

Abstract
Cyclin-dependent kinases (CDKs) 4 and 6 are important regulators of the G1 phase of the cell cycle, share 71% amino acid identity, and are expressed ubiquitously. As a result, it was assumed that each of these kinases plays a redundant role regulating normal and neoplastic proliferation. In previous reports, we have described the effects of CDK4 expression in transgenic mice, including the development of epidermal hyperplasia and increased malignant progression to squamous cell carcinoma. To study the role of CDK6 in epithelial growth and tumorigenesis, we generated transgenic mice carrying the CDK6 gene under the keratin 5 promoter (K5CDK6). Similar to K5CDK4 mice, epidermal proliferation increased substantially in K5CDK6 mice; however, no hyperplasia was observed. CDK6 overexpression also triggered keratinocyte apoptosis in interfollicular and follicular epidermis as a compensatory mechanism to override aberrant proliferation. Unexpectedly, CDK6 overexpression results in decreased skin tumor development compared with wild-type siblings. The inhibition in skin tumorigenesis was similar to that previously reported in K5-cyclin D3 mice. Furthermore, biochemical analysis of the K5CDK6 epidermis showed preferential complex formation between CDK6 and cyclin D3, suggesting that this particular complex plays an important role in tumor restraint. These studies provide in vivo evidence that CDK4 and CDK6 play a similar role as a mediator of keratinocyte proliferation but differ in apoptosis activation and skin tumor development.
Normal cell growth and differentiation require precise control of the mechanisms that govern the entry into, passage through, and exit from the cell cycle. Progress through the G1 phase of the mammalian cell cycle is mediated by D-type cyclins, which associate and activate cyclin-dependent kinases (CDKs) 4 and 6.1,2 The Retinoblastoma (pRb) family of proteins, pRb, p107, and p130, are key substrates for G1-CDK/cyclin complexes and negatively regulate the passage of cells from G1 to S phase.2 Therefore, CDK4 and CDK6 act as master integrators in the G1 phase, coupling with cell cycle mitogenic signals and their oncogenic properties in cancer cells.3–5 CDK4 and CDK6 share 71% amino acid identity, and both are expressed ubiquitously.6 As a result, it was assumed that both play a redundant function in the G1 phase and tumorigenesis. However, in the last few years, relevant differences were determined between the functional properties of these G1 kinases. Initial experiments identified CDK6 activity in T cells, suggesting that cell type–specific expression might explain the need for two G1 kinases.7 Supporting this hypothesis, CDK6-deficient mice showed a reduced number of red blood cells and lymphocytes and pronounced thymic atrophy due to decreased proliferation and blockage of differentiation.8,9 Recently, Bockstaele et al10 reported differences in CDK4 and CDK6 regulation. They showed that CDK6, but not CDK4, is regulated by CDK-Activating Kinase (CAK) and suggested a proline-directed kinase to be the main regulator of CDK4. Novel functions of CDK6 have recently been reported, for instance, residue selectivity of these kinases on the retinoblastoma protein,11 different subcellular localizations,12,13 and a specific role of CDK6 during the differentiation of a variety of cell types.13–16 Moreover, CDK6 plays a role in halting inappropriate cellular proliferation through a mechanism involving the accumulation of p53 and p130 growth-suppressing proteins,17 and the activation of CDK6 precedes CDK4 activation in T cells.18,19 Data from tumor studies also suggested similarity and differences between these kinases. For instance, both CDK4 and CDK6 have been found to be overexpressed in human gliomas.20–23 On the other hand, CDK4, but not CDK6, is specifically targeted in melanomas,24,25 whereas CDK6 activity has been found elevated in squamous cell carcinomas (SCCs)26,27 and neuroblastomas28 without alteration of CDK4 activity.
The two-stage mouse skin carcinogenesis protocol is a well-suited model for understanding the multistage nature of tumor progression. In this model, tumor initiation is accomplished through a single topical application of a carcinogen, typically, 7,12-dimethylbenz(a)anthracene (DMBA). This produces a genetic inheritable mutation in the Ha-ras oncogene. Tumor promotion takes place when the initiated cells are expanded because of multiple applications of a tumor promoter, usually 12-O-tetradecanoylphorbol-13-acetate (TPA). This stimulus induces hyperproliferation that promotes the generation of benign tumors, so-called papillomas. Finally, although papilloma regression is a common event, in some cases, malignant progression occurs and papillomas evolve to SCCs.
In the last few years, we and others have used the mouse skin model to study the role of positive and negative regulators of cell cycle in normal and neoplastic proliferation.29–40 Work from our group and other laboratories has shown that CDK4 is mechanistically involved in the development of human and experimental epidermal tumors.32,33,35,38,41,42 These studies showed that forced expression of CDK4 in the epidermis results in increased malignant progression to SCCs,33 whereas CDK4 ablation completely inhibits skin tumor development.32 On the other hand, the overexpression of CDK2 or the indirect activation of CDK2 in mouse epidermis induces keratinocyte proliferation but does not affect skin tumor development.35,36 Despite the evidences that G1-CDKs are involved in proliferation and tumorigenesis, the actual role of CDK6 in epidermis has not been established. Therefore, to study the role of CDK6 in epithelial growth, differentiation, and tumorigenesis, we generated a transgenic mouse carrying the CDK6 gene under the control of the keratin 5 promoter (K5CDK6). As expected, transgenic mice showed expression of CDK6 in the epidermal basal cell layer. Analogous to K5CDK4 mice, epidermal proliferation increased substantially in K5CDK6 mice, although no hyperplasia was observed. Interestingly, the overexpression of CDK6 also results in augmented apoptosis in interfollicular epidermis and hair follicles. Biochemical analysis of K5CDK6 epidermal tissues showed increased CDK6 kinase activity with no effect on CDK4 and CDK2 kinase activities, suggesting that sequestration of p27Kip1 and indirect activation of CDK2 is not a relevant mechanism in the K5CDK6 epidermis. We have also studied the susceptibility of K5CDK6 mice to the two-stage chemical carcinogenesis protocol. Surprisingly, we found that forced expression of CDK6 leads to decreased skin tumor development. Moreover, skin tumors from K5CDK6 mice show no progression to SCCs, as we previously observed in K5CDK4 mice. These results mimic the effect of cyclin D3 overexpression in K5–cyclin D3 mouse epidermis,40 which also showed reduced tumorigenesis, suggesting that CDK6/cyclin D3 complexes might play an important role in tumor inhibition. Supporting this hypothesis, biochemical analysis of the K5CDK6 epidermis showed preferential formation of CDK6/cyclin D3 complexes. Overall, we have established that the development of ras-induced skin tumors is diminished by overexpression of CDK6, which results in a surprisingly opposite effect to that observed in K5CDK4 mice. Thus, this model provides in vivo evidence that CDK4 and CDK6 play a similar role as mediator of keratinocyte proliferation but differ in the activated mechanisms, leading to an opposite effect in tumor development. As a result, we hypothesize that particular CDK/cyclin D complexes play different roles in epidermis homeostasis and tumor development.
Materials and Methods
Generation of Transgenic Mice
K5CDK6 transgenic mice were developed by cloning human-Cdk6 cDNA into the vector pBK5, which contains a 5.2-kb bovine keratin 5 regulatory sequence, the β-globin intron 2, and the 3′ SV40-polyadenylylation sequence. This construct was designated as pK5-CDK6. The transgene was excised from the plasmid vector by digestion with BssHII and microinjected into C57BL/6 × DBA2 hybrid embryos at the Animal Model Core, University of North Carolina School of Medicine. Several founders for K5CDK6 were obtained from the transgenic facility. Positive founders were genotyped by PCR using specific primers for the human transgene Cdk6 and β-globin intron sequence. Mice used in this study were generated by mating transgenic and wild-type animals for five to seven generations.
Transgene-Specific PCR
Genomic DNA was extracted from mouse tail clips and used for PCR detection of the transgene. We used an upstream primer CTGACCAGCAGTACGAATG and a downstream primer GAGTCCAATCACGTCCAAG specific for the β-globin intron sequence or upstream CTGACCAGCAGTACGAATG and downstream TTTCTTTGCACCTTTCCAGG primers for human CDK6. With this process, we screened all of the transgenic mice lines. The DNA amplification renders a 450-bp PCR product with β-globin primers or an 850-bp band with human-CDK6 primers. PCR was performed by denaturation at 95°C for 1 minute, followed by 32 cycles of amplification as follows: denaturation at 95°C for 30 seconds, annealing at 60°C for 40 seconds, and extension at 72°C for 45 seconds, with a final extension at 72°C for 10 minutes.
