

Abstract
Cyclooxygenase-2 (COX-2) has been associated with cell growth regulation, tissue remodeling, and carcinogenesis. Ectopic expression of COX-2 in hepatocytes constitutes a nonphysiological condition ideal for evaluating the role of prostaglandins (PGs) in liver pathogenesis. The effect of COX-2-dependent PGs in chronic liver disease, hepatitis, fibrosis, and chemical hepatocarcinogenesis, has been investigated in transgenic (Tg) mice that express human COX-2 in hepatocytes and in Tg hepatic human cell lines. We have used three different complementary approaches: i) diethylnitrosamine (DEN)-induced chemical hepatocarcinogenesis in COX-2 Tg mice, ii) DEN/phenobarbital treatment of human COX-2 Tg hepatocyte-like cells, and iii) COX-2 Tg hepatocyte-like cells implants in nude mice. The data suggest that PGs produced by COX-2 in hepatocytes promoted mild hepatitis in 60-week-old mice, as assessed by histological examination, but failed to contribute to the development of liver fibrogenesis after methionine- and choline-deficient diet treatment. Moreover, liver injury, collagen content, and hepatic stellate cell activation were equally severe in wild-type and COX-2 Tg mice. The contribution of COX-2-dependent PGs to the development of DEN-induced hepatocarcinogenesis was evaluated in Tg mice, Tg hepatocyte-like cells, and nude mice and the analysis revealed that COX-2 expression favors the development of preneoplastic foci without affecting malignant transformation. Endogenous COX-2 expression in wild-type mice is a late event in the development of hepatocellular carcinoma.
Hepatocellular carcinoma (HCC) is a neoplasm with increasing incidence worldwide.1 Epidemiological studies suggest that the major risk factors for HCC are persistent infection with hepatitis B and C virus and exposure to genotoxic and cytotoxic chemicals. All these factors cause chronic liver injury and inflammation, conditions in which hepatocytes die and resident inflammatory cells (Kupffer cells), as well as newly recruited inflammatory cells (macrophages, neutrophils, and natural killer and natural killer T cells), are activated to produce cytokines that conduct the compensatory proliferation of surviving hepatocytes.2
Cyclooxygenases 1 and 2 (COX-1 and COX-2) catalyze the first step in prostanoid biosynthesis. COX-1 is constitutively expressed in many tissues; COX-2 is induced by a variety of stimuli such as growth factors, proinflammatory stimuli, hormones, and other cellular stresses.3 Adult hepatocytes fail to induce COX-2 expression, regardless of the proinflammatory factors used; however, Kupffer, stellate, hepatoma mouse liver cells, and fetal hepatocytes retain the ability to express COX-2 on stimulation with lipopolysaccharide and pro-inflammatory cytokines.4,5 In the case of hepatocytes, and only under prolonged aggression, COX-2 is expressed as a result of the drop of C/EBPα levels, among other conditions.6 Thus, ectopic expression of COX-2 in hepatocytes constitutes an ideal condition for evaluating the role of prostaglandins (PGs) in liver pathogenesis. In this regard, we and others demonstrated that partial hepatectomy7 induced COX-2 in hepatocytes and contributed to the progression of cell cycle after partial hepatectomy.8,9 In addition to liver regeneration after partial hepatectomy or hepatotoxic agents, expression of COX-2 has been detected in animal models of cirrhosis,10 in human hepatoma cell lines,11,12 after hepatitis B virus and hepatitis C virus infection,13,14 and in human HCC.15 High levels of COX-2-derived prostaglandin E2 (PGE2) lead to a decrease in apoptosis, a modulation of the immune system and a stimulation of cell migration, proliferation, and angiogenesis.16 Nonetheless, even though COX-2 expression is elevated in the early stages of HCC, many questions remain unsolved regarding the contribution of COX-2-dependent PGs to induce tumorigenesis.
Studies with genetically altered mice have revealed many genes associated with HCC. Such genes fall into two categories: i) those that augment spontaneous HCC and ii) those that increase the susceptibility to chemically induced HCC. The HCC develops spontaneously in transgenic mice overexpressing hepatitis B virus surface antigen (HBsAg), transforming growth factor α the c-Myc proto-oncogene,17 or the IκB kinase (IKK) subunit NEMO/IKKγ.18 HCC development is also augmented in mice deficient in the Mdr2 P-glycoprotein transporter19 or carrying a lymphotoxin-transgene.20 Most of these mouse strains display enhanced susceptibility to chemical carcinogens such as diethylnitrosamine (DEN), although the molecular and cellular mechanisms involved are not well understood.
To clarify the role of COX-2 in liver tumorigenesis, we used transgenic mice expressing human COX-2 constitutively in hepatocytes under the control of the human ApoE promoter (COX-2 Tg)21 and hepatic human cell lines carrying the COX-2 transgene. We sought to examine the response of the COX-2 transgenic (Tg) mice to the DEN-induced HCC model. Our results show that COX-2 expression in hepatocytes caused mild hepatitis in aged mice, but was not involved in the development of liver fibrosis. COX-2 favored the development of preneoplastic foci and adenomas, but it was not sufficient to significantly increase the malignant transformation induced by DEN. Furthermore, endogenous COX-2 expression in wild-type (WT) mice is a late event in the development of HCC induced by DEN.
Materials and Methods
Chemicals
Antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), Sigma-Aldrich (St. Louis, MO), Cell Signaling Technology (Danvers, MA), and Cayman Chemical (Ann Arbor, MI). Reagents were from Roche Diagnostics (Mannheim, Germany) or Sigma-Aldrich. DFU [5,5-dimethyl-3(3-fluorophenyl)-4-(4-methylsulfonyl)phenyl-2(5H)-furanone] was from Merck (Darmstadt, Germany). Reagents for electrophoresis were obtained from Bio-Rad (Hercules, CA). Tissue culture dishes were from Falcon (Becton Dickinson Labware, Franklin Lakes, NJ). Tissue culture media were from Gibco (Invitrogen, Grand Island, NY).
Cell Culture
The human liver cell line CCL-13 [Chang liver (CHL)], an immortalized nontumor cell line derived from normal liver, was purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were grown on Falcon tissue culture dishes in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) and antibiotics (50 μg/ml each of penicillin, streptomycin, and gentamicin) at 37°C in a humidified, 5% CO2-enriched atmosphere.
Characterization of COX-2 Vectors
pEGFP-N1 was obtained from Clontech (Mountain View, CA) and pPyCAGIP was a gift from Dr. I. Chambers (University of Edinburgh, Edinburgh, Scotland). Human COX-2 ORF was amplified by PCR from human full-length COX-2 cDNA cloned into pcDNA1/Amp, a gift from Dr. S. Prescott (Huntsman Cancer Institute, Salt Lake City, UT). The PCR product was cloned into the XhoI-EcoRI restriction site of pEGFP-N1 vector, so that it is in lecture frame with the green fluorescent protein (GFP) coding sequence. We then subcloned the hCOX-2/GFP fusion sequence into the XhoI-NotI restriction site of pPyCAGIP vector. pPyCAGIP-GFP construct was obtained by restriction digestion and subcloning from pEGFP-N1 vector. The orientation and integrity of the constructs were determined by sequencing.
Generation of Stable hCOX-2 Cells
For plasmid transfections, attached CHL cells at 50% confluence were exposed for 24 hours to Fugene 6 reagent (Roche Applied Science, Indianapolis, IN) containing pPyCAGIP-hCOX-2-GFP or control vector pPyCAGIP-GFP. At the end of this period, the transfection medium was replaced with fresh medium containing 10% FBS; on the second day after transfection, the cells were plated at 25% confluence in medium supplemented with 3 μg/ml puromycin. Single colonies of resistant cells were detectable after approximately 12 days and were selected and subcultured for 30 days. Subsequent cultures of selected cells were grown in the presence of puromycin. Cells stably expressing hCOX-2-GFP or GFP proteins were termed CHL-C and CHL-V, respectively. At 18 hours before experiments, the culture medium was replaced with fresh medium containing 1% FBS. For the in vitro phenobarbital (PB)/DEN protocol, CHL-C and CHL-V cells were incubated with 1.5 mmol/L PB for 16 hours followed by 15 mmol/L DEN for 24 to 48 hours. These cells were also used for tumorigenesis experiments in nude mice.
Animal Experimentation
Hepatocyte COX-2 transgenic mice (25 to 30 g body weight; 4 months of age) on a C57BL6J × DBA background were used in this study, along with corresponding age-matched WT mice.21 The animals were housed on a 12-hour light/dark cycle in an air conditioned room at 25°C with food and water available ad libitum and were treated according to the Institutional Care Instructions (Bioethical Commission, Spanish National Research Council–CSIC). In some experiments, 60-week-old mice were analyzed. Liver fibrosis was induced by feeding the animals with a diet deficient in methionine and (MCD diet) (TD 90262; Harlan Laboratories, Madison, WI). The animals were sacrificed after 15 weeks of treatment, and their body weights were recorded before the liver tissues were used for analysis (n = 8 to 9).
