

Abstract
The activin receptor type IIB (ActRIIB) is a transmembrane receptor for transforming growth factor-β superfamily members, including myostatin, that are involved in the negative regulation of skeletal muscle mass. We tested the translational hypothesis that blocking ligand binding to ActRIIB for 12 weeks would stimulate skeletal muscle growth and improve muscle function in the mdx mouse. ActRIIB was targeted using a novel inhibitor comprised of the extracellular portion of the ActRIIB fused to the Fc portion of murine IgG (sActRIIB), at concentrations of 1.0 and 10.0 mg/kg−1 body weight. After 12 weeks of treatment, the 10.0 mg/kg−1 dose caused a 27% increase in body weight with a concomitant 33% increase in lean muscle mass. Absolute force production of the extensor digitorum longus muscle ex vivo was higher in mice after treatment with either dose of sActRIIB, and the specific force was significantly higher after the lower dose (1.0 mg/kg−1), indicating functional improvement in the muscle. Circulating creatine kinase levels were significantly lower in mice treated with sActRIIB, compared with control mice. These data show that targeting the ActRIIB improves skeletal muscle mass and functional strength in the mdx mouse model of DMD, providing a therapeutic rationale for use of this molecule in treating skeletal myopathies.
The muscular dystrophies are a group of progressive, hereditary primary myopathies that result in skeletal muscle degeneration and wasting.1 Duchenne's muscular dystrophy (DMD) and the milder allelic Becker form are X-linked diseases resulting from a genetic mutation in the dystrophin gene (DMD), leading to a loss or severe reduction of functional dystrophin protein.2 With the loss of dystrophin protein, the dystrophin-glycoprotein complex that forms a link between the extracellular matrix and the actin cytoskeleton is disrupted, leading to loss of muscle membrane integrity.3,4 Skeletal muscles from DMD patients have characteristic increases in inflammation, and cycles of muscle fiber degeneration and regeneration lead to collagen infiltration and muscle wasting.1,5 DMD is diagnosed in early childhood, and patients typically require wheelchairs and ventilatory assistance in their teens, demonstrating the severe progressive course of the disease.1
At present, there is no cure for DMD, and standard management for patients includes supportive measures and corticosteroid therapy (ie, prednisone, deflazacort). Although these drugs initially improve muscle function and strength, adverse effects are associated with each drug (eg, weight gain, hypertension, and reduced bone density), and there are limitations to the therapeutic value over time.6–8 Attempts to restore dystrophin protein in the muscles of DMD patients using exon skipping techniques, gene-based therapies and stem cell transplantation have shown some promise in clinical trials.9–14 Nonetheless, only a limited number of patients can benefit from these strategies, and individualized medicine may be required to take full advantage of these therapies.15 Research using animal models has also identified promising therapies that can ameliorate the dystrophic phenotype.16 The pharmacological stimulation of muscle growth, attenuation of muscle wasting, and reduction in the muscle inflammation that is characteristic of dystrophic muscle may be a viable option for patients, in conjunction with supportive measures. In the short term, if muscle mass can be increased or preserved in DMD patients, this may prolong muscle function and life expectancy. Therapeutic targets that have been investigated to stimulate muscle mass in dystrophic muscle include insulin-like growth factor-1,17–19 IL-15,20 and myostatin (MSTN).21–23
MSTN, a negative regulator of muscle mass, is a member of the transforming growth factor-β superfamily of proteins. Animals with genetic mutations in the Mstn gene, including Belgian Blue cattle,24 the Mstn knockout mouse,25 and the whippet dog,26,27 all exhibit a hypermuscled phenotype, and this has also been documented in a child.28 Endogenous MSTN protein circulates as an inactive complex that consists of a 26-kDa propeptide region and a 12-kDa active region. Release of active MSTN dimers allows binding to the activin type IIB receptor (ActRIIB), leading to subsequent phosphorylation of SMAD proteins and initiation of gene expression (reviewed by Fedoruk and Rupert).29 Experimental means to inhibit MSTN, including antibodies directed against MSTN,21,30 propeptide-mediated inhibition of MSTN,22 and DNA-based inhibition methods,31,32 have resulted in increased muscle mass in wild-type mice30 and an improvement in the dystrophic phenotype in the mdx mouse model of DMD.21–23 Additional ligands bind to ActRIIB, including activin A, NODAL, bone morphogenetic protein 2, bone morphogenetic protein-6/7, and growth/differentiation factor 11.33 Some of these ligands may act in concert with MSTN to inhibit muscle growth, as recently suggested.34
In the present study, we used a soluble form of ActRIIB (sActRIIB) as a means to suppress multiple ligands that bind to ActRIIB, and we tested the translational hypothesis that sActRIIB therapy would stimulate skeletal muscle growth and improve muscle function in the mdx mouse model of DMD. Our data demonstrate that 12 weeks of sActRIIB therapy increases body mass, muscle mass, and muscle function in the mdx mouse, providing rationale for the therapeutic use of this strategy for improving muscle wasting in skeletal myopathies such as DMD.
Materials and Methods
Mice
Male C57BL/10ScSn-Dmdmdx/J (mdx) and C57BL/10ScSn wild-type mice were purchased from Jackson Laboratories (Bar Harbor, ME) at 4 weeks of age. Mice were exposed to a 12-hour light/dark cycle and 22°C ambient temperature, and received rodent chow (Lab Diet 5001; PMI Nutrition International, Gray Summit, MO) and water ad libitum. Mice were acclimated at the animal facility at the University of Pennsylvania before initiation of experiments. The mdx mice were randomly assigned to receive either sActRIIB injections (n = 14) or sterile PBS injections (n = 13). Control and sActRIIB-treated mice were housed in separate cages during the 12-week preclinical trial. Age-matched C57BL/10ScSn mice (n = 6) were used as untreated wild-type controls. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
ActRIIB-Inhibition Strategy
Inhibition of ActRIIB signaling was achieved using a fusion protein comprised of a form of the extracellular domain of ActRIIB linked to the Fc portion of murine IgG (RAP-031; Acceleron Pharma, Cambridge, MA). Two independent drug trials were performed in mdx mice, at concentrations of 1.0 and 10.0 mg/kg−1 body weight. The mdx mice in each group were compared with a separate group of age-matched control mdx mice. Experimental mice received intraperitoneal injections of either sActRIIB or sterile PBS two times per week for 12 weeks. sActRIIB activity in vivo was verified through qPCR for expression of the transgelin gene (Tagln; synonym, Sm22) and Western immunoblotting analysis (phosphorylated Akt) analysis for markers downstream of ActRIIB, using treated and control muscles35–38 (see Supplementary Figure S1 at http://ajp.amjpathol.org).
Body Weight and Composition
The mdx mice were weighed twice per week to determine the correct volume of sActRIIB to be delivered and to plot growth curves. In the 10.0 mg/kg−1 group, body composition was determined during week 11 of treatment using nuclear magnetic resonance quantification (Echo Medical Systems), as described previously.39
Ex Vivo Physiological Assessment of Skeletal Muscle
Physiological properties, including isometric twitch force, contraction time of the twitch, half-relaxation time of the twitch, isometric tetanic force, and force drop after eccentric lengthening contractions, were quantified in freshly dissected extensor digitorum longus (EDL) muscles from 16-week-old mdx mice, as described previously.21,22 Muscle length was adjusted to obtain the maximal twitch response and this length was measured and recorded as optimal length. Three maximal twitch and tetanic contractions were performed, followed by a series of five eccentric lengthening contractions. The force drop from eccentric lengthening contractions was calculated as the percent difference in maximum force between the first and fifth eccentric lengthening contraction. Muscles were stimulated in a Ringer's solution composed of 100 mmol/L NaCl, 4.7 mmol/L KCl, 3.4 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 25 mmol/L HEPES, and 5.5 mmol/L d-glucose. At the end of the physiological assessment, EDL muscles were incubated in an oxygenated 1% w/v Procion Orange solution for 30 minutes, flash frozen in isopentane cooled to the temperature of liquid nitrogen, and stored at −80°C until used for sectioning. Muscle cross-sectional area (CSA) was calculated by dividing the muscle mass by the product of the muscle density coefficient (1.06 g · cm−3), muscle optimal length, and the fiber length coefficient for EDL muscles, as described previously40,41: CSA = EDL mass/(Lo × fiber length coefficient × 1.06 g/cm3).
