

Abstract
X-linked myotubular myopathy (XLMTM) is a congenital disorder caused by deficiency of the lipid phosphatase, myotubularin. Patients with XLMTM often have severe perinatal weakness that requires mechanical ventilation to prevent death from respiratory failure. Muscle biopsy specimens from patients with XLMTM exhibit small myofibers with central nuclei and central aggregations of organelles in many cells. It was postulated that therapeutically increasing muscle fiber size would cause symptomatic improvement in myotubularin deficiency. Recent studies have elucidated an important role for the activin-receptor type IIB (ActRIIB) in regulation of muscle growth and have demonstrated that ActRIIB inhibition results in significant muscle hypertrophy. To evaluate whether promoting muscle hypertrophy can attenuate symptoms resulting from myotubularin deficiency, the effect of ActRIIB-mFC treatment was determined in myotubularin-deficient (Mtm1δ4) mice. Compared with wild-type mice, untreated Mtm1δ4 mice have decreased body weight, skeletal muscle hypotrophy, and reduced survival. Treatment of Mtm1δ4 mice with ActRIIB-mFC produced a 17% extension of lifespan, with transient increases in weight, forelimb grip strength, and myofiber size. Pathologic analysis of Mtm1δ4 mice during treatment revealed that ActRIIB-mFC produced marked hypertrophy restricted to type 2b myofibers, which suggests that oxidative fibers in Mtm1δ4 animals are incapable of a hypertrophic response in this setting. These results support ActRIIB-mFC as an effective treatment for the weakness observed in myotubularin deficiency.
X-linked myotubular myopathy (XLMTM) is a severe form of congenital myopathy with an estimated incidence of 1 in 50,000 male births, and most often manifests with severe perinatal weakness and respiratory failure.1,2 Many patients with XLMTM die of the disease within the first year of life despite use of mechanical ventilation, and there are no US Food and Drug Administration–approved treatments for this disease. XLMTM is caused by mutations in the gene encoding myotubularin (MTM1), which is a phosphoinositide phosphatase thought to be involved in endosomal trafficking and/or maintenance of the sarcoplasmic reticulum and transverse tubular (T-tubular) system within myofibers.3–6 Muscle biopsy specimens from patients with XLMTM exhibit excessively small fibers with increased numbers of central nuclei and aggregation of organelles within the central regions of many cells.7 While the number of centrally nucleated fibers bears little relationship to prognosis, there is a clear correlation between the degree of fiber smallness and severity of the disease.8 A murine model of myotubularin deficiency, the Mtm1δ4 mouse (also referred to as Mtm1 KO in previous studies),3,9,10 demonstrates features similar to those in human beings with the disease, including severe weakness, respiratory failure, and histologic findings that include myofiber smallness and increased numbers of centrally nucleated fibers.10
The relationship between myofiber size and symptomatic severity in patients with XLMTM and Mtm1δ4 mice suggests that therapies that increase fiber size may lead to symptomatic improvement. Myostatin (formerly termed “growth differentiation factor 8”) is a protein of the transforming growth factor beta (TGF-β) superfamily that is selectively expressed in skeletal muscle, cardiac muscle, and adipose tissue during late embryogenesis and adulthood and is an important negative regulator of myofiber size.11 Myostatin binds to and signals through the activin type IIB receptor (ActRIIB) to activate the TGF-β pathway, which prevents progression through the cell cycle and down-regulates several key processes related to myofiber hypertrophy.12,13 Examples of myostatin deficiency found in sheep,14 mice,11 cattle, dogs, and one human child have all exhibited generalized muscular hypertrophy and increased strength,13 and compared with mice with intact myostatin expression, null mice for myostatin or ActRIIB demonstrate myofiber hypertrophy and hyperplasia.15,16 There seems to be myostatin-independent suppression of muscle growth that is mediated by ActRIIB. Treatment with a soluble ActRIIB-mFC increases muscle mass in MTm1δ4 mice,16 which supports the role of multiple ligands that control muscle growth postnatally and suggests that targeting ActRIIB rather than myostatin alone may provide additional therapeutic benefit. The potential for myostatin inhibition to promote muscle growth has led to development of a new class of myostatin and ActRIIB inhibitors as prospective therapeutic agents for myopathic, dystrophic, and neurologic disorders. A soluble activin-receptor type IIB fusion protein (ActRIIB-mFC) has been developed that potently binds to TGF-β family members to produce muscle fiber growth in vitro and in vivo17. Recent studies using ActRIIB-mFC in murine models of neurologic disorders including amyotrophic lateral sclerosis and spinal muscular atrophy have demonstrated variable effects on muscle strength and no effects on animal survival.18,19 The effectiveness of this agent for treatment of primary disorders of skeletal muscle is being investigated.
Myostatin and ActRIIB inhibitors are currently being evaluated for treatment of several neuromuscular disorders, but have not yet been considered for treatment of any congenital myopathies. It is believed that they hold considerable promise for treatment of XLMTM because of the direct relationship between fiber size and prognosis in affected patients. To evaluate the effectiveness of ActRIIB inhibition on myotubularin-deficient myofibers, wild-type and Mtm1δ4 mice were treated with ActRIIB-mFC, and disease progression was evaluated both behaviorally and pathologically. Treatment in Mtm1δ4 mice caused a significant extension of lifespan in this severely affected model of the disease, with transient increases in weight and forelimb grip strength. Pathologic analysis of muscles harvested while therapy was most effective revealed marked increases in muscle bulk and muscle weight, and dramatic hypertrophy of type 2b myofibers.
