of matrix deposition, and 17 molecules that control reepithelialization. Given that multiple cell types in the wound organ are capable of producing the relevant cytokines and growth factors, assigning the production of bioactive mediators in wounds specifically to macrophages and then correlating these mediators with the critical role of these cells in repair are particularly difficult. The discussion that follows focuses on TGF-β and VEGF because both are known to be produced by wound macrophages and are essential to repair.
Wound Macrophages and the Regulation of Fibrosis
Inflammation is a prerequisite for healing with a scar. Evidence linking TGF-β production by wound macrophages and scar formation is convincing. Cutaneous wounds in embryos before the development of the myeloid system do not elicit an inflammatory response, lack TGF-β, and heal without scar.43 Moreover, TGF-β signaling–deficient SMAD3 knockouts recruit macrophages poorly into skin wounds and heal without scarring.44 These observations are not restricted to cutaneous wounds. Lung injury in SMAD3 knockouts results in decreased procollagen mRNA expression and fibrosis, and the overexpression of TGF-β1 increases fibrosis in wild-type but not in SMAD3-deficient mice.45 Because wound macrophages have been demonstrated to produce and release TGF-β, this growth factor appears to fulfill the criteria of a bona fide wound macrophage-derived mediator contributing to wound healing.3
Recent results obtained from wounds in macrophage-depleted mice support the aforementioned conclusion. Mizra et al40 described decreased TGF-β1 mRNA and protein, along with reduced collagen accumulation in wounds in macrophage-deficient mice. Lucas et al24 confirmed the reduction in TGF-β1 protein by histochemistry in macrophage-depleted animals and correlated this reduction with the inability of the mice to deposit granulation tissue and to form a normal scar. The authors propose, in addition, that TGF-β1 is required for the constitution of a normal vascular network in the healing wound and support this conclusion by demonstrating alterations in animals deficient in TGF-β receptor II in myeloid cells that are strikingly similar to those in the macrophage-ablated mice.24
Goren et al,41 also using macrophage-depleted mice, found reductions in the number of myofibroblasts and in the rate of wound contraction in macrophage-depleted animals, despite normal TGF-β bioactivity. The discrepancy between findings by these authors and those reported by Lucas et al24 and Mirza et al40 on the expression of TGF-β in very similar models of macrophage depletion and injury may be explained by an uneven distribution of this growth factor in areas critically proximal to sites of myofibroblast differentiation in the wound.
The production of TGF-β by wound macrophages increases as Ly-6Clow macrophages accumulate in murine wounds, and CX3CR1 expression correlates with the extent of fibrosis in murine kidney and heart.3,5,10 Two single-nucleotide polymorphisms coding for nonsynonymous amino acid substitutions in CX3CR1 in humans are associated with increased binding of CX3CL1 to its receptor and correlate with increased hepatic fibrosis in chronic hepatitis C and with a fibrostenosing form of Crohn disease.46–48 Moreover, increased numbers of CX3CR1high monocytes/macrophages have been found in areas of cutaneous or pulmonary inflammation and fibrosis in patients with systemic sclerosis.49
Just as macrophages appear to be required for the initiation of scarring, they are also involved in matrix remodeling and in the resolution of fibrosis. Duffield et al50 reported that macrophage ablation during the phase of fibrotic development after the administration of carbon tetrachloride resulted in decreased collagen deposition and myofibroblast accumulation in the liver. In contrast, depleting macrophages during the resolution of fibrosis retarded and reduced the disappearance of scar tissue.50 Because macrophages can induce apoptosis and ingest fibroblasts, myofibroblasts, and vascular endothelial cells, they can regulate the transition between the granulation phase of wound healing and the subsequent deposition of an almost avascular scar.51,52
Wound Macrophages and Neovascularization
Evidence obtained in macrophage-deficient mice confirmed the essentiality of macrophages to the neovascularization of wounds. Mirza et al,40 Goren et al,41 and Lucas et al24 found reduced and abnormal vascularization in their models of macrophage-depleted wounds.
