

Abstract
PCR detection of chromosomal translocations and small insertion/deletion mutations is challenging when potential amplicon size varies greatly. Molecular diagnostic laboratories face such difficulties with the BCL2-IGH translocation in follicular lymphoma and with internal tandem duplication mutation of the FLT3 gene in leukemia, where breakpoints are widely distributed, mutations may be multiple, signal strength is low, and background noise is elevated. We developed a strategy, called Δ-PCR, that ensures PCR specificity and identifies individual breakpoints. Δ-PCR uses two forward primers (external and internal) and a reverse primer simultaneously. The internal primer functions as a probe with a defined distance Δ from the external primer. For follicular lymphoma, we prepared upstream, BCL2-specific primers for potential breakpoints to pair with a common, downstream VLJH primer. Multiplexed PCR amplicons are sized by capillary electrophoresis. Each of the upstream pairs has a defined interval separating them that uniquely identifies the breakpoint. The presence of two amplicons with a defined size difference confirms validity of the rearrangement and identity of the specific breakpoint, even if signal strength is low. By testing 40 follicular lymphoma and 12 control specimens from formalin-fixed, paraffin-embedded (FFPE) blocks, we showed that multiplex Δ-PCR is a simple, sensitive strategy to identify translocations with multiple breakpoints or partners. The strategy was also applied to detect minor leukemic clones with internal tandem duplication mutations and could have broader applications for other insertion/deletion and duplication mutations.
Chromosomal translocations and small insertion/deletions occur in specific hematological malignancies and solid tumors, and are often definitional genetic events.1,2 Translocations involve breakage and fusion of two or more chromosomes, and insertion/deletions (as considered in the present study) involve breakage within one chromosome. The detection of these chromosomal abnormalities by conventional cytogenetics study, standard Southern blot analysis, fluorescence in situ hybridization (FISH), PCR, and reverse-transcription PCR (RT-PCR) has been used to diagnose specific malignancies and also to monitor minimal residual cancer cells.3
Depending on the specific breakpoint and fusion, chromosomal translocations are classified into two categories.1 Breakage within genes leads to a fusion transcript producing an oncogenic chimeric protein, as in chronic myelogenous leukemia (BCR-ABL1) and synovial sarcoma (SYT-SSX1 or SYT-SSX2).4–6 In these cases, diagnostic PCR using genomic DNA is difficult because the potential breakpoint regions in the introns span large distances; however, the fusion transcript can be detected with high sensitivity and specificity by RT-PCR across the involved exon junction at the RNA level.7,8 The second mechanism of translocation-induced oncogenesis, particularly common in lymphoid malignancies, involves the approximation of an oncogene and an actively transcribed gene (such as the immunoglobulin or T-cell receptor genes) resulting in overproduction of the oncogene without a chimeric mRNA or protein. This is the case with follicular lymphoma (BCL2-IGH) and mantle cell lymphoma (CCND1-IGH).9–11 The translocation in these instances must be detected at the DNA level by PCR because of the lack of a fusion transcript,12,13 which may be technically challenging. First, malignancies often have several translocation hot spots where breakage occurs, preventing detection of all translocations using a single pair of PCR primers.9,14,15 Second, the amplicon from each individual clone varies in size, because each hot spot may be hundreds of bases in size; this necessitates validation by Southern blot analysis or real-time PCR to distinguish bona fide translocations from artifacts.12,16
For chromosomal translocations with many potential breakpoints, such as the BCL2-IGH rearrangement in follicular lymphoma, one strategy for PCR-based detection has been to combine primer sets.13 Interpretation of multiplexed assays, in particular, is complicated when signal strength is not high or when background noise is elevated, because the significance of weak bands on agarose gel electrophoresis or one or several small peaks on capillary electrophoresis is unclear. These conditions are particularly prevalent when analyzing FFPE material. Similarly, recurrent insertion/deletions, such as the internal tandem duplication (ITD) in the FLT3 oncogene in acute myeloid leukemia, may also present difficulty, because of the varied size (and sometimes varied location) of the event.17,18 Minor artifactual, background amplicons are difficult to distinguish from true genetic alterations, particularly in cases where tumor cells are a small portion of the material analyzed. In the present study, using BCL2-IGH rearrangement and ITD mutation of the FLT3 gene as the models, we have developed an approach, which we call Δ-PCR, to ensure the specificity of PCR and, at the same time, to identify the particular breakpoint from a single multiplexed PCR.
Materials and Methods
Design of Δ-PCR
For each target locus (breakpoint hot spot or insertion/deletion mutation locus), three oligonucleotide primers are designed. Two of the primers oriented in the same direction are on one side of the breakpoint/mutation locus (an external and internal couple) and the third primer is opposing (Figure 1). The primer design is analogous to semi-nested PCR, but the reaction is conducted in a single step using all three primers simultaneously. The internal primer functions as a confirmatory probe, because it was created at a defined distance Δ from the external primer. A pair of amplicons differing in size by the Δ value are indicative of a true translocation/mutation. Nonspecific products are highly unlikely to produce the appropriate pair of amplicons.
Figure 1.
