

Abstract
The release of nucleotides after injury activates purinergic receptors, leading to phosphorylation of site-specific residues on epidermal growth factor receptor (EGFR). To elucidate the differences between the injury-induced response and that induced by exogenous EGF, we examined recruitment of docking proteins, internalization of EGFR, and migration after injury. Injury induced by scratch wounds or stimulation by addition of UTP caused a brief internalization of EGFR, which paralleled the lesser association with growth factor receptor-bound protein 2 (Grb2) and phosphorylation of EGFR. The internalization caused by EGF was sustained and detected for longer than 60 minutes and correlated with phosphorylation of the receptor. The EGF caused recruitment of Grb2, phospholipase C-γ-1 (PLCγ1), Shc, and Src to EGFR. Glutathione S-transferase pull downs were performed, and glutathione S-transferase–PLCγ1 showed binding of Grb2 when stimulated with EGF but not with UTP or injury. Furthermore, UTP did not induce PLCγ1 phosphorylation, and the phosphorylation induced by EGF was attenuated by costimulation with UTP. The response to heparin-binding EGF was equivalent to that of EGF. Site-directed mutagenesis showed that phosphorylation of Y1068 and Y1086 of EGFR is required for repair. Together, our results show that injury and activation of purinergic receptors and direct activation of EGFR via EGF induce distinct downstream pathways.
Cell migration and wound repair are tightly regulated events that are critical for successful wound closure. During development, cells move as a unit to specific regions,1 and sheets of corneal epithelial cells move over the basal lamina to repair superficial wounds.2 When deeper wounds occur, cells need to move through an extensive matrix; in these cases, individual cell movement is detected with prominent actin extensions. Migration during wound repair ensures reformation of the cell–cell barrier and prevents entry of bacterial pathogens.
Epidermal growth factor (EGF) and its related family members stimulate corneal epithelial cell migration,3–5 proliferation, and synthesis of basement membrane and matrix proteins.6–9 Although EGF is not detected in the wound medium of corneal epithelial cells, EGF family members are synthesized in lacrimal gland cells and are released on activation of metalloproteinases into tear fluid.10 Furthermore, EGF has been added at high concentrations after surgery to enhance wound strength.11
Epidermal growth factor receptor (EGFR) is a cell receptor tyrosine kinase,12–15 is localized in the corneal epithelium, and is activated on injury.3,16 Epidermal growth factor receptor knockdown by antisense RNA or small-interfering RNA (siRNA) results in decreases in cell migration, phosphorylation of extracellular signal-related kinase (ERK), and phosphorylation of integrin subunit β4.17,18 In addition, inhibition of EGFR restricts both scratch and haptotactic cell migration.3,5,19 However, although EGF induces Ca2+ oscillations, pretreatment with the EGFR kinase inhibitor, AG1478, does not inhibit an injury-induced Ca2+ wave.4
Taken together, these observations indicate that there are several signaling pathways induced by injury that involve the EGFR while other responses are independent. For instance, injury- and EGF-induced ERK activation appear to be quantitatively and qualitatively different. The duration of injury-induced activation of ERK is transient (2 to 10 minutes), whereas EGF-stimulated ERK activation is long lasting (2 to 24 hours).20 Injury-induced ERK activation is linked to the release of nucleotides and the activation of purinergic receptors (P2Y and P2X).21–23 Treatment of the wound media with apyrase (an ectonucleotidase) inhibits the propagation of a Ca2+ wave and cell migration and reduces the phosphorylation of ERK. In addition, cells transfected with siRNA to the P2Y2 receptor show significant decreases in propagation of the Ca2+ wave and cell migration, indicating that the receptor and nucleotides play a critical role in wound repair.18,19,24
Cross talk between P2Y receptors and EGFR is thought to play a pivotal role in cell migration and wound closure.18 Stimulation of corneal epithelial cells via injury and release of nucleotides or with addition of exogenous nucleotides results in phosphorylation of select sites, including tyrosine residues 1068, 1086, and 1173 (but not 845). Stimulation with EGF promotes phosphorylation of multiple tyrosines, including 845, 1068, 1086, 1173, and 1148,18 indicating that nucleotides and EGF can stimulate distinct signaling mechanisms. Distinct EGFR phosphorylation responses occur with other stimuli, as indicated in the following examples. The α v integrin subunit has induced phosphorylation of 845, 1068, 1086, and 1173, whereas ionizing radiation induces phosphorylation of 992 and 1173. Betacellulin induces phosphorylation of 1068 and 1173, although the latter is markedly reduced.25–27 Heparin-binding (HB) EGF has been detected during development and after injury of several cell types.18,28–30 Phosphorylated tyrosine sites on EGFR serve as unique docking sites for a variety of EGFR-associated signaling proteins that contain the Src homology domain. We hypothesized that the rapid response elicited by injury and the later response from EGF result in the recruitment of specific or unique docking molecules and subsequent activation of specific downstream activities, such as cell migration.
In the present study, we demonstrate that the activation of purinergic receptors by injury or the addition of nucleotides caused a minimal level of phosphorylation of EGFR over time compared with the long-lasting robust phosphorylation induced by EGF. The course of internalization of EGFR was stimulus dependent. The tyrosine residues 1068 and 1086 on EGFR, which are phosphorylated in response to injury, played a critical role in wound closure; however, wound closure is not compromised when Y845 is mutated. The association of Src and phospholipase C-γ-1 (PLCγ1) with EGFR was constitutive, and growth factor receptor-bound protein 2 (Grb2) binding to EGFR was minimal and transient in response to UTP stimulation of cells. Phosphorylation of PLCγ1 was diminished in the presence of UTP, and phosphorylation induced by EGF was attenuated by costimulation with UTP. Our results indicate that the stimulation and initial activation of receptors collectively mediate the wound repair response.
Materials and Methods
Cell Culture and Primary Cell Isolation
Human corneal limbal epithelial (HCLE) cells were provided by Dr. Ilene Gipson (Harvard University, Boston, MA). Primary rabbit corneal epithelial cells were isolated as previously described.31 Briefly, corneas were removed from whole rabbit eyes (Pel-Freeze, Rogers, AR) and rinsed in Dulbecco's modified Eagle's medium (DMEM) with 100 U/ml penicillin, 100 μg/ml streptomycin (Mediatech, Manassas, VA), and 0.1% Fungizone (Invitrogen, Carlsbad, CA). The anterior third of the cornea was removed and placed in DMEM with 2.4 mg/ml Dispase II (Roche Applied Science, Indianapolis, IN) and incubated at 37°C for 1 hour.31 Epithelial sheets were removed from corneas, centrifuged, resuspended in keratinocyte serum free medium, and plated on either tissue culture–treated dishes or 22-mm2 coverslips. Cells were fed every 2 to 3 days and were not passed. All corneal cells were cultured in keratinocyte serum free medium [(Ca2+) = 0.09 mmol/L] supplemented with 30 μg/ml bovine pituitary extract, 0.032 nmol/L EGF (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin (Mediatech). The HCLE cells were supplemented with 0.3 mmol/L CaCl2.
Porcine aortic endothelial (PAE) cells and other cells (RetroPack PT67) were grown in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μmol/L nonessential amino acids (Mediatech).
Construction of Mutant EGFR
Wild-type EGFR in the retroviral pLXSN vector was a gift from David Stern (Yale University, New Haven, CT).32,33 PCR commercially available kit (QuikChange XL Kit; Stratagene, La Jolla, CA) was used to create point mutations, according to the manufacturer's instructions. Briefly, primers were designed to create tyrosine (Y) to phenylalanine (F) mutations at known EGFR phosphorylation sites. A PCR was performed with the EGFR-pLXSN template and forward and reverse mutant primers. The original template was digested for 1 hour at 37°C with DpnI (10 U/μl), and 2 μl of the DpnI-treated DNA vectors was transformed and grown in Escherichia coli (XL10 Gold). Vectors were isolated using a kit (Compass Miniprep kit; American Bioanalytical, Natick, MA) and sequenced (SeqWright DNA Technology Services, Houston, TX) to confirm the desired mutations.
A cell line (RetroPack PT67; Clontech, West Sacramento, CA) was used as a packaging line for viral production. Plasmid DNA of each mutant vector, 5 μg, was transfected into PT67 cells in serum-free media; 6 hours after transfection, 5 ml of serum-containing medium was added to cells. Transfected cells were allowed to produce virus for 48 hours. Virus-containing media were collected 48 hours after transfection and stored at 4°C. To infect the PAEs, media were replaced with virus-containing media collected from the PT67 packaging cells supplemented with 4 μg/ml polybrene. The virus media were replaced with selection media (DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mg/ml G418) 16 hours after infection. Mass spectrometry of EGFR isolated from cell lysates confirmed position and substitution.