Neovascularization during granulation tissue formation in wounds is controlled by a variety of cytokines and growth factors.42 Members of the VEGF family and their receptors have been shown to be prominently expressed in animal and human wounds and to be produced there mainly by macrophages and keratinocytes.53 Treatment with anti-VEGF A antibodies resulted in marked reductions in neovascularization and granulation tissue formation in porcine wounds,54 and an anti-VEGF antibody neutralized the angiogenic activity of human wound fluids in vitro.55
A connection between Ly-6ClowCX3CR1high wound macrophages and angiogenesis was described by Nahrendorf et al,5 who showed that these cells produce VEGF and that their elimination results in reduced neovascularization of ischemic myocardium in mice. Confirmatory results were reported by others in CX3CR1 knockout mice.7 Wounds in these mice have reduced neovessel density, as well as less VEGF mRNA and protein, than in wild-type animals. In contrast, CX3CR1-deficient animals subjected to corneal injuries exhibited increased vascularization, along with decreased local production of antiangiogenic mediators thrombospondin 1 and 2 and their activator ADAMTS-1 (a disintegrin and metalloproteinase with thrombospondin motif-1).56 It is possible that the role of wound macrophages in angiogenesis varies with injury site. It is more likely that the cells provide proangiogenic and antiangiogenic signals in wounds and that the balance of these signals regulates the early angiogenic response during granulation tissue formation and its resolution as the scar matures.
Looking Ahead
Results just discussed argue against the concept that wound macrophages express one repair phenotype, which is identical to that of the alternatively activated macrophage. A new perspective into the nature of wound macrophages has emerged from the discovery that distinct populations of circulating monocytes migrate into areas of injury in a time-restricted manner. This alone supports the view that wound macrophages change over time and express temporally restricted phenotypes that instruct other wound cells through the different phases of repair.
It is likely that a number of variables known to affect wound healing, including wound infection, comorbidities (eg, diabetes, malnutrition, and cancer), and even the age of the wounded subject, may do so by affecting the wound macrophage. Further work is needed to examine wound macrophages in these and other physiological and pathological conditions.
In addition, the phenotype of human wound macrophages has been underexplored. The substantial differences reported by this and other laboratories between human and murine macrophage phenotypes, and even between wound macrophages of mice and rats, suggest that findings in rodents may have limited parallels in human wound macrophages. The application of cutting-edge methods (eg, laser capture microdissection followed by genomic and proteomic analysis of macrophages isolated from human wound biopsies), or alternative models that allow for the recovery and study of substantial numbers of human wound macrophages (ie, wounds in humanized mice), should provide the means for the characterization of these cells.
Acknowledgments
We thank Jonathan Reichner for a very helpful critique of the manuscript and Patricia Young for excellent secretarial support.
Footnotes
Supported by National Institutes of Health grant T32GM-65085; and funds from the Department of Surgery, Rhode Island Hospital/Lifespan, and the Alpert Medical School of Brown University (S.K.B.); by National Institutes of Health grant RO1GM-42859; and by funds allocated to the Department of Surgery by Rhode Island Hospital, a Lifespan partner (J.E.A.).
None of the authors disclosed any relevant financial relationships.