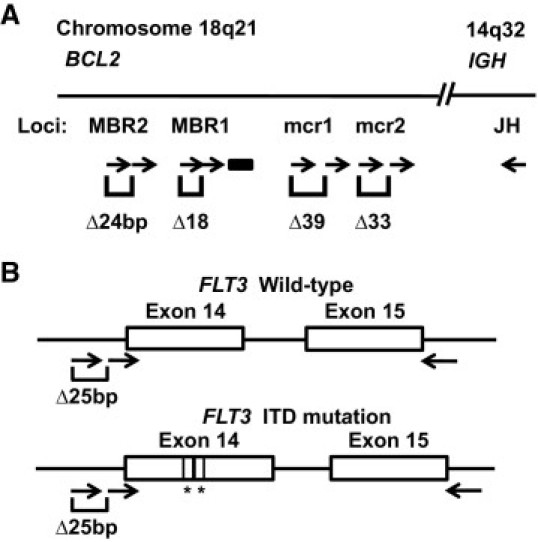
Primer design for detection of BCL2-IGH translocation (A) and internal tandem deletion mutation of the FLT3 gene (B). Sizes are not to scale. Arrows indicate primers and lines indicates location of the probe for real-time PCR. The Δ indicates the defined size difference of the paired amplicons for each breakpoint and an asterisk indicates the segment of ITD mutation.
Using the BCL2-IGH translocation of follicular lymphoma as a model, we prepared two upstream primers for each of four breakpoint hot spots of the BCL2 gene, to be paired simultaneously with a common, downstream VLJH primer in the IGH gene (Figure 1A and Table 1). The original breakpoint hot spots, MBR and mcr (including MBR1, MBR2, mcr1, and mcr2), were chosen as a proof of principle for Δ-PCR. The breakpoint primers for MBR1, MBR2, mcr1, and mcr2 were initially tested individually and then were multiplexed together. PCR products were detected by capillary electrophoresis (ABI 3100 system, Applied Biosystems, Foster City, CA). Each of the upstream pairs has a different Δ value, uniquely identifying the hot spot involved. The presence of two amplicons with a defined size difference (18 bp, 24 bp, 39 bp, and 33 bp for MBR1, MBR2, mcr1, and mcr2, respectively) confirms validity of the rearrangement, even if signal strength is low. Real-time PCR and/or direct sequencing were also performed to validate the amplicons.
Table 1.
Paired Forward Primers Used in Δ-PCR for Detection of BCL2-IGH Translocation
| Forward primer⁎ | Sequences | Primer ratio | Reference |
|---|---|---|---|
| MBR1 (18 bp) | |||
| MBR1int | 5′-CTATGGTGGTTTGACCTTTAGAGAG-3′ | 1 | 16 |
| MBR1ext | 5′-TGACCAGCAGATTCAAATCTATGG-3′ | 1 | 13† |
| MBR2 (24 bp) | |||
| MBR2int | 5′-ACTCTGTGGCATTATTGCATTATATACC-3′ | 2 | 13† |
| MBR2ext | 5′-CAGCCTTGAAACATTGATGGA-3′ | 1 | 35† |
| mcr1 (39 bp) | |||
| mcr1int | 5′-CTTGCAGGGTCTTTAAGCAGCA-3′ | 1 | |
| mcr1ext | 5′-CATAGAGCAAGCGCCCAATAAATA-3′ | 4 | 13† |
| mcr2 (33 bp) | |||
| mcr2int | 5′-GTACTTCAGAAGGAGGAACGTCCT-3′ | 1 | |
| mcr2ext | 5′-TGAATGCCATCTCAAATCCAA-3′ | 4 | 13 |
Combined with the reverse primer VLJH (5′-6FAM-GTGACCAGGTNCCTTGGCCCCAG-3′). The expected size difference between products amplified with each of the forward primers paired with the reverse primer is given in parentheses.
With modification.
A second model system was tested to validate identification of insertion/deletions. Two forward primers, an internal primer labeled with the fluorochrome FAM (blue) and an external primer labeled with HEX (green), were paired with a reverse primer for the detection of the ITD mutation of the FLT3 gene (Figure 1B). The internal primer and reverse primer have been described by Murphy et al.17 The external primer was 5′-ACTGACTCATACTTTCATCTCTGAAGC-3′. The designed Δ value between the peaks amplified from the external and internal primers is 25 bp. The amplicons were isolated by a molecular fraction collecting tool using a reconfigured ABI 310 automated sequencer as described previously,19 and the presence of the ITD mutation was confirmed by direct sequencing. Amplicons from normal control DNA samples showed a pair of peaks (blue at 327/328 bases and green at 352/353 bases) indicating the wild-type allele. A pair of peaks with size greater than 327/328 bases and a Δ value of 25 bases would indicate the presence of a bona fide ITD mutant allele.
Materials
FFPE tissues from 40 patients with a diagnosis of nodal follicular lymphoma were tested for BCL2-IGH translocation using Δ-PCR and standard real-time PCR. The diagnoses were according to standard histological and immunophenotypic criteria, including immunohistochemical studies and/or flow cytometry analysis on each case.20 FFPE tissues from six patients with follicular hyperplasia and six patients with other hematological malignancies (two with acute lymphocytic leukemia, two with chronic lymphocytic leukemia, and one each with Burkitt lymphoma, plasmablastic lymphoma, and mantle zone lymphoma) were used as negative controls. DNAs from human B-cell lymphoma cell lines SU-DHL-4, SU-DHL-6, and SU-DHL-16 (DSMZ, Braunschweig, Germany) were used as positive controls for MBR2, MBR1, and mcr2 breakpoint rearrangements, respectively. DNA was also extracted from frozen normal lymph node tissues for serial dilution with cell line DNA to test analytical sensitivity.
DNA was isolated from the peripheral blood and bone marrow white cells of three patients with acute myeloid leukemia. Detection of the ITD mutation using conventional PCR17 was done in our routine clinical diagnostic laboratory. In each case, there were multiple peaks with sizes greater than that of the normal allele, including peaks with very low relative abundance. DNA samples from four lymphoblastoid cell lines created in our laboratory (LCL53, LCL60, LCL71 and onc3286) were used as controls.