To induce HCC, DEN (25 mg/kg; Sigma-Aldrich) was injected intraperitoneally into 14-day-old mice. Mice were sacrificed at 10, 25, or 40 weeks after DEN treatment. Body and liver weights were recorded, and livers were removed and separated into individual lobes. Externally visible tumors (≥0.5 mm) were counted and measured by stereomicroscopy. It is relevant to note that DEN is a poor carcinogen on its own in mice of a C57BL6 genetic background, but the hybrid genetic background C57BL6J × DBA that we used is much more susceptible to DEN treatment.22 Liver tissues were snap-frozen in liquid nitrogen, stored at −80°C, and collected in a solution containing 30% sucrose in PBS or fixed in 10% buffered formalin. Plasma was obtained by puncture of the retro-orbital venous plexus.
Female athymic nu/nu mice (6 weeks old) were obtained from Charles River Laboratories (Wilmington, MA). The animals were kept under pathogen-free conditions and were given an autoclaved standard diet and water ad libitum. CHL-C or CHL-V cells (4 × 106) suspended in PBS were injected in one flank of the nu/nu mice, along with the corresponding control, depending on the treatment of the cells in the other flank (4 mice per condition). Tumor growth was monitored weekly by two-dimensional measurements using a vernier caliper. The formula (L/2) × (W/2) × π (where L is the maximum diameter of each tumor and W is the length at right angles to L) was used to calculate the maximal tumor surface area. After 32 days, mice were sacrificed by cervical dislocation before the tumor burden caused any obvious morbidity. The final tumor size and weights were recorded.23
RNA Isolation
Total RNA of liver biopsy samples was extracted by using TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA (1 μg) was reverse-transcribed using a Transcriptor first strand cDNA synthesis kit according to the manufacturer's instructions (Roche Applied Science). For transcriptional profiling analyses, total RNA was further cleaned using the Qiagen RNeasy mini kit (Valencia, CA; Hilden, Germany) with one step of DNase I digestion according to the manufacturer's instructions. Three micrograms RNA were used for cDNA synthesis with the RT2 First Standard kit (SuperArray Bioscience, Frederick, MD).
Quantitative Real-Time PCR Analysis
The qPCR analysis was performed with a MyiQ real-time PCR system (Bio-Rad) sequence detector using the SsoFast EvaGreen polymerase method (Bio-Rad) and d(N)6 random hexamer with the following primers: hCOX-2 F 5′-CGCAGTACAGAAAGTATCACAGGC-3′ and R 5′-GCGTTTGCGGTACTCATTAAAA-3′; 36b4 F 5′-ACTGGTCTAGGACCCGAGAAG-3′ and R 5′-TCCCACCTTGTCTCCAGTCT-3′; Ccne1 F 5′-TTCTGCAGCGTCATCCTCT-3′ and R 5′-TGGAGCTTATAGACTTCGCACA-3′; and Myc F 5′-CCTAGTGCTGCATGAGGAGA-3′ and R 5′-TCTTCCTCATCTTCTTGCTCTTC-3′.Specific primers were purchased from Invitrogen. PCR thermocycling parameters were 95°C for 30 seconds, 40 cycles of 95°C for 5 seconds, and 60°C for 10 seconds. Each sample was run in triplicate and was normalized to 36b4 RNA. The replicates were then averaged, and fold induction was determined in a ΔΔCT-based fold-change calculations.
Histochemistry Analysis
Samples of liver were fixed in 10% buffered formalin, embedded in paraffin wax, sectioned at 4-μm thickness, and stained with H&E. Histopathology analysis was performed by the Department of Medicine and Animal Surgery from Faculty of Veterinary, Madrid, Spain. Immunohistochemical staining was performed using the streptavidin-biotin-peroxidase complex method as described previously.24 Histological sections were stained following standard procedures with an antibody against the active form of caspase 3 (R&D Systems, Minneapolis, MN) or Ki-67 (Novocastra, Newcastle Upon Tyne, UK). Five entire areas of each tissue section (500 cells) were counted.
Western Blot Analysis
Extracts from tissue samples (50 to 100 mg) or cells (2 to 3 × 106) were obtained as previously described.21 For Western blot analysis, whole-cell extracts were boiled for 5 minutes in Laemmli sample buffer, and equal amounts of protein (20 to 30 μg) were separated by 10% to 15% SDS-polyacrylamide electrophoresis gel. The relative amounts of each protein were determined with the following polyclonal or monoclonal antibodies: COX-2 and COX-1 (from Cayman Chemical and Santa Cruz Biotechnology); cyclin E, PCNA, p53, and c-Jun (Santa Cruz Biotechnology); p38MAPK (Thr180/Tyr182), p38 MAPK, p-Erk (Thr202/Tyr204), Erk, p-JNK (Thr183/Thy185), JNK, p-Stat3 (Tyr705), Stat3, and cleaved caspase-3 (Cell Signaling Technology); MMP-9 (Chemicon International, Temecula, CA); and αSMA (Sigma-Aldrich) After incubation with the corresponding anti-rabbit or anti-mouse horseradish peroxidase conjugated secondary antibody, blots were developed by the enzymatic chemiluminescence protocol (Amersham–GE Healthcare, Chalfont St. Giles, UK). Target protein band densities were normalized with β-actin. The blots were revealed, and different exposition times were performed for each blot with a charged coupling device camera in a luminescent image analyzer (Gel-Doc; Bio-Rad) to ensure the linearity of the band intensities. Densitometric analysis was expressed in arbitrary units.
Cell Viability
To perform the growth rate analysis, cells were trypsinized and stained with trypan blue at the indicated times, and the cells that excluded trypan blue were counted under the microscope. Cell viability was measured using the MTT assay, 3-(4,5-dimethylthiazol-2-yl)-2,5′-diphenyltetrazolium bromide. Cells (3 × 103) were seeded on 96-well plates in Dulbecco's modified Eagle's medium supplemented with 10% FBS. After incubation with the PB/DEN-protocol, cells were treated with 10 μL of MTT solution (0.3 mg/ml) for 1 hour at 37°C. The medium was removed and dimethyl sulfoxide was added to dissolve the blue formazan residue. The optical density was measured at 550 nm.
Determination of Metabolites
PGE2 was determined in liver tissue by specific immunoassay (GE Healthcare).21 Alanine transaminase, aspartate transaminase, alkaline phosphatase, and lactate dehydrogenase were assayed spectrophotometrically in plasma at the School of Clinical Biochemistry, Faculty of Pharmacy, Complutense University, Madrid, Spain. Protein levels were determined with Bradford reagent (Bio-Rad). To quantify collagen content, hepatic hydroxyproline was assayed according to the instructions of a hydroxyproline ELISA kit (Antibodies-online, Aachen, Germany).
Measurement of Oxidative DNA Damage and Intracellular Redox State
Oxidative damage of DNA was analyzed in cultured cells under different treatments by measuring the levels of 8-hydroxy-2-deoxyguanosine (8-OH-dG) by immunoassay according to the manufacturer's instructions (StressMarq Biosciences, Victoria, BC, Canada). The oxidation-sensitive fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and the oxidative-insensitive analog carboxy-DCFH-DA (Invitrogen) were used to analyze the total intracellular content of reactive oxygen species (ROS); the analog is a nonoxidable fluorescent probe that does not change its fluorescence in the presence of ROS. The fluorescent probe monochlorobimane (Sigma-Aldrich) was used to analyze the total intracellular content of reduced glutathione. After treatment and at the indicated times, cells were incubated with 2.5 μmol/L H2DCFDA (30 minutes, 37°C), 2.5 μmol/L carboxy-DCFH-DA, or 2 mmol/L monochlorobimane (1 hour, 37°C) in Hanks' balanced salt solution without phenol red, lysed with a buffer containing 25 mmol/L HEPES pH 7.5, 60 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.2 mmol/L EDTA, and 0.1% Triton X-100 (10 minutes, 4°C), and then transferred in duplicate into a 96-well plate. Fluorescence was measured in a microplate fluorescence reader and normalized with protein content.
Soft Agar Assays
Cells (3 × 104) were suspended in 1.5 ml of Dulbecco's modified Eagle's medium containing 0.4% of low-melting-point agarose (type VII; Sigma-Aldrich) and 10% FBS, and seeded onto a coating of 1% low-melting-point agarose in complete culture medium. Every 4 days, the medium was removed and replaced with fresh medium. Cells were incubated for 21 days; the colonies containing more than 50 cells were counted after stained with 0.5% MTT for 1 hour at room temperature. The number of colonies was quantified by soft agar analysis with the software Analysis auto from Olympus (Wendenstrasse, Hamburg, Germany).