Western Blotting
The dorsal sides of the mice were shaved. After they were sacrificed, the dorsal skins were treated with a depilatory agent for 1 minute and then washed. The epidermal tissue was scraped off with a razor blade, placed into homogenization buffer [50 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 2.5 mmol/L EGTA, 1 mmol/L EDTA acid, 0.1% Tween 20, 1 mmol/L dithiothreitol, 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 0.2 U/ml of aprotinin, 10 mmol/L b-glycerophosphate, 0.1 mmol/L sodium vanadate, and 1 mmol/L NaF], and homogenized using a manual homogenizer. The epidermal homogenate was centrifuged at 14,000 × g at 4°C to collect the supernatant, which was used directly for Western blotting analysis or stored at −80°C. The protein concentration was measured with the Bio-Rad protein assay system (Bio-Rad Laboratories, Richmond, CA). Protein lysates (25 μg from each sample) were electrophoresed through 12% acrylamide gels and electrophoretically transferred onto nitrocellulose membranes. After being blocked with 5% nonfat powdered milk in Dulbecco PBS, the membranes were incubated with 1 μg/ml of specific antibodies. The following antibodies were used: polyclonal antibodies against cyclin D2 (M20), CDK4 (C22), CDK2 (M2), CDK6 (C21), pRb (M153), p107 (C18) (Santa Cruz Biotech, Santa Cruz, CA), and p53 (1C12) (Cell Signaling Tech Inc., Boston, MA), and monoclonal antibodies against cyclin D1 (DCS-6), CDK6 (DCS-83) (Santa Cruz Biotech). Secondary antibodies followed by enhanced chemiluminescence (ECL detection kit; GE Health Care, Piscataway, NJ) were used for immunoblotting detection.
Co-Immunoprecipitations and Kinase Assays
To study CDK/D-type cyclin complex formations and kinase activities, we used polyclonal antibodies against CDK4 (C-22) and CDK6 (C-21) (Santa Cruz Biotech) and a monoclonal antibody against cyclin D3 (Ab-1) (NeoMarkers, Fremont, CA) conjugated with protein A–sepharose beads (Thermo Scientific Inc., Rockford, IL) or Dynabeads Protein G (Invitrogen, Carlsbad, CA). Fresh protein lysates from epidermal tissue (500 μg) were immunoprecipitated for 1 hour at 4°C with constant rotation. After washing 3 times with extraction buffer, proteins that co-immunoprecipitated were analyzed by Western blot as described previously. Protein lysate (50 μg) was loaded as a control input. The immunoprecipitation was repeated 3 times using 250, 500, or 1000 mg of protein lysate with identical results.
To study the kinase activities, 500 μg of fresh protein was extracted and immunoprecipitated in NP-40 lysis buffer (Tris [pH 7.5], 150 mmol/L NaCl, 0.5% NP-40, 50 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L DTT, and 1 mmol/L PMSF) with precoated antibodies against CDK2, CDK4, and CDK6 for 2 hours at 4°C. Beads were washed twice each with NP-40 buffer and once with kinase buffer (50 mmol/L HEPES [pH 7], 10 mmol/L MgCl2, 5 mmol/L MnCl2). Then, 30 μL of kinase buffer, 1 μg of pRb or histone H1 (Upstate Biotechnology Inc., Charlottesville, VA) substrate, 5 μCi of [γ-32P] ATP (6000 Ci/mmol), 1 mmol/L DTT, and 5 μmol/L ATP was added to the bead pellet and incubated for 30 minutes at 30°C. SDS sample buffer was added, and each sample was boiled for 3 minutes to stop the reaction and electrophoresed through polyacrylamide gels. Western blot and kinase assay bands were quantified using UN-SCANT IT gel software for Windows.
Immunostaining
Epithelial cell proliferation was measured by intraperitoneal injection of 60 μg/g of 5-bromodeoxyuridine (BrdU) 30 minutes before the mice were sacrificed by CO2 asphyxiation. BrdU incorporation was detected by immunohistochemical staining of paraffin-embedded skin sections with a mouse anti-BrdU (ab-2) monoclonal antibody (Calbiochem; EMB Biosciences, San Diego, CA), biotin-conjugated antimouse antibody (Vector Laboratories, Burlingame, CA), and avidin-biotin Vectastain Elite peroxidase kit (Vector Laboratories) with diaminobenzidine as a chromogen. Apoptotic cells were determined by terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling assays with the FragEL DNA Fragmentation Detection Kit, Colorimetric-TdT enzyme (Calbiochem; EMB Biosciences) following the manufacturer's instructions. Briefly, the terminal deoxynucleotidyl transferase (TdT enzyme) binds to the exposed 3-OH ends of a DNA fragment generated in apoptosis progression and catalyzes the addition of biotin-labeled and unlabeled deoxynucleotides. Biotinylated nucleotides were detected using a streptavidin–horseradish peroxidase conjugate. Counterstaining with methyl green allows for quantification of normal and apoptotic cells. The numbers of apoptotic cells in the tumors were determined in sections of 250 μm2 with a reticule grid. Apoptotic keratinocytes in interfollicular and follicular epidermis were quantified in 2-cm sections. To determine the incidence of follicular apoptosis, hair follicles carrying at least 1 apoptotic cell in the bulge area were counted as a positive hair follicle. In all cases, 12 fields were counted per section on a total of 10 paraffin-embedded sections, representing 5 mice per genotype.
Mouse Experiments
Two K5-CDK6 transgenic lines were used for the two-stage carcinogenesis protocol. Mouse experiments were performed with sibling animals to reduce the influence of the genetic background. Eight mice for each transgenic and wild-type group were used (K5CDK6[H], K5CDK6[L], wild-type[H], and wild-type[L]) for a total of 16 transgenic and 16 wild-type siblings. Three-week-old K5CDK6 and wild-type mice were initiated with the topical application of 200 nmol DMBA in 200 μL of acetone on the dorsal surface of the mice. Two weeks later, mice were dosed topically twice weekly with 4 μg of TPA in 200 μL of acetone for 25 weeks. Papilloma development was tracked weekly for 25 weeks. Papillomas were counted if 1 mm or larger. Multiplicity and incidence of tumor-bearing animals were compared between K5CDK6 and wild-type mice using Fisher's exact test.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 4 Software (GraphPad Software Inc., San Diego, CA).
Results
Generation of Transgenic Mice Expressing CDK6
To study the role of CDK6 in squamous epithelial tissues, we generated transgenic mice overexpressing CDK6 driven by the regulatory sequence of the keratin 5 promoter (K5CDK6 mice). The K5CDK6 construct was made by subcloning the human CDK6 cDNA into a pBK5 vector containing the 5.2-kb fragment of the bovine keratin 5 promoter (K5), the rabbit β-globin intron 2, and the SV40 3′ polyadenylation signal (Figure 1A). The K5 promoter fragment directs transgene expression to the basal cell compartment of stratified squamous epithelia, which was shown previously.43 All of the transgenic mice were generated on the hybrid genetic background C57BL6xDBA2. The genotypes of transgenic mice were characterized by PCR analysis using primers specific for the β-globin and human CDK6 sequences. Four integration-positive mice were selected as founders based on those results and crossed with wild-type siblings to generate 4 transgenic lines (Figure 1B). A second screening to verify transgene expression was performed by Western blot analysis of epidermal preparations as described.44 We observed 5.7-, 4.7-, 2.7-, and 5.4-fold increases in CDK6 expression in the transgenic lines A through D, respectively (Figure 1C). Therefore, the transgenic lines A and C, with high and low CDK6 expression, were renamed as K5CDK6(H) and K5CDK6(L), respectively, and were used in all of the experiments presented in this report (Figure 1C, lines A and C). In addition, we performed immunofluorescence analysis of CDK6 expression in mouse epidermis from K5CDK6(H) and K5CDK6(L) mice. As expected, we observed that CDK6 expression is driven to the basal cell layer of interfollicular epidermis and hair follicle43 (Figure 1D).