The lack of expression of EGFR (ErbB1) and ErbB4 by PAE cells and expression of EGFR by E1-PAE cells were confirmed by real-time PCR using a commercially available system (ABI 7300). The gene expression assays (TaqMan) used were as follows: Hs01076078.ml for EGFR, Hs00955525.ml for ErbB4, and eukaryotic 18S ribosomal RNA endogenous control. The expression of receptors by IMR-90 cells was included as a positive control. Results are presented as relative expression normalized to 18S ribosomal RNA and were calculated using the ΔΔCT method.24
Immunoprecipitation and Western Blot Analysis
Cells were grown to confluency and placed overnight in serum-free media. Cells were washed in cold PBS and lysed in either lysis buffer (0.5% Nonidet P-40, 1% Triton X-100, 10 mmol/L Tris-HCl, and 150 mmol/L NaCl) or radioimmunoprecipitation assay buffer (150 mmol/L NaCl, 50 mmol/L Tris-HCl, and 1% Nonidet P-40) supplemented with protease and phosphatase inhibitor cocktails (Calbiochem, EMD BioSciences, San Diego, CA). Lysates were centrifuged, supernatants were transferred to a new tube, and the pellet was discarded. Protein was normalized using the bicinchoninic acid assay (Pierce, Rockford, IL). Total cell lysates were prepared in sample buffer for SDS–polyacrylamide gel electrophoresis (PAGE). For immunoprecipitation, magnetic beads (Dynabead IgG; Invitrogen) were prepared with antibody directed against EGFR, 40 μL (Santa Cruz Biotechnology, Santa Cruz, CA), according to manufacturer's instructions. Lysates were added to the antibody conjugated to the beads and incubated overnight at 4°C. Proteins were eluted, and ×2 sample buffer was added for SDS-PAGE. Protein was transferred from SDS-PAGE to a polyvinylidene difluoride membrane (PolyScreen, Pierce, Rockford, IL) for 2 hours using the semidry blotting method. Blots were blocked using Tris-buffered saline containing 7.25 mmol/L Tris hydrochloride, 2.75 mmol/L Tris base, 100 mmol/L NaCl, 0.1% Tween-20 (TBST), and 5% bovine serum albumin (BSA). Blots were incubated for either 1 hour at room temperature or overnight at 4°C. Primary antibodies directed to anti-phosphotyrosine (4G10 Platinum, Millipore, Temecula, CA) or docking proteins (Grb: BD Transduction Labs, Franklin Lakes, NJ; Src, Shc, and PLCγ1: Santa Cruz Biotechnology) were prepared in TBST containing 1% BSA and incubated for either 1 hour at room temperature or overnight at 4°C. Immunoblots were washed repeatedly in TBST and incubated in appropriate horseradish peroxidase–conjugated secondary antibody (Santa Cruz Biotechnology) for 1 hour at room temperature. The membrane was washed with TBST for 10 minutes at room temperature. Blots were exposed using Western Lightning (GE HealthCare Bio-Sciences Corp, Piscataway, NJ) and quantified using software (ImageJ).
Glutathione S-Transferase–Fusion Pull-Down Assay
Escherichia coli (DH5α) was transformed with empty vector, pGEX2T4, or pGEX2T4–Src homology–PLCγ1 constructs; and recombinant proteins were prepared as previously described.32 For the glutathione S-transferase (GST)–fusion pull-down assay, 5 μg of GST-fusion protein was used. Cell lysates were added to the beads for 3 hours at 4°C. The GST–pull-down lysates were heated at 100°C for 10 minutes, and equivalent protein was run on SDS-PAGE for Western blot analysis.
Scratch Wound Migration Assay
Cells were grown to confluency on eight-well glass-bottom chambers. The media were replaced with unsupplemented media 18 to 24 hours before experimentation. Cells were treated with either unsupplemented media or media containing various stimuli, and wounds (diameter, 200 to 300 μm) were made in each well. The cells were placed on the stage of a laser-scanning confocal microscope (Zeiss Axiovert 200M LSM 510, Zeiss, Thornwood, NY) in an environmental chamber maintained at 37°C and 5% CO2. Wounds were monitored using the multitime module in the LSM software, allowing multiple locations to be observed over time. Tiled differential contrast images were taken at each location every 20 minutes for 20 hours. The autofocus function was used. The LSM software was used to measure the wound area at various points, and percentage closure was calculated. Values were given as the mean ± SEM. Cells were selected from two independent trials, and LSM software was used to track motility and calculate the total distance traveled by each cell. Statistical comparisons were made using the Student's t-test or analysis of variance, followed by Tukey's post hoc test.
Immunohistochemistry
The HCLE cells were grown to confluency on eight-well coverslip bottom chambers, and unsupplemented media were added to the chambers 18 to 24 hours before experimentation. The cells were subjected to either unsupplemented media as the control treatment or media containing UTP, EGF, or 200- to 300-μm-diameter scratch wounds. Cultures were washed with PBS and fixed at 5, 15, 30, and 60 minutes after stimulation in 4% paraformaldehyde (ThermoFisher Scientific, Waltham, MA). The cells were permeabilized with 0.1% Triton X-100 and blocked in PBS containing 3% BSA. Cultures were incubated in the presence of a primary antibody directed against EGFR in PBS containing 1% BSA (ThermoFisher Scientific) or in PBS containing 1% BSA alone (negative control) overnight at 4°C. Cultures were washed and incubated in goat anti-rabbit (Alexa Fluor 546; Invitrogen) in PBS containing 1% BSA. Cultures were counterstained with rhodamine phalloidin and ToPro-3AM (Molecular Probes, WA). The cultures were examined using a laser-scanning confocal microscope (Zeiss Axiovert 200M LSM 510, Eugene, OR), as previously described.19,34 All secondary antibody staining was set to a negligible level, and all experimental data were imaged at the same settings. No enhancement of images was performed.
Results
Stimulation with UTP and ATP results in phosphorylation of a subset of the phosphorylation sites on EGFR compared with stimulation with EGF.17 It was previously demonstrated that injury or nucleotides induced phosphorylated EGFR (pEGFR) and phosphorylated ERK, which was inhibited with siRNA directed to the P2Y2 receptor and with CRM197.18,24
Recruitment of Cytoplasmic Proteins to EGFR in Response to EGF, HB-EGF, and UTP
The EGFR signaling pathway is mediated by recruitment of various signaling proteins (in particular, those with Src homology domains, such as Grb2, Shc, Src family kinases, and PLCγ1).35 Because these proteins bind to several different phosphorylation sites on EGFR, recruitment of these key EGFR-associated proteins was examined to determine whether the unique phosphorylation pattern18 caused differential recruitment of signaling proteins to the EGFR as a model system. Primary epithelial cells, HCLEs, and endothelial cells (PAEs) infected with the wild-type EGFR (E1-PAE) were stimulated with media change (control), injury (w), UTP, or EGF to determine recruitment of PLCγ1, Src, Shc, and Grb2 to the EGF receptor (Figure 1, A–C). The three cell types were compared to determine whether the response to the stimuli was a common cellular response. Because PAE cells do not express endogenous EGFR (ErbB1) or ErbB4, we used PAE cells that were infected with the EGFR (ErbB1) as a model for EGFR signaling.36 The PLCγ1 and Src were constitutively associated in all three cell types, and their association was not affected with EGF stimulation (Figure 1, A–C). When cells were stimulated with either UTP (25 μmol/L) or injury (w), Shc was not detected higher than control (media change) in epithelial cells. In all of the cell types tested, Grb2 was associated with EGFR when cells were treated with EGF but was only present at lower levels in response to injury or 25-μmol/L UTP. In addition, we compared the phosphorylation of EGFR of HCLE cells treated with two concentrations of EGF and a higher concentration of UTP (100 μmol/L) at two points and normalized it to EGFR. Phosphorylation was almost threefold greater when exposed to EGF compared with UTP (Figure 1D). Although 5-nmol/L EGF may be in excess of physiological concentrations, it was used as it is used in clinical situations.11,37
Figure 1.

Injury and UTP recruit specific docking proteins to the EGFR. Comparisons were made in two cell lines and primary epithelial cells. The HCLEs (A), primary corneal epithelial cells (B), and E1-PAEs (C) were stimulated with a control media change (C), injury (W), UTP (25 μmol/L), or EGF (5 nmol/L) for 5 minutes. Cells were lysed, and EGFR was immunoprecipitated from the lysates. SDS-PAGE and Western blot analyses of the immunoprecipitation were performed for EGFR, PLCγ1, Src, Shc, and Grb2. D: Phosphorylation of EGFR was determined after HCLEs were stimulated with media change (control), EGF (0.5 and 5 nmol/L), and UTP (100 μmol/L) at 5 and 60 minutes. SDS-PAGE and Western blot analyses of the immunoprecipitation were performed for EGFR. The pEGFR was detected using a phosphorylated tyrosine antibody. The pEGFR is normalized to EGFR. Data represent a minimum of three independent experiments.