Transcriptional Profiling of COX-2 Tg Liver after DEN Treatment
The mouse cancer PCR array was performed according to the manufacturer's protocol, using the Profiler PCR array system and the SYBR Green/fluorescein qPCR master mix (SuperArray Bioscience) on a MyiQ Real-Time PCR System (Bio-Rad). Gene expression was compared using a Web-based software package for the PCR array system (http://www.sabiosciences.com/pcr/arrayanalysis.php); this software automatically performs all ΔΔCT based fold-change calculations from the specific uploaded raw threshold cycle data.
Data Analysis
Statistical analyses were performed using the SPSS package version 17 (SPSS, Chicago, IL). Data are expressed as means ± SD. Statistical significance of differences between the control and transgenic groups was evaluated by the U of the Mann-Whitney U-test. The association of histological analysis was assessed using Fisher's exact test. Curves for overall survival were generated using the Kaplan-Meier method. All tests were calculated two-tailed, and the significance level was set at P < 0.05.
Results
COX-2 Tg Mice Developed Subclinical Hepatitis
hCOX-2 mRNA and protein were detected in COX-2 Tg mice, and intrahepatic PGE2 levels were threefold higher compared with WT mice. The expression of hCOX-2, COX-1, and PGE2 was similar between 16- and 60-week-old COX-2 Tg mice (Figure 1, A–C). No significant differences were observed between old WT and Tg mice in body weight (38.8 ± 2.3 g vs 41.1 ± 1.8 g), liver weight (1.9 ± 0.1 g vs 2.1 ± 0.1 g), and alanine transaminase and aspartate transaminase (Figure 1D). Nevertheless, inflammatory foci, composed mainly of lymphocytes, were seen in the livers of 60-week-old Tg mice, compared with WT mice, indicating that the expression of the COX-2 transgene causes subclinical hepatitis at 60 weeks of age (Figure 1E).
Figure 1.
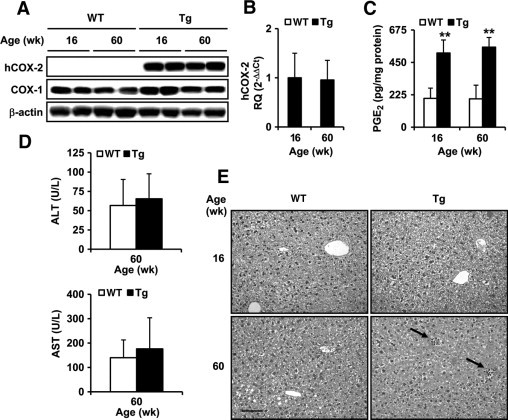
Aged COX-2 Tg mice developed subclinical hepatitis. A: Human COX-1 and COX-2 protein expression in liver homogenates from WT and Tg animals at 16 and 60 weeks of age detected by Western blot and normalized with β-actin. B: qPCR analysis of COX-2 mRNA expression. COX-2 mRNA amounts were calculated as relative quantity (RQ) and normalized to the expression of 36b4 mRNA. Values represent fold change relative to COX-2 Tg 16 weeks. C: Intrahepatic PGE2 concentration was determined by enzyme immunoassay in liver homogenates. D: Plasma levels of aspartate transaminase (AST) and alanine transaminase (ALT) were assayed spectrophotometrically E: H&E staining of liver sections of 16- and 60-week-old WT and Tg mice. Inflammatory foci are indicated with arrows. Micrographs were taken with a Color View Camera in a Vanox Olympus microscope at 20× magnification. Scale bar = 100 μm (for all images). Data are reported as means ± SD of six animals per condition. **P < 0.001 vs the corresponding matched animals. Wk, week.
COX-2 Expression in Hepatocytes Did Not Mediate the Development of Liver Fibrosis
Similar survival curves and changes in body weight were observed in WT and COX-2 Tg mice fed with the MCD diet (Figure 2, A and B). Feeding with the MCD diet for 15 weeks induced significantly higher plasma alanine transaminase levels than in the corresponding control (320 ± 20 vs 40 ± 7 UI/L and 250 ± 32 vs 25 ± 4 UI/L for WT and Tg mice, respectively). Liver histology of WT and Tg mice fed with the MCD diet (Figure 2C) showed fatty liver degeneration, although Masson's trichrome staining (collagen content) did not show significant differences between WT and Tg mice. To evaluate liver fibrosis in a quantitative manner, the levels of hydroxyproline and the amount of α-smooth muscle actin (α-SMA) and matrix metalloproteinase 9 (MMP-9) were measured as markers of hepatic stellate cell activation and liver fibrotic stage. The MCD diet induced both α-SMA and MMP-9 expression (control levels of α-SMA and MMP-9 without the MCD diet were undetectable; data not shown) and increased hydroxyproline content (Figure 2, D and E); however, COX-2 expression in Tg mice did not affect liver fibrosis. Endogenous COX-2 expression was slightly induced by the MCD diet in WT mice.
Figure 2.
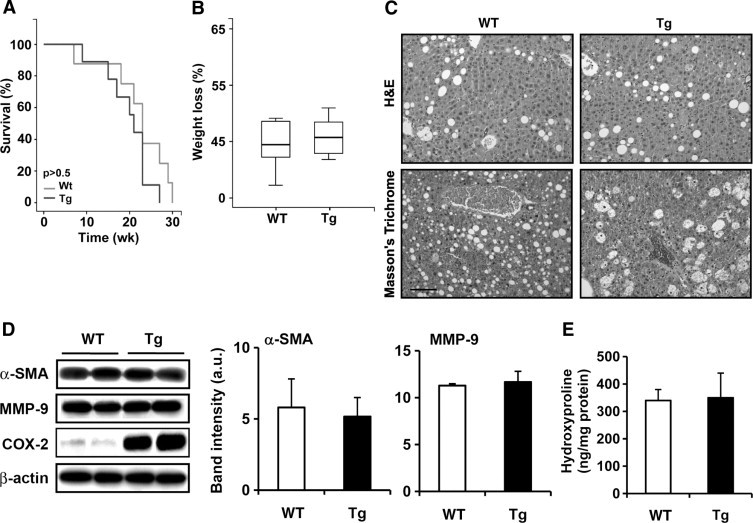
COX-2 expression in hepatocytes did not mediate the development of liver fibrosis. Survival curves (A) and the loss of body weight (B) of WT (n = 14) and Tg (n = 14) mice after the MCD diet. C: H&E and Masson's trichrome staining of liver sections of WT and Tg mice after the MCD diet and sacrificed at 15 weeks. Micrographs were taken with a Color View Camera in a Vanox Olympus microscope at 20× magnification. Scale bar = 100 μm (for all images). D and E: α-SMA, MMP-9, COX-2 protein expression measured by Western blot (D) and hydroxyproline levels analyzed by ELISA (E) in WT and Tg liver after the MCD diet. Data are reported as means ± SD of six animals per condition.
Effects of Hepatic COX-2 Expression on Tumor Development
To determine the role of COX-2 in HCC induction, WT and Tg mice were injected with DEN (25 mg/kg) at postnatal day 14 and were sacrificed at 10, 25, or 40 weeks after treatment. The survival of mice, the liver/body weight ratio, and tibial size are shown in Figure 3A and Table 1. A macroscopic view of representative WT and COX-2 livers is shown in Figure 3B. Analysis of plasma levels of liver injury markers after DEN treatment are given in Table 1. There was an important increase of aspartate transaminase, alanine transaminase, and LDH after DEN treatment in the Tg mice, but liver injury was similar in WT and Tg mice. Morphometric analysis revealed no differences in the number, size, or volume of detectable tumors (Table 2). Histological analysis confirmed that all tumors at 40 weeks after DEN treatment, both in WT and Tg mice, corresponded to liver carcinoma (Figure 3C). The histopathological analysis of the liver biopsies indicated that the incidence of preneoplastic lesions was higher in COX-2 Tg than in WT mice 10 weeks after DEN treatment (25% vs 8%), but these changes did not reach statistical significance (P = 0.053). At the end of DEN treatment (40 weeks), similar HCCs were detected in WT and Tg liver, although a high pulmonary metastatic incidence was seen in Tg mice (Table 2).
Figure 3.