Figure 1.
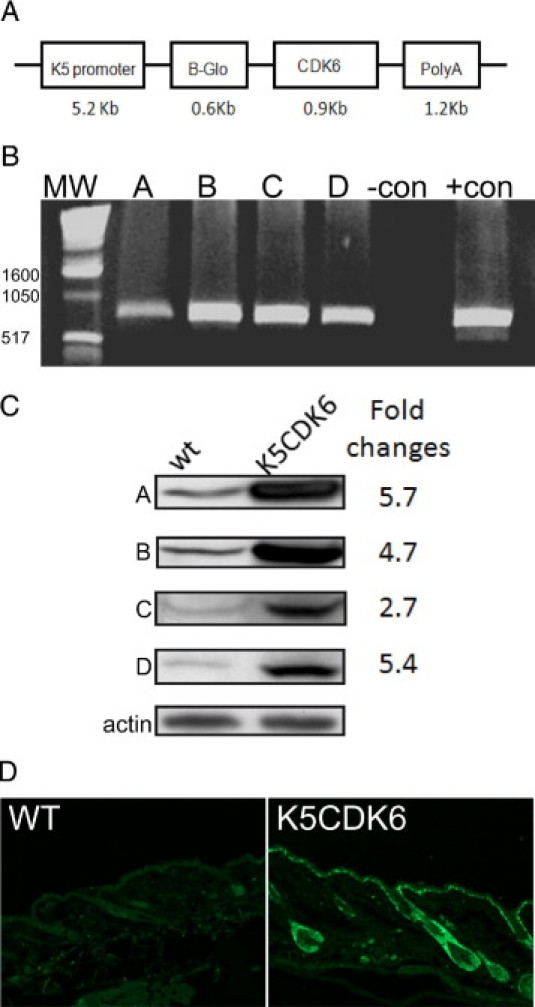
pK5-CDK6 construct and PCR screening. (A) Diagram of the K5CDK6 construct. (B) PCR amplification of DNA extracted from mouse tails. The CDK6 transgene was amplified, resulting in a 850-bp product. (C) Expression and quantification of CDK6 in each of the four mouse lines (A–D). (D) CDK6 expression was detected by immunofluorescence analysis in paraffin cross sections of wild-type (WT) and K5CDK6(H) mice. MW, molecular weight.
CDK6 Overexpression Induces Epidermal Hyperproliferation but Not Hyperplasia
The newborn K5CDK6 transgenic mice did not display any obvious developmental abnormalities, and there were no differences in size and weight compared with wild-type littermates. To determine whether the expression of the CDK6 transgene influenced the rate of keratinocyte proliferation and/or the architecture of mouse skin, we performed a histopathologic analysis of the epidermis of transgenic and wild-type siblings. Analysis of HE staining on paraffin-embedded sections showed no obvious modifications in the morphology of follicular and interfollicular epidermis between transgenic and wild-type littermates (Figure 2A). However, the proliferation status of keratinocytes, determined by BrdU incorporation, showed a twofold increase in the number of proliferating cells in the epidermis of transgenic mice compared with wild-type mice in both K5CDK6(H) (Figure 2) and K5CDK6(L) (data not shown) transgenic lines (P = 0.0005; t-test). Thus, we hypothesize that another mechanism compensates for the increase in keratinocyte proliferation observed in the K5CDK6 epidermis, resulting in no change in the epidermis structure. Quantification of the number of apoptotic cells shows a twofold increase in K5CDK6 interfollicular epidermis compared with wild-type siblings (P = 0.004; t-test) (Figure 2B). Importantly, the apoptotic cells were localized in the basal cell layer of interfollicular epidermis, suggesting that apoptosis can compensate for the increased number of proliferative keratinocytes in the K5CDK6 epidermis. It is worth mentioning that hair follicles do not contribute to the homeostasis of mouse epidermis; however, hair follicle stem cells localized in the bulge area seem to participate in epidermis homeostasis in hyperproliferative conditions, such as wound healing.45 The fact that K5CDK6 mice showed altered keratinocyte proliferation leads us to study whether apoptosis in the hair follicle can also contribute as a compensatory mechanism. Therefore, hair follicles carrying at least one apoptotic cell in the bulge were counted to determine the incidence of apoptosis in hair follicles. We observed a twofold increase in the number of hair follicles carrying apoptotic cells in K5CDK6 mice compared with wild-type siblings (P < 0.0001, t-test) (Figure 2B). Therefore, increased apoptosis in both interfollicular epidermis and hair follicles can play an important role in the K5CDK6 epidermis homeostasis.
Figure 2.
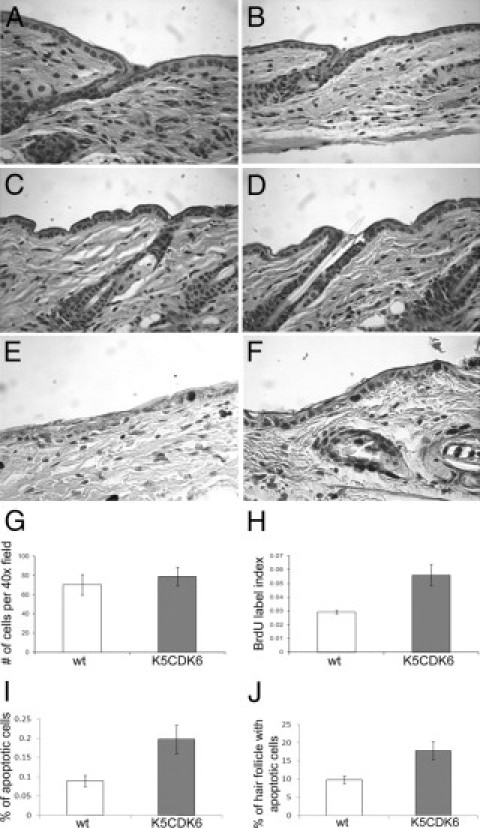
Skin phenotype of K5CDK6 transgenic mice. Representative paraffin sections of skin from high expression transgenic K5CDK6(H) (B) and low expression transgenic K5CDK6(L) (D) mice and the respective normal siblings (A, C) were stained with H&E. BrdU immunostaining of K5CDK6(H) (F) and wild-type (wt) siblings (E). Quantification of nucleated cells (G), BrdU label index (H), and apoptosis (I) in interfollicular epidermis and percentage of hair follicles with at least one apoptotic cell in the bulge area (J). Shaded bars, K5CDK6(H) transgenic mice; open bars, normal siblings (wild-type).
Moreover, we studied whether overexpression of CDK6 affects the pattern of epidermal differentiation by using keratin 5 and keratin 1 immunostaining. During epidermal differentiation there are sequential changes in the expression of the keratins. Keratin 5 and keratin 14 are the major products of basal epidermal cells, the proliferative compartment of the epidermis, whereas keratin 1 and 10 are associated with the commitment to differentiation and migration into the spinous layer.46,47 Figure 3 shows no differences in the pattern of expression of keratin 5 and 1 between K5CDK6 and wild-type siblings. The expression of keratin 5 was confined to the basal layer of interfollicular epidermis and hair follicles, whereas keratin 1 was restricted to terminally differentiated cells.
Figure 3.