Internalization and Phosphorylation of EGFR in Response to EGF and UTP
To determine the differences in downstream signaling of EGFR, we determined whether injury- or nucleotide-induced internalization of EGFR occurred in a similar manner to the well-established response to EGF. The media change control showed minimal change in localization of receptor over time. When cells were stimulated with 5-nmol/L EGF, the receptor was internalized within 5 minutes, was punctate, and remained localized as puncta for longer than 60 minutes (Figure 2). A similar pattern was seen with 0.5 nmol/L, with the exception that the puncta were decreased by 60 minutes. The addition of UTP to the epithelial cells resulted in internalization of the EGFR by 5 minutes; however, it was detected at the plasma membrane within 30 minutes (Figure 2).
Figure 2.
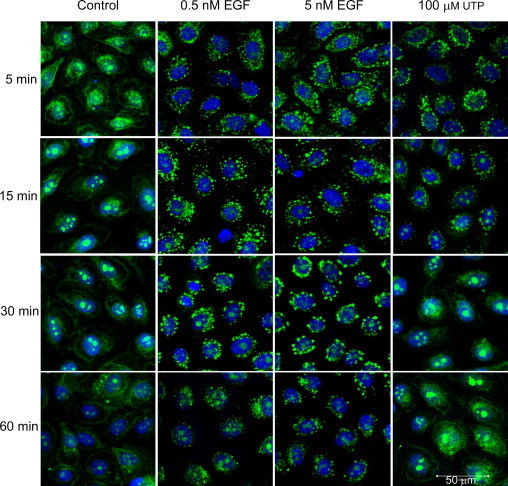
Internalization of the EGFR in response to UTP differs from EGF. The HCLEs were cultured to confluence and stimulated with medium change (control), UTP (100 μmol/L), and EGF (0.5 and 5 nmol/L) over a time course. Cells were fixed and probed with an antibody directed against EGFR, pseudocolored green, counterstained with a nuclear marker (ToPro-3AM), and pseudocolored blue. Confocal images represent single optical sections of 4 μm taken in the midregion of the cells. Scale bar = 50 μm. Images are representative of four independent experiments.
Internalization was examined in cells after a scratch injury was performed. Cells were fixed immediately after injury (time, 0 minutes) and compared over time (Figure 3). Immediately after injury, EGFR was localized along the plasma membrane and in the cytosol, agreeing with previous results of Zieske et al,3 in studies that were performed in vivo. By 5 minutes, the EGFR was internalized at the wound margin (Figure 3). Cells that were a distance from the wound edge did not display internalization; in addition, it is assumed that this is due to the limited diffusion of nucleotides from the wound (data not shown in this field). Additional control experiments performed at 4°C showed negligible internalization, as predicted (data not shown).
Figure 3.
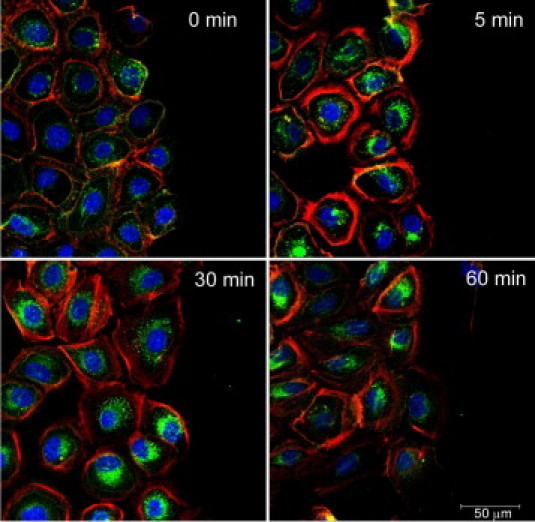
Internalization of the EGFR in response to injury changes over time. The HCLEs were cultured to confluence, subjected to a scratch wound, and monitored over time. Images were taken at the leading edge of the injury. Cells were fixed and probed with an antibody directed against EGFR, pseudocolored green, counterstained with a nuclear marker (ToPro-3AM), and pseudocolored blue. Confocal images represent single optical sections of 4 μm taken in the midregion of the cells. Scale bar = 50 μm. Images are representative of four independent experiments.
Because the time course of the internalization of the EGFR in response to injury or UTP was transient compared with that induced by EGF, the phosphorylation of EGFR was examined over a similar course. To determine whether the media change alone altered the response, cells were subject to media change and pEGFR was determined over time. At all points, phosphorylation was minimal when normalized to EGFR and decreased over time. (Figure 4A). Controls were compared with treatment with EGF (Figure 4A). Stimulation with UTP over time yielded less pEGFR relative to total EGFR compared with stimulation with EGF (Figure 4, B–D). The Shc isoforms were detected over the time course in response to EGF. Although the isoforms were detected in response to 100-μmol/L UTP compared with 25-μmol/L UTP, they did not increase over control (Figures 1D and 4D). Although association of Grb2 with EGFR increased at a concentration of 100-μmol/L UTP compared with that with 25-μmol/L UTP (Figure 1), it remained lower than when cells were stimulated with either concentration of EGF (Figure 4, B–D and F). Densitometric analysis of fold change of pEGFR and Grb2 association with EGFR are shown (Figure 4, E and F).
Figure 4.
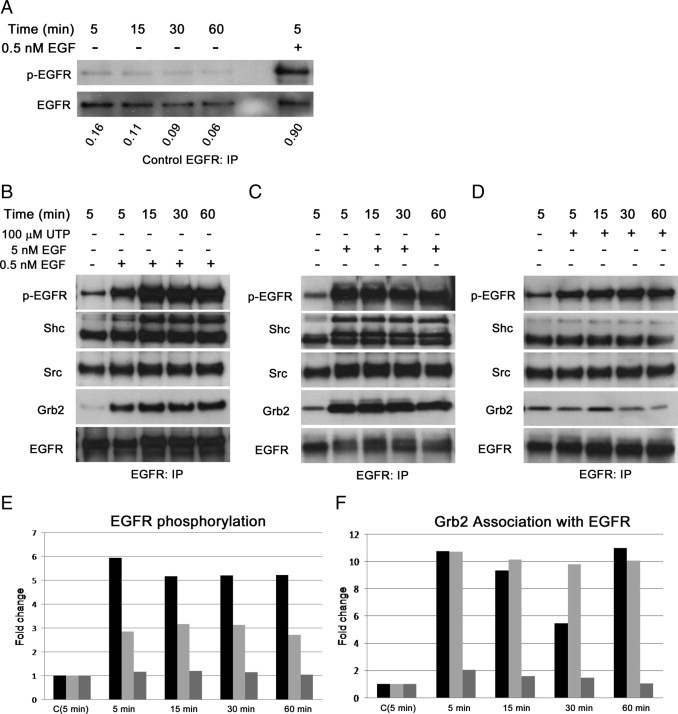
Recruitment and phosphorylation of EGFR differs in response to stimuli. A: The HCLEs were cultured to confluence and stimulated with control media change (−) over 60 minutes. The EGF (5 minutes) was used as a positive control (+). The pEGFR was detected using a phosphorylated tyrosine (p-tyr) antibody and is normalized to EGFR. B through D: The HCLEs were cultured to confluence and stimulated with EGF (0.5 and 5 nmol/L) or UTP (100 μmol/L) over 60 minutes. Confluent cells were lysed, and EGFR was immunoprecipitated from the lysates. The SDS-PAGE and Western blot analyses of the immunoprecipitation were performed for EGFR, Shc, Src, and Grb2; and probed for p-tyr to determine pEGFR. Images are representative of four experiments. E and F: Densitometric analysis [5.0-nmol/L EGF (black); 0.5-nmol/L EGF (light gray); and 100-μmol/L UTP (dark gray)]. Images are representative of four independent experiments.
We compared these results to responses induced by HB-EGF. Previously, it was shown that HB-EGF induced cell migration in a dose-dependent manner, with optimal activity at 1 nmol/L. In addition, we showed that migration was inhibited with CRM197 and that activity was restored when HB-EGF was included in the culture medium.18 In response to 1-nmol/L HB-EGF, there was a large increase in pEGFR over control media change (C) during the time course (Figures 4 and 5A). The HB-EGF induced association of Grb2 with EGFR that correlated with pEGFR (Figure 5A). The response to HB-EGF after 5-minute stimulation was compared with an equivalent molar concentration of EGF and 100-μmol/L UTP (Figure 5B). When the cells were preincubated with functional blocking antibody to HB-EGF (40 μg/ml) and then stimulated with HB-EGF, there was a 25% decrease in phosphorylation of EGFR compared with an 8% decrease in control and other stimulations with blocking antibody (Figure 5).
Figure 5.
