

Abstract
Although mitogen-activated protein kinase phosphatase-1 (MKP-1) is a key deactivator of MAP kinases, known effectors of lung vessel formation, whether it plays a role in the expression of proangiogenic vascular endothelial growth factor (VEGF) in hypoxic lung is unknown. We therefore hypothesized that MKP-1 is a crucial modulator of hypoxia-stimulated vessel development by regulating lung VEGF levels. Wild-type MKP-1+/+, heterozygous MKP-1+/−, and deficient MKP-1−/− mice were exposed to sea level (SL), Denver altitude (DA) (1609 m [5280 feet]), and severe high altitude (HYP) (∼5182 m [∼17,000 feet]) for 6 weeks. Hypoxia enhanced phosphorylation of p38 MAP kinase, a substrate of MKP-1, as well as α smooth muscle actin (αSMA) expression in vessels, respiratory epithelium, and interstitium of phosphatase-deficient lung. αSMA-positive vessel (<50 μm outside diameter) densities were markedly reduced, whereas vessel wall thickness was increased in hypoxic MKP-1−/− lung. Mouse embryonic fibroblasts (MEFs) of all three genotypes were isolated to pinpoint the mechanism involved in hypoxia-induced vascular abnormalities of MKP-1−/− lung. Sustained phosphorylation of p38 MAP kinase was observed in MKP-1-null MEFs in response to hypoxia exposure. Although hypoxia up-regulated VEGF levels in MKP-1+/+ MEFs eightfold, only a 70% increase in VEGF expression was observed in MKP-1-deficient cells. Therefore, our data strongly suggest that MKP-1 might be the key regulator of vascular densities through the regulation of VEGF levels in hypoxic lung.
Dual-specificity phosphatases are the key regulators of dephosphorylation of the ERK1/2, JNK1/2, and p38 mitogen-activated protein (MAP) kinases, and so have been designated as MAP kinase phosphatases (MKPs). MKP-1 is a member of this phosphatase family.1 MKP-1 expression in the lung is considerably higher than in other tissues, and it has been shown to protect arteries from a proinflammatory state by suppressing the activities of p38 and JNK MAP kinases in the vascular endothelium.2,3 Although MKP-1 is a hypoxia-inducible phosphatase, and hypoxia triggers angiogenesis,4,5 the role of MKP-1 in the formation and maintenance of vascular network in hypoxic lung remains unexplored.
Vascular endothelial growth factor (VEGF) is crucial for the formation of new blood vessels and plays a central role in the development and maturation of a healthy vasculature, as well as in vascular pathophysiological conditions.5 Chronic hypoxia exposure leads to increased VEGF expression in the lung and subsequent angiogenesis.6–8 Recent studies have demonstrated that VEGF receptor pathway induces MKP-1 transcription in endothelial cells9,10; however, the role of MKP-1 in hypoxia-induced VEGF expression in the lung is unknown.
Although MKP-1-deficient mice seem to be largely normal, these animals have an exaggerated innate immune response to lipopolysaccharide (LPS) and exhibit increased serum levels of various cytokines, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interferon-γ, IL-10, IL-12, and monocyte chemotactic protein-1 (MCP-1; also known as C-C motif chemokine 2 protein, or CCL2).11–13 MKP-1-null mice are lean and resistant to diet-induced obesity.14 Activation levels of ERK1/2, JNK1/2, and p38 MAP kinase are elevated in skeletal muscle and white adipose tissue of these mice. MKP-1 has also been shown to play a role in the maintenance of bone mass by negatively regulating MAP kinase-dependent osteoclast signaling.15 In LPS-treated MKP-1−/− mice, the lungs manifest edema, thickening of the alveolar septa, and infiltration by leukocytes in the interstitial space.13 Jin et al16 recently reported that mice deficient in MKP-1 develop more severe pulmonary hypertension, with lower levels of nitric oxide synthase and greater arginase levels, after 4 weeks in hypoxia than do wild-type mice. However, little information exists regarding the vascular status of the lungs of MKP-1-deficient mice challenged with chronic exposure to hypoxia. Therefore, using MKP-1-deficient mice, we tested the hypothesis that MKP-1 functions as a regulator of the maintenance and development of vascular network in hypoxic lung by controlling VEGF levels.