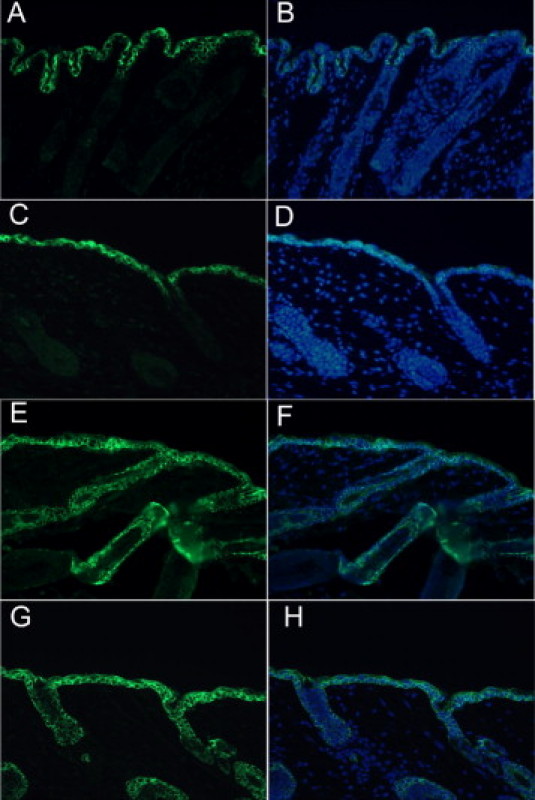
Keratin expression in K5CDK6 transgenic and normal sibling mice. Expression of keratin 1 (K1) and keratin 5 (K5) was determined on representative paraffin sections of skin from K5CDK6 transgenic (A and E) and wild-type siblings (C and G). Specific antibodies against keratin 5 (E and G) and keratin 1 (A and C). B–H: Merge of CDK6 expression (green) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) of the respective image on the left.
Therefore, these results suggest that forced expression of CDK6 in epidermis does not affect the morphology of epidermis or epidermal differentiation as a result of the counteraction between both increased proliferation and elevated apoptosis.
Biochemical Analysis of the K5CDK6 Mouse Epidermis
To study whether the expression of CDK6 affects associated cell-cycle regulators, we assessed the protein levels of CDKs, cyclin-dependent kinase inhibitors, and cyclins. CDK4 and CDK6 have common functional and biochemical properties; thus, we analyzed whether CDK4 expression is affected as a compensatory mechanism for the increased expression of CDK6. However, Western blot analysis showed no changes in protein expression for CDK4 and CDK2 (Figure 4A). As regulatory subunits of CDK4 and CDK6, D-type cyclins are rate-limiting controllers of G1 phase progression, but again no changes in protein levels of cyclin D1, cyclin D2, and cyclin D3 were observed (Figure 4A). The pRb family members are negative regulators that act in the G1 phase and are the main substrates of CDK4,6/D-type cyclin complexes; however, we did not detect changes in protein levels or mobility consistent with phosphorylation (Figure 4A).
Figure 4.
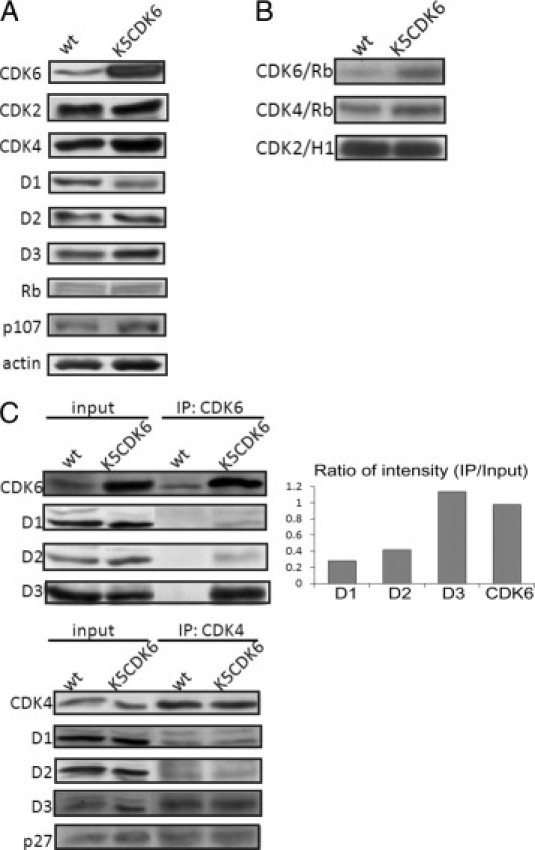
Biochemical analysis of cell-cycle regulators in epidermis of K5CDK6(H) transgenic and wild-type (wt) mice. A: Epidermal lysates were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. Primary antibodies against CDK6, CDK2, CDK4, cyclin D1, D2, D3, pRb, and p107 were used for immunoblot analysis. Actin was used as a loading control. B: Kinase activity of CDK6, CDK4 and CDK2 from K5CDK6(H) and wild-type mice. Fresh epidermal lysates were immunoprecipitated (IP) with specific antibodies against CDKs, and in vitro kinase assays were performed with pRb or histone H1 peptides as substrates. C: Epidermis lysates from K5CDK6(H) and wild-type siblings were immunoprecipitated with CDK6 or CDK4 antibodies and blotted with antibodies against cyclins D1, D2, D3, CDK6, CDK4, and p27Kip1. Protein lysates from the wild-type and the K5CDK6 epidermis were loaded as input. Bands of CDK6, cyclin D1, cyclin D2, and cyclin D3 were quantified in the input and immunoprecipitation, and the ratio for each individual protein was calculated as immunoprecipitation/input.
Modification of the protein levels of D-type cyclins or CDKs changes the kinetics of complex formations in mice epidermis. In fact, we have previously demonstrated that CDK4 binds preferentially to cyclin D1, whereas CDK6 binds to both cyclin D1 and D3 in transgenic mice overexpressing cyclin D3.44 In addition, epidermis from K5–cyclin D3 transgenic mice show elevated CDK6 and CDK4 kinase activities, mainly associated with CDK4,6/cyclin D1 and CDK6/cyclin D3 complexes.40 Therefore, we analyzed D-type cyclin/CDKs complex formations and in vitro associated kinase activities. Epidermal lysates from K5CDK6 and wild-type mice were immunoprecipitated with antibodies against CDK6 and CDK4 followed by Western blot analysis to determine associations with D-type cyclins. We found that forced expression of CDK6 resulted in elevated CDK6/cyclin D3 complex formation with minimum binding to cyclin D1 and D2 (Figure 4C). We quantified the expression levels of CDK6, cyclin D1, cyclin D2, and cyclin D3 in protein lysates (input) and in immunoprecipitated samples to determine the ratio of input to immunoprecipitation (Figure 4C). Therefore, we established that most of the cyclin D3 proteins bind to CDK6, whereas 2% to 4% of the cyclin D1 and cyclin D2 bind to CDK6 in the K5CDK6 epidermis. The fact that CDK6 is not expressed at high levels in wild-type epidermis does not allow verification of the ratio of D-type cyclin/CDK6 complex formation in wild-type mice.
We also established that the overexpression of CDK6 does not modify CDK4/D-type cyclin or CDK4/p27Kip1 complex formation, both of which are similar between K5CDK6 and wild-type littermates (Figure 4C). The preferential binding of CDK6 and cyclin D3 was confirmed by reverse co-immunoprecipitation assay in which cyclin D3 immunoprecipitation was followed by Western blot analysis to detect CDK6 (data not shown). To study whether the overexpression of CDK6 resulted in functional changes in the CDKs, we performed an in vitro analysis of CDK6, CDK4, and CDK2 kinase activities in epidermis lysates from transgenic and wild-type mice using pRb and histone H1 as substrates. As expected, CDK6 activities increased 2.5-fold in K5CDK6 transgenic mice compared with wild-type siblings, whereas no modification in the level of CDK4 and CDK2 activities was observed (Figure 4B). We conclude that forced expression of CDK6 does not lead to changes in other G1 phase CDKs or their regulatory subunits. Therefore, the effect of CDK6 expression in keratinocyte proliferation and apoptosis is not due to modification of protein levels or kinase activities of G1 phase other than CDK6.
CDK6 Overexpression Reduces Skin Tumor Development
According to the current model of cell proliferation, aberrant levels of a positive regulator of cell cycle provides a growth advantage that can result in increased tumor development. Supporting this model, we showed that forced expression of CDK4 results in increased malignant progression to skin SCCs in a two-stage carcinogenesis protocol.33 This protocol induces skin papillomas development by a single application of a carcinogen followed by biweekly treatment with a tumor promoter causing a selection of cells bearing Ha-ras mutations. To evaluate the role of CDK6 in skin tumorigenesis, we assessed the response of K5CDK6(H) and K5CDK6(L) transgenic mice to the two-stage carcinogenesis protocol.