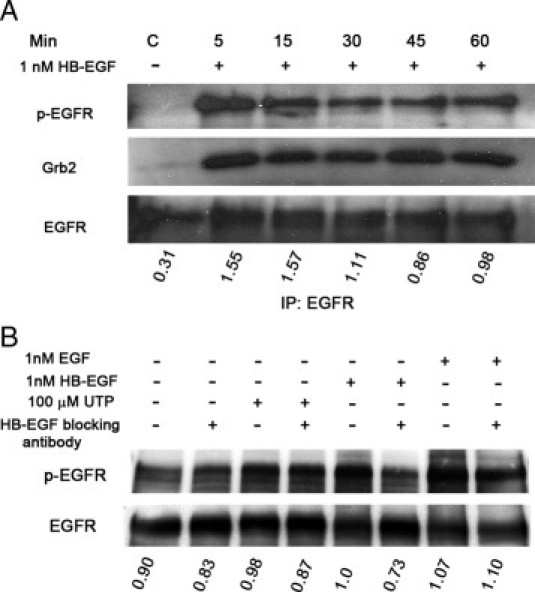
The HCLEs were stimulated with HB-EGF. A: The HCLEs were cultured to confluence and stimulated with HB-EGF (1 nmol/L) over 60 minutes. The EGFR was immunoprecipitated from the lysates, and SDS-PAGE and Western blot analyses of the immunoprecipitation were performed. Antibodies directed against EGFR and Grb2 and an antibody to p-tyr were used. The pEGFR was normalized to EGFR. B: The HCLEs were cultured to confluence, preincubated for 20 minutes with a functional blocking antibody to HB-EGF (40 μg/ml) or control media (−), and stimulated with media change (−), EGF (1 nmol/L), HB-EGF (1 nmol/L), or UTP (100 μmol/L) for 5 minutes. Lysates were extracted, and EGFR was immunoprecipitated. The EGFR and p-tyr antibodies were used, and pEGFR was normalized to EGFR.
UTP Inhibits PLCγ1 Phosphorylation and Its Association with Grb2
To examine if there were other differences that occurred in response to injury or UTP, HCLEs were stimulated and the phosphorylation of PLCγ1 was examined. The GST pull downs were performed using PLCγ1 and Grb2 (Figure 6A). Consistent with other experiments, GST-PLCγ1 showed binding of Grb2 with a strong band when stimulated with either 0.5- or 5-nmol/L EGF. However, Grb2 was not detected over control levels in response to UTP. There was also no Grb2 in the injury-stimulated cells (data not shown). When cells were stimulated with EGF, PLCγ1 was phosphorylated rapidly (Figure 6B). However, the phosphorylation was attenuated when cells were costimulated with UTP (Figure 6B). Phosphorylation of PLCγ1 in response to UTP alone was only detected at background levels. To verify that PLCγ1 was not labile, cross-linking experiments were performed with glutaraldehyde, and there was no detectable difference in results. Furthermore, when cells were immunoprecipitated with EGFR and blotted for PLCγ1 or Grb2, we found that EGF stimulated the association, whereas costimulation with UTP (or injury) either inhibited or did not facilitate binding (Figure 6C). Costimulation experiments performed during a time course showed phosphorylation after an extended period (data not shown). Taken together, UTP antagonizes the EGF-induced phosphorylation of PLCγ1 and its ability to interact with EGFR and adaptor protein, Grb2.
Figure 6.
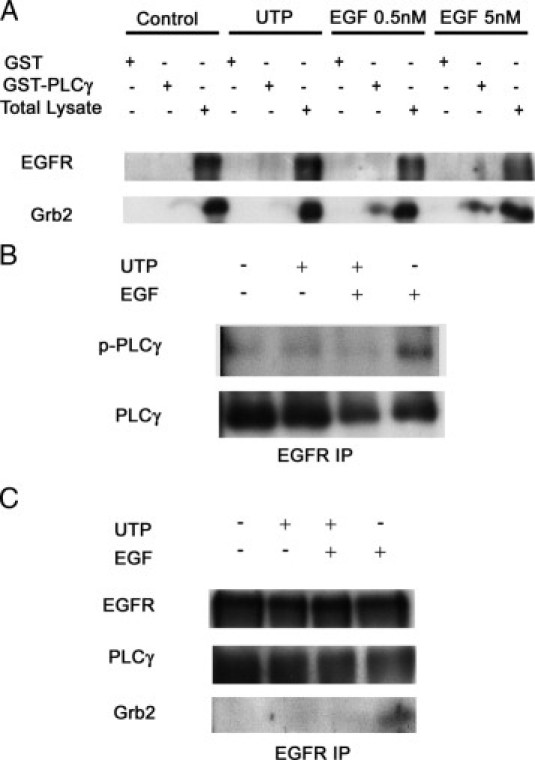
Injury, EGF, and UTP have unique roles in associating adaptor molecules PLCγ1 and Grb2. A: The HCLEs were lysed, and in vitro GST-fusion protein assays were performed. Cells were stimulated with UTP and EGF (0.5 and 5 nmol/L). The GST-PLCγ–Src homology domains were used to bind the cell lysates. An SDS-PAGE Western blot analysis for binding of EGFR and Grb2 was performed. B: The HCLE cells were stimulated with a control media change (−), 25-μmol/L UTP, 25-μmol/L UTP followed by 5-nmol/L EGF, or 5-nmol/L EGF for 5 minutes. Lysates were immunoprecipitated by EGFR antibody and run on SDS-PAGE, and Western blot analysis was performed for p-PLCγ and total PLCγ. C: Cells were stimulated with media change, UTP, EGF, or both UTP and EGF. The EGFR was immunoprecipitated from the lysates, and Western blot analysis was performed. Blots are representative of three independent experiments.
Effect of UTP on Cell Migration
Migration assays were performed to examine the role of EGFR in nucleotide-mediated cell migration. In the first set of experiments, HCLE cells were preincubated in the presence or absence of AG1478, an EGFR kinase inhibitor, and stimulated with UTP, EGF, or media change (control); wounds were made. Images along the wound margin were taken every 20 minutes. Optimization experiments established a wound size for which proliferation was not required to close the wound.5 The absence of proliferation during corneal epithelial wound migration has been well documented by several investigators.3,16,38–40
Wound closure of 30% was detected in unstimulated cultures, whereas cells stimulated with either UTP or EGF attained an average 65% to 70% wound closure (Figure 7A). Cells pretreated with AG1478 demonstrated a significant reduction in the groups stimulated with EGF or UTP (P < 0.05). When HCLEs were stimulated by EGF, the rate of migration was greater than the rate for other conditions for the first 5 hours (Figure 7C). In contrast, cultures stimulated with UTP showed a constant migration rate over time (Figure 7C). When cultures were pretreated with the kinase inhibitor, the cells migrated at a slower rate over the entire distance whether stimulated with EGF or UTP. Analysis of movies revealed that epithelial cells back from the leading edge migrated into the wound region to fill the void. The HCLEs stimulated with EGF or UTP showed that cells at the leading edge possessed rapid movement toward the center of the wound and then continued to move in a swirling pattern (Figure 7, E–G). Cells preincubated with AG1478 retained the ability to be motile but lost the ability to detect which direction to travel to close the wound (data not shown).
Figure 7.
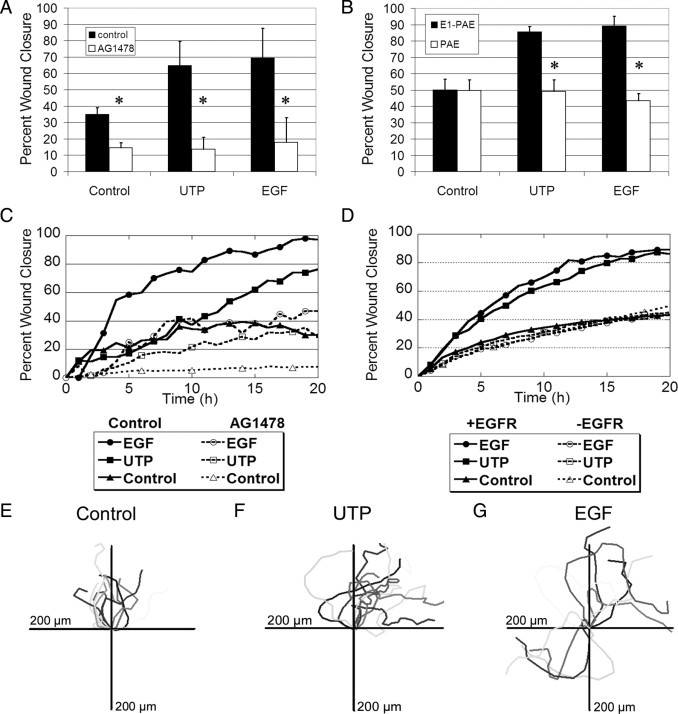
The EGFR is necessary for nucleotide-induced migration. The HCLEs were incubated in the presence or absence of AG1478 (10 nmol/L) and stimulated with UTP (100 μmol/L), EGF (0.5 nmol/L), or medium alone (control) before injury. Confluent cultures in 8-well chamber slides were incubated overnight in medium lacking growth factors. Each slide was placed on a heated microscope stage, wounded, and incubated in an environmental chamber at 37°C and 5% CO2 for 20 hours. The wounds were demarcated, and contiguous regions were tiled and imaged every 20 minutes using autofocus. A: The percentage wound closure at 20 hours for HCLEs after injury (t-test, *P < 0.05). B: The percentage wound closure at 20 hours for E1-PAEs and PAEs after injury. The E1-PAEs and PAEs (cells lacking the EGFR) were stimulated with UTP, EGF, or medium alone (control) before injury (t-test, *P < 0.05). C: Migration time course of a representative run using HCLEs after injury and stimulation. D: Migration time course of a representative run using E1-PAEs and PAEs after injury and stimulation. E–G: Cell tracking data for control and UTP- and EGF-stimulated cells. The HCLE cells were tracked every 20 minutes using proprietary software (Zeiss) and placed on an axis on which all cells start at the origin. Data represent a minimum of three independent experiments for each cell line.