In the present study, we found that chronic exposure to hypoxia induces exaggerated p38 MAP kinase activation and αSMA expression in lung of MKP-1-null mice. Most importantly, marked reduction in vessel densities and remodeling of the vascular wall were observed in lung of hypoxia-exposed MKP-1-deficient mice. Using cultured mouse embryonic fibroblasts (MEFs), we demonstrated that hypoxia-stimulated up-regulation of VEGF expression is defective on deletion of the MKP-1 gene, suggesting that MKP-1 is a crucial phosphatase for the regulation of VEGF levels and vessel densities in hypoxic lung.
Materials and Methods
Animal Studies
All experiments with mice were approved by the University of Colorado Denver Institutional Animal Care and Use Committee. MKP-1 heterozygous mice were provided by Lexicon Pharmaceuticals (The Woodlands, TX). Male mice, 4 to 5 weeks old, of the three genotypes MKP-1+/+, MKP-1+/−, and MKP-1−/− were divided into three groups and exposed to sea level (SL), ambient Denver (DA) (1609 m [5280 feet]), or simulated severe high altitude (HYP) (5182 m [17,000 feet]) conditions for 6 weeks, according to a previously described method.17,18 Briefly, the first group was placed in a hyperbaric chamber in which the pressure was increased to simulate sea-level barometric pressure (PB = 760 mmHg). The second group was kept at Denver's altitude (PB = 630 mmHg). The third group was placed in a hypobaric chamber and exposed to simulated severe high altitude (PB = 410 mmHg). The chambers were continuously flushed with room air to prevent accumulation of CO2, NH3, and H2O. Both hyperbaric and hypobaric exposures of mice were 24 hours/day, except when the chambers were opened for 10 to 15 minutes every 2 days to clean cages and replenish food and water. All mice were exposed to a 12:12 hours light-dark cycle and were allowed free access to standard chow and water. After 6 weeks, mice were anesthetized with tribromoethanol (500 mg/kg, i.p.), weighed, and subjected to whole-body perfusion with EDTA-containing heparinized saline. Lungs were inflated with 1% agarose, solidified on ice, fixed in formalin overnight, embedded in paraffin, and then sectioned.
Genotyping of Mouse
For genotype analysis, DNA from mouse tail piece was isolated using a DNeasy kit (Qiagen, Valencia, CA). PCR analysis was performed using primer sets specific for MKP-1 and neomycin: MKP-1 forward: 5′-CTTCTCGGAAGGATATGCTTGACG-3′, reverse: 5′-TCAGTTCAACTGAGTCTCAGTGAGG-3′ and neomycin forward: 5′-GCAGCGCATCGCCTTCTATC-3′, reverse: 5′GGCTCCTCCCAAGCTTTGTATATCC-3′.
Isolation of Mouse Embryonic Fibroblasts
Pregnant (11 to 13 days) female mice were sacrificed with pentobarbital (200 mg/kg i.p.) followed by cervical dislocation. The uterine horns were removed and the embryos were dissected in sterile PBS. Fetal membranes were used for genotyping, and embryos minus primordial organs were mechanically dispersed, strained, and plated on 0.1% gelatin-coated Petri dishes with Dulbecco's modified Eagle's medium/Ham's F-12 medium (DMEM/F-12) containing 15% fetal bovine serum. Confluent cells were detached with Accutase (Innovative Cell Technologies, San Diego, CA), counted with a hemocytometer, and plated for experiments.