Muscle Histology and Morphology
Frozen muscle sections (10 μm thickness) were cut at the midbelly of the muscle and fixed in ice-cold methanol for 5 minutes. Diaphragm and tibialis anterior sections were processed for histological examination with H&E and Masson's trichrome stains. For morphometric analyses, EDL muscle sections were stained with an anti-laminin monoclonal antibody (Sigma-Aldrich, St. Louis, MO), incubated with Alexa 488 goat anti-mouse secondary antibody (Invitrogen, Carlsbad, CA), incubated with the DNA-binding dye Hoechst 33825, and visualized using epi-fluorescence illumination on an Olympus BX51 microscope at an objective magnification of 10× or 40×. Digital images were acquired using an Olympus Magnafire digital camera. Morphometric measurements were made using ImageJ image processing software version 1.43 (NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/). Morphometric measurements included single-fiber CSA, total number of fibers, percentage of centrally nucleated fibers, and number of Procion Orange-positive fibers.
Serum Creatine Kinase Quantification
Blood was collected from all experimental mice via cardiac puncture and was allowed to clot for 30 minutes at room temperature. Serum was collected after centrifugation for 10 minutes at 4.0 relative centrifugal force. Creatine kinase (CK) levels were quantified using a kit as per manufacturer's instructions (catalog no. 2910-430; Stanbio Laboratories, Boerne, TX). In addition to serum samples, normal and abnormal serum standards were included in all analyses to validate the assay (catalog no. G427, G428; Stanbio Laboratories).
Hydroxyproline Content
The content of the amino acid hydroxyproline has been used as a measure of the extent of fibrosis in dystrophic skeletal muscle.42 The tibialis anterior muscle and the diaphragm were used for hydroxyproline quantification (AAA Laboratory, Mercer Island, WA).
qPCR Gene Expression Analysis
Total RNA was isolated from gastrocnemius muscles as described previously.43 RNA quantity and integrity was accessed using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE); the 260/280 ratio for all samples used was between 1.9 and 2.1. Two micrograms of total RNA was reverse transcribed to make cDNA using a high-capacity cDNA reverse transcription kit according to the manufacturer's instructions (Applied Biosystems, Foster City, CA). The wells of a 96-well optical reaction plate were loaded with a 20-μL volume consisting of 10× PCR master mix (Applied Biosystems, Foster City, CA), cDNA diluted in sterile H2O, and either a primer mix for the gene of interest or the housekeeping gene 18S (Rn18s).The relative quantification of each gene was calculated using the ΔΔCt method, with one control B6129 cDNA sample used as the assay calibrator.
Western Immunoblotting
Quadriceps tissue was homogenized in lysis buffer containing 50 mmol/L Tris · HCl (pH 7.4), 250 mmol/L mannitol, 50 mmol/L NaF, 1 mmol/L sodium pyrophosphate, 1 mmol/L benzamidine, and 1 mmol/L phenylmethylsulfonyl fluoride with 0.5% (w/v) Triton X-100, supplemented with complete protein inhibition cocktail tablet from Roche (Penzberg, Germany), as described previously.36 Protein extracts (50 μg) were separated using 4% to 12% NuPAGE Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes with wet transfer cells (Bio-Rad Laboratories, Hercules, CA). After 1 hour of blocking with Tris-buffered saline with 0.1% (v/v) Tween 20 containing 10% (w/v) nonfat dried milk, membranes were incubated with a polyclonal antibody against phosphorylated (Ser473) Akt, Akt1/2/3 (Santa Cruz), and GAPDH (Cell Signaling Technology, Danvers, MA). Blots were washed with the Tris-buffered saline with Tween 20 and were incubated with anti-rabbit IgG conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA). The signals were detected with enhanced enzymatic chemiluminescence (Amersham-GE Healthcare, Chalfont St Giles, UK).
Statistical Analysis
Student's t-test was used to analyze the data from the two independent trials, using independent control groups for comparisons with the two doses of sActRIIB used. Data were analyzed using the GraphPad Prism 4 statistical software package (GraphPad Software, La Jolla, CA), with statistical significance set at P < 0.05. Data are reported as means ± SEM.
Results
sActRIIB Treatment Increases Body Mass and Lean Muscle Mass in Vivo in mdx Mice
To test the effects of sActRIIB therapy, two independent preclinical drug trials were performed in which 4-week-old mdx mice received intraperitoneal injections twice weekly for 12 weeks at a dose of 1.0 mg/kg−1 or 10.0 mg/kg−1 body weight and were compared with trial-matched control mdx mice that received equal volumes of sterile PBS. Mice treated with the 10.0 mg/kg−1 dose had a significant increase in body weight, which began after only 3.5 weeks of treatment (ie, seven injections; Figure 1A). This resulted in a 27% increase in body weight at the termination of the trial (Figure 1B). In contrast, the body weight of mdx mice that received the 1.0 mg/kg−1 dose was not significantly different from control (Figure 1, A and B). Nuclear magnetic resonance quantification revealed that treatment with the 10.0 mg/kg−1 dose produced a selective increase in muscle mass. Skeletal muscle mass was 33% greater in treated mdx mice, compared with control mdx mice, with no changes in the amount of adipose tissue mass (Figure 1C). The increases in lean mass were clearly visible in the gross muscle anatomy of mice treated with the 10.0 mg/kg−1 dose, compared with control. The upper and lower limb skeletal muscles of treated mice, as well as the muscles along the caudal region, were all visibly larger than in control mice, reminiscent of the dumbbell shape characteristic of Mstn-null mice (Figure 1D).25
Figure 1.
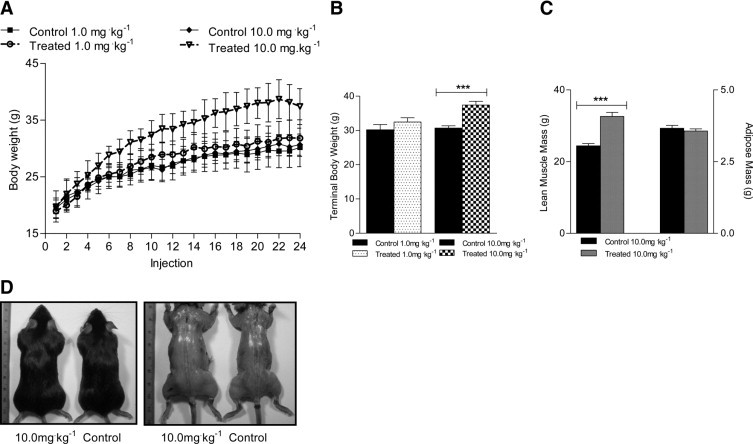
Effects of sActRIIB therapy on body weight, lean muscle, skeletal muscle, and adipose tissue mass. A: Growth curves were plotted to show the changes in body weight of control mdx mice and mdx mice treated with 1.0 mg/kg−1 or 10.0 mg/kg−1 sActRIIB. B: Body weight at the termination of the trial was significantly greater for mdx mice treated with the higher dose, but not the lower dose, compared with control mdx mice. C: Lean muscle mass but not adipose tissue mass (nuclear magnetic resonance quantification) was significantly greater in mdx mice treated with 10.0 mg/kg−1 sActRIIB, compared with control mdx mice. D: Gross anatomical examination revealed increases in skeletal muscle mass in mdx mice treated with the 10.0 mg/kg−1 dose of sActRIIB, compared with control mdx mice. ***P < 0.0001.