PCR
DNA used for detection of the BCL2-IGH translocation was extracted from tissue blocks using xylene/ethanol deparaffinization followed by proteinase K digestion, heat inactivation at 97°C for 10 minutes,21 and further cleanup using a QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA). DNA used for detection of the ITD mutation was extracted by a QIAamp DNA Blood Mini Kit. PCR reactions were performed in a 20-μL total volume containing 5 pmol of forward primer, 5 pmol reverse primer, 1.5 mmol/L MgCl2, 0.2 mmol/L each deoxyribonucleotide, 0.5 units AmpliTaq Gold DNA polymerase, and 2.0 μL buffer (Applied Biosystems). Samples containing 20 or 50 ng DNA were subjected to 40 cycles of denaturation (95°C, 30 seconds), annealing (57°C for translocation detection using DNA samples extracted from FFPE tissues, 62°C for translocation detection using DNA samples extracted from frozen lymph node tissues, and 56°C for ITD detection, 30 seconds), and extension (72°C, 60 seconds). The total amount of each set of paired forward primers used for multiplex Δ-PCR was 5 pmol; the ratio of the paired forward primers is given in Table 1. Each DNA sample extracted from paraffin was tested for quality using PCR primers designed for human β-globin (amplicon of 264 bases determined by capillary gel electrophoresis) as described previously.22
Capillary Electrophoresis
One microliter PCR products, 0.5 μL ROX size standard, and 8.5 μL deionized formamide were mixed according to the manufacturer's protocol (Applied Biosystems), heated at 95°C for 2 minutes, and placed on ice for at least 1 minute before electrokinetic injection (45 seconds for translocation detection and 20 seconds for ITD detection) into the ABI 3100 genetic analyzer. The size and positions of peaks on the electropherogram were analyzed using GeneMapper v4.1 analysis software (Applied Biosystems).
Real-Time PCR
Primers and probes were designed to detect BCL2-IGH translocations with a breakpoint in MBR1. Primers were MBR1int (forward, Table 1) and VLJH (reverse, Table 1); the probe was 5′-6FAM-CTGTTTCAACACAGACCCA-MGBNFQ-3′ (Applied Biosystems, modified from Ladetto et al).16 Real-time PCR reactions were performed in a 15-μL total volume containing 3 pmol each primer, 1.5 pmol probe, 2.5 mmol/L MgCl2, 0.2 mmol/L each deoxyribonucleotide, 0.5 units AmpliTaq Gold DNA polymerase, and 1.5 μL buffer (Applied Biosystems). Samples containing 20 ng DNA were subjected to 45 cycles of denaturation (95°C, 30 seconds), annealing (57°C, 30 seconds), and extension (72°C, 60 seconds). The fluorescent signal was detected by the 7900HT Sequence Detection System (Applied Biosystems).
DNA Sequencing
PCR products were purified using QIAquick columns (Qiagen) and were cycle sequenced using ABI BigDye 3 according to the manufacturer's protocol and resolved on an ABI 3700 system at the Synthesis and Sequencing Facility, Johns Hopkins University. Sequences were analyzed using Sequencher 4.8 software (Gene Codes Corporation, Ann Arbor, MI).
Results
Detection of BCL2-IGH Translocation by Conventional PCR
The individual forward primers along with the universal reverse primer VLJH (MBR1ext/VLJH and MBR1int/VLJH for MBR1, MBR2ext/VLJH and MBR2int/VLJH for MBR2, mcr1ext/VLJH and mcr1int/VLJH for mcr1, and mcr2ext/VLJH and mcr2int/VLJH for mcr2) were first tested using DNA from cell lines SU-DHL-4, -6, and -1616,23 carrying breakpoints in MBR2, MBR1, and mcr2, respectively. The monoplexed capillary electrophoretic analysis of SU-DHL-6 revealed only 219-bp and 237-bp PCR products amplified by MBR1int/VLJH primers and MBR1ext/VLJH primers, respectively (data not shown). The 18-bp difference between the PCR products confirmed that the breakpoint of SU-DHL-6 is in MBR1. Similarly, monoplexed capillary electrophoresis also showed the expected size differences between PCR products amplified from SU-DHL-4 using MBR2ext/VLJH and MBR2int/VLJH primers (24 bp difference), and SU-DHL-16 using mcr2ext/VLJH and mcr2int/VLJH primers (33 bp difference). There is no readily available cell line control for translocations involving the mcr1 hot spot; however, one of our clinical cases (case 19) does have such a translocation. Analysis of this case by Δ-PCR for the mcr1 breakpoint using mcr1ext/VLJH and mcr1int/VLJH primers revealed 220 bp and 260 bp PCR products (with the anticipated 39 ± 1 bp difference). Sequencing of the amplicons also confirmed the BCL2-IGH translocation breakpoints of cell lines SU-DHL-4, -6, and -1616,23 and clinical case 19 at MBR2, MBR1, mcr 2, and mcr1, respectively (data not shown).
Analytic Sensitivity of Multiplex Δ-PCR for Detection of BCL2-IGH Translocation
The paired forward primers were mixed with reverse primer VLJH for Δ-PCR and tested at relative ratios of the paired forward primers from 1:4 to 4:1 to produce two expected amplicons of approximately the same peak heights (Table 1). The sensitivity for each set of Δ-PCR primers designed to detect breakpoints at MBR1, MBR2, and mcr2 was at least 40–50 cell equivalents using the appropriate cell line diluted into distilled water or at least 5 × 10−3 using the appropriate cell line diluted into DNA samples extracted from frozen normal lymph node tissues (data not shown).