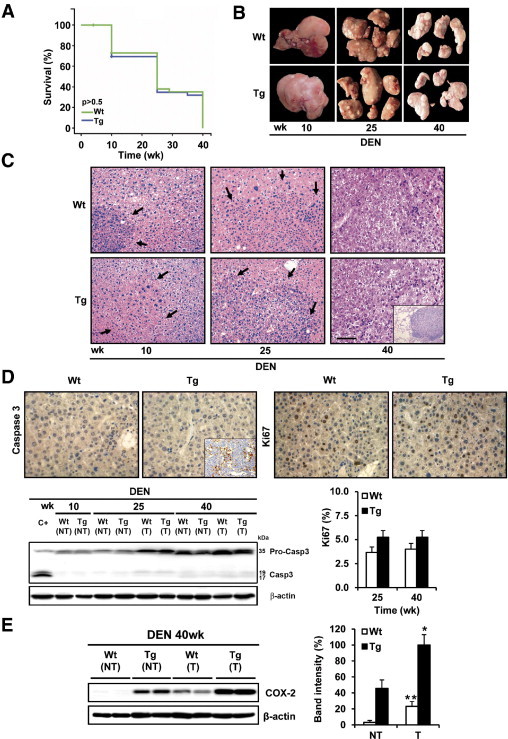
Effects of hepatic COX-2 expression on tumor development. A: Survival curves of WT and Tg mice injected with DEN (25 mg/kg) at 2 weeks of age (n = 36). B: Gross liver features after 10, 25, and 40 weeks of DEN treatment in WT and Tg mice. C: H&E staining of liver sections in WT and Tg mice after 10, 25, and 40 weeks of DEN treatment. Lesions are indicated by arrows. Inset: Lung metastases. Micrographs were taken with a Color View Camera in a Vanox Olympus microscope at 20× magnification. Scale bar = 100 μm (for all images in C). D: Cell proliferation and apoptosis were analyzed in liver sections from WT and Tg mice after DEN treatment by Ki-67 and active caspase 3 staining. Inset: Positive control of murine thymic lymphoma section. A representative Western blot of cleaved caspase 3 is shown. C+ is a liver extract from WT mice treated with lipopolysaccharide (35 μg/kg)/D-Galn (1g/kg). The percentage of mitotic cells was quantified at 25 and 40 weeks after DEN treatment. Data are reported as means ± SD of five sections per animal (n = 3). E: Endogenous COX-2 expression was detected in tumor tissue from WT liver at 40 weeks after DEN treatment. Data are reported as means ± SD of 12 animals per condition. *P < 0.05 and **P < 0.001 tumor vs paired control tissue.
Table 1.
Body and Liver Weight, Tibia Length, and Plasma Levels of Liver Injury Markers in WT and COX-2 Tg Mice after DEN Treatment
| Treatment | Body weight (g) | Liver weight (g) | Tibia length (cm) | Liver/body ratio | AST⁎ (UI/L) | ALT⁎ (UI/L) | LDH⁎ (UI/L) |
|---|---|---|---|---|---|---|---|
| WT | |||||||
| DEN10 | 32.6 ± 4.1 | 1.6 ± 0.2 | 2.2 ± 0.1 | 0.05 | 240 ± 120 | 95 ± 60 | 500 ± 260 |
| DEN25 | 41.8 ± 2.8 | 2.7 ± 0.7 | 2.3 ± 0.1 | 0.07 | 215 ± 70 | 100 ± 30 | 1015 ± 310 |
| DEN40 | 36.7 ± 0.7 | 5.3 ± 1.4 | 2.3 ± 0.1 | 0.15 | 160 ± 50 | 160 ± 30 | 630 ± 220 |
| Tg | |||||||
| DEN10 | 30.5 ± 2.7 | 1.5 ± 0.1 | 2.2 ± 0.1 | 0.05 | 300 ± 180 | 83 ± 60 | 470 ± 290 |
| DEN25 | 42.7 ± 6.1 | 2.5 ± 0.7 | 2.3 ± 0.1 | 0.06 | 230 ± 140 | 97 ± 40 | 1080 ± 580 |
| DEN40 | 37.1 ± 9.4 | 4.2 ± 0.8 | 2.3 ± 0.1 | 0.11 | 210 ± 130 | 195 ± 140 | 1140 ± 750 |
Both WT and Tg mice were given diethylnitrosamine starting at 2 weeks of age for 10, 25, or 40 weeks. Hepatocyte COX-2 Tg mice on a C57BL6J × DBA background were used, along with corresponding age-matched WT mice.21 Results are reported as mean ± SD of 10 to 12 animals per condition. ALT, alanine transaminase; AST, aspartate transaminase; DEN, diethylnitrosamine; LDH, lactate dehydrogenase; Tg, transgenic; WT, wild type.
Plasma levels of AST, ALT, and LDH were assayed spectrophotometrically at the indicated times after DEN treatment. The control levels of AST, ALT, and LDH of WT vs Tg mice were 84 ± 16 vs. 71 ± 12 UI/L, 39 ± 7 vs. 22 ± 4 UI/L, and 47 ± 9 vs. 33 ± 6 UI/L, respectively.
Table 2.
Morphometric Analysis and Incidence of Pathological Signs in Livers of WT and COX-2 Tg Male Mice after DEN Treatment
| Treatment | Sample size | Tumors, ≥0.5 mm (no.) | Tumor area (%) | Maximal size (mm) | Preneoplastic lesions, no. (%) | Adenomas, no. (%) | Hepatocellular carcinomas, no. (%) | Lung metastasis, no. (%) |
|---|---|---|---|---|---|---|---|---|
| WT | ||||||||
| DEN10 | 12 | 0 | 0 | 0 | 1 (8) | 0 (0) | 0 (0) | 0 (0) |
| DEN25 | 12 | 42.5 ± 9.4 | 8.0 ± 2.6 | 6.8 ± 1.5 | 1 (8) | 11 (92) | 0 (0) | 0 (0) |
| DEN40 | 12 | 38.7 ± 0.5 | 43.7 ± 3.9 | 19.3 ± 2.4 | 0 (0) | 0 (0) | 12 (100) | 3 (25) |
| Tg | ||||||||
| DEN10 | 12 | 0 | 0 | 0 | 3 (25) | 0 (0) | 0 (0) | 0 (0) |
| DEN25 | 12 | 59.2 ± 11.8 | 8.8 ± 2.38 | 5.5 ± 1.1 | 0 (0) | 12 (100) | 0 (0) | 0 (0) |
| DEN40 | 12 | 43.0 ± 6.3 | 43.5 ± 6.0 | 14.7 ± 0.7 | 0 (0) | 0 (0) | 12 (100) | 8 (67)⁎ |
P < 0.05 vs. WT.
Next, we measured the apoptotic rate in WT and Tg liver at 25 and 40 weeks after DEN treatment by quantifying the percentage of cells with active caspase 3 and the mitotic index measured by Ki-67 immunohistochemistry. Caspase 3 staining was not detected, nor cleaved caspase 3 (measured by Western blotting), and Ki-67 was similar in both WT and Tg liver (Figure 3D). DEN treatment has been reported to decrease CYP2E1 levels,25 but in the present study the expression was similar between WT and Tg mice, suggesting that COX-2 does not affect DEN bioactivation (data not shown). Of note, endogenous COX-2 expression was detected in tumors of WT mice at 40 weeks after DEN treatment (Figure 3E).
COX-2 Tg Liver Tumors Showed an Increase in Cyclin E1 and c-Myc
To better understand how COX-2 accelerates the initial events of liver tumorigenesis induced by DEN, cell cycle regulators and tumor suppressors in tumor and nontumor samples were analyzed. No changes were observed in the protein levels of p53, PCNA, cyclin E1, and c-Jun at 10 weeks after treatment (Figure 4A). At 25 weeks, significant differences were observed in cyclin E levels in tumor versus normal tissue. Furthermore, cyclin E levels were significantly increased in Tg versus WT liver. At 40 weeks after DEN treatment, a significant decrease of p53 and a significant increase of PCNA, cyclin E, and c-Jun levels were detected in tumor versus paired control tissue from both WT and Tg liver and in Tg liver versus WT (Figure 4, A and B).
Figure 4.
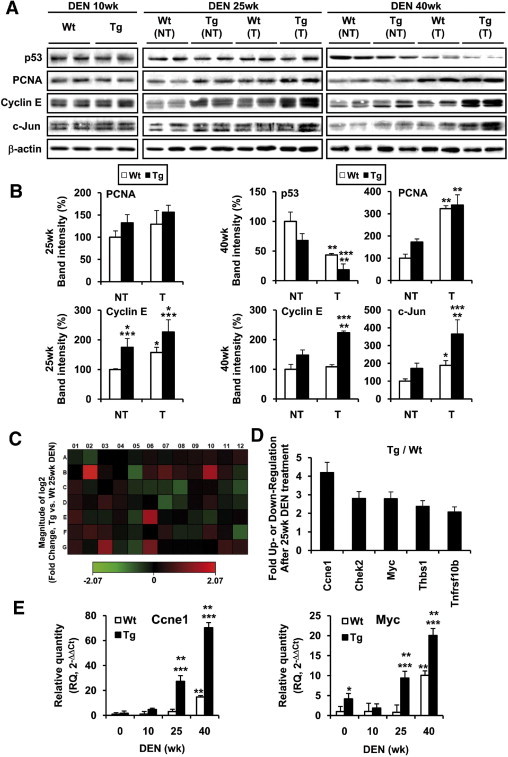
COX-2 Tg liver tumors showed an increase in cyclin E1 and c-Myc. A: Nontumor liver (NT) and HCC (T) were analyzed for cell cycle and tumor suppressors associated proteins. Representative Western blots showing the expression of p53, PCNA, cyclin E and c-Jun in WT and Tg mice. B: Densitometric analysis of the expression of the proteins in WT and Tg mice after DEN treatment. Data are reported as means ± SD of 12 animals per condition. *P < 0.05 and **P < 0.001 tumor vs paired control tissue. ***P < 0.05 COX-2 Tg vs WT mice treated under identical conditions. C and D: Colorimetric diagram of 84 cancer-related genes (C) and fold change (D) for genes whose transcription is up- or down-regulated twofold in COX-2 Tg mice at 25 weeks after DEN treatment. Data are reported as means of four animals per condition. E: Validated array results from qPCR analysis. mRNA amounts were calculated as RQ and were normalized to the expression of 36b4 mRNA. *P < 0.05 and **P < 0.001 for COX-2 Tg or WT after DEN treatment vs WT mice without treatment. ***P < 0.001 for COX-2 Tg vs WT mice treated under identical conditions.