The absolute weight of gastrocnemius, tibialis anterior, and quadriceps skeletal muscles from mdx mice treated with the 10.0 mg/kg−1 dose were all 30% to 45% larger at the end of the 12-week trial, with marginal increases in muscle size at the 1.0 mg/kg−1 dose (Figure 2, A–C). When muscle weights were normalized to body weight, the tibialis anterior muscle from mice treated with the 10.0 mg/kg−1 dose was 16.6% greater than control (Figure 2E). In contrast, the tibialis anterior and quadriceps muscles from mice treated with the 1.0 mg/kg−1 dose were 16.4% and 26.4% less, compared with control, respectively (Figure 2, E and F). These data demonstrate the ability of sActRIIB to stimulate increases in body weight and specifically lean muscle mass in the mdx mouse, although a dose-dependent effect was clearly evident.
Figure 2.
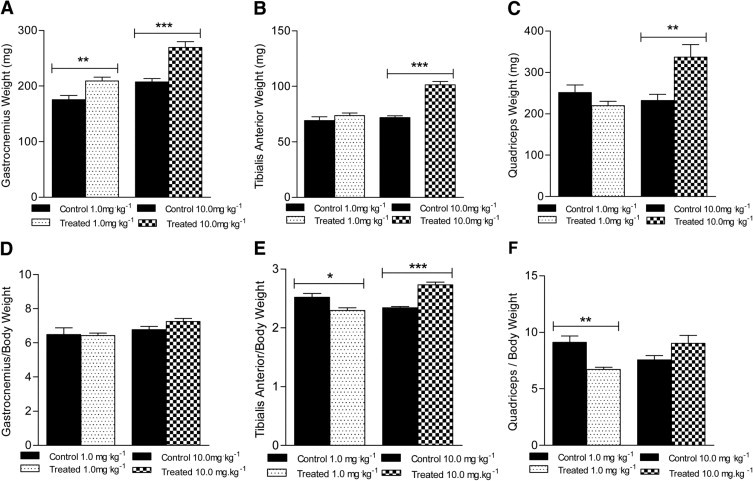
Effects of sActRIIB therapy on skeletal muscle mass in control mdx mice and mdx mice treated with 1.0 mg/kg−1 or 10.0 mg/kg−1 sActRIIB. Significant increases in absolute skeletal muscle mass were observed at one or both doses for gastrocnemius (A), tibialis anterior (B), and quadriceps (C) muscles, compared with control mdx mice. Normalized to body weight, however, no significant change in skeletal muscle mass was observed for gastrocnemius muscle (D) at either dose, compared with control mice. A significant decrease was observed at the lower dose for tibialis anterior (E) and quadriceps (F) muscles; at the higher dose, significant increase was observed in the tibialis anterior (E) muscle. *P < 0.05; **P < 0.001; ***P < 0.0001.
sActRIIB Treatment Increases the Mass and Force Generating Capacity of the EDL Muscle
Previous studies have shown that MSTN blockade strategies in both wild-type and mdx mice improves muscle mass and force-generating capacity after treatment,21,23,30 but absolute force production in muscle from Mstn-null mice is not significantly different from control mice.25 To directly determine the effects of sActRIIB treatment on muscle contractile properties, the EDL muscles from mdx mice were dissected and analyzed ex vivo after the 12-week preclinical trials. The EDL muscles from mdx mice treated with the 1.0 mg/kg−1 dose were 7% greater than control mice, whereas EDL muscles from mice treated with the 10.0 mg/kg−1 dose were 42% greater, compared with control mice (Figure 3A). Normalized to body weight, the EDL muscles from mice treated with the 10 mg/kg−1 dose were 21.1% greater than control mice, with no differences noted for the 1.0 mg/kg−1 dose (Figure 3B). There was a corresponding 41% increase in the EDL whole-muscle CSA in the mice treated with the 10.0 mg/kg−1 dose, compared with control mice, but no differences were noted in muscle CSA from mice treated with the 1.0 mg/kg−1 dose (Figure 3C). The increase in whole-muscle CSA was verified when muscle sections were stained with an anti-laminin fluorescent antibody to measure single-fiber CSA. The single-fiber CSA histogram of the EDL muscles from mice treated with the 10.0 mg/kg−1 dose was shifted to the right, compared with control mice, indicating a greater number of larger sized muscle fibers (Figure 3D). No such shift was observed in the muscles from mice treated with the 1.0 mg/kg−1 dose (Figure 3E). The total number of muscle fibers from EDL muscles was not significantly different between the sActRIIB-treated mdx mice and the respective control mdx mice (Table 1), suggesting that the greater EDL mass was due to muscle fiber hypertrophy and not to hyperplasia.
Figure 3.
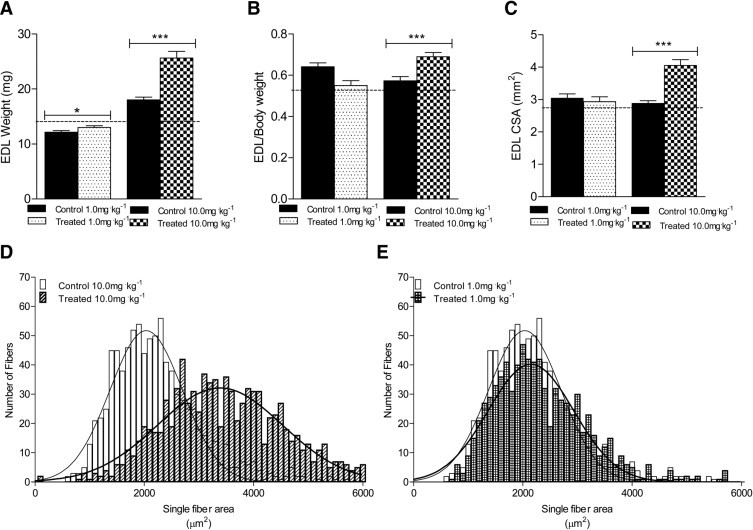
Effects of sActRIIB therapy on EDL weight and CSA in control mdx mice and in mdx mice treated with 1.0 mg/kg−1 or 10.0 mg/kg−1 sActRIIB. Significant increases were observed at both doses in absolute EDL weight (A) and at the higher dose in normalized EDL weight (B), and EDL whole-muscle cross-sectional area (CSA) (C). The dashed line (A–C)indicates values for untreated, age-matched wild-type BL/10 mice. D and E: Histograms from single-fiber CSA analysis reveal a shift to the right for the higher dose (D). *P < 0.05; ***P < 0.001.
Table 1.