The dorsal skin of K5CDK6 transgenic mice and wild-type littermates were topically treated with a subcarcinogenic dose of the genotoxic carcinogen DMBA and later promoted with TPA for 25 weeks. The incidence and multiplicity of papillomas were scored in each group for 35 weeks. Papilloma development was delayed 2 to 3 weeks in both K5CDK6 transgenic lines compared with the respective wild-type siblings (Figure 5A). The incidence of papilloma formation reached a plateau of 100% at approximately 17 to 19 weeks in wild-type mice. In contrast, K5CDK6(L) mice reached 90% papilloma incidence and only 60% of K5CDK6(H) mice developed papillomas, showing that high level of CDK6 expression correlates with increased inhibition of tumor development (Figure 5A). Tumor multiplicity (mean number of tumors per mice) clearly shows a reduced number of tumors in both K5CDK6 transgenic lines compared with their respective control littermates throughout the experiment. On an interesting note, we established an inverse correlation between CDK6 expression and the number of skin tumors. Although 25% of the high expression transgenic line (K5CDK6[H]) developed skin tumors (a 75% reduction compared with wild-type siblings), the low expression transgenic line (K5CDK6[L]) showed a 50% decrease in the number of papillomas per mouse (Figure 5A).
Figure 5.
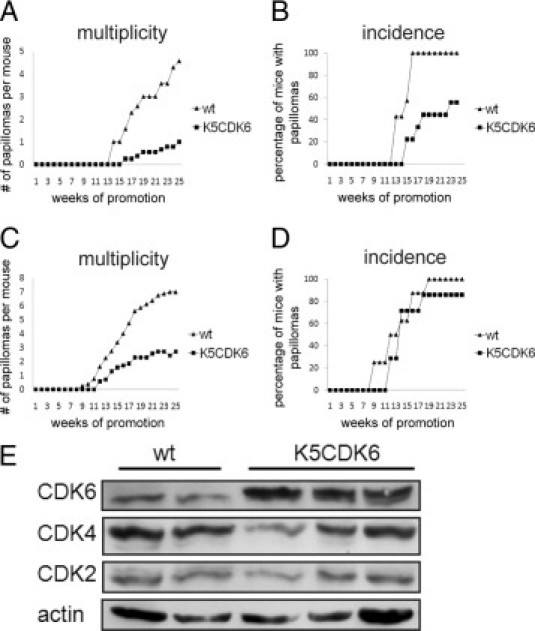
Kinetics of papilloma formation and biochemical analysis of K5-CDK6 tumors. K5CDK6 transgenic and wild-type (wt) sibling mice were initiated with DMBA and promoted with multiple applications of TPA on dorsal mouse skin. Average number of papillomas per mouse (multiplicity) as a function of weeks of study in K5CDK6(H) (A) and K5CDK6(L) (C). Percentage of mice with at least one papilloma as a function of weeks of study (incidence) in K5CDK6(H) (B) and K5CDK6(L) (D). E: Immunoblot analysis of wild-type and K5CDK6(H) papilloma lysates developed with antibodies against CDK6, CDK4, CDK2, and actin as a loading control.
Biochemical analysis of skin tumors indicates that overexpression of CDK6 is maintained during tumorigenesis (Figure 5B). Although certain heterogeneity was observed in the CDKs protein levels, no effect on CDK4 and CDK2 expression was observed in transgenic versus wild-type tumors. It has been shown that CDK6 plays a role halting cellular proliferation through accumulation of growth suppressors p53 and p130.17 Nevertheless, immunoblot analysis of p53 and p130 in epidermis and skin papillomas did not show differences between K5-CDK6 and wild-type siblings (data not shown). These data are consistent with the observations of Ruggeri et al48 that show that the p53 gene appeared normal in all papillomas and early well-differentiated carcinomas, whereas p53 alterations were observed in SCCs.
Histopathologic analyses were performed on skin tumors that had undergone 25 weeks of promotion. No differences were observed between K5CDK6 and wild-type tumors, and all of them were classified as well-differentiated papillomas with no atypia in the basal layers. However, immunostaining analysis showed a twofold increase in keratinocyte proliferation in papillomas from K5CDK6 mice compared with wild-type littermates (P < 0.0001, t-test) (Figure 6). Moreover, we also observed a 1.5-fold increase in the number of apoptotic cells in K5CDK6 papillomas compared with wild-type tumors (P = 0.001, t-test) (Figure 6). Therefore, forced expression of CDK6 increases keratinocyte proliferation in skin tumors but similar to normal epidermis CDK6 expression also induces apoptosis.
Figure 6.
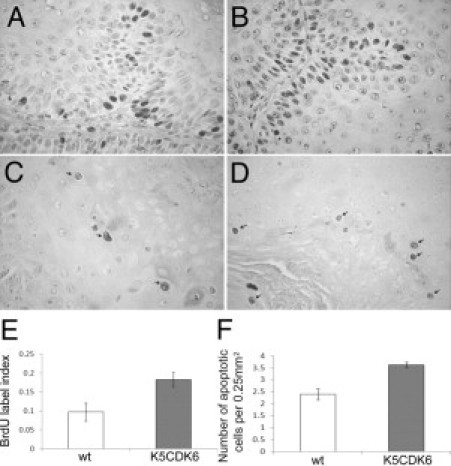
Increased apoptosis and keratinocyte proliferation in mouse skin tumors. BrdU incorporation in papillomas from wild-type (wt) (A) and K5CDK6 (B) siblings. Apoptotic keratinocytes in skin papillomas from wild-type (C) and K5CDK6 (D) mice. Quantification of BrdU label index (E) and apoptosis (F) in skin papillomas.
Collectively, these observations show that, although counterintuitive, overexpression of CDK6 does not result in advantages leading to increased carcinogenesis or increased malignant progression but rather decreases in papilloma development in ras-dependent tumorigenesis.
Discussion
For more than two decades, the pRb/p16/Cdk/cyclin pathway has been implicated in proliferation and tumorigenesis. The fact that both CDK4 and CDK6 have the same substrates, bind to D-type cyclins, and share 71% of amino acid identity leads to the assumption that they play a redundant role in the G1 phase of the cell cycle. However, in vivo studies only partly support a redundant function for these kinases. For instance, CDK4−/− mice show growth retardation, reproductive dysfunction associated with defects in seminiferous tubules and corpus luteum, and insulin-deficient diabetes due to a reduction in β-islet pancreatic cells.49,50 None of these phenotypes were observed in CDK6-deficient mice, which show pronounced thymic atrophy because of the reduction in cell proliferation and the reduced susceptibility to lymphomagenesis.9 These results demonstrated that a lack of CDK6 or CDK4 affected a different spectrum of tissues and argue against a redundant and compensatory function in those organs. On the other hand, Malumbres et al8 showed that mice lacking both CDK4 and CDK6 died during embryonic development, supporting the hypothesis of functional compensation between these proteins. Studies performed with experimental and human tumors also suggested similarities and differences between these kinases. For instance, both CDK4 and CDK6 are overexpressed in human gliomas.20–23 In contrast, CDK4, but not CDK6, is specifically targeted in melanomas,24,25 whereas CDK6, but not CDK4, activity has been found elevated in SCCs and neuroblastomas.26–28
Role of CDK6 in Keratinocyte Proliferation, Differentiation, and Apoptosis
Several groundbreaking works in cell culture and in in vivo models have shown that activation of CDK4 and CDK6 is essential for responses to extracellular mitogenic signaling and the progression beyond the restriction point in the G1 phase. Our earliest studies have shown that G1-CDKs are differently regulated in mouse epidermis. Whereas CDK4 and CDK2 remain at constant levels, CDK6 is up-regulated in mouse epidermis on TPA-induced proliferation, leading to an increase in CDK6/cyclin 3 and CDK4/cyclin D1 complexes.44 In this report, we show that similar to K5CDK4 transgenic mice, forced expression of CDK6 in mouse epidermis results in increased keratinocyte proliferation, but contrary to K5CDK4, no epidermal hyperplasia was observed. Interestingly, CDK6 expression resulted in an elevated number of apoptotic keratinocytes in interfollicular and follicular epidermis, suggesting that apoptosis behaves as a compensatory mechanism for unrestricted proliferation. This compensatory mechanism was only elicited in K5CDK6 keratinocytes because neither K5CDK4 nor K5CDK2 epidermis showed an increased number of apoptotic cells.36,37 CDK6 overexpression does not result in increased CDK2 activities as was determined in epidermis and papillomas from K5CDK4 mice.33,37 Therefore, the increased apoptosis observed in the K5CDK6 mouse epidermis seems to be independent of the role of CDK2-mediated apoptosis.51–54
Here, we have determined that increased CDK6 activities were associated with elevated CDK6/cyclin D3 complex formations. Thus, one might speculate that this particular complex plays an important role in epidermis homeostasis by keeping inappropriate proliferation in check. Therefore, we hypothesize that the CDK6/cyclin D3 complex inactivates pRb and/or p107, inducing apoptosis by a similar mechanism exhibited by the lack of pRb.55,56 Although we did not observe changes in pRb or p107 phosphorylation status in epidermal extract from K5CDK6 mice, in vitro kinase assays clearly showed increased CDK6 activity against a pRb peptide. Taken together, these results suggest that the forced expression of CDK6 induces apoptosis as a compensatory mechanism for keratinocyte hyperproliferation. The molecular mechanism by which CDK6, but not CDK4, stimulates apoptosis in mouse epidermis remains unclear and is beyond the scope of this study but suggests that CDK6 may play a role in halting cellular growth when proliferation is inappropriate.