To determine the role of EGFR, PAE cells lacking the EGFR and PAE cells containing the wild-type EGFR (E1-PAE) were injured and treated with control media, UTP, or EGF; and the percentage wound closure was determined (Figure 7B). The unstimulated wounds closed by approximately 50% in cells with or without the EGFR. When E1-PAE cells were stimulated with UTP or EGF, the wound closed 85% and 89%, respectively, both significantly more than PAE cells (Figure 7B, P < 0.05). When the rate of closure was examined, cultures that were stimulated with EGF or UTP and contained the EGFR migrated faster and the rate did not plateau until after 10 hours. In the control cultures (without EGFR), the samples did not show greater wound closure or motility than cells lacking the receptor (Figure 7D).
Expression and localization of EGFR in E1-PAE cells were demonstrated using real-time PCR. Cells infected with EGFR expressed EGFR (ErbB1) and not ErbB4, whereas the PAE cells did not express either EGFR or ErbB4 (Figure 8A). Expression was graphed as relative quantitation with IMR-90 cells set to one for both EGFR and ErbB4. Although the expression of EGFR mRNA is high, both E1-PAE and HCLE cells display comparable levels of EGFR protein when normalized to total protein (Figure 8B). To demonstrate that the pattern of internalization is similar to HCLEs and can be used as a model for cell migration, E1-PAEs were stimulated with media change (control), EGF, or UTP; and internalization was evaluated after 5 minutes and was localized as puncta in cells stimulated with UTP and EGF (Figure 8C). Cells subjected to a media change displayed diffuse staining lacking the puncta. Secondary antibody controls were obtained and were not fluorescent at the laser settings used for the experimental conditions (data not shown). Together, these data indicate that the expression of EGFR protein and internalization of the receptor are similar to those of HCLEs (Figure 2). In addition, the localization of the receptor is similar to that seen in primary cells5 and in vivo.3
Figure 8.
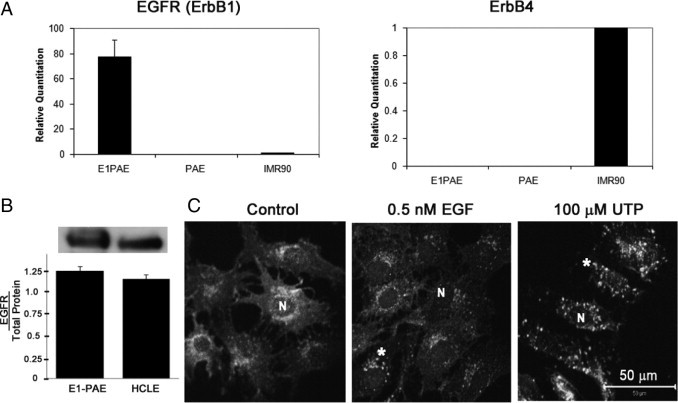
Localization and expression of EGFR in E1-PAE cells. A: Real-time PCR showed no expression of EGFR (ErbB1) or ErbB4 mRNA in PAEs. The E1-PAEs (cells infected with EGFR) express EGFR mRNA. (IMR-90 cells, which express both ErbB1 and ErbB4, were used as positive control for both EGFR and ErbB4; and expression was set to 1). Results presented as relative expression were normalized to 18S RNA and were calculated using the ΔΔCt method. B: Protein expression: E1-PAEs were lysed, equivalent amounts of protein were loaded, and SDS-PAGE and Western blot analyses were performed and probed for EGFR. Densitometric analysis was performed, and the blot is representative of three experiments. C: Localization of EGFR: E1-PAEs were cultured and stimulated with medium change (control), UTP (100 μmol/L), and EGF (0.5 nmol/L) for 5 minutes. Cells were fixed and probed with an antibody directed against EGFR (asterisk) and counterstained with ToPro-3AM (N). Confocal images are presented as merged images in black and white and represent single optical sections of 3 μm taken in the midregion of the cells. Scale bar = 50 μm. Images are representative of three independent experiments.
Role of Y1068 and Y1086 of EGFR in Cell Migration
Because Grb2 was not recruited by EGFR when cells were injured or stimulated with UTP, we determined whether the tyrosine residues 1068 and 1086 played a role in wound repair because previous results indicated that injury resulted in site-specific phosphorylation at those sites.18 Cell migration of cells lacking 1068 or 1086 was compared with cells lacking tyrosine residue 845, which was not phosphorylated with injury and is not a binding site for Grb2. However, Y845 is phosphorylated when cells are stimulated with EGF. Scratch wound assays were performed on the EGFR mutants Y845F, Y1068F, and Y1086F in the presence of either 5-nmol/L EGF or 100-μmol/L UTP (Figure 9, A–C). The mutant unstimulated control cultures did not exhibit differences in percentage wound closure after 20 hours, and the rate of migration was similar to E1-PAE–unstimulated cultures (Figures 9A and 7, B and D). The E1-PAE Y845F and Y1068F cultures showed significant migration over control in response to both UTP and EGF. The absence of Y845 did not alter migration. Although E1-PAE, Y845F, Y1068F, and Y1086F cultures showed an increase in migration on stimulation with EGF, the percentage wound closure of the E1-PAE Y1086F cultures was attenuated. With the absence of Y1086, the cells did not demonstrate enhanced migration to UTP (Figure 9A).
Figure 9.
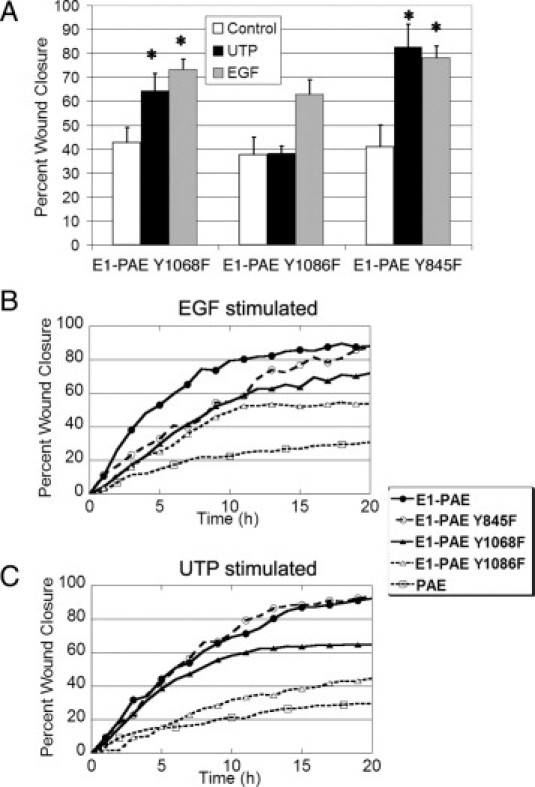
Specific EGFR tyrosine residues play a role in nucleotide-induced cell migration. The PAEs, E1-PAE, and E1 PAE mutants (ie, Y1068F, Y1086F, and Y845F) were cultured in eight-well chamber slides and incubated overnight in medium lacking growth factors. The slides were placed on a heated microscope stage, wounded, and incubated in an environmental chamber at 37°C and 5% CO2 for 20 hours. Cells were stimulated with UTP or EGF and compared with medium alone (control). The wounds were demarcated, and contiguous regions were tiled and imaged every 20 minutes using autofocus. A: The percentage wound closure at 20 hours. B: Course of migration of mutants in response to EGF. Representative runs stimulated with 5-nmol/L EGF are shown. C: Course of migration in response to UTP. Representative runs stimulated with 100-μm UTP are shown. Significance was determined by a one-way analysis of variance, followed by Tukey's post hoc test (*P < 0.05). Data represent a minimum of three independent experiments.
Representative time courses between cells with mutant receptors were compared to examine whether differences in migration rates changed over time (Figure 9B). When the three mutant cultures were stimulated with EGF, they migrated at a similar rate for the first 10 hours (Figure 9B). However, the migration rate of E1-PAE Y1068F cultures slowed after 10 hours and plateaued in the E1-PAE Y1086F cultures. When cells were stimulated with UTP, the E1-PAE Y845F cultures migrated at a rate similar to the E1-PAE cultures. In contrast, the E1-PAE Y1068F cultures began to plateau before 10 hours and demonstrated a markedly slower rate for the second 10 hours (Figure 9C). The E1-PAE Y1086F cultures demonstrated a slope similar to the PAE cells, which lacked the EGFR (Figure 9C). In summary, the E1-PAE Y1068F and Y1086F cultures showed altered wound closure, which appeared to be, in part, their ability to respond to UTP and, to a lesser degree, EGF.