Contractile and Morphometric Properties of the Extensor Digitorum Longus Muscle Following sActRIIB Therapy in the mdx Mouse Model of Duchenne Muscular Dystrophy
| Variable | 1.0 mg/kg−1 Treatment⁎ |
10.0 mg/kg−1 Treatment⁎ |
||||
|---|---|---|---|---|---|---|
| Control mdx | Treated mdx | P value | Control mdx | Treated mdx | P value | |
| Contraction time (ms) | 34.0 ± 1.6 | 37.5 ± 1.6 | 0.1545 | 41.0 ± 2.3 | 38.0 ± 1.3 | 0.2790 |
| ½ Relaxation time (ms) | 37.5 ± 1.3 | 38.6 ± 2.1 | 0.6583 | 47.0 ± 2.5 | 44.5 ± 1.4 | 0.3924 |
| ECC force drop (%) | 22.8 ± 5.6 | 15.5 ± 1.4 | 0.2438 | 27.9 ± 2.4 | 33.3 ± 4.7 | 0.2938 |
| Total fibers (no.) | 769.7 ± 38.6 | 633.3 ± 46.0 | 0.0638 | 742.0 ± 28.1 | 842.4 ± 71.4 | 0.1910 |
| CN fibers (%) | 52.3 ± 4.6 | 62.3 ± 5.9 | 0.2079 | 52.2 ± 5.6 | 43.2 ± 6.0 | 0.3066 |
| PO-positive fibers (%) | 6.9 ± 2.1 | 8.7 ± 3.8 | 0.6754 | 11.9 ± 2.3 | 11.1 ± 1.1 | 0.7544 |
CN, centrally nucleated; ECC, eccentric contraction; ms, milliseconds; PO, Procion Orange.
Treatment: Inhibition of activin receptor type IIB (ActRIIB) signaling was achieved using a fusion protein comprised of a form of the extracellular domain of ActRIIB linked to the Fc portion of murine IgG (RAP-031; Acceleron Pharma, Cambridge, MA).
The absolute force generated by EDL muscles in response to a single twitch was 27% greater (1.0 mg/kg−1) and 52% greater (10.0 mg/kg−1), compared with control mice (Figure 4, A and B). When absolute force was normalized to muscle CSA; however, the specific twitch force was 33% greater in the 1.0 mg/kg−1 group, but did not differ from control mice in the 10.0 mg/kg−1 group (Figure 4C). The absolute force from a tetanic contraction was 31% greater (1.0 mg/kg−1) and 39% greater (10.0 mg/kg−1), compared with control mice (Figure 4, D and E). Similarly, specific tetanic force was 45% greater in the EDL muscles from mice treated with 1.0 mg/kg−1, compared with control mice, but did not differ from control mice in the 10.0 mg/kg−1 group (Figure 4F). The contraction time and half-relaxation time of maximal twitch contractions were not different in EDL muscles from treated or control mice (Table 1). Additionally, no differences were observed in the eccentric contraction-induced force drop or in the percentage of centrally nucleated fibers (Table 1). The increases in specific force of the EDL muscle during twitch and tetanic contractions in the 1.0 mg/kg−1 group provide evidence for a functional improvement in the EDL muscles of mdx mice at this dose of sActRIIB that cannot be explained solely by an increase in muscle size. In addition, the lack of a difference in the contraction time and half-relaxation time after treatment suggests that sActRIIB administration did not alter any of the calcium-related proteins related to the speed of contraction and relaxation (ie, sarco/endoplasmic reticulum Ca2+ ATPase isoforms).
Figure 4.
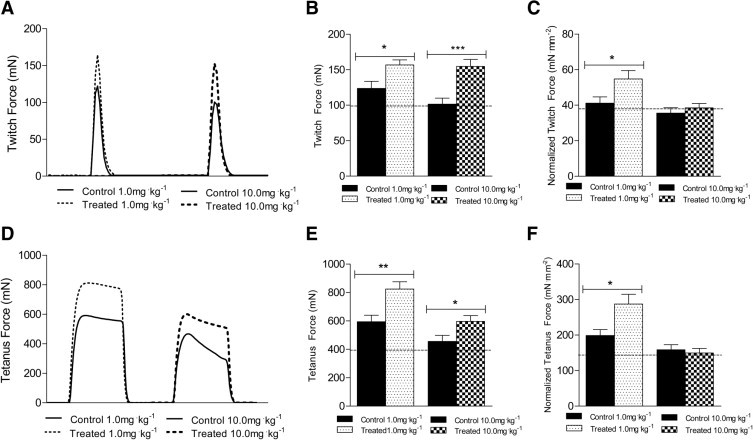
Effects of sActRIIB therapy on ex vivo contraction force of the EDL muscle. A: Representative twitch force traces from EDL muscles of control mdx mice and mdx mice treated with 1.0 mg/kg−1 or 10.0 mg/kg−1 sActRIIB. B: At both doses, absolute twitch force was significantly greater in mice treated with sActRIIB. C: Normalized twitch force was greater in EDL muscle only at the lower dose. D: Representative tetanic force traces from EDL muscles of control mdx mice and mdx mice treated with 1.0 mg/kg−1 or 10.0 mg/kg−1 sActRIIB. E: At both doses, absolute twitch force was significantly greater in mice treated with sActRIIB. F: Normalized tetanic force was greater in EDL muscles only at the lower dose. The dashed line (B, C, E, and F) indicates values for untreated, age-matched, wild-type BL/10 mice. *P < 0.05; **P < 0.001; ***P < 0.0001.
mdx Mice Treated with sActRIIB Have Reduced Circulating Creatine Kinase Levels
Creatine kinase is a muscle enzyme that is used as a biomarker of muscle damage and muscle pathology; it is consistently elevated in the blood of DMD patients, because of loss of muscle membrane integrity. Improvements (ie, decreases) in the amount of CK circulating in the blood suggest an improvement in muscle membrane integrity and thus in disease pathology. After 12 weeks of sActRIIB treatment, the amount of CK measured in the serum of mdx mice that received the 10.0 mg/kg−1 dose was significantly lower, compared with trial-matched control mice (Figure 5). This reduction occurred in the absence of differences in the percentage of Procion Orange-positive fibers among the groups (Table 1). A trend for lower CK values in the 1.0 mg/kg−1 group was observed, but this was not statistically significant because of a relatively high level of variation in the trial-matched control mice (Figure 5).
Figure 5.
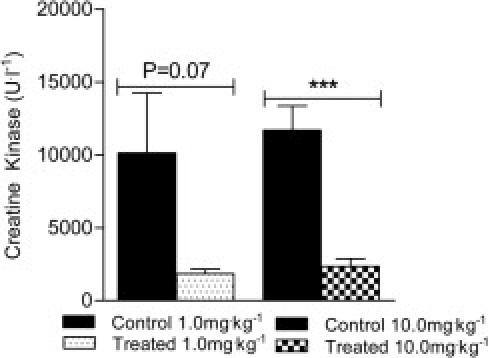
Effects of sActRIIB therapy on circulating CK levels. The mdx mice treated with the 10.0 mg/kg−1 dose of sActRIIB had a significantly lower concentration of serum CK compared with control mdx mice. ***P < 0.0001.