In the last few years, several alternative roles for CDK6 blocking cell differentiation and/or inducing cell proliferation have been described.56–59 For instance, CDK6 disrupts the C/EBP-Runx1 interaction and Runx1 DNA binding, leading to blocked myeloid differentiation.60 Moreover, bone morphogenetic protein 2–induced osteoblast differentiation requires down-regulation of CDK6.15 The ability of CDK6 to interfere with Runx-C/EBP cooperation and control terminal differentiation might apply to several cell types. Thus, we hypothesized that overexpression of CDK6 can alter the pattern of keratinocyte differentiation. However, immunofluorescence analysis of keratin 5/keratin 1 distribution in basal and suprabasal cell layers of K5CDK6 mice showed no modifications in the pattern of expression, suggesting no alterations in keratinocyte differentiation. Consistent with our data, changes in keratinocytes differentiation or proliferation by the ablation of CDK6 in mouse models have not been reported.8,9 Interestingly, Hoi et al61 have recently reported that Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Therefore, whether CDK6 disrupts the Runx1 role on hair follicle stem cells, leading to changes in keratinocyte proliferation and a reduction in formation, warrants further investigation.
CDK6 in Tumor Development
Our earlier studies established that CDK4 and CDK6 remain at constant levels in mouse papillomas and increased expression of D-type cyclin drive cyclin/CDK complex formation and CDK activity in skin tumorigenesis.30 In vivo studies demonstrated that transgenic expression of CDK4 results in increased epidermal proliferation, epidermal hyperplasia, and enhanced malignant progression to SCCs in a two-stage chemical carcinogenesis model.33,37 We also showed that the role of CDK4 in skin carcinogenesis partly depends on CDK2 activation through sequestration of p27Kip1 and p21Cip1 by CDK4.35,37 An important role of CDK4 in mouse epidermal tumorigenesis was further supported by the fact that ablation of CDK4 leads to reduction of skin tumorigenesis.32 These results and the convincing evidence showing that disabling the pRb pathway is essential for tumor formation led us to hypothesize that elevated CDK6 activity would also enhance ras-mediated tumor development. Surprisingly, we found that elevated CDK6 kinase activity did not increase tumor development or affect malignant progression to SCC. In fact, K5CDK6 transgenic mice developed a lower number of tumors per mouse, and an increased fraction of the K5CDK6 mice remained refractory to tumor development. Importantly, the inhibitory action of CDK6 was dose dependent because the high expression K5CDK6(H) transgenic line showed enhanced resistance to papilloma development compared with the K5CDK6(L) transgenic line. We also analyzed the rate of malignant conversion to SCC, but changes were not observed in K5CDK6 mice compared with wild-type siblings. These results are clearly opposite of the increased malignant progression observed in K5CDK4 mice under the same protocol.33 Similar to the K5CDK6 model, ablation of pRb in mouse epidermis results in a reduced number of papillomas and increased apoptosis.55 The similar effect in skin carcinogenesis noted in K5CDK6 and pRb−/− mice contrasted with that of K5CDK4 mice. Thus, we hypothesize that CDK6- or CDK4-mediated phosphorylation and inactivation of pRb family members are responsible for the different effects in papilloma development. Therefore, the following hypotheses merit future investigations to understand the different effects of CDK4 and CDK6 expression in mouse epidermis. First, differences in residue selectivity for pRb phosphorylation by CDK6 and CDK4 might induce a different rate of pRb inactivation, leading to increased apoptosis and, consequently, reduction in tumorigenesis in K5CDK6 mice.11 Second, CDK6 might preferentially phosphorylate pRb but not p107, which in turn will inhibit tumor development. Remarkably, reduced expression of p107 on pRb−/− p107+/− compound mice led to partial restoration in the incidence, number, and size of tumors.62 Another alternative hypothesis to be tested is pRb-independent mechanisms. It is known that chromosome replication is a highly regulated mechanism that seems to be mainly regulated by the assembly of Mcm2-7 complexes onto replication origins. This mechanism depends on the CDK levels to allow licensing DNA duplication; however, the roles of each member of the CDK family have not been clearly established.63,64 Importantly, Braden et al65 have recently shown that CDK4 and CDK6 activities are critical determinants of prereplication complex assembly by allowing the accumulation of the licensing factors cdc6 and cdt1. Therefore, determining whether G1-CDKs play a unique or shared role during DNA duplication will allow us to establish whether CDK6 up-regulation affects DNA duplication and further triggers the apoptotic pathway.
It is worth mentioning that overexpression of cyclin D3 also results in a reduced number of skin papillomas. Interestingly, cyclin D3 preferentially binds to CDK6 in K5–cyclin D3 mouse epidermis.40 Thus, it is tempting to hypothesize that CDK6/cyclin D3 complexes play a unique role in blocking tumor development in mouse epidermis. Supporting a specific role for cyclin D3 complexes in inhibition of cell proliferation, Wang et al66 have shown that cyclin D3/CDK4,6 maintains the growth inhibitory activity of C/EBPα. Whether different CDKs/D-type cyclin complexes play unique roles in proliferation and apoptosis and whether these effects are tissue specific remain to be determined. However, our results strongly suggest that modulation of the levels of D-type cyclins and/or G1-CDKs are useful in planning cancer therapies.
Acknowledgments
We thank Dr. Paula Miliani de Marval for her critical review of the paper. We thank the Laboratory Animal Resources and the Histology Core at the College of Veterinary Medicine, North Carolina State University, for helping with skin and tumor samples.
Footnotes
Supported by grant RO1 CA116328 from the National Cancer Institute, National Institutes of Health.