The mutation of Y845 did not hinder the motility of cells and wound closure persistence (Figure 10). In contrast, the lack of Y1068 caused impaired cell motility in unstimulated cultures (Figure 10). When these cells were stimulated with EGF, migration was enhanced compared with the positive control E1-PAEs, but motility continued after the two cell margins met. When the same cultures were stimulated with UTP, the result was the excessive migration was not detected (Figure 10). Although the mutation of Y1086 did not inhibit the cells' ability to migrate compared with unstimulated E1-PAE controls (indicated by the mean total length of distance traveled), the cells did not travel in a straight line to the opposing wound margin (Figure 10). In response to UTP, the Y1086 cells traveled erratically and were often observed backtracking on each other, indicating that they did not detect environmental cues. Together, this analysis demonstrated that for proper wound closure, Y1068 and Y1086 are required.
Figure 10.
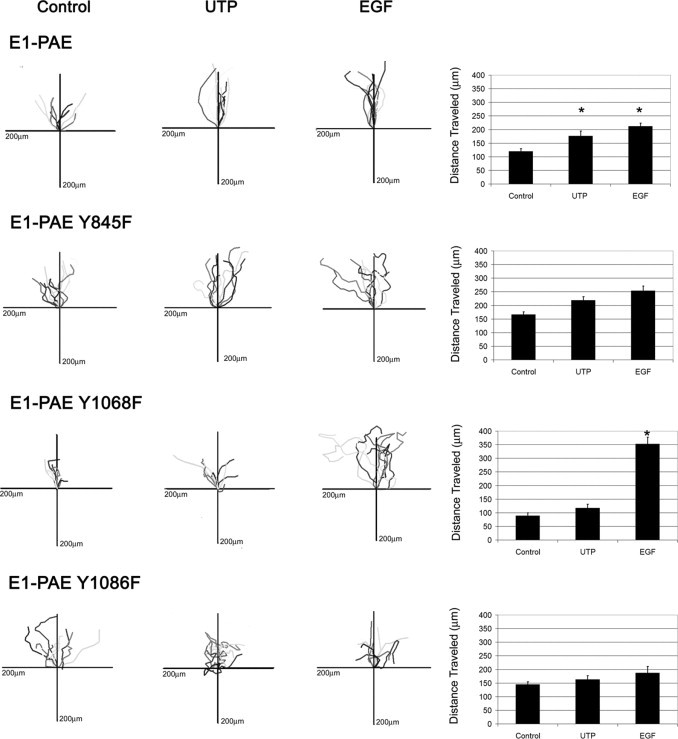
Migration is altered in cells with mutated EGFR tyrosine residues (ie, E1-PAE, E1-PAE Y1068F, E1-PAE Y1086F, and E1-PAE Y845F). Cells were tracked every 20 minutes using proprietary software (Zeiss) and placed on an axis where all cells start at the origin. The total distance traveled is presented for each condition in bar graphs. Significance was determined by a one-way analysis of variance, followed by Tukey's post hoc test (*P < 0.05). Bar graphs represent a minimum of three independent experiments.
Discussion
Increasingly, evidence has indicated that activation of EGFR, either indirectly through injury and nucleotide stimulation or directly by EGF as a ligand, leads to two distinct mechanisms underlying wound repair. Site-specific tyrosine residues are phosphorylated in response to injury, supporting the differences detected in downstream signaling.18 Although the underlying mechanisms are not fully understood, we do know that both injury and nucleotides, which are released at injury, initiate a rapid Ca2+ wave to neighboring cells, whereas EGF initiates a distinct oscillatory response. Although the injury-induced Ca2+ wave is not inhibited with tyrosine kinase inhibitors, such as AG1478, it is inhibited when cells are transfected with P2Y2 receptor siRNA, supporting the idea that injury-induced calcium release is independent of the EGFR system.4,24 To understand further how injury and nucleotides modulate a different response from EGF, we analyzed the association of cytoplasmic signaling proteins with EGFR, EGFR internalization, and wound closure. Primary corneal epithelial cells and an epithelial cell line (HCLE) were used, and the results were compared with PAE cells in which the EGFR was either absent or wild-type or mutant receptors (ie, Y845F, Y1068F, or Y1086F) were expressed.
There is evidence that signaling by the EGFR from the cell surface prevents the induction of cell death.3 This may occur in communication between purinergic and EGFR signaling, especially in corneal epithelium, where apoptosis is rare. Other researchers have shown that stimulation of kidney epithelial cells with pro–HB-EGF protects cells from anoikis41; in epithelial cells, HB-EGF may play a role in nucleotide-induced ERK1/2 signaling and migration.18 In addition, when cells that express high levels of EGFR are stimulated with EGF, the enhanced levels of activated EGFR may be accompanied by an accumulation of the receptors within the cell because of a decrease in the rate of receptor degradation.42,43 This is hypothesized to occur in the breast cancer epithelial cell line (MDA-MB-468 cells) in which endocytic trafficking is slow and cell growth is mediated by the localization of the receptor and not its prolonged activation.44
In HCLE cells, pEGFR is greater in response to EGF than UTP, but there is a difference in internalization over time. The intracellular localization of the receptor that is detected under control conditions has been reported previously in primary corneal epithelial cells and in vivo.3,5 The EGF causes an accumulation of receptor within the cell. In contrast, UTP appears to induce a rapid recycling of the receptor. These results are confirmed in E1-PAE cells, which only express EGFR. In addition, HB-EGF causes a rapid and sustained pEGFR with associated Grb2. Although control conditions (media change) do not cause increased phosphorylation of pEGFR, there is a minimal level present that may explain the intracellular localization. This has been reported to be due to autocrine activation, which has been reported in human keratinocytes in which autocrine phosphorylation is correlated with the appearance of the soluble ligand, amphiregulin.45
When we determined whether recruitment of docking molecules was time dependent, we found no difference in binding of PLCγ1 or Src in either epithelial cells or E1-PAE. Cells stimulated with both concentrations of EGF showed internalization and enhanced binding of Grb2 at the longer time points. In contrast, when primary epithelial cells were stimulated by exogenous nucleotides or injury, internalization was present, but the time course was attenuated. Together, these results indicate that, although several ligands and molecules, via cross-activation, may cause phosphorylation of the EGFR, the endocytosis may depend on the recruitment of docking proteins. Although some investigators15,46 indicated that tyrosine phosphorylation of EGFR was not necessary for internalization, others47,48 found that specific protein–protein interactions were responsible for receptor-mediated endocytosis. In the latter experiments, Shc was proposed to mediate ligand-induced internalization of EGFR. Our results indicate that all isoforms of Shc were not associated with EGFR unless higher concentrations of UTP were used. Failure to bind Grb2 and internalization of EGFR in clathrin-coated pits was thought to depend on phosphorylation of Y1068 and Y1086 and was dependent on whether one or both sites were mutated.49 More recently, the binding of the ligand to EGFR has accelerated its internalization and receptor down–regulation.50 Our results suggest a more complex regulation. Previously, when cells were stimulated by either injury or treatment of nucleotides, phosphorylation occurred on tyrosine residues on EGFR, including 1068 and 1086.18 In this study, we demonstrated that EGFR internalization has a distinct course in response to nucleotide release and activation of purinergic receptors, followed by transactivation and minimal phosphorylation of EGFR. In addition, Grb2 is only minimally recruited by EGFR higher than control. The minimal binding of Grb2 may be sufficient to cause the transient nature induced by nucleotides.
In epithelial cells, the initial rate of nucleotide-induced cell migration was slower than that of EGF, but the slope remained the same over time, whereas EGF-stimulated cells began to plateau at 10 hours. The two different rates of migration achieved 80% wound closure for UTP and almost complete closure in response to EGF (Figure 7, B and C). To test the role of the EGFR in nucleotide-induced cell migration, we measured migration of PAE and E1-PAE cells. When the EGFR was present, cells stimulated with either EGF or UTP achieved 80% wound closure compared with unstimulated conditions, which resulted in less than 50% wound closure (Figure 7). When E1-PAE Y1068F and E1-PAE Y1086F cultures were examined after injury, both demonstrated enhanced migration on EGF or UTP stimulation. However, although mutation of Y1068F did not alter the UTP-stimulated rate of migration for the first 10 hours, its rate plateaus dramatically at later points (Figure 9C). Furthermore, in examining individual cell movement in wound closure, unstimulated E1-PAE Y1068F cells showed minimal total migration, whereas EGF-stimulated cells lack wound closure persistence compared with E1-PAEs (Figures 9 and 10). Likewise, mutation of Y1086F inhibited both cell migration of E1-PAE cells and wound closure persistence (Figures 9 and 10). In contrast, in E1-PAE Y845F cells, there was no significant difference between EGF- and UTP-stimulated cell migration (Figure 10). Interestingly, treatment of cells with AG1478 inhibited cell migration and wound closure persistence. Therefore, the path that cells take to achieve the final point might depend on differential phosphorylation of EGFR and its ability to recruit signaling proteins. Recent studies49 in endothelial cells suggest that specific inputs modulate different cell migratory outputs, such as directed migration, single-cell motility, and cell–cell coordination. Each of these behaviors may compose a separate signaling module; we expect that the different phosphorylation sites mediate the different modules. The changes in cell migration might well be because of differences in cell adhesion to the substrate.51,52 The latter statement is supported by previous findings53 demonstrating that epithelial cell morphological features and the expression of integrins change with substrate chemistry. This hypothesis will be addressed in future studies.