Treatment with sActRIIB Results in Minimal Effects on Muscle Histopathology in the Young Adult mdx Mouse
The diaphragm muscle in the mdx mouse most closely resembles the characteristic histopathology observed in DMD patients,44 showing the characteristic evidence of diffuse degeneration, fibrosis, and inflammation, whereas peripheral muscles show more localized areas of pathology.44,45 Previous studies have shown that MSTN blockade can result in improvements in the presence of degenerative and regenerative muscle fibers, inflammatory foci, and collagen infiltration in the diaphragm muscle.21,22 In the present study, histopathology of the diaphragm and tibialis anterior muscles were analyzed by H&E and Masson's trichrome stains, and hydroxyproline content was assessed as a marker of connective tissue infiltration. In these young adult mdx mice, there was minimal evidence of fibrosis, but diaphragm muscle pathology showed the presence of degenerative fibers, regenerative fibers, and inflammatory cell infiltration, evident after histological staining. sActRIIB treatment for 12 weeks did not appreciably alter the diaphragm muscle histopathology evident in 4-month-old mdx mice after either the 1.0 mg/kg−1 dose or the 10.0 mg/kg−1 dose (Figure 6, A–C). Similarly, sActRIIB treatment had minimal effects on histopathology in the tibialis anterior muscle after either dose (Figure 6, D and E). Hydroxyproline quantification in diaphragm (Figure 6F) and tibialis anterior muscles (Figure 6G) supported the histological evidence, with no significant differences between control and treatment groups. These data suggest that, despite functional improvements in limb skeletal muscles after sActRIIB treatment, the muscle histopathology characteristic in 4-month-old mdx mice was not significantly improved with treatment in the present study. This is surprising, given the role of MSTN in promoting fibrosis, and we speculate that longer treatment or initiating therapy at an earlier age may produce a more beneficial effect in reducing muscle histopathology.
Figure 6.
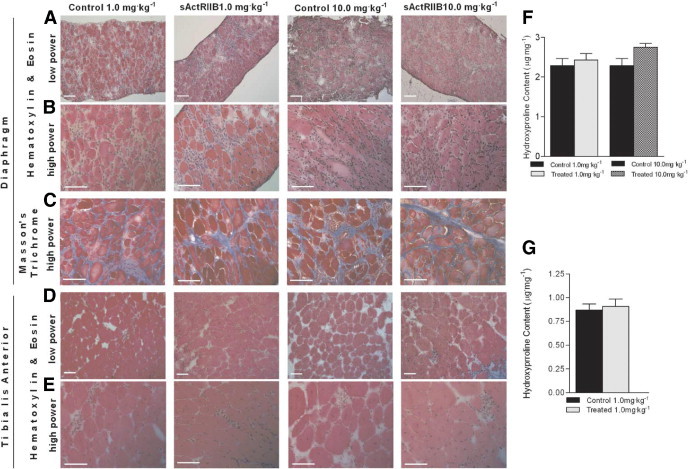
Effects of sActRIIB therapy on histopathology and hydroxyproline content of diaphragm and tibialis anterior muscle. Limited effects on diaphragm and tibialis anterior histopathology were observed in mdx mice treated with 1.0 mg/kg−1 or 10.0 mg/kg−1 sActRIIB, compared with mdx control mice. Representative diaphragm sections, H&E stain (A and B) and Mason's trichrome stain (C). Representative tibialis anterior sections H&E stain (D and E). No significant difference was observed in the hydroxyproline content of the diaphragm (F) and the tibialis anterior muscle (G). Original magnification, 10× (A and D); 40× (B, C, and E). Scale bars = 100 μm (all images).
Discussion
A definitive cure for DMD remains elusive, and until therapies are developed to treat the primary defect of this myopathy (ie, dystrophin deficiency), minimizing muscle wasting and/or maintaining muscle function may be a viable option for patients. In fact, the necessity for therapies to spare existing muscle mass in patients with muscular dystrophies has been suggested, with continued efforts to develop DNA-based therapies aimed at introducing and/or stimulating dystrophin expression in muscle.46,47 Therapies aimed at sparing existing muscle mass or stimulating increases in muscle mass would be beneficial as adjuncts to DNA-based methodologies, which require intact muscle fibers for translation of functional dystrophin protein.
Preclinical trials in experimental animals provide a means to relatively quickly screen drugs and small molecules for therapeutic efficacy.48,49 With respect to DMD, the mdx mouse has been used as a model because it has been shown to recapitulate certain aspects of DMD disease progression. Specifically, the diaphragm muscle undergoes significant muscle fiber degeneration, inflammatory cell infiltration, and reduced functional capacity, and significant increases in circulating CK are evident in serum from these mice.44,45 Thus, preclinical studies in this mouse strain that improve these dystrophic impairments can potentially lead to further studies in nonhuman primates and eventually in humans.
MSTN is a negative regulator of skeletal muscle mass, and blockade of MSTN activity in vivo has been reported in numerous mouse models.21–23 Researchers from our laboratory, and others, have reported improvements in muscle mass and function in wild-type mice in the dystrophic-phenotype mdx mice, using both an antibody directed against MSTN and the MSTN propeptide.21,22,30 The sActRIIB molecule has recently been shown to improve muscle function in a mouse model of amyotrophic lateral sclerosis,50 increase muscle mass and reduce adiposity in mice fed a high-fat diet,51 and attenuate muscle dysfunction in wild-type mice exposed to normobaric hypoxia.52 The therapeutic efficacy of sActRIIB observed during hypoxic exposure is especially pertinent to DMD pathology, because respiratory impairments in patients result in the need for ventilatory assistance at a young age. The advantages of sActRIIB therapy may lie in the ability to bind to multiple ActRIIB ligands, including growth/differentiation factor 8 (MSTN) and growth/differentiation factor 11, with similar binding affinities to activin A, a known high-affinity ligand for this receptor.53 As previously demonstrated, treatment of Mstn-null mice with a similar soluble activin receptor molecule led to additional modest increases in muscle mass, demonstrating that multiple ActRIIB ligands can act in concert to limit muscle growth in vivo.34
In the present study, we hypothesized that sActRIIB therapy would stimulate skeletal muscle growth and improve muscle function in the mdx mouse model of DMD by inhibiting multiple members of the transforming growth factor-β superfamily, in addition to MSTN. This therapeutic protocol produced a rapid increase in body weight and skeletal muscle mass of mdx mice treated with the 10.0 mg/kg−1 dose of sActRIIB; the difference in body weight was less dramatic in response to the 1.0 mg/kg−1 dose. At the 10.0 mg/kg−1 dose, we observed a statistically significant increase in body weight over control mice after just seven injections (3.5 weeks). With the 10.0 mg/kg−1 dose in wild-type C57BL/10 mice, we also observed a significant 16% increase in body weight after 2 weeks (four injections) of sActRIIB.52 These observations are in contrast to the smaller degree of body weight changes we had observed in mdx mice treated with antibodies directed against MSTN and the MSTN propeptide.21,22 Mice treated with the high dose of sActRIIB also developed pronounced upper and lower limb musculature, with lean body mass being 33% greater than control mice as determined using nuclear magnetic resonance. These mice exhibited a dumbbell body shape similar to that reported in MSTN knockout mice,25 and the absolute weight of the gastrocnemius, tibialis anterior, and quadriceps muscles from mice treated with the 10.0 mg/kg−1 dose were between 30% and 45% greater than trial-matched control mice. These data demonstrate that sActRIIB therapy has the ability to rapidly stimulate improvements in lean muscle mass, and may be beneficial therapeutically in muscle wasting conditions, although dose-dependent effects were evident.