Materials and Methods
Live Animal Studies
All studies were performed with approval from the institutional animal care and utilization committee at Children's Hospital Boston (Boston, MA). MTM1/HSA mice20 were a gift from Anna Buj-Bello and colleagues at the Université de Strasbourg, Collège de France (Illkirch, France), and were subsequently back-crossed onto a C57BL6 background. Genotyping of these MTM1/C57BL6 mice (Mtm1δ4) was performed as previously described.20 Male wild-type and Mtm1δ4 mice were injected semiweekly beginning at 14 days of life with ActRIIB-mFC (also called RAP-031; Acceleron Pharma Inc., Cambridge, MA) at a dose of 20 mg/kg. This dosage was chosen because it was above the level at which muscle hypertrophy plateaued in wild-type mice (data not shown). Vehicle-treated animals were injected with an equivalent volume of Tris-buffered saline solution. Treatment was continued until Mtm1δ4 animals were judged to be at end stage, and wild-type animals were sacrificed at equivalent time points. Behavior of wild-type and Mtm1δ4 animals was evaluated throughout the treatment period, and they were considered at end stage when they had either lost 20% of their highest body mass measurement or demonstrated complete inability to use their hindlimbs. The age at which Mtm1δ4 animals reached end stage was closely tracked to enable construction of Kaplan-Meier survival curves using commercially available software (Prism 4; GraphPad Software, Inc., San Diego, CA). Animals were weighed five times per week during the treatment period. For statistical analysis, running averages of the animals' weight over 3 days were calculated to provide daily measurements of animal weight. Forelimb grip strength was measured weekly using a Chatillon grip force meter (Columbus Instruments International, Columbus, OH) by placing the animal on a horizontal grid and allowing it to pull away from the experimenter by using only its forelimbs. The average of three independent measurements, with a 1-minute recovery period between measurements, was used for subsequent statistical analysis. Antigravity hanging performance was tested three times weekly by placing the animals on a rigid mesh surface, inverting the surface at approximately 40 cm above their cage, and recording the amount of time necessary for the animal to fall back into the cage. Animals that did not fall within 60 seconds were lowered back into their cages. The maximum of three independent measurements, with a 1-minute recovery period between measurements, was used for subsequent statistical analysis. Footprint analysis was performed weekly by immersion of an animal's feet in nontoxic ink and allowing the animal to walk across an 8.5 × 11-inch piece of paper contained in an acrylic safety glass (Plexiglas) walkway. Footsteps and foot drags were counted, and the ratio of foot drags to foot steps was calculated for subsequent statistical analysis.21
Statistical Analysis
Statistical analysis was performed using commercially available software (Prism 4; GraphPad, Inc.). For statistical analysis of animal weight, forelimb grip strength, antigravity hanging performance, and foot drag, analysis of variance was performed, with Bonferroni posttests. For measurement of muscle weight and mean myofiber diameter, one-way analysis of variance was performed, with Bonferroni posttests. For survival data, statistical significance was evaluated using a log-rank test.
Western Blot
Tissues from the quadriceps, gastrocnemius, and triceps muscles and from elsewhere in the forelimbs and hindlimbs were frozen at necropsy and stored at −80°C until analysis. Protein isolation and Western blot procedures were performed as previously described.22 Transferred proteins were probed with antibodies against myostatin (MAB788, 1:250 dilution; R&D Systems, Inc., Minneapolis, MN) and GAPDH [glyceraldehyde-3-phosphate dehydrogenase (6C5), 1:10,000 dilution; Abcam PLC, Cambridge, MA] and visualized using enhanced chemiluminescence. Adequacy of transfer was determined using Ponceau S staining. Quantification of protein levels normalized to GAPDH was performed using the program QuantityOne, version 4.2.1 (Bio-Rad Laboratories, Inc., Hercules, CA) on an Image Station 440 (Kodak DS; Eastman Kodak Co., Rochester, NY).
Pathologic Evaluation and Tissue Collection
Animals were euthanized using CO2 followed by cervical dislocation, per the regulations of the institutional animal care and utilization committee at Children's Hospital Boston. Animals were photographed externally and after removal of the skin from the torso and limbs. The quadriceps, gastrocnemius, triceps, soleus, extensor digitorum longus, tibialis anterior, and diaphragm muscles were removed and weighed. For subsequent ultrastructural studies, a small portion of the quadriceps muscle was fixed in 5% glutaraldehyde, 2.5% paraformaldehyde, and 0.06% picric acid in 0.2 mmol/L of cacodylate buffer, pH 7.4.
Histologic Evaluation
Eight-micrometer cross sections of isopentane-frozen quadriceps muscle were obtained midway down the length of the muscle and stained with H&E for evaluation using an Eclipse 50i microscope (Nikon Instruments Inc., Melville, NY). Light microscopic images were captured using a SPOT Insight 4 Meg FW Color Mosaic camera and SPOT 4.5.9.1 software (Diagnostic Instruments Inc., Sterling Heights, MI).
For immunofluorescence studies, 8-μm frozen transverse sections of quadriceps muscle were double stained with rabbit antidystrophin antibodies (ab15277, 1:100; Abcam PLC) and mouse monoclonal antibodies against myosin heavy chain type 1 (Skeletal, Slow, clone NOQ7.5.4D, 1:100 dilution; Sigma Aldrich, St. Louis, MO) or types 2a (clone SC-71, 1:50 dilution) or 2b (clone BF-F3, 1:50 dilution; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City). Secondary antibodies included fluorescein isothiocyanate–conjugated anti-mouse IgG or IgM (both 1:100; Sigma-Aldrich) and AlexaFluor-conjugated anti-rabbit IgG (1:50; Molecular Probes, Carlsbad, CA). Staining was evaluated using a Nikon Eclipse 90i microscope using NIS-Elements AR software (Nikon Instruments Inc.). For morphometric evaluation and estimation of fiber number, nonoverlapping fields of muscle immunostained for dystrophin were photographed using a Nikon Plan Fluor 4×/0.13 objective (Nikon Instruments Inc.). Fibers that stained positive were individually selected using NIS-Elements AR software, and fibers were automatically measured with respect to their minimum Feret (MinFeret) diameter. The MinFeret diameter is the smallest diameter across an ellipse, which corresponds to the “greatest distance between the opposite sides of the narrowest aspect of the fiber,”23 which is a measurement commonly used in morphometric analyses. An adequate number of fibers were counted to ensure measurements representative of the overall specimen, which involved a larger number of fibers counted in Mtm1δ4 animals (mean, 755; range, 238–1294 fibers) than in wild-type animals (mean, 340; range, 230–416 fibers) because of the small fiber size and the regional variability seen in the Mtm1δ4 animals. All MinFeret diameters for a given specimen were pooled for generation of frequency histograms and estimation of total number of fibers within the quadriceps muscle. In addition, the mean fiber MinFeret diameter for each specimen was calculated for subsequent statistical analysis.
For electron microscopy, fixed tissue was subjected to osmication, stained using uranyl acetate, dehydrated in alcohols, and embedded in TAAB Epon (Marivac Ltd., Halifax, Nova Scotia, Canada). Subsequently, 1-μm scout sections were stained with toluidine blue, and evaluated and photographed as described. Areas of interest were cut at 95-nm thickness using an ultracut microtome (Leica Camera AG, Solms, Germany), picked up on 100-m formvar-coated copper grids, stained with 0.2% lead citrate, and viewed and imaged using a Tecnai BioTwin Spirit Electron Microscope (FEI Co., Hillsboro, OR).