Immunohistochemistry
The following antibodies were used for staining of the lung sections: αSMA (1:100) (Sigma-Aldrich, St. Louis, MO)19,20; phosphoP38 (1:50), phosphoJNK (1:100), and phosphoERK (1:100) (Cell Signaling Technology, Danvers, MA)21,22; Ki-67 (1:100) (Epitomics, Burlingame, CA)23,24; and VEGF (1:100) (Novus Biologicals, Littleton, CO).25 Immunohistochemical staining was performed according to our previously described method.26 Briefly, a universal avidin-biotin-peroxidase kit (Vector Laboratories, Burlingame, CA) was used for immunohistochemical staining. Peroxidase activity was visualized with diaminobenzidine (DAB) (Vector Laboratories). Sections were lightly counterstained with hematoxylin, coverslipped, and mounted. A universal negative control (DakoCytomation, Glostrup, Denmark) was used for staining control slides. Murine kidney, brain, heart, and spleen were also stained with the above-mentioned antibodies and were used as positive controls for immunohistochemical staining of lung sections. Images were captured at ×40 with Spot Advanced software v4.0.6 (Diagnostic Instruments, Sterling Heights, MI) and a Nikon Eclipse E800 microscope.
Determination of Density and Wall Thickness of Lung Vasculature
Vascular density and wall thickness in the lung of MKP-1+/+, MKP-1+/−, and MKP-1−/− mice were measured according to previously described methods.27–29 Briefly, lung sections were stained with αSMA antibody and lightly counterstained with hematoxylin. The number of αSMA-positive vessels [between 10 and 50 μm outside diameter (OD)] and alveoli were counted in αSMA-stained sections by an investigator who was unaware of the experimental conditions; 5 to 12 vessels were counted in each of the three lobes of lung in every animal. Vessel density was expressed as the ratio of vessels to alveoli.
For the evaluation of lung vascular wall thickness, Zeiss AxioVision software v2.05 was used. Diameter and wall thickness of muscular arteries were measured in αSMA-positive vessels (between 10 and 50 μm OD). Vascular wall thickness was expressed as the ratio of wall width to vessel width. Each lung section was assessed at five randomly captured fields at ×20, and at least 50 vessels were analyzed per animal.
Western Blot Analysis
Cell lysates harvested from MEFs were separated by immunoblot analysis for the evaluation of hypoxia-induced phosphorylation of MAP kinases and levels of αSMA protein according to our previously described method.30
Activation of MAP Kinases
MEFs were exposed to hypoxic (1% O2) conditions for 0, 30, 60, and 120 minutes and then were harvested with cell lysis buffer. Protein concentrations were measured using a Bradford protein assay (Bio-Rad, Hercules, CA). Equal amounts of total protein from each experimental condition were separated on Bis-Tris gels (NuPage; Invitrogen, Carlsbad, CA) and were transferred onto polyvinylidene difluoride membranes, blocked with 5% nonfat milk in Tris-buffered saline–Tween for 1 hour at room temperature, and incubated overnight with antibodies against phosphoP38 (1:500), phosphoJNK (1:500), and phosphoERK1/2 (1:1000) (Cell Signaling Technology). After washing in Tris-buffered saline–Tween solution, membranes were incubated with the alkaline phosphatase-conjugated anti-rabbit IgG (1:10,000) (Upstate; Millipore, Billerica, MA) for 1 hour at room temperature. Blots were developed using Lumi-Phos reagent (Pierce Biotechnology, Rockford, IL), and exposed to film. Protein bands were quantified using NIH Image J software v1.58.30
Expression of αSMA
MEFs were preincubated with inhibitors of MAP kinase pathways (U0126, SB203580, and SP600125, and with dimethyl sulfoxide as control) for 1 hour and then exposed to either normoxia or hypoxia for 24 hours. Antibody against αSMA (1:20,000) (Sigma-Aldrich) was used for overnight incubation at 4°C. Alkaline phosphatase-conjugated anti-mouse IgG 1:10,000 (Santa Cruz Biotechnology, Santa Cruz, CA) was used for 1 hour of incubation at room temperature.