References
- 1.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 2.Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daley J.M., Brancato S.K., Thomay A.A., Reichner J.S., Albina J.E. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auffray C., Sieweke M.H., Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 5.Nahrendorf M., Swirski F.K., Aikawa E., Stangenberg L., Wurdinger T., Figueiredo J.L., Libby P., Weissleder R., Pittet M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geissmann F., Jung S., Littman D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 7.Ishida Y., Gao J.L., Murphy P.M. Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J Immunol. 2008;180:569–579. doi: 10.4049/jimmunol.180.1.569. [DOI] [PubMed] [Google Scholar]
- 8.Seki E., de Minicis S., Inokuchi S., Taura K., Miyai K., van Rooijen N., Schwabe R.F., Brenner D.A. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50:185–197. doi: 10.1002/hep.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L., Huang L., Sung S.S., Vergis A.L., Rosin D.L., Rose C.E., Jr, Lobo P.I., Okusa M.D. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;74:1526–1537. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furuichi K., Gao J.L., Murphy P.M. Chemokine receptor CX3CR1 regulates renal interstitial fibrosis after ischemia-reperfusion injury. Am J Pathol. 2006;169:372–387. doi: 10.2353/ajpath.2006.060043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gharaee-Kermani M., McCullumsmith R.E., Charo I.F., Kunkel S.L., Phan S.H. CC-chemokine receptor 2 required for bleomycin-induced pulmonary fibrosis. Cytokine. 2003;24:266–276. doi: 10.1016/j.cyto.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Gumley T.P., McKenzie I.F., Sandrin M.S. Tissue expression, structure and function of the murine Ly-6 family of molecules. Immunol Cell Biol. 1995;73:277–296. doi: 10.1038/icb.1995.45. [DOI] [PubMed] [Google Scholar]
- 13.Jaakkola I., Merinen M., Jalkanen S., Hanninen A. Ly6C induces clustering of LFA-1 (CD11a/CD18) and is involved in subtype-specific adhesion of CD8 T cells. J Immunol. 2003;170:1283–1290. doi: 10.4049/jimmunol.170.3.1283. [DOI] [PubMed] [Google Scholar]
- 14.Dipietro L.A., Reintjes M.G., Low Q.E., Levi B., Gamelli R.L. Modulation of macrophage recruitment into wounds by monocyte chemoattractant protein-1. Wound Repair Regen. 2001;9:28–33. doi: 10.1046/j.1524-475x.2001.00028.x. [DOI] [PubMed] [Google Scholar]
- 15.DiPietro L.A., Burdick M., Low Q.E., Kunkel S.L., Strieter R.M. MIP-1alpha as a critical macrophage chemoattractant in murine wound repair. J Clin Invest. 1998;101:1693–1698. doi: 10.1172/JCI1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eming S.A., Krieg T., Davidson J.M. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127:514–525. doi: 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- 17.Albina J.E., Mills C.D., Henry W.L., Jr, Caldwell M.D. Temporal expression of different pathways of 1-arginine metabolism in healing wounds. J Immunol. 1990;144:3877–3880. [PubMed] [Google Scholar]
- 18.Bellingan G.J., Caldwell H., Howie S.E.M., Dransfield I., Haslett C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J Immunol. 1996;157:2577–2585. [PubMed] [Google Scholar]
- 19.Harmsen A.G., Muggenburg B.A., Snipes M.B., Bice D.E. The role of macrophages in particle translocation from lungs to lymph nodes. Science. 1985;230:1277–1280. doi: 10.1126/science.4071052. [DOI] [PubMed] [Google Scholar]
- 20.Randolph G.J., Inaba K., Robbiani D.F., Steinman R.M., Muller W.A. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 21.Randolph G.J. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr Opin Lipidol. 2008;19:462–468. doi: 10.1097/MOL.0b013e32830d5f09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dominguez P.M., Ardavin C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev. 2010;234:90–104. doi: 10.1111/j.0105-2896.2009.00876.x. [DOI] [PubMed] [Google Scholar]