Results
Myostatin Expression in Mtm1δ4 Mice
The relative levels of myostatin were investigated in untreated wild-type and Mtm1δ4 animals to ensure that the dosage would be adequate to inhibit the circulating myostatin in Mtm1δ4 mice. Similar amounts of myostatin protein are produced in Mtm1δ4 and wild-type mice (Figure 1) at 43 days of life, although the levels observed in individual wild-type mice varied markedly. The presence of similar levels of myostatin in wild-type and Mtm1δ4 mice suggests that ActRIIB-mFC could induce effective myofiber hypertrophy in Mtm1δ4 animals at dosages that are effective in wild-type mice.17,18
Figure 1.
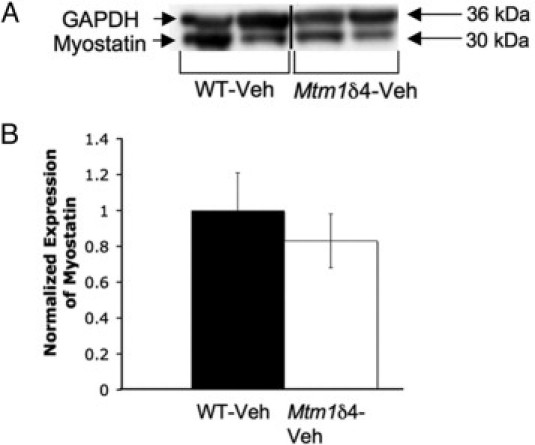
Myostatin expression in wild-type and Mtm1δ4 mice. A: Western blot analysis performed on the triceps muscle of 43 day-old animals demonstrates similar levels of myostatin expression in Mtm1δ4 mice and their wild-type littermates. These are nonadjacent lanes from the same immunoblot and using the same exposure time, as denoted by the vertical line. B: Expression of myostatin normalized to GAPDH expression in vehicle-treated wild-type and Mtm1δ4 mice at 43 days of life. Numbers are shown in reference to the mean value for the wild-type animals, and reflect the mean (SEM) value of 3 animals for each genotype.
Weight Studies
In animals that received semiweekly injections of Tris-buffered saline solution (the ActRIIB-mFC vehicle), Mtm1δ4 animals were distinguishable from age-matched wild-type animals on the basis of weight at 20 days of life (P < 0.05) (Figure 2A). These differences increased with age because of continued weight gain in wild-type animals in comparison with the plateau observed after 34 days of life in Mtm1δ4 animals. Compared with vehicle-treated mice, wild-type animals treated with semiweekly injections of ActRIIB-mFC, 20 mg/kg beginning at 14 days of life, showed significant weight gain after 36 days of life (P < 0.05), and continued to gain weight with age. ActRIIB-mFC treated Mtm1δ4 animals initially exhibited a modest sustained weight gain, reaching a maximum of 124% at 53 days of life; however, the weight of these animals plateaued quickly and did not increase as observed in wild-type animals.
Figure 2.
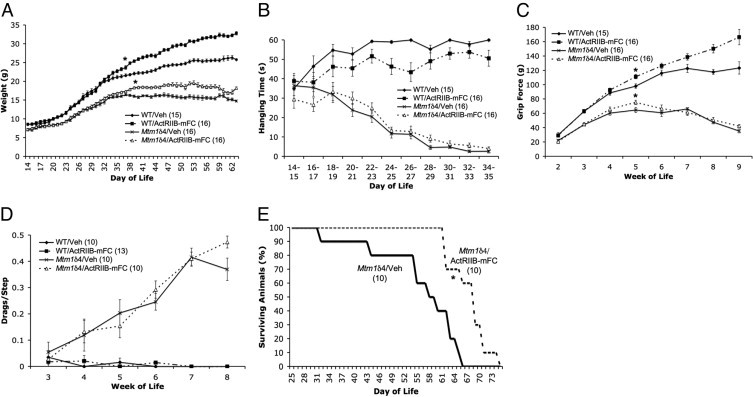
Behavioral findings in vehicle- and ActRIIB-mFC–treated mice. A: Body weight of vehicle- and ActRIIB-mFC–treated mice. Measurements are the running mean (SEM) of (n) animals in each treatment group. B: Antigravity hanging performance of vehicle- and ActRIIB-mFC–treated mice. C: Forelimb grip force of vehicle- and ActRIIB-mFC–treated mice. D: Measurements are mean (SEM) values of (n) animals in each treatment group. E: Kaplan-Meier survival curves for vehicle- (Veh) and ActRIIB-mFC–treated Mtm1δ4 mice. *P < 0.05.
Antigravity Hanging Performance
At antigravity hanging assay, in which animals are suspended from a mesh grid until they either drop into the cage or have been hanging for 60 seconds, wild-type mice were able to hang for up to 60 seconds starting from 3 weeks of life (Figure 2B). Treatment of wild-type mice with ActRIIB-mFC led to a slight decrease in antigravity hanging performance, which was statistically significant only at 26 to 27 days of life (P < 0.05). In contrast, vehicle-treated Mtm1δ4 animals exhibited impaired hanging performance at 2 weeks of life, which subsequently degenerated into nearly complete inability to remain suspended against gravity by 28 to 29 days of life. Treatment with ActRIIB-mFC did not measurably improve the antigravity hanging performance of Mtm1δ4 mice.
Forelimb Grip Strength
Mtm1δ4 animals exhibited consistently lower forelimb grip strength measurements at all ages tested (P < 0.001) (Figure 2C). Forelimb grip force measurements in vehicle-treated wild-type animals showed consistent gains in grip strength as the animals aged, whereas the grip force of Mtm1δ4 animals was greatest at 5 weeks of life and then decreased as the disease progressed. Compared with their vehicle-treated counterparts, ActRIIB-mFC–treated wild-type animals also demonstrated increased grip strength as the treatment period progressed, with grip force up to 135% greater at 9 weeks of life, the latest time point tested. In ActRIIB-mFC–treated Mtm1δ4 animals, grip strength was transiently improved by 116% at 5 weeks of life (P < 0.05), after which grip strength declined in both vehicle- and ActRIIB-mFC–treated Mtm1δ4 animals.
Footprint Analysis
Abnormalities of gait can be used to differentiate wild-type from Mtm1δ4 animals. Mtm1δ4 mice experience hindlimb weakness with disease progression, which can be visualized and quantified as foot drags on a footprinting assay. While foot drags are extremely uncommon in wild-type mice, the number of drags observed in Mtm1δ4 mice steadily increased after 3 weeks of life (P < 0.05) (Figure 2D). Treatment with ActRIIB-mFC did not have any effect on the number of foot drags observed at footprint analysis.