References
- 1.Sherr C.J. D-type cyclins. Trends Biochem Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 2.Weinberg R.A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 3.Blain S.W., Montalvo E., Massague J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem. 1997;272:25863–25872. doi: 10.1074/jbc.272.41.25863. [DOI] [PubMed] [Google Scholar]
- 4.Sherr C.J., McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 5.Sherr C.J., Roberts J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;10:1491–1502. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 6.Meyerson M., Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14:2077–2086. doi: 10.1128/mcb.14.3.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lucas J.J., Szepesi A., Modiano J.F., Domenico J., Gelfand E.W. Regulation of synthesis and activity of the PLSTIRE protein (cyclin-dependent kinase 6 (cdk6)), a major cyclin D-associated cdk4 homologue in normal human T lymphocytes. J Immunol. 1995;154:6275–6284. [PubMed] [Google Scholar]
- 8.Malumbres M., Sotillo R., Santamaria D., Galan J., Cerezo A., Ortega S., Dubus P., Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Hu M.G., Deshpande A., Enos M., Mao D., Hinds E.A., Hu G.F., Chang R., Guo Z., Dose M., Mao C., Tsichlis P.N., Gounari F., Hinds P.W. A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009;69:810–818. doi: 10.1158/0008-5472.CAN-08-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bockstaele L., Bisteau X., Paternot S., Roger P.P. Differential regulation of cyclin-dependent kinase 4 (CDK4) and CDK6, evidence that CDK4 might not be activated by CDK7, and design of a CDK6 activating mutation. Mol Cell Biol. 2009;29:4188–4200. doi: 10.1128/MCB.01823-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takaki T., Fukasawa K., Suzuki-Takahashi I., Semba K., Kitagawa M., Taya Y., Hirai H. Preferences for phosphorylation sites in the retinoblastoma protein of D-type cyclin-dependent kinases: Cdk4 and Cdk6, in vitro. J Biochem. 2005;137:381–386. doi: 10.1093/jb/mvi050. [DOI] [PubMed] [Google Scholar]
- 12.Grossel M.J., Baker G.L., Hinds P.W. cdk6 can shorten G(1) phase dependent upon the N-terminal INK4 interaction domain. J Biol Chem. 1999;274:29960–29967. doi: 10.1074/jbc.274.42.29960. [DOI] [PubMed] [Google Scholar]
- 13.Ericson K.K., Krull D., Slomiany P., Grossel M.J. Expression of cyclin-dependent kinase 6, but not cyclin-dependent kinase 4, alters morphology of cultured mouse astrocytes. Mol Cancer Res. 2003;1:654–664. [PubMed] [Google Scholar]
- 14.Matushansky I., Radparvar F., Skoultchi A.I. CDK6 blocks differentiation: coupling cell proliferation to the block to differentiation in leukemic cells. Oncogene. 2003;22:4143–4149. doi: 10.1038/sj.onc.1206484. [DOI] [PubMed] [Google Scholar]
- 15.Ogasawara T., Kawaguchi H., Jinno S., Hoshi K., Itaka K., Takato T., Nakamura K., Okayama H. Bone morphogenetic protein 2-induced osteoblast differentiation requires Smad-mediated down-regulation of Cdk6. Mol Cell Biol. 2004;24:6560–6568. doi: 10.1128/MCB.24.15.6560-6568.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogasawara T., Katagiri M., Yamamoto A., Hoshi K., Takato T., Nakamura K., Tanaka S., Okayama H., Kawaguchi H. Osteoclast differentiation by RANKL requires NF-kappaB-mediated downregulation of cyclin-dependent kinase 6 (Cdk6) J Bone Miner Res. 2004;19:1128–1136. doi: 10.1359/jbmr.2004.19.7.1128. [DOI] [PubMed] [Google Scholar]
- 17.Nagasawa M., Gelfand E.W., Lucas J.J. Accumulation of high levels of the p53 and p130 growth-suppressing proteins in cell lines stably over-expressing cyclin-dependent kinase 6 (cdk6) Oncogene. 2001;20:2889–2899. doi: 10.1038/sj.onc.1204396. [DOI] [PubMed] [Google Scholar]
- 18.Lucas J.J., Terada N., Szepesi A., Gelfand E.W. Regulation of synthesis of p34cdc2 and its homologues and their relationship to p110Rb phosphorylation during cell cycle progression of normal human T cells. J Immunol. 1992;148:1804–1811. [PubMed] [Google Scholar]
- 19.Lucas J.J., Szepesi A., Domenico J., Tordai A., Terada N., Gelfand E.W. Differential regulation of the synthesis and activity of the major cyclin-dependent kinases, p34cdc2, p33cdk2, and p34cdk4, during cell cycle entry and progression in normal human T lymphocytes. J Cell Physiol. 1995;165:406–416. doi: 10.1002/jcp.1041650222. [DOI] [PubMed] [Google Scholar]
- 20.Costello J.F., Plass C., Arap W., Chapman V.M., Held W.A., Berger M.S., Su Huang H.J., Cavenee W.K. Cyclin-dependent kinase 6 (CDK6) amplification in human gliomas identified using two-dimensional separation of genomic DNA. Cancer Res. 1997;57:1250–1254. [PubMed] [Google Scholar]
- 21.Ichimura K., Schmidt E.E., Goike H.M., Collins V.P. Human glioblastomas with no alterations of the CDKN2A (p16INK4A. MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13:1065–1072. [PubMed] [Google Scholar]
- 22.Lam P.Y., Di Tomaso E., Ng H.K., Pang J.C., Roussel M.F., Hjelm N.M. Expression of p19INK4d: CDK4, CDK6 in glioblastoma multiforme. Br J Neurosurg. 2000;14:28–32. doi: 10.1080/02688690042870. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt E.E., Ichimura K., Reifenberger G., Collins V.P. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54:6321–6324. [PubMed] [Google Scholar]
- 24.Zuo L., Weger J., Yang Q., Goldstein A.M., Tucker M.A., Walker G.J., Hayward N., Dracopoli N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12:97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 25.Wölfel T., Hauer M., Schneider J., Serrano M., Wölfel C., Klehmann-Hieb E., De Plaen E., Hankeln T., Meyer zum Büschenfelde K.H., Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 26.Timmermann S., Hinds P.W., Munger K. Elevated activity of cyclin-dependent kinase 6 in human squamous cell carcinoma lines. Cell Growth Differ. 1997;8:361–370. [PubMed] [Google Scholar]
- 27.Piboonniyom S.O., Timmermann S., Hinds P., Munger K. Aberrations in the MTS1 tumor suppressor locus in oral squamous cell carcinoma lines preferentially affect the INK4A gene and result in increased cdk6 activity. Oral Oncol. 2002;38:179–186. doi: 10.1016/s1368-8375(01)00042-2. [DOI] [PubMed] [Google Scholar]
- 28.Easton J., Wei T., Lahti J.M., Kidd V.J. Disruption of the cyclin D/cyclin-dependent kinase/INK4/retinoblastoma protein regulatory pathway in human neuroblastoma. Cancer Res. 1998;58:2624–2632. [PubMed] [Google Scholar]
- 29.Zhang S.-Y., Liu S.-C., Goodrow T., Morris R., Klein-Szanto A.J.P. Increased expression of G1 cyclins and cyclin-dependent kinases during tumor progression of chemically induced mouse skin neoplasms. Mol Carcinog. 1997;18:142–152. [PubMed] [Google Scholar]
- 30.Rodriguez-Puebla M.L., LaCava M., Gimenez-Conti I.B., Johnson D.G., Conti C.J. Deregulated expression of cell-cycle proteins during premalignant progression in SENCAR mouse skin. Oncogene. 1998;17:2251–2258. doi: 10.1038/sj.onc.1202131. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Puebla M.L., LaCava M., Conti C.J. Cyclin D1 overexpression in mouse epidermis increases cyclin-dependent kinase activity and cell proliferation in vivo but does not affect skin tumor development. Cell Growth Differ. 1999;10:467–472. [PubMed] [Google Scholar]