The altered migration detected when specific residues are mutated is likely to be caused by differential recruitment of docking molecules to the EGFR after EGF or injury/nucleotide stimulation. We have shown that the ligand causes a difference in the recruitment of docking proteins. Interestingly, preliminary experiments indicate that there is less recruitment of Grb2 when cells lack specific tyrosine residues. In addition, treatment of corneal epithelial cells with either a functional blocking antibody to HB-EGF or CRM19754 attenuated the phosphorylation of EGFR. In other experiments, the addition of HB-EGF to CRM197 rescued epithelial cell migration,18 suggesting that the release of nucleotides and cleavage of pro–HB-EGF may be critical in mediating the injury response. However, the system is complex in primary cells and HCLEs because HB-EGF may bind to EGFR, ErbB4, or heparan sulfate proteoglycans, the latter of which are up-regulated with corneal injury.55
Communication between the purinergic and EGFR signaling pathways was demonstrated in experiments in which UTP appears to inhibit the phosphorylation of PLCγ1. Our results provide evidence that, although phosphorylation of EGFR appears to be critical to wound closure, it can be achieved by direct ligand stimulation or other cell surface signaling pathways. When it occurs through this secondary route, recruitment of signaling molecules is modified, allowing for differences in EGFR internalization, recruitment, and subsequent cell migration. Additional studies exploring different ligands present in biological fluids with injury and the molecules that are recruited are being undertaken and will provide insight into the complex regulation of wound repair.
Acknowledgments
We thank the critical comments of and discussions with Drs. James Zieske, Matthew Nugent, and Karen Symes.
Footnotes
Supported by funds from (grant EY06000 to V.T.-R.) and departmental grants from the Massachusetts Lions Eye Research Fund and the New England Corneal Transplant Fund.
I.B. and A.K. contributed equally to this work.
References
- 1.Tahinci E., Symes K. Distinct functions of Rho and Rac are required for convergent extension during Xenopus gastrulation. Dev Biol. 2003;259:318–335. doi: 10.1016/s0012-1606(03)00206-9. [DOI] [PubMed] [Google Scholar]
- 2.Danjo Y., Gipson I.K. Specific transduction of the leading edge cells of migrating epithelia demonstrates that they are replaced during healing. Exp Eye Res. 2002;74:199–204. doi: 10.1006/exer.2001.1115. [DOI] [PubMed] [Google Scholar]
- 3.Zieske J.D., Takahashi H., Hutcheon A.E., Dalbone A.C. Activation of epidermal growth factor receptor during corneal epithelial migration. Invest Ophthalmol Vis Sci. 2000;41:1346–1355. [PubMed] [Google Scholar]
- 4.Klepeis V.E., Cornell-Bell A., Trinkaus-Randall V. Growth factors but not gap junctions play a role in injury-induced Ca2+ waves in epithelial cells. J Cell Sci. 2001;114:4185–4195. doi: 10.1242/jcs.114.23.4185. [DOI] [PubMed] [Google Scholar]
- 5.Song Q.H., Singh R.P., Trinkaus-Randall V. Injury and EGF mediate the expression of alpha6beta4 integrin subunits in corneal epithelium. J Cell Biochem. 2001;80:397–414. doi: 10.1002/1097-4644(20010301)80:3<397::aid-jcb140>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 6.Schultz G., Chegini N., Grant M., Khaw P., MacKay S. Effects of growth factors on corneal wound healing. Acta Ophthalmol Suppl. 1992;(202):60–66. doi: 10.1111/j.1755-3768.1992.tb02170.x. [DOI] [PubMed] [Google Scholar]
- 7.Bennett N.T., Schultz G.S. Growth factors and wound healing: biochemical properties of growth factors and their receptors. Am J Surg. 1993;165:728–737. doi: 10.1016/s0002-9610(05)80797-4. [DOI] [PubMed] [Google Scholar]
- 8.Gipson I.K., Inatomi T. Extracellular matrix and growth factors in corneal wound healing. Curr Opin Ophthalmol. 1995;6:3–10. doi: 10.1097/00055735-199508000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Wilson S.E. EGF, basic FGF, and TGF beta-1 messenger RNA production in rabbit corneal epithelial cells. Invest Ophthalmol Vis Sci. 1992;33:1987–1995. [PubMed] [Google Scholar]
- 10.Dartt D.A. Interaction of EGF family growth factors and neurotransmitters in regulating lacrimal gland secretion. Exp Eye Res. 2004;78:337–345. doi: 10.1016/s0014-4835(03)00202-1. [DOI] [PubMed] [Google Scholar]
- 11.Leibowitz H.M., Morello S., Jr, Stern M., Kupferman A. Effect of topically administered epidermal growth factor on corneal wound strength. Arch Ophthalmol. 1990;108:734–737. doi: 10.1001/archopht.1990.01070070120048. [DOI] [PubMed] [Google Scholar]
- 12.Huang F., Kirpatrick D., Jiang X., Gygi S., Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination with in the kinase domain. Mol Cell. 2006;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q., Villeneuve G., Wang Z. Control of epidermal growth factor endocytosis by receptor dimerization, rather than receptor kinase activation. EMBO Rep. 2005;6:942–948. doi: 10.1038/sj.embor.7400491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpenter G. Properties of the receptor for epidermal growth factor. Cell. 1984;37:357–358. doi: 10.1016/0092-8674(84)90365-9. [DOI] [PubMed] [Google Scholar]
- 15.Chen W.S., Lazar C.S., Poenie M., Tsien R.Y., Gill G.N., Rosenfeld M.G. Requirement for intrinsic protein tyrosine kinase in the immediate and late actions of the EGF receptor. Nature. 1987;328:820–823. doi: 10.1038/328820a0. [DOI] [PubMed] [Google Scholar]
- 16.Zieske J.D., Wasson M. Regional variation in distribution of EGF receptor in developing and adult corneal epithelium. J Cell Sci. 1993;106(pt 1):145–152. doi: 10.1242/jcs.106.1.145. [DOI] [PubMed] [Google Scholar]
- 17.Song Q.H., Gong H., Trinkaus-Randall V. Role of epidermal growth factor and epidermal growth factor receptor on hemidesmosome complex formation and integrin subunit beta4. Cell Tissue Res. 2003;312:203–220. doi: 10.1007/s00441-002-0693-x. [DOI] [PubMed] [Google Scholar]
- 18.Boucher I., Yang L., Mayo C., Klepeis V., Trinkaus-Randall V. Injury and nucleotides induce phosphorylation of epidermal growth factor receptor: MMP and HB-EGF dependent pathway. Exp Eye Res. 2007;85:130–141. doi: 10.1016/j.exer.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klepeis V.E., Weinger I., Kaczmarek E., Trinkaus-Randall V. P2Y receptors play a critical role in epithelial cell communication and migration. J Cell Biochem. 2004;93:1115–1133. doi: 10.1002/jcb.20258. [DOI] [PubMed] [Google Scholar]
- 20.Yang L., Cranson D., Trinkaus-Randall V. Cellular injury induces activation of MAPK via P2Y receptors. J Cell Biochem. 2004;91:938–950. doi: 10.1002/jcb.10774. [DOI] [PubMed] [Google Scholar]
- 21.Burnstock G. The past, present and future of purine nucleotides as signaling molecules. Neuropharmacology. 1997;36:1127–1139. doi: 10.1016/s0028-3908(97)00125-1. [DOI] [PubMed] [Google Scholar]
- 22.Abbracchio M.P., Burnstock G. Purinergic signalling: pathophysiological roles. Jpn J Pharmacol. 1998;78:113–145. doi: 10.1254/jjp.78.113. [DOI] [PubMed] [Google Scholar]
- 23.Neary J.T., Kang Y., Bu Y., Yu E., Akong K., Peters C.M. Mitogenic signaling by ATP/P2Y purinergic receptors in astrocytes: involvement of a calcium-independent protein kinase C, extracellular signal-regulated protein kinase pathway distinct from the phosphatidylinositol-specific phospholipase C/calcium pathway. J Neurosci. 1999;19:4211–4220. doi: 10.1523/JNEUROSCI.19-11-04211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boucher I., Rich C., Lee A., Marcincin M., Trinkaus-Randall V. The P2Y2 receptor mediates the epithelial injury response and cell migration. Am J Physiol Cell Physiol. 2010;299:C411–C421. doi: 10.1152/ajpcell.00100.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moro L., Dolce L., Cabodi S., Bergatto E., Erba E.B., Smeriglio M., Turco E., Retta S.F., Giuffrida M.G., Venturino M., Godovac-Zimmerman J., Conti A., Schaefer E., Beguinot L., Tacchetti C., Gaggini P., Silengo L., Tarone G., Defilippi P. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem. 2002;227:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- 26.Sturla L., Amorino G., Alexander M., Mikkelsen R., Valeriw K., Schmidt-Ullrich R. Requirement of Tyr-992 and Tyr-1173 in phosphorylation of the epidermal growth factor receptor by ionizing radiation and modulation by SHP2. J Biol Chem. 2005;280:14597–14604. doi: 10.1074/jbc.M413287200. [DOI] [PubMed] [Google Scholar]