Survival
Similar to the first published reports using Mtm1δ4 mice on the HSA background, which stated that the survival of Mtm1δ4 animals ranged from 6 to 12 weeks (mean, 59 days), in the present study, untreated and vehicle-treated Mtm1δ4 animals demonstrated a maximum lifespan of approximately 8 to 9 weeks (mean, 56.1 days; range, 32–65 days). In comparisons of vehicle- and ActRIIB-mFC–treated Mtm1δ4 mice, treatment with ActRIIB-mFC significantly lengthened this lifespan by 17% (mean, 67.6 days; range, 61–74 days), with a shift in median survival from 58 days to 68 days (P < 0.05) (Figure 2E). This survival benefit was due to both a decrease in the number of early deaths and delayed death in the oldest treated animals. This survival benefit also seems to be dose-dependent; reducing the dose to 5 mg/kg of ActRIIB-mFC in a pilot study of six animals resulted in an increase in median survival to 65 days (data not shown), which is still a significant improvement over that in the vehicle-treated animals (P < 0.05).
Gross Evaluation
Mtm1δ4 mice with late-stage disease can be easily differentiated from wild-type mice at gross and histologic examination. Mtm1δ4 mice are much smaller than age-matched wild-type mice and have proportionately smaller muscles (Figure 3A). At 43 days of life, when treated and untreated mice of both genetic backgrounds are easily distinguishable from their vehicle-treated counterparts on the basis of weight, ActRIIB-mFC treatment resulted in larger muscles in both wild-type and Mtm1δ4 mice (Figure 3B). Necropsy performed in treated animals at 8 to 10 weeks of life, which corresponded to the end-stage in treated and untreated Mtm1δ4 mice, revealed dramatic increases in muscle size in ActRIIB-mFC–treated wild-type animals. In contrast, vehicle- and ActRIIB-mFC–treated Mtm1δ4 mice were grossly indistinguishable from one another at end stage; both groups of mice were severely emaciated (Figure 3A). It should be noted, however, that the survival benefit produced by ActRIIB-mFC therapy prevented age-matching of these mice for this comparison. While the end-stage appearance of treated and untreated Mtm1δ4 mice was similar, the vehicle-treated mice were approximately 10 days younger than the ActRIIB-mFC–treated mice when the disease progressed to end stage.
Figure 3.
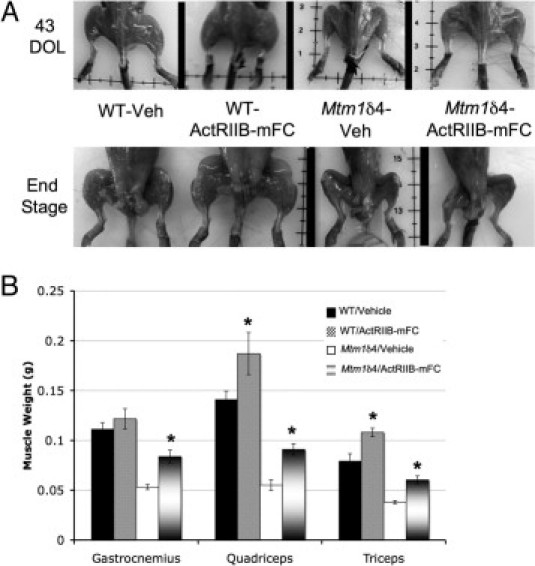
Gross evaluation of vehicle- and ActRIIB-mFC–treated mice. A: Gross examination of skinned animals at 43 days of life (DOL) or at end stage (∼9 weeks) reveals increased muscle bulk in wild-type animals as a result of treatment with ActRIIB-mFC. Treatment-related increase in muscle bulk was apparent in Mtm1δ4 mice at 43 days of life; however, muscle atrophy in the terminal phase of disease prevents differentiation of vehicle- and ActRIIB-mFC–treated Mtm1δ4 animals at end stage. B: Weights of individual muscles in treated and untreated animals at 43 days of life. *P < 0.05.
Histologic Examination
At histologic analysis, quadriceps muscle fibers of vehicle-treated Mtm1δ4 mice were dramatically smaller than those of age-matched wild-type mice, with mean fiber diameter of 19 and 30 mm, respectively, by 43 days of life (P < 0.001) (Figure 4A and Figure 5). Although central nucleation is not a primary pathologic feature in this animal model, compared with muscles from wild-type mice, quadriceps muscles of Mtm1δ4 mice contained increased numbers of centrally nucleated fibers (P < 0.05) (Figure 4B). The number of centrally nucleated fibers in Mtm1δ4 mice increased with age, irrespective of treatment. The greatest number of central nuclei was observed in ActRIIB-mFC–treated Mtm1δ4 mice, which may have been a consequence of their extended lifespan compared with that of vehicle-injected animals. Treatment of wild-type animals with ActRIIB-mFC produced an increase in the size of most fibers by 43 days of life, as evidenced by a 123% increase in mean fiber diameter and an increased percentage of large fibers in the quadriceps muscle (Figure 4A). Immunostaining for oxidative (type 1 or 2a) or glycolytic (type 2b) myosin subtypes in these muscles revealed that all types of muscle fibers experienced equivalent degrees of hypertrophy in response to ActRIIB-mFC treatment (Figure 5). Similar findings were also observed in wild-type mice that were sacrificed at end stage. ActRIIB-mFC–treated Mtm1δ4 animals also exhibited marked hypertrophy of a subpopulation of muscle fibers when compared with their vehicle-treated counterparts, with a 132% increase in mean fiber diameter overall. In contrast to the hypertrophy observed across all fiber types in wild-type animals, which was recently reported elsewhere,17 hypertrophy in treated Mtm1δ4 mice was noted only in the glycolytic type 2b fibers, and there was a marked increase (149%) in mean type 2b fiber diameter in treated animals (P < 0.001) at 35 and 43 days of life (Figure 5). Evaluation of animals with end-stage disease revealed small myofibers in both vehicle- and ActRIIB-mFC–treated animals, preventing histologic distinction between these two groups of mice at this time point. The percentage of type 1, 2a, and 2b fibers was similar between animals irrespective of genotype or treatment group (Figure 5B). These results are consistent with a transient histologic improvement in Mtm1δ4 mice after ActRIIB-mFC treatment, which corresponds to the observed increases in animal and muscle weight at the same stage. In addition, counts of myofibers within the quadriceps muscle revealed no significant difference between wild-type and Mtm1δ4 mice or between vehicle- and ActRIIB-mFC–treated mice of either genetic background. Diaphragm muscles collected from all treatment groups at 35 days of life were indistinguishable from one another (Figure 4C) despite clear histologic differences between treatment groups in the quadriceps muscle at this time point. These findings suggest that the diaphragm is not significantly affected in this disease model and that the cause of death in these animals may be related to dehydration or nutritional issues rather than to respiratory insufficiency.