Proliferation Assay
MEFs were plated in 96-well plates with 10% fetal bovine serum-containing media and then were growth-arrested for 72 hours according to our previously described method.31 Quiescent cells were preincubated for 1 hour with or without inhibitors (U0126, SB203580, and SP600125, and with dimethyl sulfoxide as control) and then were exposed to either normoxia or hypoxia for 72 hours. At the end of the experimental period, proliferation was measured with CellTiter 96 AQueous One Solution reagent according to the manufacturer's protocol (Promega, Madison, WI).
VEGF Enzyme-Linked Immunosorbent Assay
For the quantitation of intracellular VEGF levels, growth-arrested MEFs were exposed to either normoxia or hypoxia in the presence of various antagonists targeting members of the MAP kinase family for 24 hours. Cells were harvested at the end of exposure and enzyme-linked immunosorbent assay was performed with cell lysates using a mouse VEGF enzyme-linked immunosorbent assay kit (RayBiotech, Norcross, GA) according to the manufacturer's protocol. Briefly, equal amounts of protein from cell lysates were added in the wells of 96-well plates, which were precoated with mouse-specific anti-VEGF antibody, incubated overnight at 4°C, washed, and again incubated with biotinylated anti-mouse VEGF antibody for 1 hour at room temperature. After washing, horseradish peroxidase-conjugated streptavidin solution was added to each well and incubated for 45 minutes. 3,3′,5,5′-tetramethylbenzidine one-step substrate reagent was added to the wells after washing and incubated for 30 minutes. Stop solution was added to the wells at the end of the incubation and plates were immediately read at 450 nm.
Statistical Analysis
All data are expressed as arithmetic means ± SEM. For animal studies, the sample number n represents the number of mice per experimental group. For experiments with cultured cells, the n represents the number of cell populations, each isolated from an individual embryo, or the number of replicate wells per test condition in representative experiments. One-way analysis of variance and then Student-Newman-Keuls multiple comparison tests were conducted for comparisons within and between groups of data points. Data were considered significantly different if P < 0.05.
Results
Hypoxia Inhibits Weight Gain in MKP-1−/− Mice
We first genotyped MKP-1+/+, MKP-1+/−, and MKP-1−/− mice by PCR (Figure 1A) and exposed them to SL, DA, and HYP conditions for 6 weeks. The SL exposure was used as a control group, because mice were bred and maintained at DA (1609 m higher than SL), and this elevated altitude significantly affects the physiological responses of the animals. Hypoxia-exposed MKP-1−/− mice had a significantly greater reduction in total body weight gain, compared with MKP-1-deficient mice in SL and DA conditions, after 6 weeks (Figure 1B). By contrast, total body weight gain was not affected by altitude in MKP-1+/+ and MKP-1+/− mice (Figure 1B). Therefore, the MKP-1 gene might be a critical regulator of body weight under hypoxic conditions.
Figure 1.
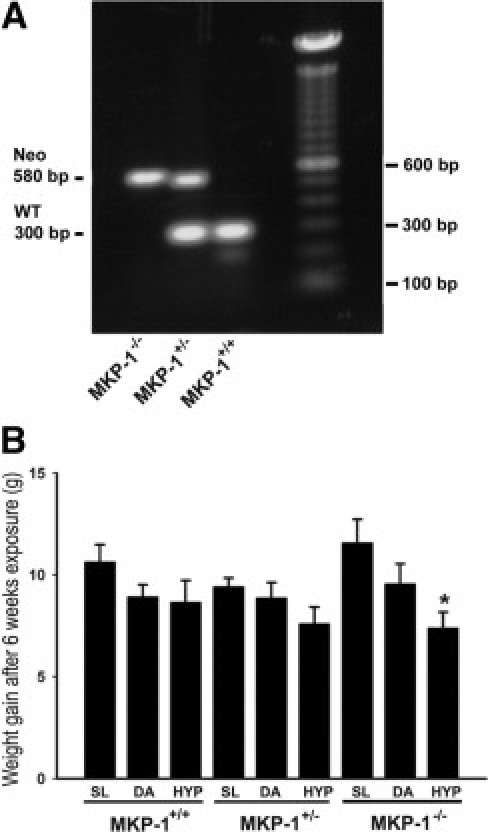
Chronic hypoxia exposure interferes with total body weight gain in MKP-1-deficient mice. A: Genotyping of MKP-1+/+, MKP-1+/−, and MKP-1−/− mice by PCR. Mouse tail DNA amplified by PCR shows a MKP-1 band of 300 bp and a neomycin band of 580 bp. B: Weight gain in mice in the absence of MKP-1 expression is attenuated by hypoxia exposure (n = 8 to 15 animals/group). MKP-1−/− mice had marked decrease in gain of body weight compared with MKP-1+/+ and MKP-1+/− mice after 6 weeks exposure to a simulated altitude of ∼5182 m (∼17,000 feet). *P < 0.05 compared with SL group of MKP-1-deficient mice. SL, sea level; DA, Denver altitude; HYP, high-altitude hypoxia.