- 27.Saito T., Okada S., Ohshima K., Yamada E., Sato M., Uehara Y., Shimizu H., Pessin J.E., Mori M. Differential activation of epidermal growth factor (EGF) receptor downstream signaling pathways by betacellulin and EGF. Endocrinology. 2004;145:4232–4243. doi: 10.1210/en.2004-0401. [DOI] [PubMed] [Google Scholar]
- 28.Fischer O.M., Hart S., Gschwind V., Prensel N., Ullrich A. Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol Cell Biol. 2004;24:5172–5183. doi: 10.1128/MCB.24.12.5172-5183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Block E.R., Matela V., Sundar Raj N., Iszkula E.R., Klarlund J. Wounding induces motility in sheets of corneal epithelial cells through loss of spatial constraints. J Biol Chem. 2004;279:24307–24312. doi: 10.1074/jbc.M401058200. [DOI] [PubMed] [Google Scholar]
- 30.Stoll S.W., Johnson J.L., Bhasin A., Johnston A., Gudjonsson J.E., Rittié L., Elder J.T. Metalloproteinase-mediated, context-dependent function of amphiregulin and HB-EGF in human keratinocytes and skin. J Invest Dermatol. 2010;130:295–304. doi: 10.1038/jid.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trinkaus-Randall V., Gipson I.K. A technique for obtaining basal corneal epithelial cells. Invest Ophthalmol Vis Sci. 1985;26:233–237. [PubMed] [Google Scholar]
- 32.Riese D.J., 2nd, van Raaij T.M., Plowman G.D., Andrews G.C., Stern D.F. The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol. 1995;15:5770–5776. doi: 10.1128/mcb.15.10.5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer R.D., Latz C., Rahimi N. Recruitment and activation of phospholipase C gamma1 by vascular endothelial growth factor receptor-2 are required for tubulogenesis and differentiation by endothelial cells. J Biol Chem. 2003;278:16347–16355. doi: 10.1074/jbc.M300259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monis G., Schultz C., Ren R., Eberhard J., Costello C., Connors L., Skinner M., Trinkaus-Randall V. Role of endocytic inhibitory drugs on internalization of amyloidogenic light chains by cardiac fibroblasts. Am J Pathol. 2006;169:1939–1952. doi: 10.2353/ajpath.2006.060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olayioye M.A., Graus-Porta D., Beerli R.R., Rohrer J., Gay B., Hynes N.E. ErbB-1 and ErbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol Cell Biol. 1998;18:5042–5051. doi: 10.1128/mcb.18.9.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh A.J., Meyer R.D., Band H., Rahimi N. The carboxyl terminus of VEGFR-2 is required for PKC-mediated down-regulation. Mol Biol Cell. 2005;16:2106–2118. doi: 10.1091/mbc.E04-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheardown H., Cheng Y.L. Tear EGF concentration following corneal epithelial wound creation. J Ocul Pharmacol Ther. 1996;12:239–243. doi: 10.1089/jop.1996.12.239. [DOI] [PubMed] [Google Scholar]
- 38.Chung E.-H., Hutcheon A.E.K., Joyce N.C., Zieske J.D. Synchronization of the G1/S transition in response to corneal debridement. Invest Ophthalmol Vis Sci. 1999;40:1952–1958. [PubMed] [Google Scholar]
- 39.Hutcheon A.E.K., Guo X.Q., Stepp M.A., Simon K.J., Weinreb P.H., Violette S.M., Zieske J.D. Effect of wound type on Smad 2 and 4 translocation. Invest Ophthalmol Vis Sci. 2005;46:2362–2368. doi: 10.1167/iovs.04-0759. [DOI] [PubMed] [Google Scholar]
- 40.Zieske J.D., Francesconi C.M., Guo X. Cell cycle regulators at the ocular surface. Exp Eye Res. 2004;78:447–456. doi: 10.1016/s0014-4835(03)00205-7. [DOI] [PubMed] [Google Scholar]
- 41.Singh A.B., Sugimoto K., Harris R.C. Juxtacrine activation of epidermal growth factor (EGF) receptor by membrane-anchored heparin-binding EGF-like growth factor protects epithelial cells from anoikis while maintaining an epithelial phenotype. J Biol Chem. 2007;282:32890–32901. doi: 10.1074/jbc.M702677200. [DOI] [PubMed] [Google Scholar]
- 42.Hyatt D.C., Ceresa B.P. Cellular localization of the activated EGFR determines its effect on cell growth in MDA-MB-468 cells. Exp Cell Res. 2008;314:3415–3425. doi: 10.1016/j.yexcr.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiley H.S. Anomalous binding of epidermal growth factor to A431 cells is due to the effect of high receptor densities and a saturable endocytic system. J Cell Biol. 1988;107:801–810. doi: 10.1083/jcb.107.2.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan V.H., Tamama K., Au A., Littrell R., Richardson L.B., Wright J.W., Wells A., Griffith L.G. Tethered epidermal growth factor provides a survival advantage to mesenchymal stem cells. Stem Cells. 2007;25:1241–1251. doi: 10.1634/stemcells.2006-0320. [DOI] [PubMed] [Google Scholar]
- 45.Kansra S., Stoll S.W., Johnson J.L., Elder J.T. Autocrine extracellular signal-regulated kinase (ERK) activation in normal human keratinocytes: metalloproteinase-mediated release of amphiregulin triggers signalling from ErbB1 to ERK. Mol Biol Cell. 2004;15:4299–4309. doi: 10.1091/mbc.E04-03-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang C.P., Kao J.P., Lazar C.S., Walsh B.J., Wiley H.S., Gill G.N., Rosenfeld M.G. Ligand-induced internalization and increased cell calcium are mediated via distinct structural elements in the carboxyl terminus of the epidermal growth factor receptor. J Biol Chem. 1991;266:23467–23470. [PubMed] [Google Scholar]
- 47.Soubeyran P., Kowanetz K., Szymkiewicz I., Langdon W.Y., Dikic I. Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature. 2002;416:183–187. doi: 10.1038/416183a. [DOI] [PubMed] [Google Scholar]
- 48.Sakaguchi K., Okabayashi Y., Kasuga M. Shc mediates ligand-induced internalization of epidermal growth factor receptors. Biochem Biophys Res Commun. 2001;282:1154–1160. doi: 10.1006/bbrc.2001.4680. [DOI] [PubMed] [Google Scholar]
- 49.Jiang X., Huang F., Marusyk A., Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell. 2003;14:858–870. doi: 10.1091/mbc.E02-08-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sorkin A., Goh L.K. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 51.Vitorino P., Meyer T. Modular control of endothelial sheet migration. Genes Dev. 2008;22:3268–3281. doi: 10.1101/gad.1725808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu X., Trinkaus-Randall V. The expression of integrin subunits alpha 6 and beta 4 by corneal epithelial cells on modified hydrogel surfaces. J Biomed Mater Res. 1997;37:166–175. doi: 10.1002/(sici)1097-4636(199711)37:2<166::aid-jbm4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 53.Xu W.X., Kodama A., Fuchs E. ACF7 regulates cytoskeletal-focal adhesion dynamics and migration and has ATPase activity. Cell. 2008;135:137–148. doi: 10.1016/j.cell.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kageyama T., Ohishi M., Miyamoto S., Mizushima H., Iwamoto R., Mekada E. Diphtheria toxin mutant Crm197 possesses weak EF2-ADP-ribosyl activity that potentiates its anti-tumorogenic activity. J Biochem. 2007;142:95–104. doi: 10.1093/jb/mvm116. [DOI] [PubMed] [Google Scholar]
- 55.Brown C.T., Applebaum E., Banwatt R., Trinkaus-Randall V. Synthesis of stromal glycosaminoglycans in response to injury. J Cell Biochem. 1995;59:57–68. doi: 10.1002/jcb.240590108. [DOI] [PubMed] [Google Scholar]