To gain further insight into the response to DEN in WT and Tg liver, a specific mouse cancer microarray was used. The magnitude of changes and the corresponding statistical significance of the 84 genes analyzed are summarized in Figure 4, C and D, and in Table 3. We sorted the candidate genes whose expression threshold after DEN treatment was altered more than twofold (Figure 4D), including Ccne1 (cyclin E1), Chek2, Myc, Tnfrsf10b, and Thbs1 in Tg mice at 25 weeks after DEN treatment, all of them related with cell proliferation. These results were validated by qPCR (Figure 4E).
Table 3.
Heat Map Data for Gene Expression
| Layout | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | Akt1 | Akt2 | Angpt1 | Apaf1 | Atm | Bad | Bax | Bcl2 | Bcl2l1 | Birc5 | Brca1 | Casp8 |
| −1.16 | −1.42 | −1.01 | 1.02 | −1.09 | 1.17 | −1.08 | 1.14 | 1.20 | 1.27 | 1.12 | 1.32 | |
| B | Ccnd1 | Ccne1 | Cdc25a | Cdh1 | Cdk2 | Cdk4 | Cdkn1a | Cdkn2a | Cflar | Chek2 | Col18a1 | Ctnnb1 |
| 1.00 | 4.19 | 1.15 | 1.03 | −1.68 | 1.38 | 1.75 | 1.36 | 1.23 | 2.8 | 1.28 | −1.27 | |
| C | E2f1 | Egfr | Ets2 | Fas | Fgf1 | Fgfr2 | Figf | Fos | Grb2 | Hgf | Ifnb1 | Igf1 |
| 1.35 | −1.13 | −1.01 | 1.22 | −1.26 | −1.31 | −1.29 | −1.64 | 1.09 | 1.10 | −1.20 | −1.50 | |
| D | Itga2 | Itga3 | Itga4 | Itgav | Itgb1 | Itgb3 | Jun | Kiss1 | Map2k1 | Mcam | Mdm2 | Met |
| 1.27 | 1.10 | 1.43 | 1.20 | −1.08 | 1.05 | −1.61 | −1.28 | 1.08 | 1.29 | −1.12 | −1.22 | |
| E | Mmp2 | Mmp9 | Mta1 | Mta2 | Muc1 | Myc | Ncam1 | Nfkb1 | Nfkbia | Nme4 | Pdgfa | Pdgfb |
| 1.52 | −1.15 | −1.18 | 1.15 | −1.37 | 2.78 | −1.04 | 1.07 | −1.17 | −1.31 | −1.04 | 1.09 | |
| F | Pik3r1 | Plau | Plaur | Pten | Raf1 | Rb1 | S100a4 | Serpinb2 | Serpine1 | Syk | Tek | Tert |
| 1.30 | 1.52 | 1.38 | −1.07 | −1.17 | 1.03 | 1.30 | −1.20 | −1.19 | 1.42 | 1.12 | −1.66 | |
| G | Tgfb1 | Tgfbr1 | Thbs1 | Timp1 | Tnf | Tnfrsf10b | Tnfrsf1a | Trp53 | Twist1 | Vegfa | Vegfb | Vegfc |
| 1.38 | −1.06 | 2.37 | −1.20 | −1.45 | 2.07 | 1.05 | −1.03 | −1.03 | 1.03 | 1.48 | 1.36 |
Gene expression between Tg and WT mice at 25 weeks after DEN treatment was compared using RT2 Profiler version 3.2, a Web-based software package for the Profiler PCR array system (SABiosciences-Qiagen, Frederick, MD; http://www.sabiosciences.com/pcr/arrayanalysis.php); this software automatically performs all ΔΔCT-based fold-change calculations from the specific uploaded raw threshold cycle data.
Effect of DEN on CHL Cells Expressing a COX-2 Transgene
To investigate more deeply the molecular mechanisms triggered by COX-2/PGE2 in the chemical carcinogenesis induced by DEN, a human CHL cell line stably transfected with an hCOX-2-GFP expression vector or control vector and treated with an in vitro PB/DEN induction protocol. The hCOX-2 was expressed only in CHL-C cells (Figure 5, A and B). Moreover, CHL-C cells released significantly greater quantities of PGE2 into the culture medium than did CHL-V (Figure 5C). The contribution of COX-2 to this process was confirmed by the inhibition of PGE2 synthesis after treating the cells with 5 μmol/L DFU. The CHL-C cell number after 72 hours of culture in 10% (v/v) FBS was greater (150%) than that of CHL-V (Figure 5D). COX-2/PGE2 levels were not modified by the effect of PB/DEN treatment (data not shown).
Figure 5.
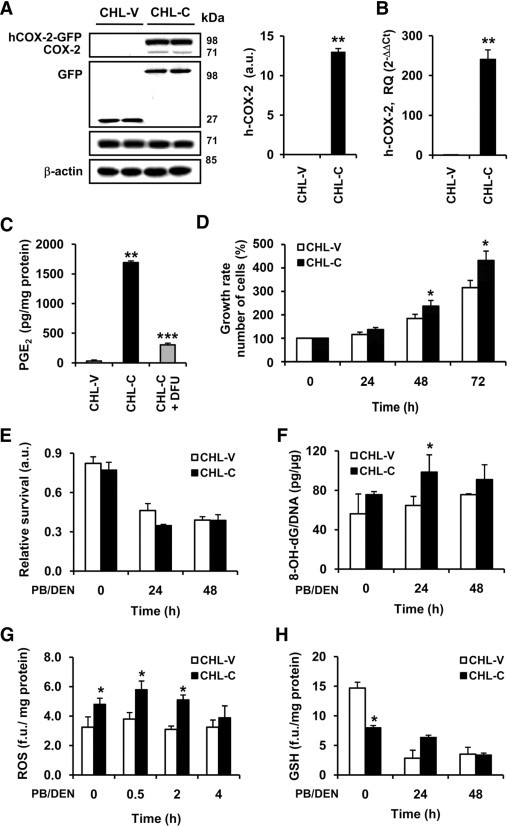
Characterization of CHL cells expressing COX-2 after DEN treatment. CHL cells were stably transfected with empty pPyCAGIP-GFP vector (CHL-V) or pPyCAGIP-hCOX-2-GFP encoding human COX-2 (CHL-C). For in vitro PB/DEN protocol, CHL-C and CHL-V cells were incubated with 1.5 mmol/L PB for 16 hours following by 15 mmol/L DEN for 24 to 48 hours. A: hCOX-2-GFP and COX-1 expression in whole cell extracts were detected by Western blot and normalized with β-actin. B: qPCR analysis of COX-2 mRNA expression. COX-2 mRNA amounts were calculated as RQ and normalized to the expression of 36b4 mRNA. Values represent fold change relative to CHL-V. C: PGE2 production determined by enzyme immunoassay in the culture medium of CHL-V and CHL-C cells. D: Growth rate of CHL-V and CHL-C cells. E: Cell viability was measured after PB/DEN treatment at 24 and 48 hours as indicated in Materials and Methods using the MTT assay. F: Oxidative damage of DNA was analyzed in cultured cells under PB/DEN treatment by measuring the levels of 8-OH-dG by immunoassay. Intracellular content of ROS (G) and glutathione (GSH) (H) were measured by using the fluorescent probes H2DCFDA and monochlorobimane, respectively. Data are reported as means ± SD of four independent experiments. *P < 0.05 and **P < 0.001 vs the CHL-V control cells treated under identical conditions. ***P < 0.05 vs CHL-C cells without DFU treatment.