ERK1/2 and p38 MAP Kinases Are Activated in Lung of MKP-1+/− and MKP-1−/− Mice
To evaluate the activation status of substrates of MKP-1 (ERK1/2, JNK1/2, and p38 MAP kinase), lung sections were immunostained with the antibodies against phosphoERK1/2, phosphoJNK1/2, and phosphoP38 MAP kinase. The strongest staining intensities of activated ERK1/2 were observed in the vessel wall, alveoli, and airway of SL-exposed lung of MKP-1+/− and MKP-1−/− mice (Figure 2A). There was no positive staining of phosphorylated ERK1/2 in MKP-1+/+ lung at any altitude condition (Figure 2A). We did not detect any differences in the staining patterns of phosphoJNK1/2 in lung of all three genotypes (data not shown).
Figure 2.
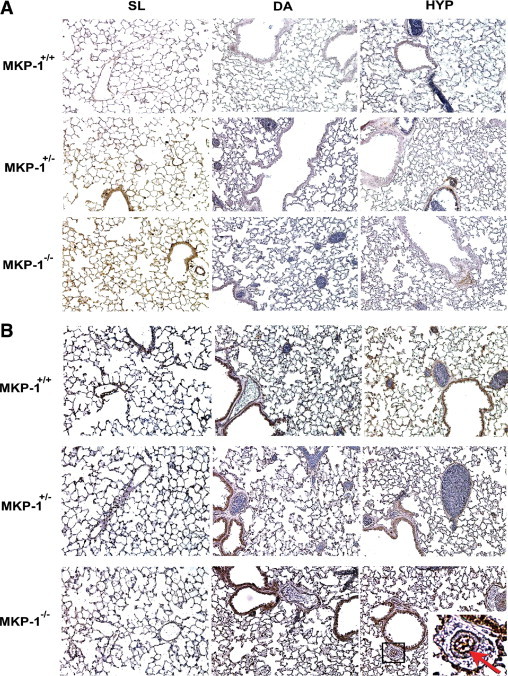
Heightened phosphorylation of ERK1/2 and p38 MAP kinase is observed in the lung of MKP-1-deficient mice. A: Sea-level (SL)-exposed lung of MKP-1+/− and MKP-1−/− mice have the greatest phosphorylated ERK1/2 levels. Positive signal of immunostaining is represented as brown (DAB) in lung section (n = 3 to 4 mice/group). B: Staining intensity of phosphoP38 MAP kinase is strongest in lung of MKP-1−/− mice both at DA and HYP conditions (n = 3 to 4 animals/group). In MKP-1−/− lung section of HYP group, the small box marks a vessel, magnified in the inset; the red arrow indicates the endothelial cell layer of the vessel. Original magnification ×400; ×1200 (inset).