Multiplex Δ-PCR was performed by mixing all eight forward primers together with the reverse primer. Each control cell line yielded a pair of peaks with the expected size difference and little background noise. The analytic sensitivity of multiplex Δ-PCR was also at least 40–50 cell equivalents (Figure 2) or 5 × 10−3. The average peak heights of the two amplicons tested in five replicate reactions with 45-second injections were 1529 and 3678 relative fluorescent units (RFU) (range, 738–2216 and 1530–5472) for MBR1, 1328 and 2654 RFU (range, 836–1779 and 1184–3791) for MBR2, and 522 and 790 RFU (range, 371–800 and 593–1259) for mcr2.
Figure 2.
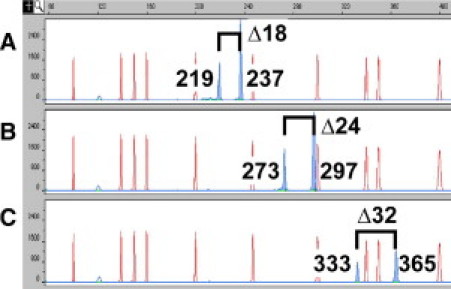
Analytic sensitivity of Δ-PCR for BCL2-IGH translocation. Genomic DNA (250 pg, or 40–50 cell equivalents) from cell lines SU-DHL-6 (A), SU-DHL-4 (B), and SU-DHL-16 (C) (positive control for MBR1, MBR2, and mcr2 breakpoints, respectively) was subjected to multiplex Δ-PCR and analyzed by electrophoresis (ABI 3100 system, Applied Biosystems, Foster City, CA) with 45-second electrokinetic injection. Blue peaks are Δ-PCR products (Δ numbers indicate amplicon size difference in base pairs) and red peaks are size markers. The presence of two peaks with size difference of Δ18 ± 1 bp, Δ24 ± 1 bp, and Δ33 ± 1 bp indicates BCL2-IGH translocation with breakpoints at MBR1, MBR2, and mcr2, respectively.
To further demonstrate that our multiplex Δ-PCR has an acceptable analytic sensitivity, we compared conventional PCR (forward primer MBR1int and reverse primer VLJH) and multiplex Δ-PCR (mixture of eight forward primers and reverse primer VLJH) using real-time PCR. The cycle threshold Ct values (mean of triplicates) of cell line SU-DHL-6 (10 ng) and of three clinical cases (cases 8, 12, and 22) with an MBR1 breakpoint (20 ng) were similar: 30.7, 31.4, 32.2, and 32.6 for conventional PCR and 31.9, 31.6, 32.4, and 32.9 for multiplex Δ-PCR.
Detection and Localization of Breakpoints in Patients with Follicular Lymphoma
We tested 40 FFPE specimens from follicular lymphoma cases using multiplex Δ-PCR (Table 2). Half of the cases carried breakpoints at the MBR1 (20 cases); the mcr1 (1 case, 2.5%) and mcr2 (1 case, 2.5%) breakpoints were much less common (Table 2 and Figure 3). No cases with the BCL2-IGH MBR2 translocation were identified. Sequencing of the amplicon from case 30 confirmed the breakpoint at mcr2 and, as already indicated, sequence confirmation of the mcr1 translocation in case 19 was obtained. Although the peak heights of the two amplicons from case 4 were very low, at 492 and 277 RFU, the presence of two peaks (245 bp and 263 bp) with a size difference of 18 bp verified the validity of the breakpoint at MBR1 (data not shown). All 12 control cases were negative for BCL2-IGH translocation at all four breakpoints.
Table 2.
Results of Multiplex Δ-PCR on Clinical Formalin-Fixed, Paraffin-Embedded Samples
| Breakpoint | Follicular lymphoma (n = 40) | Negative control (n = 12)⁎ |
|---|---|---|
| MBR1 | 20 | 0 |
| MBR2 | 0 | 0 |
| mcr1 | 1 | 0 |
| mcr2 | 1 | 0 |
6 cases of lymphoid hyperplasia and 6 cases of hematological malignancies other than follicular lymphoma.
Figure 3.
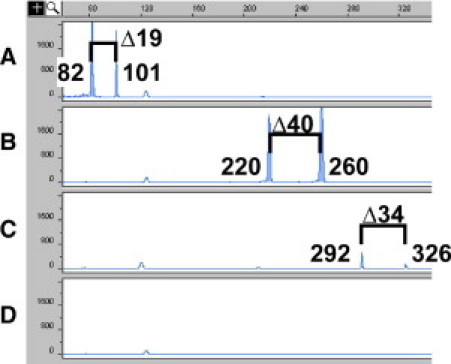
Representative cases with MBR1, mcr1, and mcr2 breakpoints detected by Δ-PCR. DNA (20 ng) extracted from cases 36 (A), 19 (B), and 30 (C) and from a case with lymphoid hyperplasia (D) was subjected to multiplex Δ-PCR and analyzed by capillary electrophoresis with 45-second electrokinetic injection. Cases 36, 19, and 30 were positive for MBR1 (Δ18 ± 1 bp), mcr1 (Δ39 ± 1 bp), and mcr2 (Δ33 ± 1 bp), respectively, based on the size difference of the two amplicon peaks.