Next, we evaluated the contribution of COX-2 activity to cell viability after PB/DEN treatment, assessed by the MTT reduction assay. No significant differences were found between CHL-V and CHL-C cells, although DEN treatment decreased cell survival by approximately 50% (Figure 5E). Similar results were obtained when LDH activity was measured (data not shown). Oxidative damage of DNA was analyzed by measuring the 8-OH-dG levels, and CHL-C cells exhibited a higher content (1.6 fold) of 8-OH-dG than CHL-V after DEN treatment (Figure 5F). ROS production was analyzed in CHL-V and CHL-C cells after DEN treatment, and ROS production was significantly increased in CHL-C cells, compared with CHL-V without DEN treatment (1.5 fold) (Figure 5G). No differences in fluorescence between cells were found when carboxy-DCFH-DA was used (0.52 vs 0.48 fluorescence units/mg protein for CHL-V and CHL-C, respectively). As expected, the levels of glutathione were lower in the CHL-C cells without treatment (Figure 5H). To identify signaling pathways that affect hepatocyte proliferation, we examined the phosphorylation of several protein kinases. An increase in the p-JNK1/JNK1, p-Erk/Erk, p-p38/p-38, and p-Stat3/Stat3 ratios was observed in both CHL-V and CHL-C cells after 2 to 4 hours of PB/DEN treatment, with some significant differences (Figure 6, A and B).
Figure 6.
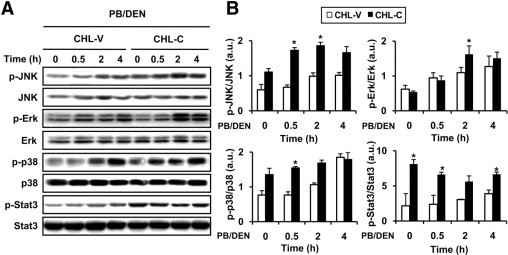
Signaling pathways implicated in proliferation in CHL cells expressing COX-2 after DEN treatment. A: Representative Western blots showing the phosphorylation of JNK, Erk, p38, and Stat3 in CHL-V and CHL-C cells after PB/DEN treatment at the indicated times. The expression of target proteins was normalized to that of β-actin. B: Densitometric analysis of the expression of the ratios p-JNK/JNK, p-Erk/Erk, p-p38/p38, and p-Stat3/Stat3 in CHL-V and CHL-C cells after DEN treatment. Data are reported as means ± SD of four independent experiments. *P < 0.05 vs the CHL-V control cells treated under identical conditions.
Transforming Activity and Growth of CHL Cells Implants in Nude Mice
The proliferation of CHL-V and CHL-C cells with and without PB/DEN treatment was compared by soft-agar colony formation assays. The number and size of the colonies formed by CHL-C cells treated with PB/DEN were similar to those formed by CHL-V (Figure 7, A–C). Finally, we evaluated the growth of transplanted CHL-V and CHL-C cells with and without PB/DEN treatment in nude mice. PB/DEN treatment increased the tumor area at the end of the treatment by 1.7 and 3.2 fold in CHL-V and CHL-C, respectively (Figure 7D). Of note, a significant increase in tumor area due to the effect of COX-2-dependent PGs was observed (2.2 fold) (Figure 7E).
Figure 7.
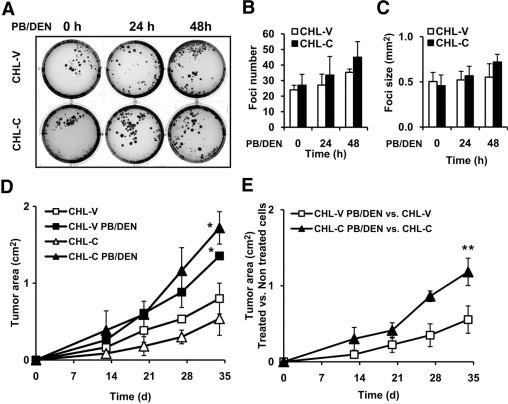
Soft agar colony formation and growth of CHL-V and CHL-C cells implants in nude mice. A: CHL-V and CHL-C cells without or with PB/DEN treatment protocol were maintained 72 hours with 10% FBS, plated in soft agar as described in Materials and Methods and were incubated for 21 days. A representative experiment is shown. The number (B) and size (C) of colonies were quantified by soft agar analysis. D: CHL-V and CHL-C cells without or with PB/DEN treatment were injected in nude mice as described in experimental procedures and tumor area was measured. E: The ratio between treated vs untreated CHL-V or CHL-C cells, related to tumor area. Data are reported as means ± SD of three different experiments or four animals per conditions. *P < 0.05 vs the CHL-V or the CHL-C cells without treatment. **P < 0.05 vs CHL-V control cells.
Discussion
In the present study, we analyzed the role of COX-2 in the development of chronic liver disease, hepatitis, fibrosis, and chemical hepatocarcinogenesis, by using both in vivo and in vitro models of constitutive COX-2 expression and elevated PGE2 synthesis in the hepatocyte. Our results demonstrate that aged COX-2 Tg mice develop mild hepatitis, but COX-2 does not appear to mediate the development of liver fibrosis. COX-2 favors early assessment of preneoplastic foci, but it is not sufficient to enhance malignant transformation and HCC induced by DEN.
Ectopic expression of COX-2 in hepatocytes constitutes a nonphysiopathological condition26 and, therefore, the generation and characterization of COX-2 genetically altered mice models may help to clarify the phenotypic changes related to COX-2 activity in normal hepatocytes and might facilitate the identification of the mechanisms by which COX-2 contributes to liver pathology. In previous work, we generated and partially characterized the first model of Tg mice carrying human hCOX-2 under the control of the human Apo E promoter.21 Prostaglandin synthesis in hepatocytes exerted an efficient protection against acute liver injury by antiapoptotic/antinecrotic mechanisms and by accelerating early hepatocyte proliferation.27 Recently, two other models of COX-2 Tg mice have been published, in which COX-2 is expressed under the control of the transthyretin promoter28 or the albumin enhancer promoter29; on challenge with proinflammatory stimuli, the animals behave very differently. Expression of COX-2 under the albumin-enhancer promoter produces very low levels of PGE2, and animals show increased injury after endotoxin challenge. Moreover, in the same model, this COX-2 transgene accelerates endotoxin-induced acute liver failure29 but prevents Fas-induced liver injury.30 The expression of hCOX-2 under the transthyretin promoter produced the highest levels of intrahepatic PGE2 and induced hepatitis in animals more than 12 months of age, characterized by the activation of NF-κB, stimulation of the secretion of proinflammatory cytokines, and enhanced macrophage and lymphocyte infiltration.28 These data are in agreement with the results we observed in aged animals.
Chronic liver injury can promote fibrosis, characterized by proliferation of hepatic stellate cells and their change to myofibroblast-like cells, leading to extracellular matrix and collagen accumulation. Activated hepatic stellate cells but not quiescent hepatic stellate cells express COX-2, suggesting that this pathway has an active role in hepatic fibrogenesis. Nonetheless, data regarding the role of COX-2 on liver fibrotic process are controversial, with reports describing both exacerbation of fibrogenesis31 and amelioration of fibrogenesis, through the induction of celecoxib-mediated apoptosis of hepatic stellate cells.32 Our results, however, and those of others33 using COX-2 Tg mice clearly demonstrate that COX-2 expression did not influence liver fibrogenesis even after feeding with the MCD diet.
Several lines of evidence suggest that COX-2 signaling is implicated in hepatocarcinogenesis and that COX-2 inhibitors prevent HCC cell growth in vitro and in animal models.23 Increased COX-2 expression has been found in human HCC, in which COX-2 is highly expressed at early stages but is down-regulated in advanced stages, and with changes in the areas of expression in tumor versus nontumor adjacent tissue.15 To clarify the contribution of COX-2 to the development of HCC, we used three complementary approaches: i) DEN-induced chemical hepatocarcinogenesis in COX-2 Tg mice, ii) PB/DEN treatment of human COX-2 Tg hepatocyte-like cells, and iii) COX-2 Tg hepatocyte-like cells implants in nude mice.
Both p53 and JNK are well known markers of the genotoxic and cytotoxic effects induced by DEN.2 Of note, both WT and COX-2 Tg mice exhibited an important decrease of p53 (involved in G1 cell arrest) and an increase of c-Jun levels after DEN treatment. COX-2 has been reported to promote tumorigenesis by functional inactivation of p53 under genotoxic stress,34 and p53 expression declined notably in hepatocytes expressing COX-2.35 The levels of p53 declined in our models with DEN treatment, but COX-2 expression did not modify these changes.