- 32.Rodriguez-Puebla M.L., Miliani de Marval P.L., LaCava M., Moons D.S., Kiyokawa H., Conti C.J. Cdk4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am J Pathol. 2002;161:405–411. doi: 10.1016/S0002-9440(10)64196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miliani de Marval P.L., Macias E., Conti C.J., Rodriguez-Puebla M.L.R. Enhanced malignant tumorigenesis in Cdk4 transgenic miceR. Oncogene. 2004;23R:1863–1873. doi: 10.1038/sj.onc.1207309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rojas P., Benavides F., Blando J., Perez C., Cardenas K., Richie E., Knudsen E.S., Johnson D.G., Senderowicz A.M., Rodriguez-Puebla M.L., Conti C.J. Enhanced skin carcinogenesis and lack of thymus hyperplasia in transgenic mice expressing human cyclin D1b (CCND1b) Mol Carcinog. 2009;48:508–516. doi: 10.1002/mc.20489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macias E., Kim Y., Miliani de Marval P.L., Klein-Szanto A., Rodriguez-Puebla M.L. Cdk2 deficiency decreases ras/CDK4-dependent malignant progression, but not myc-induced tumorigenesis. Cancer Res. 2007;67:9713–9720. doi: 10.1158/0008-5472.CAN-07-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macias E., Miliani de Marval P.L., De Siervi A., Conti C.J., Senderowicz A.M., Rodriguez-Puebla M.L. CDK2 activation in mouse epidermis induces keratinocyte proliferation but does not affect skin tumor development. Am J Pathol. 2008;173:526–535. doi: 10.2353/ajpath.2008.071124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miliani de Marval P.L., Gimenez-Conti I.B., LaCava M., Martinez L.A., Conti C.J., Rodriguez-Puebla M.L. Transgenic expression of cyclin-dependent kinase 4 results in epidermal hyperplasia, hypertrophy, and severe dermal fibrosis. Am J Pathol. 2001;159:369–379. doi: 10.1016/S0002-9440(10)61703-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miliani de Marval P.L., Macias E., Rounbehler R., Sicinski P., Kiyokawa H., Johnson D.G., Conti C.J., Rodriguez-Puebla M.L. Lack of cyclin-dependent kinase 4 inhibits c-myc tumorigenic activities in epithelial tissues. Mol Cell Biol. 2004;24:7538–7547. doi: 10.1128/MCB.24.17.7538-7547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez-Puebla M.L., LaCava M., Miliani De Marval P.L., Jorcano J.L., Richie E.R., Conti C.J. Cyclin D2 overexpression in transgenic mice induces thymic and epidermal hyperplasia whereas cyclin D3 expression results only in epidermal hyperplasia. Am J Pathol. 2000;157:1039–1050. doi: 10.1016/S0002-9440(10)64616-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rojas P., Cadenas M.B., Lin P.C., Benavides F., Conti C.J., Rodriguez-Puebla M.L. Cyclin D2 and cyclin D3 play opposite roles in mouse skin carcinogenesis. Oncogene. 2007;26:1723–1730. doi: 10.1038/sj.onc.1209970. [DOI] [PubMed] [Google Scholar]
- 41.Yu Q., Sicinska E., Geng Y., Ahnstrom M., Zagozdzon A., Kong Y., Gardner H., Kiyokawa H., Harris L.N., Stal O., Sicinski P. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. doi: 10.1016/j.ccr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 42.Zou X., Ray D., Aziyu A., Christov K., Boiko A.D., Gudkov A.V., Kiyokawa H. Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to Arf/p53-independent senescence. Genes Dev. 2002;16:2923–2934. doi: 10.1101/gad.1033002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramirez A., Bravo A., Jorcano J., Vidal M. Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacZ gene in the adult and during development. Differentiation. 1994;58:53–64. doi: 10.1046/j.1432-0436.1994.5810053.x. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Puebla M.L., Robles A.I., Johnson D.G., LaCava M., Conti C.J. Synchronized proliferation induced by 12-O-tetradecanoylphorbol-13-acetate treatment of mouse skin: an in vivo model for cell cycle regulation. Cell Growth Differ. 1998;9:31–39. [PubMed] [Google Scholar]
- 45.Ito M., Liu Y., Yang Z., Nguyen J., Liang F., Morris R.J., Cotsarelis G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med. 2005;11:1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 46.Woodcock-Mitchell J., Eichner R., Nelson W.G., Sun T. Immunolocalization of keratin polypeptides in human epidermis using monoclonal antibodies. J Cell Biol. 1982;95:580–588. doi: 10.1083/jcb.95.2.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roop D., Hawley-Nelson P., Cheng C.K., Yuspa S.H. Keratin gene expression in mouse epidermis and cultured epidermal cells. Proc Natl Acad Sci U S A. 1983;80:716–720. doi: 10.1073/pnas.80.3.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruggeri B., Caamano J., Goodrow T., DiRado M., Bianchi A., Trono D., Conti C.J., Klein-Szanto A.J. Alterations of the p53 tumor suppressor gene during mouse skin tumor progression. Cancer Res. 1991;51:6615–6621. [PubMed] [Google Scholar]
- 49.Rane S.G., Dubus P., Mettus R.V., Galbreath E.J., Boden G., Reddy E.P., Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 50.Tsutsui T., Hesabi B., Moons D.S., Pandolfi P.P., Hansel K.S., Koff A., Kiyokawa H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–7019. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hakem A., Sasaki T., Kozieradzki I., Penninger J.M. The cyclin-dependent kinase Cdk2 regulates thymocyte apoptosis. J Exp Med. 1999;189:957–968. doi: 10.1084/jem.189.6.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi K.S., Eom Y.W., Kang Y., Ha M.J., Rhee H., Yoon J.W., Kim S.J. Cdc2 and Cdk2 kinase activated by transforming growth factor-beta1 trigger apoptosis through the phosphorylation of retinoblastoma protein in FaO hepatoma cells. J Biol Chem. 1999;274:31775–31783. doi: 10.1074/jbc.274.45.31775. [DOI] [PubMed] [Google Scholar]
- 53.Maddika S., Ande S.R., Wiechec E., Hansen L.L., Wesselborg S., Los M. Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. J Cell Sci. 2008;121:979–988. doi: 10.1242/jcs.009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gil-Gomez G., Berns A., Brady H.J. A link between cell cycle and cell death: bax and Bcl-2 modulate Cdk2 activation during thymocyte apoptosis. EMBO J. 1998;17:7209–7218. doi: 10.1093/emboj/17.24.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruiz S., Santos M., Lara M.F., Segrelles C., Ballestin C., Paramio J.M. Unexpected roles for pRb in mouse skin carcinogenesis. Cancer Res. 2005;65:9678–9686. doi: 10.1158/0008-5472.CAN-05-1853. [DOI] [PubMed] [Google Scholar]
- 56.Ruiz S., Santos M., Paramio J.M. Is the loss of pRb essential for the mouse skin carcinogenesis. Cell Cycle. 2006;5:625–629. doi: 10.4161/cc.5.6.2580. [DOI] [PubMed] [Google Scholar]
- 57.Nerlov C. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007;17:318–324. doi: 10.1016/j.tcb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 58.Grossel M.J., Hinds P.W. From cell cycle to differentiation: an expanding role for cdk6. Cell Cycle. 2006;5:266–270. doi: 10.4161/cc.5.3.2385. [DOI] [PubMed] [Google Scholar]
- 59.Grossel M.J., Hinds P.W. Beyond the cell cycle: a new role for Cdk6 in differentiation. J Cell Biochem. 2006;97:485–493. doi: 10.1002/jcb.20712. [DOI] [PubMed] [Google Scholar]
- 60.Fujimoto T., Anderson K., Jacobsen S.E., Nishikawa S.I., Nerlov C. Cdk6 blocks myeloid differentiation by interfering with Runx1 DNA binding and Runx1-C/EBPalpha interaction. EMBO J. 2007;26:2361–2370. doi: 10.1038/sj.emboj.7601675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoi C.S., Lee S.E., Lu S.Y., McDermitt D.J., Osorio K.M., Piskun C.M., Peters R.M., Paus R., Tumbar T. Runx1 directly promotes proliferation of hair follicle stem cells and epithelial tumor formation in mouse skin. Mol Cell Biol. 2010;30:2518–2536. doi: 10.1128/MCB.01308-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santos M., Ruiz S., Lara M.F., Segrelles C., Moral M., Martinez-Cruz A.B., Ballestin C., Lorz C., Garcia-Escudero R., Paramio J.M. Susceptibility of pRb-deficient epidermis to chemical skin carcinogenesis is dependent on the p107 allele dosage. Mol Carcinog. 2008;47:815–821. doi: 10.1002/mc.20426. [DOI] [PubMed] [Google Scholar]
- 63.Blow J.J., Hodgson B. Replication licensing–defining the proliferative state. Trends Cell Biol. 2002;12:72–78. doi: 10.1016/s0962-8924(01)02203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Porter A.C. Preventing DNA over-replication: a Cdk perspective. Cell Div. 2008;3:3. doi: 10.1186/1747-1028-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braden W.A., McClendon A.K., Knudsen E.S. Cyclin-dependent kinase 4/6 activity is a critical determinant of pre-replication complex assembly. Oncogene. 2008;27:7083–7093. doi: 10.1038/onc.2008.319. [DOI] [PubMed] [Google Scholar]
- 66.Wang G.L., Shi X., Salisbury E., Sun Y., Albrecht J.H., Smith R.G., Timchenko N.A. Cyclin D3 maintains growth-inhibitory activity of C/EBPalpha by stabilizing C/EBPalpha-cdk2 and C/EBPalpha-Brm complexes. Mol Cell Biol. 2006;26:2570–2582. doi: 10.1128/MCB.26.7.2570-2582.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]