Real-Time PCR
All cases were subjected to MBR1-specific real-time PCR to confirm breakpoints at MBR1. The results of real-time PCR from control cases and follicular lymphoma cases were consistent with those from multiplex Δ-PCR, except for lymphoma case 36 (Table 3). This case showed no amplification by real-time PCR but had two relatively small peaks at 82 and 101 bp amplified by multiplex Δ-PCR (Figure 3A). Amplicons from all other cases of follicular lymphoma with breakpoints at MBR1 were larger (ranges, 109–245 bp and 127–263 bp for amplicons amplified from primers MBR1int/VLJH and MBR1ext/VLJH, respectively). This suggested that case 36 had a breakpoint close to one of the primers. Sequencing of the amplicons confirmed that this was true: the breakpoint was located 9 bp 5′ to the real-time PCR probe and thus prevented annealing of the probe (data not shown). These results indicate that the clinical diagnostic sensitivity at MBR1 using multiplex Δ-PCR is at least as good as real-time PCR, and the diagnostic specificity with regard to reactive lesions, leukemias, and lymphomas other than follicular lymphoma is 100% (95% CI, 70%–100%) (proportional confidence interval with continuity correction, http://faculty.vassar.edu/lowry/VassarStats.html, accessed on Aug. 11, 2010).
Table 3.
Comparison between Real-Time PCR and Multiplex Δ-PCR
| Real-time PCR | Multiplex Δ-PCR |
|||
|---|---|---|---|---|
| Follicular lymphoma |
Negative control |
|||
| MBR1 (+) | MBR1 (−) | MBR1 (+) | MBR1 (−) | |
| Positive | 19 | 0 | 0 | 0 |
| Negative | 1⁎ | 20 | 0 | 12 |
Discrepancy between real-time PCR and multiplex Δ-PCR.
Detection of Multiple Minor Leukemic Clones with ITD Mutation by Δ-PCR
Apparent multiple ITD mutations in acute myeloid leukemia patients are occasionally detected using conventional PCR in our routine clinical laboratory. DNA samples from three such cases were subjected to Δ-PCR. Multiple ITD mutations, likely representing individual subclones, were confirmed by each pair of peaks with a size difference of defined Δ, 25 bases in this design (Figure 4). The sensitivity for detection of minor clones is <1% of the wild type. The minor clones were isolated by a molecular fraction collecting tool using a reconfigured ABI 310 automated sequencer, and the presence of different ITD mutations was further confirmed by sequencing (data not shown).
Figure 4.
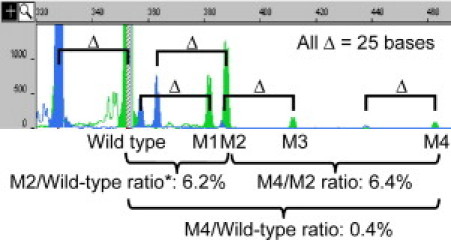
Detection of multiple minor leukemic clones with internal tandem duplication mutation by Δ-PCR. The two wild-type peaks were 328 bases (blue) and 353 bases (green). Four mutant clones (M1, M2, M3, and M4) were detected. Each clone has one blue peak and one green peak, with a Δ value of 25 bases. The peak height ratio of M4/M2 was 6.4%, ratio of M2/wild-type was 6.2%, and the ratio of M4/wild-type was 0.4% (6.4% × 6.2%). *The red striped line within the 353-base wild-type peak indicates off-scale height. The PCR product was therefore diluted fivefold and reinjected into the ABI 3100 genetic analyzer (data not shown) to calculate the ratio of M2/wild-type.
Discussion
PCR and RT-PCR can be extremely sensitive and specific assays for the detection of tumor-specific chromosomal translocations. Several variants of PCR have been designed to improve sensitivity, specificity, and throughput. The development of real-time PCR was a significant advance for molecular diagnostic laboratories.24–26 The combination of a fluorochrome-labeled probe and the real-time detection of fluorescence after each cycle of PCR increases not only the sensitivity and throughput but also the specificity of the PCR. In addition to its widespread application in the quantitative detection of microorganisms and minimal residual cancer cells,27,28 real-time PCR has become an alternative to traditional Southern blot analysis for the detection of translocations with varied sizes of PCR products amplified from different tumor clones, such as BCL2-IGH in follicular lymphoma.29
Several probes and paired primers are needed for translocations with more than one common breakpoint or for assays using small DNA targets, such as fragmented templates extracted from FFPE tissues. Furthermore, the number of multiplexed probes is limited by the fluorochrome detection channels of the real-time machine. In the present study, we developed a new strategy, Δ-PCR, using two upstream primers for each potential breakpoint hot spot, to be paired simultaneously with a common, downstream primer. The presence of two peaks with the expected size difference (when analyzed by capillary electrophoresis) indicates the specific amplification of PCR products from the defined breakpoint locus. Because only a single fluorochrome-labeled oligonucleotide is needed, the assays can be easily multiplexed to detect multiple breakpoints in a single PCR reaction.
The t(14;18) rearrangement resulting in fusion of the BCL2 and IGH genes is detectable in 80% to 90% of follicular lymphoma by a variety of methods.30–33 Several molecular assays, including nested PCR, Southern blot analysis using probe hybridization, multiplex PCR, and real-time PCR have been developed to identify the BCL2-IGH translocation.13,16,29,34,35 Nested PCR and Southern blot analysis are not desirable in molecular diagnostic laboratories, however, because manipulation of the first PCR product could be a serious source of contamination, and probe hybridization is time-consuming. Real-time PCR using a TaqMan-based (Applied Biosystems) or LightCycler-based assay has been a very useful tool for the detection of minimal residual follicular lymphoma cells carrying the BCL2-IGH translocation.16,29,36,37 Although real-time PCR can be applied as a diagnostic assay to confirm the presence of BCL2-IGH translocation, multiple probes and paired primers are needed, because of the multiple and widely separated loci of the breakpoint in the BCL2 gene.13 Multiplex PCR is an alternative strategy for detecting most breakpoints of BCL2-IGH translocations. The BIOMED-2 protocol was developed to detect BCL2-IGH translocation with breakpoints at MBR1, MBR2, 3′MBR1, 3′MBR2, 3′MBR3, or 3′MBR4, as well as 5′mcr, mcr1, or mcr2 loci using multiplexed PCR reactions in three separate tubes.9,13–15 The accompanying nonspecific PCR bands may complicate interpretation of the results, however. In the present study, we have shown that Δ-PCR can provide both high specificity and a multiplexed format.