Analysis of cell cycle proteins in WT and COX-2 Tg liver showed a significant difference of cyclin E levels. These results suggest that the presence of COX-2 accelerated early hepatocyte proliferation in this in vivo model of HCC, corroborating previous results, and that COX-2 expression could be related to its anti-apoptotic effect and inflammatory phenomena in the early phases of HCC.27
Enhancement of liver tumorigenesis by DEN administration was seen after hepatocyte-specific ablation in IKKβ, p38α, and IKKγ/NEMO deficient mice.18 It was proposed that enhanced liver damage and hepatocyte death driven by ROS accumulation could explain augmented HCC development in all these mouse genetic models.36 In agreement with these data, in the present study COX-2 Tg hepatocyte-like cells exhibited elevated oxidative stress and ROS accumulation after DEN administration, together with an important decrease in the levels of glutathione. Indeed, ROS production may cause oxidative DNA damage, reflected by a higher level of anti-8-OHdG reactivity in COX-2 Tg cells.
The JNKs are activated by stress signals, pro-inflammatory stimuli, and some mitogenic signals and are required for hepatocyte proliferation after partial hepatectomy.37 JNK1-deficient mice are much less susceptible to DEN-induced HCC development, whereas prolonged activation of JNK1 is a principal mechanism by which the absence of hepatocyte IKKβ results in increased susceptibility to DEN-induced hepatocarcinogenesis.38 Erk, JNK/SAPK, and p38 contribute to COX-2 expression, and the anti-inflammatory activity of many compounds is a function of COX-2 inhibition via JNK and p38 MAPK inactivation.39 The effect of COX-2-dependent PGs on these signaling pathways have been recently studied using cell lines stably overexpressing COX-2.40 Our data in COX-2 Tg hepatocyte-like cells verify moderate activation of JNK, Erk, and p38 in CHL-C cells.
How hepatocyte death promotes HCC development is not clear, but it was proposed that necrotic hepatocytes release IL-1α, which induces IL-6 production by Kupffer cells, and that this response drives the compensatory proliferation of surviving hepatocytes, involving Stat3 activation.36 He et al,41 using liver-specific Stat3 knockout mice, found that this transcriptional target for IL-6 is required for HCC development and that hepatocyte NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven Stat3 activation. Furthermore, expression of IL-6, a major Stat3 activator in the liver, is elevated in cirrhosis and in HCC.42 Moreover, activation of COX-2/PGE2 enhances Stat3 phosphorylation through EP-1 receptor-induced activation of c-Src and EGFR in human cholangiocarcinoma cells and in non-small-cell lung cancer.43 We observed a moderate increase of Stat3 phosphorylation in CHL-C cells, in agreement with an elevated oxidative stress and ROS accumulation, together with a significant decrease in the levels of glutathione even without PB/DEN treatment.
Targets of PGs through their EP receptors are the PI3K and the Akt/PKB (protein kinase B) pathways. Akt improves cellular energy stores by accelerating cell proliferation and size, and induces anti-apoptotic effects by phosphorylation and inactivation of the proapoptotic protein Bad and up-regulation of Bcl-2 and Mcl-1.44 Our previous studies showed that, in COX-2 Tg hepatocyte-like cells and in COX-2 Tg liver, Akt phosphorylation is enhanced, compared with WT cells, indicating a reinforcement of survival pathways.27,35 The present data show that COX-2 moderately increased the growth of CHL cell implants in nude mice, which may be due to the modulation of Akt and JNK-c-Jun pathways.23
Tumor initiation occurred in the present study through DEN-induced mutagenesis, which induces genetic changes that cause proto-oncogene activation (c-Myc) and/or loss of tumor suppressors (p53). After initiation, tumor promotion is needed for expansion of altered cells into premalignant lesions that progress into malignant tumors. Our results show that, although COX-2 induces changes in different signaling pathways implicated in the initiation, promotion, and expansion of liver cells, other genes besides the COX-2 gene are needed to enhance liver tumorigenesis caused by DEN. Furthermore, COX-2 was detected in WT tumor tissue at the end of DEN treatment, indicating that endogenous COX-2 expression is a late event in HCC development. These results, together with the high pulmonary metastatic incidence of HCC in Tg mice and the promotion and migration of HCC cells induced by PGE2,12 suggest that COX-2 might be implicated in the expansion and metastatic phase of HCC.
In conclusion, our results demonstrate that COX-2 Tg mice develop subclinical hepatitis, but that COX-2 does not appear to mediate the development of liver fibrosis in the absence of a broad proinflammatory onset. COX-2 expression is not sufficient to enhance malignant transformation induced by DEN, and it is a late event in the development of HCC.
Acknowledgments
We thank Carlos Grau for the morphometric analysis and Verónica Terrón for technical assistance.
Footnotes
Supported by grants SAF2007-60551, SAF2009-12602, and BFU2008-02161 from the Ministry of Science and Innovation (MICINN) and grant no. S-BIO-0283/2006 from the Community of Madrid (Comunidad de Madrid). The Biomedical Research Centre Network of Hepatic and Digestive Diseases (CIBERehd) is funded by National Institute of Health Carlos III (Instituto de Salud Carlos III).
C.L.I. and R.M. contributed equally to the present work.
Contributor Information
Marta Casado, Email: mcasado@ibv.csic.es.
Paloma Martín-Sanz, Email: pmartins@iib.uam.es.
References
- 1.Llovet J.M., Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maeda S., Kamata H., Luo J.L., Leffert H., Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 3.Simmons D.L., Botting R.M., Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Ledwith B.J., Pauley C.J., Wagner L.K., Rokos C.L., Alberts D.W., Manam S. Induction of cyclooxygenase-2 expression by peroxisome proliferators and non-tetradecanoylphorbol 12,13-myristate-type tumor promoters in immortalized mouse liver cells. J Biol Chem. 1997;272:3707–3714. doi: 10.1074/jbc.272.6.3707. [DOI] [PubMed] [Google Scholar]
- 5.Martín-Sanz P., Callejas N.A., Casado M., Díaz-Guerra M.J., Boscá L. Expression of cyclooxygenase-2 in foetal rat hepatocytes stimulated with lipopolysaccharide and pro-inflammatory cytokines. Br J Pharmacol. 1998;125:1313–1319. doi: 10.1038/sj.bjp.0702196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Callejas N.A., Boscá L., Williams C.S., Du B.R., Martín-Sanz P. Regulation of cyclooxygenase 2 expression in hepatocytes by CCAAT/enhancer-binding proteins. Gastroenterology. 2000;119:493–501. doi: 10.1053/gast.2000.9374. [DOI] [PubMed] [Google Scholar]
- 7.Pham H., Eibl G., Vincenti R., Chong B., Tai H.H., Slice L.W. 15-Hydroxyprostaglandin dehydrogenase suppresses K-RasV12-dependent tumor formation in Nu/Nu mice. Mol Carcinog. 2008;47:466–477. doi: 10.1002/mc.20404. [DOI] [PubMed] [Google Scholar]
- 8.Casado M., Callejas N.A., Rodrigo J., Zhao X., Dey S.K., Boscá L., Martín-Sanz P. Contribution of cyclooxygenase 2 to liver regeneration after partial hepatectomy. FASEB J. 2001;15:2016–2018. doi: 10.1096/fj.01-0158fje. [DOI] [PubMed] [Google Scholar]
- 9.Rudnick D.A., Perlmutter D.H., Muglia L.J. Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proc Natl Acad Sci USA. 2001;98:8885–8890. doi: 10.1073/pnas.151217998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto H., Kondo M., Nakamori S., Nagano H., Wakasa K., Sugita Y., Chang-De J., Kobayashi S., Damdinsuren B., Dono K., Umeshita K., Sekimoto M., Sakon M., Matsuura N., Monden M. JTE-522, a cyclooxygenase-2 inhibitor, is an effective chemopreventive agent against rat experimental liver fibrosis1. Gastroenterology. 2003;125:556–571. doi: 10.1016/s0016-5085(03)00904-1. [DOI] [PubMed] [Google Scholar]
- 11.Kern M.A., Haugg A.M., Koch A.F., Schilling T., Breuhahn K., Walczak H., Fleischer B., Trautwein C., Michalski C., Schulze-Bergkamen H., Friess H., Stremmel W., Krammer P.H., Schirmacher P., Müller M. Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res. 2006;66:7059–7066. doi: 10.1158/0008-5472.CAN-06-0325. [DOI] [PubMed] [Google Scholar]