The differences in contractile parameters observed in the present study between the 10.0 mg/kg−1 dose and the 1.0 mg/kg−1 dose of sActRIIB were significant, and they demonstrate that the lower dose can improve muscle function. Despite the limited effects on overall body size, the lower dose of sActRIIB caused a significant improvement in specific force of the EDL muscle when stimulated ex vivo. In our laboratory, an antibody directed against MSTN and the MSTN propeptide were each able to improve absolute contractile force of EDL muscles from mdx mice.21,22 In addition, the MSTN propeptide improved specific force of the EDL muscle, providing evidence for an improvement in muscle mechanics independent of any increases in muscle mass.22 The greater degree of improvement in muscle physiology in response to the MSTN propeptide treatment, compared with the MSTN antibody, may have been due to the higher binding affinity of the propeptide for MSTN than for the antibody.22 In a similar fashion, we hypothesize that the binding affinities of the sActRIIB molecule for MSTN and growth/differentiation factor 11 contributed to the improvement in EDL specific force. This hypothesis is intriguing, given the conclusions of a phase I/II clinical trial using an antibody directed against MSTN (MYO-029). The study authors supported the use of MSTN inhibitory drugs to treat muscular dystrophies, although they also concluded that drugs with more potent inhibitory actions should be evaluated.54 Recently, a humanized version of sActRIIB (ACE-031), composed of the extracellular portion of the human ActRIIB fused to a human Fc, has been reported to produce similar effects (ie, improved muscle mass and strength) in wild-type mice.55
In addition to the effects noted of sActRIIB on muscle mass and function, sActRIIB therapy resulted in lower circulating CK levels, compared with saline-treated control mice. The reduced CK levels occurred despite no clear improvements in skeletal muscle histopathology, nor any changes in sarcolemmal integrity as accessed via Procion Orange uptake. Previous research has shown that performing exercise at the same absolute and relative intensity after 16 weeks of exercise training designed to improve muscle hypertrophy results in a blunted CK response.56 In addition, when trained weightlifters were compared with untrained individuals, the serum CK response to a strenuous resistance training session was lower in the weightlifters.57 These data demonstrate that hypertrophied muscles can perform exercise without the same CK response as untrained muscles. In the present study, skeletal muscle size was on average 30% to 45% greater with the 10.0 mg/kg−1 dose of sActRIIB, with a concomitant 80% lower level of circulating CK. Additionally, skeletal muscle size was less affected with the 1.0 mg/kg−1 dose of sActRIIB, with a nonsignificant 82% lower CK value. Thus, the reductions in circulating CK observed in the mice treated with the 10.0 mg/kg−1 dose of sActRIIB may have occurred as a result of the significant increases in muscle hypertrophy and/or strength in the mdx mouse. The reduced levels of circulating CK observed after sActRIIB therapy are clinically meaningful, based on statistical and effect size analyses indicating serum CK as an effective marker for determining preclinical drug efficacy.49
Previous studies using MSTN blockade therapies in mdx mice have reported improvements in diaphragm muscle histopathology.21,22 In vitro studies localize MSTN and ActRIIB directly on fibroblasts, supporting the idea that MSTN is a profibrotic factor that interacts directly with fibroblasts to mediate its effects.58 In vivo, MSTN-coated beads implanted into skeletal muscle increased fibrotic tissue, and this effect was inhibited when the endogenous MSTN inhibitor follistatin was also present on the beads.58 Diaphragmatic fibrosis was reduced by 44% in Mstn−/− mdx mice at 6 months of age, compared with Mstn+/+ mdx mice, but was still elevated compared with the wild type.23 These data support MSTN involvement in fibrosis, but also demonstrate that it is not absolutely required, because fibrosis can develop in the absence of MSTN. It was indeed surprising that we observed no improvements in diaphragm or limb muscle histopathology or hydroxyproline content in the present study with sActRIIB therapy, given that both antibody-mediated and propeptide-mediated MSTN inhibition did improve histopathology in mdx diaphragm muscle.21,22 A possible explanation for the minimal effects on diaphragm histopathology after sActRIIB therapy in the present study is that a relatively low level of histopathological damage or evidence of damage at the time the tissues were analyzed (ie, at 4 months of age) limited the possibility of identifying any therapeutic effect. In addition, the recent observation of histopathological improvements in dystrophic muscle with early (ie, before active disease onset) MSTN blockade therapy suggests that the presence of active muscle pathology may also limit therapeutic efficacy.59
In conclusion, 12 weeks of treatment with sActRIIB at a dose of 1.0 mg/kg−1 caused increases in specific force of the EDL muscle in mdx mice, indicating a functional improvement in the muscle, whereas the 10.0 mg/kg−1 dose led to significant increases in overall body mass and lean muscle mass. Circulating CK levels were also significantly lower after sActRIIB therapy at the 10.0 mg/kg−1 dose, although improvements in muscle histopathology were limited in the young adult mdx mice used in the present study. These data demonstrate that pharmacologically targeting the ActRIIB can improve skeletal muscle mass and strength in the mdx mouse model of DMD, providing a therapeutic rationale for use of this small molecule in treating the muscle-wasting characteristic of skeletal myopathies.
Footnotes
Supported by a research grant from the World Anti-Doping Agency (WADA) (T.K.) and a National Institutes of Health Training Grant in Muscle Biology (AR053461 to E.E.P.) through the Pennsylvania Muscle Institute.
J.L. and J.S. are paid employees of Acceleron Pharma. E.E.P. and T.K. have been paid as consultants to Acceleron Pharma.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2010.11.071.
Current address of S.B.: Department of Ophthalmology and Visual Sciences, University of Kentucky College of Medicine, Lexington, Kentucky.
Supplementary data
Supplementary Figure S1.
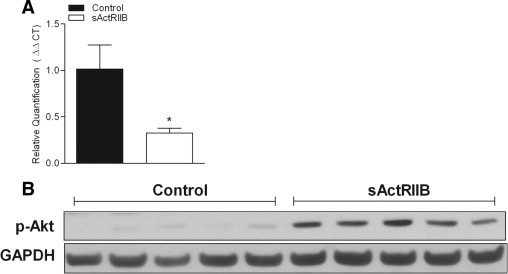
Validation of sActRIIB activity in vivo. Biological activity of sActRIIB therapy in vivo was determined through gene and protein quantification of targets downstream of ActRIIB. (A) Expression of the transgelin gene (Tagln; synonym, Sm22) is regulated by nuclear translocation of phosphorylated Smad after ActRIIB activation. sActRIIB therapy in mdx mice resulted in lower Tagln gene expression. (B) Levels of phosphorylated Akt were significantly greater in muscles after sActRIIB therapy. *P < 0.05.