Figure 4.
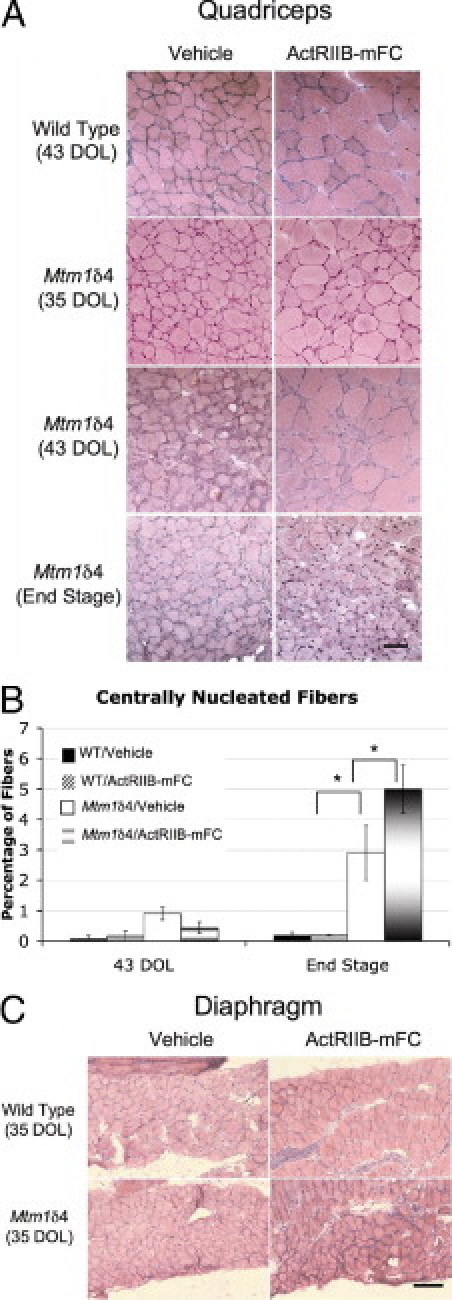
Histologic evaluation of vehicle- and ActRIIB-mFC–treated mice. A: H&E-stained transverse sections from the quadriceps muscles of vehicle- and ActRIIB-mFC–treated wild-type and Mtm1δ4 mice. Histologic findings in wild-type mice were similar at all three time points. B: Counts of centrally nucleated fibers in quadriceps muscles from mice at 43 days of life (DOL) and at end stage. C: H&E-stained transverse sections from the diaphragm muscles of vehicle- and ActRIIB-mFC–treated wild-type and Mtm1δ4 mice. Scale bars: 100 μm (A); 200 μm (C). *P < 0.05.
Figure 5.
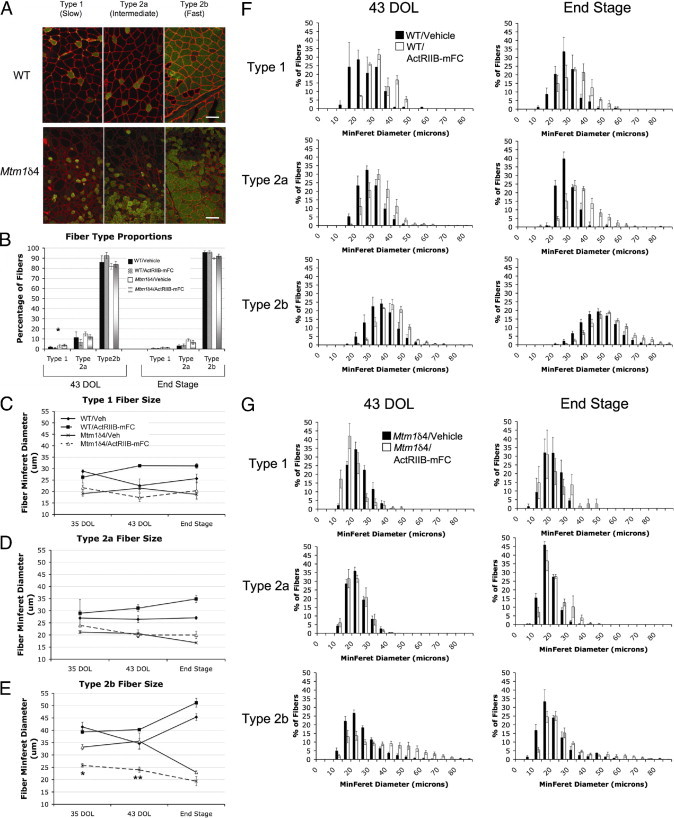
Histologic analysis according to fiber type. A: Immunostaining of transverse sections from the quadriceps muscles of an ActRIIB-mFC–treated wild-type or Mtm1δ4 mouse at 43 days of life for dystrophin (red) and either myosin heavy chain type 1, type 2a, or type 2b fibers (green). Scale bar = 100 μm. Staining was performed in all treatment groups at 35 and 43 days of life and at end stage (determined by complete hindlimb paralysis or a >20% loss in body weight), and myofiber size was quantitated according to fiber type. B: Percentages of type 1, 2a, and 2b fibers were counted. C–E: Muscles from animals at 35 days of life, 43 days of life, and end stage were analyzed. Mean MinFeret diameter measurements for type 1 (C), type 2a (D), and type 2b (E) fibers are shown. The legend for panel C also applies to panels D and E. F and G: Frequency histograms demonstrate the distribution of fiber sizes in the quadriceps muscles of vehicle- and ActRIIB-mFC–treated wild-type (F) and Mtm1δ4 (G) mice. *P < 0.01. **P < 0.0001.