In SL-exposed lung of all three genotypes, either minimal or no positive reaction was observed with antibody against phosphoP38 MAP kinase (Figure 2B). However, activated p38 MAP kinase was detected by immunoperoxidase staining in DA-exposed lung of all genotypes (Figure 2B). The strongest signal for phosphoP38 MAP kinase was observed in HYP-exposed lung (Figure 2B). MKP-1−/− lung had maximal levels of p38 MAP kinase phosphorylation in the vascular wall, alveoli, and airways in response to chronic hypoxia exposure (Figure 2B). Interestingly, phosphorylated p38 MAP kinase might be localized in the intimal endothelial cells of the vascular wall in lung of MKP-1−/− mice (Figure 2B, arrow in inset). Taken together, these data suggest that activation of ERK1/2 (SL) and p38 MAP kinase (HYP) is greatest in lung of MKP-1−/− mice.
MKP-1−/− Mice Have Altered Expression of αSMA and VEGF in the Lung
To examine the role of MKP-1 in the development and maintenance of vessels, lung sections were stained with anti-αSMA and anti-VEGF antibodies. Mouse lung of all three genotypes expressed αSMA under all three altitude conditions. However, MKP-1−/− mice had greatest levels of αSMA expression in lung at both DA and HYP conditions (Figure 3A). αSMA was expressed in the vessels, airways, and interstitial cells of MKP-1-deficient lung (Figure 3A). Exaggerated αSMA expression in the vascular wall of MKP-1-null lung (Figure 3A, arrows in insets, SL and HYP groups) suggests that chronic exposure to hypoxia might trigger structural remodeling of the wall.
Figure 3.
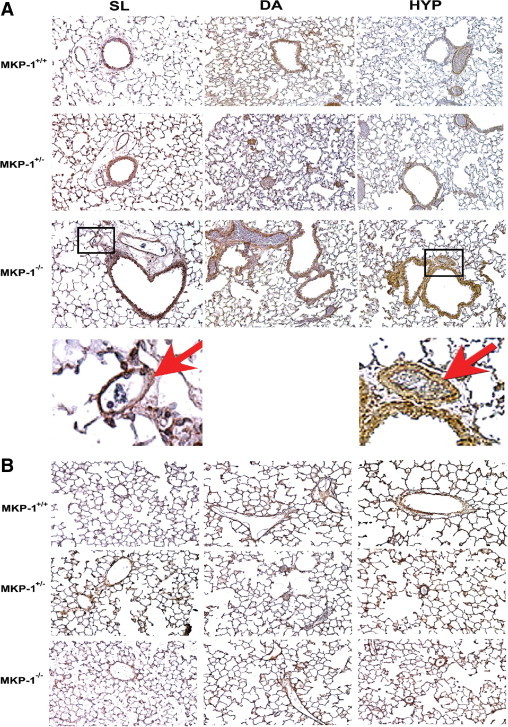
Deletion of MKP-1 affects the expression of αSMA and VEGF in the lung. A: αSMA expression is up-regulated in hypoxia-exposed MKP-1-deficient lung (n = 3 to 4 animals/group). Insets identify areas from MKP-1−/− lung of SL and HYP groups that are magnified in the row below, with red arrows indicating the vessels positive for αSMA staining. B: VEGF expression is increased in hypoxic lung of wild-type mice (n = 3 to 4 animals/group). Original magnification ×400; ×1600 (insets).
We then evaluated lung vasculature of these mice by VEGF immunostaining (Figure 3B). There were no striking differences in the staining patterns of VEGF in lung of MKP-1+/− and MKP-1−/− mice across the three altitude conditions examined (Figure 3B). However, VEGF levels were up-regulated in HYP-exposed MKP-1+/+ lung, because the staining intensity was enhanced (Figure 3B). Therefore, these data suggest that αSMA and VEGF expression in the lung might be affected by the absence of MKP-1 expression.
Hypoxia Induces Reduction in and Remodeling of αSMA-Positive Vessels in Lung of MKP-1+/− and MKP-1−/− Mice
We then counted αSMA-positive vessels (<50 μm OD) and alveoli in αSMA-stained lung sections. In MKP-1+/+ lung, vessel numbers were markedly decreased in the DA and HYP groups, compared with those of the SL condition (Figure 4A). There was a trend toward an increase in the vessel/alveoli ratio in hypoxic lung of these mice, compared with those under DA conditions (Figure 4A); however, the difference was not statistically significant. In the case of MKP-1+/− and MKP-1−/− mice, the vessel/alveoli ratio was sequentially reduced with increasing altitude throughout the lung (Figure 4A). In MKP-1-deficient lung, vessel densities were greatest under SL conditions and least under HYP conditions (Figure 4A), suggesting that MKP-1 might be an important regulator of the maintenance of vessel density in hypoxic lung.