References
- 1.Adams R.D., Victor M. ed 4. McGraw-Hill; New York: 1989. Principles of Neurology; pp. 1117–1132. [Google Scholar]
- 2.Hoffman E.P., Brown R.H., Jr, Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 3.Bogdanovich S., Perkins K.J., Krag T.O., Khurana T.S. Therapeutics for Duchenne muscular dystrophy: current approaches and future directions. J Mol Med. 2004;82:102–115. doi: 10.1007/s00109-003-0484-1. [DOI] [PubMed] [Google Scholar]
- 4.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engel A.G., Ozawa E. Dystrophinopathies. In: Engel A.G., Franzini-Armstrong C., editors. Myology. ed 3. vol 2. McGraw-Hill; New York: 2004. pp. 961–1026. ch 34. [Google Scholar]
- 6.Ames W.A., Hayes J.A., Crawford M.W. The role of corticosteroids in Duchenne muscular dystrophy: a review for the anesthetist. Paediatr Anaesth. 2005;15:3–8. doi: 10.1111/j.1460-9592.2005.01424.x. [DOI] [PubMed] [Google Scholar]
- 7.Angelini C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle Nerve. 2007;36:424–435. doi: 10.1002/mus.20812. [DOI] [PubMed] [Google Scholar]
- 8.Bonifati M.D., Ruzza G., Bonometto P., Berardinelli A., Gorni K., Orcesi S., Lanzi G., Angelini C. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23:1344–1347. doi: 10.1002/1097-4598(200009)23:9<1344::aid-mus4>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 9.Torrente Y., Belicchi M., Marchesi C., Dantona G., Cogiamanian F., Pisati F., Gavina M., Giordano R., Tonlorenzi R., Fagiolari G., Lamperti C., Porretti L., Lopa R., Sampaolesi M., Vicentini L., Grimoldi N., Tiberio F., Songa V., Baratta P., Prelle A., Forzenigo L., Guglieri M., Pansarasa O., Rinaldi C., Mouly V., Butler-Browne G.S., Comi G.P., Biondetti P., Moggio M., Gaini S.M., Stocchetti N., Priori A., D'Angelo M.G., Turconi A., Bottinelli R., Cossu G., Rebulla P., Bresolin N. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007;16:563–577. doi: 10.3727/000000007783465064. [DOI] [PubMed] [Google Scholar]
- 10.Peltz S.W., Welch E.M., Jacobson A., Trotta C.R., Naryshkin N., Sweeney H.L., Bedwell D.M. Nonsense suppression activity of PTC124 (ataluren) Proc Natl Acad Sci USA. 2009;106(25):E64. doi: 10.1073/pnas.0901936106. author reply E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welch E.M., Barton E.R., Zhuo J., Tomizawa Y., Friesen W.J., Trifillis P., Paushkin S., Patel M., Trotta C.R., Hwang S., Wilde R.G., Karp G., Takasugi J., Chen G., Jones S., Ren H., Moon Y.C., Corson D., Turpoff A.A., Campbell J.A., Conn M.M., Khan A., Almstead N.G., Hedrick J., Mollin A., Risher N., Weetall M., Yeh S., Branstrom A.A., Colacino J.M., Babiak J., Ju W.D., Hirawat S., Northcutt V.J., Miller L.L., Spatrick P., He F., Kawana M., Feng H., Jacobson A., Peltz S.W., Sweeney H.L. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 12.Skuk D., Goulet M., Roy B., Chapdelaine P., Bouchard J.P., Roy R., Dugré F.J., Sylvain M., Lachance J.G., Deschênes L., Senay H., Tremblay J.P. Dystrophin expression in muscles of Duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol. 2006;65:371–386. doi: 10.1097/01.jnen.0000218443.45782.81. [DOI] [PubMed] [Google Scholar]
- 13.Kinali M., Arechavala-Gomeza V., Feng L., Cirak S., Hunt D., Adkin C., Guglieri M., Ashton E., Abbs S., Nihoyannopoulos P., Garralda M.E., Rutherford M., McCulley C., Popplewell L., Graham I.R., Dickson G., Wood M.J., Wells D.J., Wilton S.D., Kole R., Straub V., Bushby K., Sewry C., Morgan J.E., Muntoni F. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Deutekom J.C., Janson A.A., Ginjaar I.B., Frankhuizen W.S., Aartsma-Rus A., Bremmer-Bout M., den Dunnen J.T., Koop K., van der Kooi A.J., Goemans N.M., de Kimpe S.J., Ekhart P.F., Venneker E.H., Platenburg G.J., Verschuuren J.J., van Ommen G.J. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 15.Odom G.L., Gregorevic P., Chamberlain J.S. Viral-mediated gene therapy for the muscular dystrophies: successes, limitations and recent advances. Biochim Biophys Acta. 2007;1772:243–262. doi: 10.1016/j.bbadis.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banks G.B., Chamberlain J.S. The value of mammalian models for Duchenne muscular dystrophy in developing therapeutic strategies. Curr Top Dev Biol. 2008;84:431–453. doi: 10.1016/S0070-2153(08)00609-1. [DOI] [PubMed] [Google Scholar]
- 17.Gehrig S.M., Ryall J.G., Schertzer J.D., Lynch G.S. Insulin-like growth factor-I analogue protects muscles of dystrophic mdx mice from contraction-mediated damage. Exp Physiol. 2008;93:1190–1198. doi: 10.1113/expphysiol.2008.042838. [DOI] [PubMed] [Google Scholar]
- 18.Schertzer J.D., Gehrig S.M., Ryall J.G., Lynch G.S. Modulation of insulin-like growth factor (IGF)-I and IGF-binding protein interactions enhances skeletal muscle regeneration and ameliorates the dystrophic pathology in mdx mice. Am J Pathol. 2007;171:1180–1188. doi: 10.2353/ajpath.2007.070292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schertzer J.D., van der Poel C., Shavlakadze T., Grounds M.D., Lynch G.S. Muscle-specific overexpression of IGF-I improves E-C coupling in skeletal muscle fibers from dystrophic mdx mice. Am J Physiol Cell Physiol. 2008;294:C161–C168. doi: 10.1152/ajpcell.00399.2007. [DOI] [PubMed] [Google Scholar]
- 20.Harcourt L.J., Holmes A.G., Gregorevic P., Schertzer J.D., Stupka N., Plant D.R., Lynch G.S. Interleukin-15 administration improves diaphragm muscle pathology and function in dystrophic mdx mice. Am J Pathol. 2005;166:1131–1141. doi: 10.1016/S0002-9440(10)62333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 22.Bogdanovich S., Perkins K.J., Krag T.O., Whittemore L.A., Khurana T.S. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005;19:543–549. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- 23.Wagner K.R., McPherron A.C., Winik N., Lee S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 24.McPherron A.C., Lee S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amthor H., Macharia R., Navarrete R., Schuelke M., Brown S.C., Otto A., Voit T., Muntoni F., Vrbóva G., Partridge T., Zammit P., Bunger L., Patel K. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA. 2007;104:1835–1840. doi: 10.1073/pnas.0604893104. [Erratum appeared in Proc Natl Acad Sci USA 2007, 104:4240] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosher D.S., Quignon P., Bustamante C.D., Sutter N.B., Mellersh C.S., Parker H.G., Ostrander E.A. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3:e79. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shelton G.D., Engvall E. Gross muscle hypertrophy in whippet dogs is caused by a mutation in the myostatin gene. Neuromuscul Disord. 2007;17:721–722. doi: 10.1016/j.nmd.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Schuelke M., Wagner K.R., Stolz L.E., Hübner C., Riebel T., Komen W., Braun T., Tobin J.F., Lee S.J. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 29.Fedoruk M.N., Rupert J.L. Myostatin inhibition: a potential performance enhancement strategy? Scand J Med Sci Sports. 2008;18:123–131. doi: 10.1111/j.1600-0838.2007.00759.x. [DOI] [PubMed] [Google Scholar]
- 30.Whittemore L.A., Song K., Li X., Aghajanian J., Davies M., Girgenrath S., Hill J.J., Jalenak M., Kelley P., Knight A., Maylor R., O'Hara D., Pearson A., Quazi A., Ryerson S., Tan X.Y., Tomkinson K.N., Veldman G.M., Widom A., Wright J.F., Wudyka S., Zhao L., Wolfman N.M. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun. 2003;300:965–971. doi: 10.1016/s0006-291x(02)02953-4. [DOI] [PubMed] [Google Scholar]