Ultrastructural Examination
Ultrastructural tests of vehicle- and ActRIIB-mFC–treated mice of both genetic backgrounds were performed to evaluate the degree of structural organization in hypertrophied myofibers. At the ultrastructural level, the intracellular contents of vehicle- and ActRIIB-mFC–treated wild-type mice were indistinguishable from one another, with well-organized contractile filaments surrounded by pockets of mitochondria and stereotypically oriented triads consisting of T-tubules and sarcoplasmic reticulum (Figure 6). Rare fibers from vehicle- and ActRIIB-mFC–treated Mtm1δ4 mice contained centrally located nuclei surrounded by aggregations of mitochondria and other disorganized organelles; however, these fibers were too uncommon to quantify accurately. Comparisons between vehicle- and ActRIIB-mFC–treated Mtm1δ4 mice revealed that some large fibers were present in ActRIIB-mFC–treated mice at 43 days of life; however, there was no difference in the organization of the contractile filaments between the two groups. The organization of the triads, which are composed of two elements of sarcoplasmic reticulum oriented perpendicular to a T-tubule (insets, Figure 6, A and B), was abnormal in the Mtm1δ4 mice because of loss of the appropriate orientation between these elements and an apparent increase in the number of stacks of sarcoplasmic reticulum between contractile elements. These abnormalities of the triads were unchanged by ActRIIB-mFC therapy. A subpopulation of degenerating atrophic myofibers with organellar and myofibrillar disorganization was observed in all Mtm1δ4 mice (Figure 6E). Some fibers of ActRIIB-mFC–treated Mtm1δ4 mice demonstrated ruffling and redundancy of the basement membrane (Figure 6F) consistent with atrophy of hypertrophic fibers.
Figure 6.

Ultrastructural evaluation of vehicle- and ActRIIB-mFC–treated mice. A–D: Ultrastructural examination of gastrocnemius muscle from vehicle- and ActRIIB-mFC–treated wild-type animals reveals no differences in the organization of the contractile apparatus between treated and untreated mice. Similarly, vehicle- and ActRIIB-mFC–treated Mtm1δ4 mice exhibit appropriate organization of contractile elements, but also exhibit abnormalities of T-tubule and sarcoplasmic reticulum architecture (insets), as previously reported.4,41E: Myofiber degeneration in Mtm1δ4 mice is characterized by disorganization of organelles and abnormalities in the contractile apparatus, which are unaffected by treatment with ActRIIB-mFC. F: Atrophic fibers, identifiable by folding and redundancy of the basal lamina (inset), are observed in Mtm1δ4 animals treated with ActRIIB-mFC. Scale bars: 500 nm (A–D); 2 μm (E and F).
Discussion
XLMTM is a currently untreatable severe congenital myopathy that is pathologically characterized by small myofibers and increased numbers of centrally nucleated myofibers. Based on the relationship between small fiber size and poor prognosis, the present study evaluated the therapeutic efficacy of ActRIIB-mFC, an agent that induces myofiber hypertrophy through inhibition of myostatin, in the Mtm1δ4 mouse. After treatment with ActRIIB-mFC, Mtm1δ4 mice exhibited increases in weight, muscle weight, and myofiber size, which behaviorally corresponded to a transient increase in forelimb grip strength and a 17% increase in lifespan.
Preclinical trials using myostatin loss or inhibition have been effective in ameliorating symptoms of weakness in some animal models of muscle injury, atrophy, and disease. Myostatin-deficient animals demonstrate less fibrosis and improved muscle regeneration after laceration injury24 and experience less muscle atrophy in murine models of age-related sarcopenia25 and cancer-related cachexia.26,27 Similarly, a recent study using ActRIIB-mFC in the SODG93A model of amyotrophic lateral sclerosis described muscle growth and delay in symptomatic onset in treated animals that was greater than that observed in SODG93A/Mstn−/− mice.18 These studies indicate a role for myostatin inhibitors in conditions in which the primary disease process occurs outside of the myofiber. In addition, several myostatin inhibition strategies have improved muscle disease and/or function in the mdx mouse model of Duchenne muscular dystrophy,28–32 which suggests a therapeutic benefit in primary myopathies in which the basic defect involves myofiber structure and function. The only reported human trial using myostatin inhibition, which used antimyostatin antibodies in patients with adult muscular dystrophies (Becker muscular dystrophy, facioscapulohumeral dystrophy, and limb-girdle muscular dystrophy), did not demonstrate significant improvements in muscle strength or size.33 These results may reflect an impaired ability of adult patients with dystrophy to respond to myostatin inhibition, which was recently demonstrated in mdx mice,29 or may simply reflect poor efficacy of that particular agent. Alternatively, additional ligands of ActRIIB may prevent myostatin inhibition alone from being therapeutically effective in human beings, which provides a strong rationale for pursuing studies of ActRIIB inhibition rather than myostatin inhibition in human beings.
In contrast to other conditions in which myostatin and ActRIIB inhibition has been tested, congenital myopathies represent a class of disease in which the primary disease process occurs within myofibers and in which the primary pathologic process causes weakness owing to the insufficient size and abnormal organization of elements within the myofiber. Thus, there is great potential for targeting of ActRIIB to be effective in patients with congenital myopathy, provided that the diseased muscle is capable of growth that causes functional improvement. The present data support the possibility of therapeutic benefits of myostatin inhibitors in patients with congenital myopathy, in particular because the Mtm1δ4 model of myotubular myopathy has a much more severe clinical course than that observed in most patients with congenital myopathy. While patients with XLMTM are usually born with significant weakness, the weakness is rarely progressive. In contrast, Mtm1δ4 mice are grossly normal at birth but undergo considerable symptomatic deterioration during the second month of life. In patients and animal models that do not undergo the catastrophic degeneration seen in Mtm1δ4 mice at approximately 2 months of life, greater or more prolonged benefits can be expected than what is describe herein in Mtm1δ4 mice.
These studies provide evidence that ActRIIB inhibition can extend lifespan in diseased animals, and represent the first description of a therapy that extends lifespan in Mtm1δ4 animals. Recent studies using ActRIIB-mFC in the SODG93A model of amyotrophic lateral sclerosis18 and the hSMN2/delta7SMN/mSmn−/− model of severe spinal muscular atrophy19 described symptomatic improvements without extension of lifespan. It should be noted, however, that an extension of lifespan was observed after follistatin therapy in this mouse model, which, to our knowledge, is the only other published report of lifespan extension while manipulating the myostatin pathway.34 The extension of lifespan observed in the present study was due to delay in the point at which animals experience weight loss and/or complete hindlimb paralysis. This prolongation of survival may correspond to a greater extension of lifespan in patients with XLMTM because of the availability of supportive care and the greater degree of symptomatic variability observed in these patients.