We have tested FFPE specimens from 40 cases of follicular lymphoma with previously unknown t(14;18) status, using multiplex Δ-PCR to detect breakpoints at the MBR1 (20 cases), MBR2 (0 cases), mcr1 (1 case), and mcr2 (1 case) loci of the BCL2-IGH translocation. The analytic sensitivity of Δ-PCR is at least 40–50 cell equivalents or 5 × 10−3 for the breakpoints at MBR1, MBR2, and mcr2. The clinical diagnostic sensitivity for the detection of breakpoints at MBR1 using multiplex Δ-PCR is at least as good as real-time PCR, and the diagnostic specificity is 100% (95% CI, 70%–100%). The overall results in the present study were consistent with studies of larger patient populations. Weinberg et al38 reported BCL2-IGH translocation of 236 patients with histologically confirmed follicular lymphoma using real-time PCR; translocation within MBR and mcr was found in 50% and 5% of patients, respectively. Van Dongen et al13 studied a total 124 patient of t(14;18)-positive follicular lymphoma and found that 67% and 9% had a breakpoint within the MBR and mcr, respectively. In principle, Δ-PCR for other BCL2-IGH breakpoint loci could be developed.
The strategy of Δ-PCR will also be useful for other translocations with a few widely separated breakpoints, such as the CCND1-IGH translocation in mantle cell lymphoma,11,13 and for insertion/deletion mutations of varied size, such as the ITD of the FLT3 gene and small insertion/deletion and duplication mutations of the transcription factor gene GATA1 and Wilms' tumor 1 (WT1) gene in leukemia or myelodysplastic syndrome.17,18,39–43
In our molecular diagnostic laboratory, we have seen cases of FLT3 mutation with multiple small peaks in addition to a major ITD mutant peak. In the present study, we applied the Δ-PCR strategy to these cases and showed that these small peaks are indeed minor clones with different ITD mutations, rather than artifacts. Δ-PCR has a great advantage over real-time PCR in this situation, because most clinical samples have wild-type alleles that interfere with mutation detection. Using the strategy described here, multiplex Δ-PCR can be used to detect and distinguish, simultaneously, the small insertion/deletion mutations of the FLT3, GATA1, and WT1 genes of a leukemic clone.
With some modification, the Δ-PCR method may be more broadly applicable, such as in analyzing the species-specific length polymorphisms in the internal transcribed spacer 2 (ITS2) region of the rRNA genes for rapid identification of fungal infection. For samples with suspected infection but low fungal loads, such as peripheral blood and cerebrospinal fluid, Δ-PCR can be useful in distinguishing low-frequency amplicons from background nonspecific amplification.44–46
In summary, we have developed the strategy of Δ-PCR to determine the specific breakpoint of BCL2/IGH translocation in a single multiplexed PCR reaction with high analytic sensitivity and detection specificity and sensitivity. The addition of more primers can expand the multiplex to include other, rarer loci of known BCL2/IGH translocations without a loss in specificity. The strategy of Δ-PCR can also be applied to other translocations or insertion/deletion mutations.
Footnotes
Supported in part by award R21HG004315 from the National Human Genome Research Institute, National Institutes of Health.
CME Disclosure: The authors did not disclose any relevant financial relationships.
References
- 1.Rabbitts T.H. Chromosomal translocations in human cancer. Nature. 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 2.Look A.T. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 3.Rowley J.D. Cytogenetic and molecular analysis of pediatric neoplasms: diagnostic and clinical implications. Pediatr Pathol. 1994;14:167–176. doi: 10.3109/15513819409022036. [DOI] [PubMed] [Google Scholar]
- 4.Rowley J.D. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 5.de Klein A., van Kessel A.G., Grosveld G., Bartram C.R., Hagemeijer A., Bootsma D., Spurr N.K., Heisterkamp N., Groffen J., Stephenson J.R. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765–767. doi: 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 6.Clark J., Rocques P.J., Crew A.J., Gill S., Shipley J., Chan A.M., Gusterson B.A., Cooper C.S. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p112;q112) translocation found in human synovial sarcoma. Nat Genet. 1994;7:502–508. doi: 10.1038/ng0894-502. [DOI] [PubMed] [Google Scholar]
- 7.Lee M.S., LeMaistre A., Kantarjian H.M., Talpaz M., Freireich E.J., Trujillo J.M., Stass S.A. Detection of two alternative bcr/abl mRNA junctions and minimal residual disease in Philadelphia chromosome positive chronic myelogenous leukemia by polymerase chain reaction. Blood. 1989;73:2165–2170. [PubMed] [Google Scholar]
- 8.Kawai A., Woodruff J., Healey J.H., Brennan M.F., Antonescu C.R., Ladanyi M. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998;338:153–160. doi: 10.1056/NEJM199801153380303. [DOI] [PubMed] [Google Scholar]
- 9.Bakhshi A., Jensen J.P., Goldman P., Wright J.J., McBride O.W., Epstein A.L., Korsmeyer S.J. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 10.Weiss L.M., Warnke R.A., Sklar J., Cleary M.L. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med. 1987;317:1185–1189. doi: 10.1056/NEJM198711053171904. [DOI] [PubMed] [Google Scholar]