- 31.Magee T.R., Artaza J.N., Ferrini M.G., Vernet D., Zuniga F.I., Cantini L., Reisz-Porszasz S., Rajfer J., Gonzalez-Cadavid N.F. Myostatin short interfering hairpin RNA gene transfer increases skeletal muscle mass. J Gene Med. 2006;8:1171–1181. doi: 10.1002/jgm.946. [DOI] [PubMed] [Google Scholar]
- 32.Bartoli M., Poupiot J., Vulin A., Fougerousse F., Arandel L., Daniele N., Roudaut C., Noulet F., Garcia L., Danos O., Richard I. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not alpha-sarcoglycan deficiency. Gene Ther. 2007;14:733–740. doi: 10.1038/sj.gt.3302928. [DOI] [PubMed] [Google Scholar]
- 33.de Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15:1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Lee S.J., Reed L.A., Davies M.V., Girgenrath S., Goad M.E., Tomkinson K.N., Wright J.F., Barker C., Ehrmantraut G., Holmstrom J., Trowell B., Gertz B., Jiang M.S., Sebald S.M., Matzuk M., Li E., Liang L.F., Quattlebaum E., Stotish R.L., Wolfman N.M. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu P., Feng X.H., Li L. Interaction of Smad3 and SRF-associated complex mediates TGF-beta1 signals to regulate SM22 transcription during myofibroblast differentiation. J Mol Cell Cardiol. 2003;35:1407–1420. doi: 10.1016/j.yjmcc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Goncalves M.D., Pistilli E.E., Balduzzi A., Birnbaum M.J., Lachey J., Khurana T.S., Ahima R.S. Akt deficiency attenuates muscle size and function but not the response to ActRIIB inhibition. PLoS One. 2010;5:e12707. doi: 10.1371/journal.pone.0012707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S., Kulik M., Lechleider R.J. Smad proteins regulate transcriptional induction of the SM22alpha gene by TGF-beta. Nucleic Acids Res. 2003;31:1302–1310. doi: 10.1093/nar/gkg224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amirouche A., Durieux A.C., Banzet S., Koulmann N., Bonnefoy R., Mouret C., Bigard X., Peinnequin A., Freyssenet D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150:286–294. doi: 10.1210/en.2008-0959. [DOI] [PubMed] [Google Scholar]
- 39.Wong T., Hildebrandt M.A., Thrasher S.M., Appleton J.A., Ahima R.S., Wu G.D. Divergent metabolic adaptations to intestinal parasitic nematode infection in mice susceptible or resistant to obesity. Gastroenterology. 2007;133:1979–1988. doi: 10.1053/j.gastro.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks S.V., Faulkner J.A. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lynch G.S., Hinkle R.T., Chamberlain J.S., Brooks S.V., Faulkner J.A. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol. 2001;535:591–600. doi: 10.1111/j.1469-7793.2001.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gargioli C., Coletta M., De Grandis F., Cannata S.M., Cossu G. PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat Med. 2008;14:973–978. doi: 10.1038/nm.1852. [DOI] [PubMed] [Google Scholar]
- 43.Pistilli E.E., Siu P.M., Alway S.E. Interleukin-15 responses to aging and unloading-induced skeletal muscle atrophy. Am J Physiol Cell Physiol. 2007;292:C1298–C1304. doi: 10.1152/ajpcell.00496.2006. [DOI] [PubMed] [Google Scholar]
- 44.Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 45.Coulton G.R., Morgan J.E., Partridge T.A., Sloper J.C. The mdx mouse skeletal muscle myopathy: I: A histological, morphometric and biochemical investigation. Neuropathol Appl Neurobiol. 1988;14:53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 46.Tidball J.G., Wehling-Henricks M. Evolving therapeutic strategies for Duchenne muscular dystrophy: targeting downstream events. Pediatr Res. 2004;56:831–841. doi: 10.1203/01.PDR.0000145578.01985.D0. [DOI] [PubMed] [Google Scholar]
- 47.Danièle N., Richard I., Bartoli M. Ins and outs of therapy in limb girdle muscular dystrophies. Int J Biochem Cell Biol. 2007;39:1608–1624. doi: 10.1016/j.biocel.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Nagaraju K., Willmann R., TREAT-NMD Network. Wellstone Muscular Dystrophy Cooperative Research Network Developing standard procedures for murine and canine efficacy studies of DMD therapeutics: report of two expert workshops on “Pre-clinical testing for Duchenne dystrophy”: Washington DC: October 27th–28th 2007 and Zurich, June 30th–July 1st 2008. Neuromuscul Disord. 2009;19:502–506. doi: 10.1016/j.nmd.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spurney C.F., Gordish-Dressman H., Guerron A.D., Sali A., Pandey G.S., Rawat R., Van Der Meulen J.H., Cha H.J., Pistilli E.E., Partridge T.A., Hoffman E.P., Nagaraju K. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrison B.M., Lachey J.L., Warsing L.C., Ting B.L., Pullen A.E., Underwood K.W., Kumar R., Sako D., Grinberg A., Wong V., Colantuoni E., Seehra J.S., Wagner K.R. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2009;217:258–268. doi: 10.1016/j.expneurol.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 51.Akpan I., Goncalves M.D., Dhir R., Yin X., Pistilli E.E., Bogdanovich S., Khurana T.S., Ucran J., Lachey J., Ahima R.S. The effects of a soluble activin type IIB receptor on obesity and insulin sensitivity. Int J Obes (Lond) 2009;33:1265–1273. doi: 10.1038/ijo.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pistilli E.E., Bogdanovich S., Mosqueira M., Lachey J., Seehra J., Khurana T.S. Pretreatment with a soluble activin type IIB receptor/Fc fusion protein improves hypoxia-induced muscle dysfunction. Am J Physiol Regul Integr Comp Physiol. 2010;298:R96–R103. doi: 10.1152/ajpregu.00138.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sako D., Grinberg A.V., Liu J., Davies M.V., Castonguay R., Maniatis S., Andreucci A.J., Pobre E.G., Tomkinson K.N., Monnell T.E., Ucran J.A., Martinez-Hackert E., Pearsall R.S., Underwood K.W., Seehra J., Kumar R. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type IIb. J Biol Chem. 2010;285:21037–21048. doi: 10.1074/jbc.M110.114959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wagner K.R., Fleckenstein J.L., Amato A.A., Barohn R.J., Bushby K., Escolar D.M., Flanigan K.M., Pestronk A., Tawil R., Wolfe G.I., Eagle M., Florence J.M., King W.M., Pandya S., Straub V., Juneau P., Meyers K., Csimma C., Araujo T., Allen R., Parsons S.A., Wozney J.M., Lavallie E.R., Mendell J.R. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 55.Cadena S.M., Tomkinson K.N., Monnell T.E., Spaits M.S., Kumar R., Underwood K.W., Pearsall R.S., Lachey J.L. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J Appl Physiol. 2010;109:635–642. doi: 10.1152/japplphysiol.00866.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hurley B.F., Redmond R.A., Pratley R.E., Treuth M.S., Rogers M.A., Goldberg A.P. Effects of strength training on muscle hypertrophy and muscle cell disruption in older men. Int J Sports Med. 1995;16:378–384. doi: 10.1055/s-2007-973024. [DOI] [PubMed] [Google Scholar]
- 57.Vincent H.K., Vincent K.R. The effect of training status on the serum creatine kinase response, soreness and muscle function following resistance exercise. Int J Sports Med. 1997;18:431–437. doi: 10.1055/s-2007-972660. [DOI] [PubMed] [Google Scholar]
- 58.Li Z.B., Kollias H.D., Wagner K.R. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem. 2008;283:19371–19378. doi: 10.1074/jbc.M802585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murphy K.T., Ryall J.G., Snell S.M., Nair L., Koopman R., Krasney P.A., Ibebunjo C., Holden K.S., Loria P.M., Salatto C.T., Lynch G.S. Antibody-directed myostatin inhibition improves diaphragm pathology in young but not adult dystrophic mdx mice. Am J Pathol. 2010;176:2425–2434. doi: 10.2353/ajpath.2010.090932. [DOI] [PMC free article] [PubMed] [Google Scholar]