- 23.Stout R.D., Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucas T., Waisman A., Ranjan R., Roes J., Krieg T., Muller W., Roers A., Eming S.A. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 25.Albina J.E., Abate J.A., Mastrofrancesco B. Role of ornithine as a proline precursor in healing wounds. J Surg Res. 1993;55:97–102. doi: 10.1006/jsre.1993.1114. [DOI] [PubMed] [Google Scholar]
- 26.Pesce J.T., Ramalingam T.R., Mentink-Kane M.M., Wilson M.S., El Kasmi K.C., Smith A.M., Thompson R.W., Cheever A.W., Murray P.J., Wynn T.A. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009;5:e1000371. doi: 10.1371/journal.ppat.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munder M., Mollinedo F., Calafat J., Canchado J., Gil-Lamaignere C., Fuentes J.M., Luckner C., Doschko G., Soler G., Eichmann K., Muller F.M., Ho A.D., Goerner M., Modolell M. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 2005;105:2549–2556. doi: 10.1182/blood-2004-07-2521. [DOI] [PubMed] [Google Scholar]
- 28.Raes G., Van den Bergh R., De Baetselier P., Ghassabeh G.H., Scotton C., Locati M., Mantovani A., Sozzani S. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol. 2005;174:6561. doi: 10.4049/jimmunol.174.11.6561. [DOI] [PubMed] [Google Scholar]
- 29.Debats I.B., Wolfs T.G., Gotoh T., Cleutjens J.P., Peutz-Kootstra C.J., van der Hulst R.R. Role of arginine in superficial wound healing in man. Nitric Oxide. 2009;21:175–183. doi: 10.1016/j.niox.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Jude E.B., Boulton A.J., Ferguson M.W., Appleton I. The role of nitric oxide synthase isoforms and arginase in the pathogenesis of diabetic foot ulcers: possible modulatory effects by transforming growth factor beta 1. Diabetologia. 1999;42:748–757. doi: 10.1007/s001250051224. [DOI] [PubMed] [Google Scholar]
- 31.Spector E.B., Rice S.C., Kern R.M., Hendrickson R., Cederbaum S.D. Comparison of arginase activity in red blood cells of lower mammals, primates, and man: evolution to high activity in primates. Am J Hum Genet. 1985;37:1138–1145. [PMC free article] [PubMed] [Google Scholar]
- 32.Metchnikoff E. Masson; Paris: 1892. Leçons sur la pathologie comparée de l'inflammation. [Google Scholar]
- 33.Meszaros A.J., Reichner J.S., Albina J.E. Macrophage phagocytosis of wound neutrophils. J Leukoc Biol. 1999;65:35–42. doi: 10.1002/jlb.65.1.35. [DOI] [PubMed] [Google Scholar]
- 34.Meszaros A.J., Reichner J.S., Albina J.E. Macrophage-induced neutrophil apoptosis. J Immunol. 2000;165:435–441. doi: 10.4049/jimmunol.165.1.435. [DOI] [PubMed] [Google Scholar]
- 35.Savill J., Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 36.Peng Y., Latchman Y., Elkon K.B. Ly6C(low) monocytes differentiate into dendritic cells and cross-tolerize T cells through PDL-1. J Immunol. 2009;182:2777–2785. doi: 10.4049/jimmunol.0803172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters T., Sindrilaru A., Hinz B., Hinrichs R., Menke A., Al-Azzeh E.A., Holzwarth K., Oreshkova T., Wang H., Kess D., Walzog B., Sulyok S., Sunderkotter C., Friedrich W., Wlaschek M., Krieg T., Scharffetter-Kochanek K. Wound-healing defect of CD18(-/-) mice due to a decrease in TGF-beta1 and myofibroblast differentiation. EMBO J. 2005;24:3400–3410. doi: 10.1038/sj.emboj.7600809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daley J.M., Reichner J.S., Mahoney E.J., Manfield L., Henry W.L., Jr, Mastrofrancesco B., Albina J.E. Modulation of macrophage phenotype by soluble product(s) released from neutrophils. J Immunol. 2005;174:2265–2272. doi: 10.4049/jimmunol.174.4.2265. [DOI] [PubMed] [Google Scholar]
- 39.Leibovich S.J., Ross R. The role of the macrophage in wound repair: a study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71–100. [PMC free article] [PubMed] [Google Scholar]