- 11.Tsujimoto Y., Yunis J., Onorato-Showe L., Erikson J., Nowell P.C., Croce C.M. Molecular cloning of the chromosomal breakpoint of B-cell lymphomas and leukemias with the t(11;14) chromosome translocation. Science. 1984;224:1403–1406. doi: 10.1126/science.6610211. [DOI] [PubMed] [Google Scholar]
- 12.Crescenzi M., Seto M., Herzig G.P., Weiss P.D., Griffith R.C., Korsmeyer S.J. Thermostable DNA polymerase chain amplification of t(14;18) chromosome breakpoints and detection of minimal residual disease. Proc Natl Acad Sci USA. 1988;85:4869–4873. doi: 10.1073/pnas.85.13.4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Dongen J.J., Langerak A.W., Brüggemann M., Evans P.A., Hummel M., Lavender F.L., Delabesse E., Davi F., Schuuring E., García-Sanz R., van Krieken J.H., Droese J., González D., Bastard C., White H.E., Spaargaren M., González M., Parreira A., Smith J.L., Morgan G.J., Kneba M., Macintyre E.A. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98–3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 14.Cleary M.L., Smith S.D., Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 15.Buchonnet G., Lenain P., Ruminy P., Lepretre S., Stamatoullas A., Parmentier F., Jardin F., Duval C., Tilly H., Bastard C. Characterisation of BCL2-JH rearrangements in follicular lymphoma: pCR detection of 3′ BCL2 breakpoints and evidence of a new cluster. Leukemia. 2000;14:1563–1569. doi: 10.1038/sj.leu.2401889. [DOI] [PubMed] [Google Scholar]
- 16.Ladetto M., Sametti S., Donovan J.W., Ferrero D., Astolfi M., Mitterer M., Ricca I., Drandi D., Corradini P., Coser P., Pileri A., Gribben J.G., Tarella C. A validated real-time quantitative PCR approach shows a correlation between tumor burden and successful ex vivo purging in follicular lymphoma patients. Exp Hematol. 2001;29:183–193. doi: 10.1016/s0301-472x(00)00651-2. [DOI] [PubMed] [Google Scholar]
- 17.Murphy K.M., Levis M., Hafez M.J., Geiger T., Cooper L.C., Smith B.D., Small D., Berg K.D. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5:96–102. doi: 10.1016/S1525-1578(10)60458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakao M., Yokota S., Iwai T., Kaneko H., Horiike S., Kashima K., Sonoda Y., Fujimoto T., Misawa S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. [PubMed] [Google Scholar]
- 19.Lin M.T., Rich R.G., Shipley R.F., Hafez M.J., Tseng L.H., Murphy K.M., Gocke C.D., Eshleman J.R. A molecular fraction collecting tool for the ABI 310 automated sequencer. J Mol Diagn. 2007;9:598–603. doi: 10.2353/jmoldx.2007.070022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris N.H., Swerdlow S.H., Jaffe E.S., Ott G., Nathwani B.N., de Jong D., Yoshino T., Spagnolo D. Follicular lymphoma: WHO classification of tumors of haematopoietic and lymphoid tissues. In: Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H., Thiele J., Vardiman J.W., editors. IARC; Lyon: 2008. pp. 220–226. [Google Scholar]
- 21.Berg K.D., Glaser C.L., Thompson R.E., Hamilton S.R., Griffin C.A., Eshleman J. Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J Mol Diagn. 2000;2:20–28. doi: 10.1016/S1525-1578(10)60611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S.C., Berg K.D., Racke F.K., Griffin C.A., Eshleman J.R. Pseudo-spikes are common in histologically benign lymphoid tissues. J Mol Diagn. 2000;2:145–152. doi: 10.1016/S1525-1578(10)60630-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ngan B.Y., Nourse J., Cleary M.L. Detection of chromosomal translocation t(14;18) within the minor cluster region of bcl-2 by polymerase chain reaction and direct genomic sequencing of the enzymatically amplified DNA in follicular lymphomas. Blood. 1989;73:1759–1762. [PubMed] [Google Scholar]
- 24.Holland P.M., Abramson R.D., Watson R., Gelfand D.H. Detection of specific polymerase chain reaction product by utilizing the 5′→3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci USA. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heid C.A., Stevens J., Livak K.J., Williams P.M. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 26.Lie Y.S., Petropoulos C.J. Advances in quantitative PCR technology: 5′ nuclease assays. Curr Opin Biotechnol. 1998;9:43–48. doi: 10.1016/s0958-1669(98)80082-7. [DOI] [PubMed] [Google Scholar]
- 27.Gabert J., Beillard E., van der Velden V.H., Bi W., Grimwade D., Pallisgaard N., Barbany G., Cazzaniga G., Cayuela J.M., Cavé H., Pane F., Aerts J.L., De Micheli D., Thirion X., Pradel V., González M., Viehmann S., Malec M., Saglio G., van Dongen J.J. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—a Europe Against Cancer program. Leukemia. 2003;17:2318–2357. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- 28.Espy M.J., Uhl J.R., Sloan L.M., Buckwalter S.P., Jones M.F., Vetter E.A., Yao J.D., Wengenack N.L., Rosenblatt J.E., Cockerill F.R., 3rd, Smith T.F. Real-time PCR in clinical microbiology: applications for routine laboratory testing [Erratum appears in Clin Microbiol Rev 2006;19:595] Clin Microbiol Rev. 2006;19:165–256. doi: 10.1128/CMR.19.1.165-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luthra R., McBride J.A., Cabanillas F., Sarris A. Novel 5′ exonuclease-based real-time PCR assay for the detection of t(14;18)(q32;q21) in patients with follicular lymphoma. Am J Pathol. 1998;153:63–68. doi: 10.1016/S0002-9440(10)65546-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cleary M.L., Galili N., Sklar J. Detection of a second t(14;18) breakpoint cluster region in human follicular lymphomas. J Exp Med. 1986;164:315–320. doi: 10.1084/jem.164.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yunis J.J., Frizzera G., Oken M.M., McKenna J., Theologides A., Arnesen M. Multiple recurrent genomic defects in follicular lymphoma. A possible model for cancer N Engl J Med. 1987;316:79–84. doi: 10.1056/NEJM198701083160204. [DOI] [PubMed] [Google Scholar]