Recent studies have established that postnatal inhibition of the myostatin pathway produces myofiber hypertrophy28,35–37 without the myocyte hyperplasia observed in myostatin knockout animals.11 While there is some evidence that myostatin inhibition during development can produce increased numbers of type 2b fibers in skeletal muscle,38 treatment of wild-type and Mtm1δ4 mice with ActRIIB-mFC did not change the number or percentage of type 1, 2a, and 2b fibers in the quadriceps muscles. These findings, which are consistent with a recent report of the effects of ActRIIB-mFC treatment in wild-type mouse muscle,17 support the theory that myocyte hyperplasia or changes in fiber type distribution do not occur with postnatal inhibition of myostatin or ActRIIB. In addition, while satellite cell behavior in response to treatment was not specifically examined, no evidence of satellite cell fusion or myoblast proliferation was observed at histologic evaluation of ActRIIB-mFC–treated muscles.
In contrast to the hypertrophy observed in all fiber types in treated wild-type animals,17 ActRIIB-mFC–treated Mtm1δ4 animals did not demonstrate hypertrophy in the oxidative type 1 and 2a myofibers at 43 days of life. The degree to which hypertrophy was observed in type 2b fibers, however, markedly exceeded the degree of hypertrophy observed in treated wild-type animals. These results suggest that myotubularin deficiency interferes with the hypertrophic response to myostatin inhibition in oxidative fibers. The growth pathways in type 1 and 2a myofibers are calcium-dependent, whereas hypertrophy in type 2b fibers occurs through the relatively calcium-independent Akt pathway (reviewed in 39, 40). Recent studies of myotubularin-deficient mice,9 fish,5 and dogs41 have described abnormalities in T-tubule and sarcoplasmic reticulum architecture and abnormalities in intracellular calcium handling.9 These abnormalities in intracellular calcium may interfere with activation of hypertrophic pathways in oxidative fibers and, thereby, prevent ActRIIB-mFC–induced hypertrophy in these fibers. The Akt pathway may not be affected by abnormalities of calcium homeostasis in myotubularin-deficient myofibers, which could explain the marked fiber type-specific hypertrophy observed in Mtm1δ4 animals. However, myotubularin seems to have some critical role in type 2b myofibers because these fibers eventually become atrophic between 43 days of life and disease end stage.
In conclusion, the present study provides evidence that myostatin inhibition via treatment with ActRIIB-mFC is therapeutically beneficial to Mtm1δ4 mice according to findings of behavioral and pathologic studies. Furthermore, the selective hypertrophy of type 2b myofibers in Mtm1δ4 animals may provide important information about the biological processes in which myotubularin is involved. Although these findings may provide the first potential therapy for XLMTM in children, the selective nature of the response to treatment provides interesting possibilities for improving therapeutic responses once the role of myotubularin in oxidative myofibers is more completely understood.
Acknowledgments
We thank Drs. Vandana Gupta, Ph.D., Pankaj Agrawal, M.D., and Behzad Moghadaszadeh, Ph.D., for scientific advice, and Ms. Samantha Lundeen for technical assistance with the Western blot analyses.
This study is dedicated to the memory of Joshua Miles Frase, who provided tremendous inspiration to children with congenital myopathy, their families, and researchers around the world.
Footnotes
Supported by the grants L40 AR057721-01 and K08 AR059750-01 (to M.W.L.) and P50 NS040828 and R01 AR044345 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the Joshua Frase Foundation, and the Lee and Penny Anderson Family Foundation (to A.H.B.).
J.L.L. and J.S.S. are employees of and have equity in Acceleron Pharma, Inc. M.W.L. and A.H.B. have filed a US provisional patent application (61/330,011) entitled “Method for Treating Congenital Myopathy” based on this work.
References
- 1.Heckmatt J.Z., Sewry C.A., Hodes D., Dubowitz V. Congenital centronuclear (myotubular) myopathy: a clinical, pathological and genetic study in eight children. Brain. 1985;108(Pt 4):941–964. doi: 10.1093/brain/108.4.941. [DOI] [PubMed] [Google Scholar]
- 2.Jungbluth H., Wallgren-Pettersson C., Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buj-Bello A., Fougerousse F., Schwab Y., Messaddeq N., Spehner D., Pierson C.R., Durand M., Kretz C., Danos O., Douar A.M., Beggs A.H., Schultz P., Montus M., Denefle P., Mandel J.L. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum Mol Genet. 2008;17:2132–2143. doi: 10.1093/hmg/ddn112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao C., Backer J.M., Laporte J., Bedrick E.J., Wandinger-Ness A. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol Biol Cell. 2008;19:3334–3346. doi: 10.1091/mbc.E08-04-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowling J.J., Vreede A.P., Low S.E., Gibbs E.M., Kuwada J.Y., Bonnemann C.G., Feldman E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5:e1000372. doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A., Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 7.Pierson C.R., Tomczak K., Agrawal P., Moghadaszadeh B., Beggs A.H. X-linked myotubular and centronuclear myopathies. J Neuropathol Exp Neurol. 2005;64:555–564. doi: 10.1097/01.jnen.0000171653.17213.2e. [DOI] [PubMed] [Google Scholar]
- 8.Pierson C.R., Agrawal P.B., Blasko J., Beggs A.H. Myofiber size correlates with MTM1 mutation type and outcome in X-linked myotubular myopathy. Neuromuscul Disord. 2007;17:562–568. doi: 10.1016/j.nmd.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Qusairi L., Weiss N., Toussaint A., Berbey C., Messaddeq N., Kretz C., Sanoudou D., Beggs A.H., Allard B., Mandel J.L., Laporte J., Jacquemond V., Buj-Bello A. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc Natl Acad Sci USA. 2009;106:18763–18768. doi: 10.1073/pnas.0900705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buj-Bello A., Laugel V., Messaddeq N., Zahreddine H., Laporte J., Pellissier J.F., Mandel J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci USA. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McPherron A.C., Lawler A.M., Lee S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 12.McCroskery S., Thomas M., Maxwell L., Sharma M., Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joulia-Ekaza D., Cabello G. Myostatin regulation of muscle development: molecular basis, natural mutations, physiopathological aspects. Exp Cell Res. 2006;312:2401–2414. doi: 10.1016/j.yexcr.