- 40.Mirza R., DiPietro L.A., Koh T.J. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175:2454–2462. doi: 10.2353/ajpath.2009.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goren I., Allmann N., Yogev N., Schurmann C., Linke A., Holdener M., Waisman A., Pfeilschifter J., Frank S. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol. 2009;175:132–147. doi: 10.2353/ajpath.2009.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Werner S., Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 43.Buchanan E.P., Longaker M.T., Lorenz H.P. Fetal skin wound healing. Adv Clin Chem. 2009;48:137–161. doi: 10.1016/s0065-2423(09)48006-5. [DOI] [PubMed] [Google Scholar]
- 44.Ashcroft G.S., Roberts A.B. Loss of Smad3 modulates wound healing. Cytokine Growth Factor Rev. 2000;11:125–131. doi: 10.1016/s1359-6101(99)00036-2. [DOI] [PubMed] [Google Scholar]
- 45.Bonniaud P., Kolb M., Galt T., Robertson J., Robbins C., Stampfli M., Lavery C., Margetts P.J., Roberts A.B., Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 46.Daoudi M., Lavergne E., Garin A., Tarantino N., Debre P., Pincet F., Combadiere C., Deterre P. Enhanced adhesive capacities of the naturally occurring Ile249-Met280 variant of the chemokine receptor CX3CR1. J Biol Chem. 2004;279:19649–19657. doi: 10.1074/jbc.M313457200. [DOI] [PubMed] [Google Scholar]
- 47.Wasmuth H.E., Zaldivar M.M., Berres M.L., Werth A., Scholten D., Hillebrandt S., Tacke F., Schmitz P., Dahl E., Wiederholt T., Hellerbrand C., Berg T., Weiskirchen R., Trautwein C., Lammert F. The fractalkine receptor CX3CR1 is involved in liver fibrosis due to chronic hepatitis C infection. J Hepatol. 2008;48:208–215. doi: 10.1016/j.jhep.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Brand S., Hofbauer K., Dambacher J., Schnitzler F., Staudinger T., Pfennig S., Seiderer J., Tillack C., Konrad A., Goke B., Ochsenkuhn T., Lohse P. Increased expression of the chemokine fractalkine in Crohn's disease and association of the fractalkine receptor T280M polymorphism with a fibrostenosing disease phenotype. Am J Gastroenterol. 2006;101:99–106. doi: 10.1111/j.1572-0241.2005.00361.x. [DOI] [PubMed] [Google Scholar]
- 49.Hasegawa M., Sato S., Echigo T., Hamaguchi Y., Yasui M., Takehara K. Up regulated expression of fractalkine/CX3CL1 and CX3CR1 in patients with systemic sclerosis. Ann Rheum Dis. 2005;64:21–28. doi: 10.1136/ard.2003.018705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duffield J.S., Forbes S.J., Constandinou C.M., Clay S., Partolina M., Vuthoori S., Wu S., Lang R., Iredale J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Desmouliere A., Redard M., Darby I., Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 52.Diez-Roux G., Lang R.A. Macrophages induce apoptosis in normal cells in vivo. Development. 1997;124:3633–3638. doi: 10.1242/dev.124.18.3633. [DOI] [PubMed] [Google Scholar]
- 53.Brown L.F., Yeo K.T., Berse B., Yeo T.K., Senger D.R., Dvorak H.F., van de Water L. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med. 1992;176:1375–1379. doi: 10.1084/jem.176.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howdieshell T.R., Callaway D., Webb W.L., Gaines M.D., Procter C.D., Jr, Sathyanarayana, Pollock J.S., Brock T.L., McNeil P.L. Antibody neutralization of vascular endothelial growth factor inhibits wound granulation tissue formation. J Surg Res. 2001;96:173–182. doi: 10.1006/jsre.2001.6089. [DOI] [PubMed] [Google Scholar]
- 55.Nissen N.N., Polverini P.J., Koch A.E., Volin M.V., Gamelli R.L., DiPietro L.A. Vascular endothelial growth factor mediates angiogenic activity during the proliferative phase of wound healing. Am J Pathol. 1998;152:1445–1452. [PMC free article] [PubMed] [Google Scholar]
- 56.Lu P., Li L., Liu G., van Rooijen N., Mukaida N., Zhang X. Opposite roles of CCR2 and CX3CR1 macrophages in alkali-induced corneal neovascularization. Cornea. 2009;28:562–569. doi: 10.1097/ICO.0b013e3181930bcd. [DOI] [PubMed] [Google Scholar]