- 12.Mayoral R., Fernández-Martinez A., Boscá L., Martin-Sanz P. Prostaglandin E2 promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis. 2005;26:753–761. doi: 10.1093/carcin/bgi022. [DOI] [PubMed] [Google Scholar]
- 13.Cheng A.S., Yu J., Lai P.B., Chan H.L., Sung J.J. COX-2 mediates hepatitis B virus X protein abrogation of p53-induced apoptosis. Biochem Biophys Res Commun. 2008;374:175–180. doi: 10.1016/j.bbrc.2008.06.098. [DOI] [PubMed] [Google Scholar]
- 14.Núñez O., Fernández-Martínez A., Majano P.L., Apolinario A., Gómez-Gonzalo M., Benedicto I., López-Cabrera M., Boscá L., Clemente G., García-Monzón C., Martín-Sanz P. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: role of viral core and NS5A proteins. Gut. 2004;53:1665–1672. doi: 10.1136/gut.2003.038364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cusimano A., Fodera D., Lampiasi N., Azzolina A., Notarbartolo M., Giannitrapani L., D'Alessandro N., Montalto G., Cervello M. Prostaglandin E2 receptors and COX enzymes in human hepatocellular carcinoma: role in the regulation of cell growth. Ann N Y Acad Sci. 2009;1155:300–308. doi: 10.1111/j.1749-6632.2009.03701.x. [DOI] [PubMed] [Google Scholar]
- 16.Cha Y.I., DuBois R.N. NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med. 2007;58:239–252. doi: 10.1146/annurev.med.57.121304.131253. [DOI] [PubMed] [Google Scholar]
- 17.Calvisi D.F., Factor V.M., Ladu S., Conner E.A., Thorgeirsson S.S. Disruption of beta-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology. 2004;126:1374–1386. doi: 10.1053/j.gastro.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 18.Luedde T., Beraza N., Kotsikoris V., van Loo G., Nenci A., De Vos R., Roskams T., Trautwein C., Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 19.Pikarsky E., Porat R.M., Stein I., Abramovitch R., Amit S., Kasem S., Gutkovich-Pyest E., Urieli-Shoval S., Galun E., Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 20.Haybaeck J., Zeller N., Wolf M.J., Weber A., Wagner U., Kurrer M.O., Bremer J., Iezzi G., Graf R., Clavien P.A., Thimme R., Blum H., Nedospasov S.A., Zatloukal K., Ramzan M., Ciesek S., Pietschmann T., Marche P.N., Karin M., Kopf M., Browning J.L., Aguzzi A., Heikenwalder M. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16:295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casado M., Mollá B., Roy R., Fernández-Martínez A., Cucarella C., Mayoral R., Boscá L., Martín-Sanz P. Protection against Fas-induced liver apoptosis in transgenic mice expressing cyclooxygenase 2 in hepatocytes. Hepatology. 2007;45:631–638. doi: 10.1002/hep.21556. [DOI] [PubMed] [Google Scholar]
- 22.Goldsworthy T.L., Fransson-Steen R. Quantitation of the cancer process in C57BL/6J, B6C3F1 and C3H/HeJ mice. Toxicol Pathol. 2002;30:97–105. doi: 10.1080/01926230252824770. [DOI] [PubMed] [Google Scholar]
- 23.Kern M.A., Schoneweiss M.M., Sahi D., Bahlo M., Haugg A.M., Kasper H.U., Dienes H.P., Kaferstein H., Breuhahn K., Schirmacher P. Cyclooxygenase-2 inhibitors suppress the growth of human hepatocellular carcinoma implants in nude mice. Carcinogenesis. 2004;25:1193–1199. doi: 10.1093/carcin/bgh110. [DOI] [PubMed] [Google Scholar]
- 24.De la Cueva E., García-Cao I., Herranz M., López P., García-Palencia P., Flores J.M., Serrano M., Fernández-Piqueras J., Martín-Caballero J. Tumorigenic activity of p21Waf1/Cip1 in thymic lymphoma. Oncogene. 2006;25:4128–4132. doi: 10.1038/sj.onc.1209432. [DOI] [PubMed] [Google Scholar]
- 25.Freeman J.E., Stirling D., Russell A.L., Wolf C.R. cDNA sequence, deduced amino acid sequence, predicted gene structure and chemical regulation of mouse Cyp2e1. Biochem J. 1992;281:689–695. doi: 10.1042/bj2810689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martín-Sanz P., Mayoral R., Casado M., Boscá L. COX-2 in liver, from regeneration to hepatocarcinogenesis: what we have learned from animal models? World J Gastroenterol. 2010;16:1430–1435. doi: 10.3748/wjg.v16.i12.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mayoral R., Mollá B., Flores J.M., Boscá L., Casado M., Martín-Sanz P. Constitutive expression of cyclo-oxygenase 2 transgene in hepatocytes protects against liver injury. Biochem J. 2008;416:337–346. doi: 10.1042/BJ20081224. [DOI] [PubMed] [Google Scholar]
- 28.Yu J., Hui A.Y., Chu E.S., Cheng A.S., Go M.Y., Chan H.L., Leung W.K., Cheung K.F., Ching A.K., Chui Y.L., Chan K.K., Sung J.J. Expression of a cyclo-oxygenase-2 transgene in murine liver causes hepatitis. Gut. 2007;56:991–999. doi: 10.1136/gut.2006.097923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han C., Li G., Lim K., DeFrances M.C., Gandhi C.R., Wu T. Transgenic expression of cyclooxygenase-2 in hepatocytes accelerates endotoxin-induced acute liver failure. J Immunol. 2008;181:8027–8035. doi: 10.4049/jimmunol.181.11.8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li G., Han C., Xu L., Lim K., Isse K., Wu T. Cyclooxygenase-2 prevents fas-induced liver injury through up-regulation of epidermal growth factor receptor. Hepatology. 2009;50:834–843. doi: 10.1002/hep.23052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hui A.Y., Leung W.K., Chan H.L., Chan F.K., Go M.Y., Chan K.K., Tang B.D., Chu E.S., Sung J.J. Effect of celecoxib on experimental liver fibrosis in rat. Liver Int. 2006;26:125–136. doi: 10.1111/j.1478-3231.2005.01202.x. [DOI] [PubMed] [Google Scholar]
- 32.Paik Y.H., Kim J.K., Lee J.I., Kang S.H., Kim D.Y., An S.H., Lee S.J., Lee D.K., Han K.H., Chon C.Y., Lee S.I., Lee K.S., Brenner D.A. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut. 2009;58:1517–1527. doi: 10.1136/gut.2008.157420. [DOI] [PubMed] [Google Scholar]
- 33.Yu J., Wu C.W., Chu E.S., Hui A.Y., Cheng A.S., Go M.Y., Ching A.K., Chui Y.L., Chan H.L., Sung J.J. Elucidation of the role of COX-2 in liver fibrogenesis using transgenic mice. Biochem Biophys Res Commun. 2008;372:571–577. doi: 10.1016/j.bbrc.2008.05.069. [DOI] [PubMed] [Google Scholar]
- 34.Choi E.M., Kim S.R., Lee E.J., Han J.A. Cyclooxygenase-2 functionally inactivates p53 through a physical interaction with p53. Biochim Biophys Acta. 2009;1793:1354–1365. doi: 10.1016/j.bbamcr.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 35.Fernández-Martínez A., Mollá B., Mayoral R., Boscá L., Casado M., Martín-Sanz P. Cyclo-oxygenase 2 expression impairs serum-withdrawal-induced apoptosis in liver cells. Biochem J. 2006;398:371–380. doi: 10.1042/BJ20060780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakurai T., He G., Matsuzawa A., Yu G.Y., Maeda S., Hardiman G., Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwabe R.F., Bradham C.A., Uehara T., Hatano E., Bennett B.L., Schoonhoven R., Brenner D.A. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 38.Sakurai T., Maeda S., Chang L., Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci USA. 2006;103:10544–10551. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanabe T., Tohnai N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002;68–69:95–114. doi: 10.1016/s0090-6980(02)00024-2. [DOI] [PubMed] [Google Scholar]
- 40.Kim Y.M., Park S.Y., Pyo H. Cyclooxygenase-2 (COX-2) negatively regulates expression of epidermal growth factor receptor and causes resistance to gefitinib in COX-2-overexpressing cancer cells. Mol Cancer Res. 2009;7:1367–1377. doi: 10.1158/1541-7786.MCR-09-0004. [DOI] [PubMed] [Google Scholar]
- 41.He G., Yu G.Y., Temkin V., Ogata H., Kuntzen C., Sakurai T., Sieghart W., Peck-Radosavljevic M., Leffert H.L., Karin M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trikha M., Corringham R., Klein B., Rossi J.F. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. 2003;9:4653–4665. [PMC free article] [PubMed] [Google Scholar]
- 43.Han C., Demetris A.J., Stolz D.B., Xu L., Lim K., Wu T. Modulation of Stat3 activation by the cytosolic phospholipase A2alpha and cyclooxygenase-2-controlled prostaglandin E2 signaling pathway. J Biol Chem. 2006;281:24831–24846. doi: 10.1074/jbc.M602201200. [DOI] [PubMed] [Google Scholar]
- 44.Schulze-Bergkamen H., Brenner D., Krueger A., Suess D., Fas S.C., Frey C.R., Dax A., Zink D., Büchler P., Müller M., Krammer P.H. Hepatocyte growth factor induces Mcl-1 in primary human hepatocytes and inhibits CD95-mediated apoptosis via Akt. Hepatology. 2004;39:645–654. doi: 10.1002/hep.20138. [DOI] [PubMed] [Google Scholar]