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Clop A., Marcq F., Takeda H., Pirottin D., Tordoir X., Bibe B., Bouix J., Caiment F., Elsen J.M., Eychenne F., Larzul C., Laville E., Meish F., Milenkovic D., Tobin J., Charlier C., Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 15.Lee S.J., McPherron A.C. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S.J., Reed L.A., Davies M.V., Girgenrath S., Goad M.E., Tomkinson K.N., Wright J.F., Barker C., Ehrmantraut G., Holmstrom J., Trowell B., Gertz B., Jiang M.S., Sebald S.M., Matzuk M., Li E., Liang L.F., Quattlebaum E., Stotish R.L., Wolfman N.M. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cadena S.M., Tomkinson K.N., Monnell T.E., Spaits M.S., Kumar R., Underwood K.W., Pearsall R.S., Lachey J.L. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type [published online ahead of print May 13, 2010] J Appl Physiol. 2010;109:635–642. doi: 10.1152/japplphysiol.00866.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrison B.M., Lachey J.L., Warsing L.C., Ting B.L., Pullen A.E., Underwood K.W., Kumar R., Sako D., Grinberg A., Wong V., Colantuoni E., Seehra J.S., Wagner K.R. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;217:258–268. doi: 10.1016/j.expneurol.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 19.Sumner C.J., Wee C.D., Warsing L.C., Choe D.W., Ng A.S., Lutz C., Wagner K.R. Inhibition of myostatin does not ameliorate disease features of severe SMA mice. Hum Mol Genet. 2009;18:3145–3152. doi: 10.1093/hmg/ddp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buj-Bello A., Furling D., Tronchere H., Laporte J., Lerouge T., Butler-Browne G.S., Mandel J.L. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum Mol Genet. 2002;11:2297–2307. doi: 10.1093/hmg/11.19.2297. [DOI] [PubMed] [Google Scholar]
- 21.Crawley J.N. 2nd ed. Wiley-Liss; Hoboken, NJ: 2007. What's Wrong with My Mouse?: Behavioral Phenotyping of Transgenic and Knockout Mice; p. 368. [Google Scholar]
- 22.Wattanasirichaigoon D., Swoboda K.J., Takada F., Tong H.Q., Lip V., Iannaccone S.T., Wallgren-Pettersson C., Laing N.G., Beggs A.H. Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology. 2002;59:613–617. doi: 10.1212/wnl.59.4.613. [DOI] [PubMed] [Google Scholar]
- 23.Brooke M.H., Engel W.K. The histographic analysis of human muscle biopsies with regard to fiber types. 4: Children's biopsies. Neurology. 1969;19:591–605. doi: 10.1212/wnl.19.6.591. [DOI] [PubMed] [Google Scholar]
- 24.Zhu J., Li Y., Shen W., Qiao C., Ambrosio F., Lavasani M., Nozaki M., Branca M.F., Huard J. Relationships between transforming growth factor-beta1, myostatin, and decorin: implications for skeletal muscle fibrosis. J Biol Chem. 2007;282:25852–25863. doi: 10.1074/jbc.M704146200. [DOI] [PubMed] [Google Scholar]
- 25.Siriett V., Salerno M.S., Berry C., Nicholas G., Bower R., Kambadur R., Sharma M. Antagonism of myostatin enhances muscle regeneration during sarcopenia. Mol Ther. 2007;15:1463–1470. doi: 10.1038/sj.mt.6300182. [DOI] [PubMed] [Google Scholar]
- 26.Liu C.M., Yang Z., Liu C.W., Wang R., Tien P., Dale R., Sun L.Q. Myostatin antisense RNA-mediated muscle growth in normal and cancer cachexia mice. Gene Ther. 2008;15:155–160. doi: 10.1038/sj.gt.3303016. [DOI] [PubMed] [Google Scholar]
- 27.Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA: Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 391:1548–1554 [DOI] [PubMed]
- 28.Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 29.Murphy KT, Ryall JGM, Snell SM, Nair L, Koopman R, Krasney PA, Ibebunjo C, Holden KS, Loria PM, Salatto CT, Lynch GS: Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am J Pathol 176:2425–2434 [DOI] [PMC free article] [PubMed]
- 30.Qiao C., Li J., Zheng H., Bogan J., Yuan Z., Zhang C., Bogan D., Kornegay J., Xiao X. Hydrodynamic limb vein injection of adeno-associated virus serotype 8 vector carrying canine myostatin propeptide gene into normal dogs enhances muscle growth. Hum Gene Ther. 2009;20:1–10. doi: 10.1089/hum.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuchida K. Myostatin inhibition by a follistatin-derived peptide ameliorates the pathophysiology of muscular dystrophy model mice. Acta Myol. 2008;27:14–18. [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner K.R., McPherron A.C., Winik N., Lee S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 33.Wagner K.R., Fleckenstein J.L., Amato A.A., Barohn R.J., Bushby K., Escolar D.M., Flanigan K.M., Pestronk A., Tawil R., Wolfe G.I., Eagle M., Florence J.M., King W.M., Pandya S., Straub V., Juneau P., Meyers K., Csimma C., Araujo T., Allen R., Parsons S.A., Wozney J.M., Lavallie E.R., Mendell J.R. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 34.Rose F.F., Jr., Mattis V.B., Rindt H., Lorson C.L. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2009;18:997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grobet L., Poncelet D., Royo L.J., Brouwers B., Pirottin D., Michaux C., Menissier F., Zanotti M., Dunner S., Georges M. Molecular definition of an allelic series of mutations disrupting the myostatin function and causing double-muscling in cattle. Mamm Genome. 1998;9:210–213. doi: 10.1007/s003359900727. [DOI] [PubMed] [Google Scholar]
- 36.Whittemore L.A., Song K., Li X., Aghajanian J., Davies M., Girgenrath S., Hill J.J., Jalenak M., Kelley P., Knight A., Maylor R., O'Hara D., Pearson A., Quazi A., Ryerson S., Tan X.Y., Tomkinson K.N., Veldman G.M., Widom A., Wright J.F., Wudyka S., Zhao L., Wolfman N.M. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun. 2003;300:965–971. doi: 10.1016/s0006-291x(02)02953-4. [DOI] [PubMed] [Google Scholar]
- 37.Zhu X., Hadhazy M., Wehling M., Tidball J.G., McNally E.M. Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett. 2000;474:71–75. doi: 10.1016/s0014-5793(00)01570-2. [DOI] [PubMed] [Google Scholar]
- 38.Girgenrath S., Song K., Whittemore L.A. Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve. 2005;31:34–40. doi: 10.1002/mus.20175. [DOI] [PubMed] [Google Scholar]
- 39.Bassel-Duby R., Olson E.N. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- 40.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–170. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 41.Beggs A.H., Bohm J., Snead E., Kozlowski M., Maurer M., Minor K., Childers M.K., Taylor S.M., Hitte C., Mickelson J.R., Guo L.T., Mizisin A.P., Buj-Bello A., Tiret L., Laporte J., Shelton G.D. MTM1 Mutation associated with X-linked myotubular myopathy in Labrador retrievers. Proc Natl Acad Sci USA. 2010;107:14697–14702. doi: 10.1073/pnas.1003677107. [DOI] [PMC free article] [PubMed] [Google Scholar]