- 32.Vaandrager J.W., Schuuring E., Raap T., Philippo K., Kleiverda K., Kluin P. Interphase FISH detection of BCL2 rearrangement in follicular lymphoma using breakpoint-flanking probes. Genes Chromosomes Cancer. 2000;27:85–94. [PubMed] [Google Scholar]
- 33.Aster J.C., Longtine J.A. Detection of BCL2 rearrangements in follicular lymphoma. Am J Pathol. 2002;160:759–763. doi: 10.1016/S0002-9440(10)64897-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ladanyi M., Wang S. Detection of rearrangements of the BCL2 major breakpoint region in follicular lymphomas: Correlation of polymerase chain reaction results with Southern blot analysis. Diagn Mol Pathol. 1992;1:31–35. doi: 10.1097/00019606-199203000-00005. [DOI] [PubMed] [Google Scholar]
- 35.Gribben J.G., Freedman A., Woo S.D., Blake K., Shu R.S., Freeman G., Longtine J.A., Pinkus G.S., Nadler L.M. All advanced stage non-Hodgkin's lymphomas with a polymerase chain reaction amplifiable breakpoint of bcl-2 have residual cells containing the bcl-2 rearrangement at evaluation and after treatment. Blood. 1991;78:3275–3280. [PubMed] [Google Scholar]
- 36.Chang C.C., Bredeson C., Juckett M., Logan B., Keever-Taylor C.A. Tumor load in patients with follicular lymphoma post stem cell transplantation may correlate with clinical course. Bone Marrow Transplant. 2003;32:287–291. doi: 10.1038/sj.bmt.1704130. [DOI] [PubMed] [Google Scholar]
- 37.Martin S., Fischer C., Free M., Kurreck B., Stockinger H., Fenk R., Arnold C., Kliszewski S., Meckenstock G., Haas R., Kronenwett R. LightCycler-based quantitative real-time PCR monitoring of patients with follicular lymphoma receiving rituximab in combination with conventional or high-dose cytotoxic chemotherapy. Eur J Haematol. 2005;74:282–292. doi: 10.1111/j.1600-0609.2004.00391.x. [DOI] [PubMed] [Google Scholar]
- 38.Weinberg O.K., Ai W.Z., Mariappan M.R., Shum C., Levy R., Arber D.A. “Minor” BCL2 breakpoints in follicular lymphoma: frequency and correlation with grade and disease presentation in 236 cases. J Mol Diagn. 2007;9:530–537. doi: 10.2353/jmoldx.2007.070038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yokota S., Kiyoi H., Nakao M., Iwai T., Misawa S., Okuda T., Sonoda Y., Abe T., Kahsima K., Matsuo Y., Naoe T. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines Leukemia. 1997;11:1605–1609. doi: 10.1038/sj.leu.2400812. [DOI] [PubMed] [Google Scholar]
- 40.Wechsler J., Greene M., McDevitt M.A., Anastasi J., Karp J.E., Le Beau M.M., Crispino J.D. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002;32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 41.Cabelof D.C., Patel H.V., Chen Q., van Remmen H., Matherly L.H., Ge Y., Taub J.W. Mutational spectrum at GATA1 provides insights into mutagenesis and leukemogenesis in Down syndrome. Blood. 2009;114:2753–2763. doi: 10.1182/blood-2008-11-190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King-Underwood L., Renshaw J., Pritchard-Jones K. Mutations in the Wilms' tumor gene WT1 in leukemias. Blood. 1996;87:2171–2179. [PubMed] [Google Scholar]
- 43.Paschka P., Marcucci G., Ruppert A.S., Whitman S.P., Mrózek K., Maharry K., Langer C., Baldus C.D., Zhao W., Powell B.L., Baer M.R., Carroll A.J., Caligiuri M.A., Kolitz J.E., Larson R.A., Bloomfield C.D. Wilms' tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2008;26:4595–4602. doi: 10.1200/JCO.2007.15.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turenne C.Y., Sanche S.E., Hoban D.J., Karlowsky J.A., Kabani A.M. Rapid identification of fungi by using the ITS2 genetic region and an automated fluorescent capillary electrophoresis system [Erratum appears in J Clin Microbiol 2000;38:944] J Clin Microbiol. 1999;37:1846–1851. doi: 10.1128/jcm.37.6.1846-1851.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y.C., Eisner J.D., Kattar M.M., Rassoulian-Barrett S.L., LaFe K., Yarfitz S.L., Limaye A.P., Cookson B.T. Identification of medically important yeasts using PCR-based detection of DNA sequence polymorphisms in the internal transcribed spacer 2 region of the rRNA genes. J Clin Microbiol. 2000;38:2302–2310. doi: 10.1128/jcm.38.6.2302-2310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landlinger C., Basková L., Preuner S., Willinger B., Buchta V., Lion T. Identification of fungal species by fragment length analysis of the internally transcribed spacer 2 region. Eur J Clin Microbiol Infect Dis. 2009;28:613–622. doi: 10.1007/s10096-008-0683-3. [DOI] [PubMed] [Google Scholar]