Figure 4.
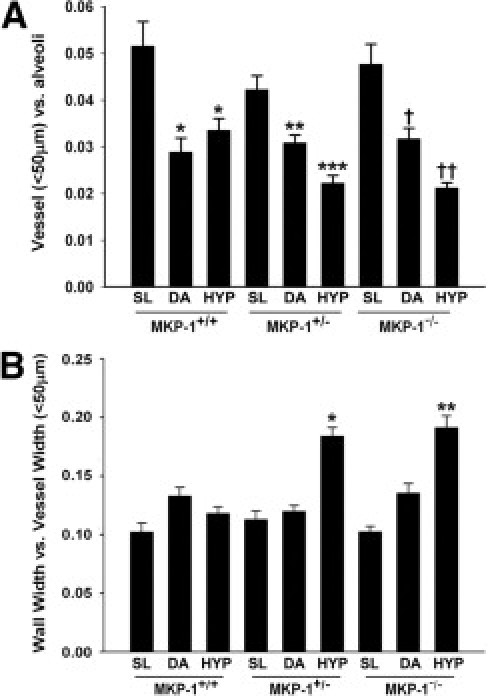
Hypoxia induces loss and structural remodeling of αSMA-positive vessels (<50 μm OD) in MKP-1-deficient lung. A: αSMA-positive vessel densities are markedly decreased in hypoxic lung of MKP-1+/− and MKP-1−/− mice. Data reported as mean ± SEM, 3 to 5 animals per group. *P < 0.001 compared with SL-exposed MKP-1+/+ lung. **P < 0.001 compared with SL-exposed MKP-1+/− lung. ***P < 0.01 compared with SL- and DA-exposed MKP-1+/− mice and HYP-exposed MKP-1+/+ mice. †P < 0.001 compared with SL-exposed MKP-1−/− mice. ††P < 0.05 compared with SL- and DA-exposed MKP-1−/− mice and HYP-exposed MKP-1+/+ mice. n = 3 to 5 animals/group; 5 to 12 vessels in each lung/animal were counted. B: Hypoxia induces structural remodeling of the vascular wall (wall width to vessel width ratio) in lung of MKP-1+/− and MKP-1−/− mice. *P < 0.001 compared with SL- and DA-exposed MKP-1+/− and HYP-exposed MKP-1+/+ mice. **P < 0.001 compared with SL- and DA-exposed MKP-1−/− and HYP-exposed MKP-1+/+ mice. n = 3 to 5 animals/group; at least 50 vessels/animal were analyzed.
Structural remodeling of the vessels (<50 μm OD) was evaluated by measuring the ratio of vessel wall width to vessel diameter. Altitude did not have any effect on the vascular wall thickness in the lung of wild-type mice. However, the ratio was markedly increased in HYP-exposed lung of MKP-1+/− and MKP-1−/− mice, indicating that the vessel wall underwent relative thickening with respect to the overall vessel diameter (Figure 4B). Remodeled vessels were distributed throughout the lungs of MKP-1-deficient mice. Taken together, these data suggest that MKP-1 is a crucial phosphatase for the development and maintenance of vessels (<50 μm OD) in the lung under hypoxic conditions.
Ki-67 Labeling Is Greatest in Hypoxia-Exposed Lung of Wild-Type Mice
To evaluate whether vascular wall remodeling in hypoxic MKP-1+/− and MKP-1−/− lung is due to cell proliferation, lung sections were stained with antibody against Ki-67, a proliferative marker. Ki-67-positive vessels were not detectable in SL- or in DA-exposed lung of all three genotypes (Figure 5); in the HYP-exposed lung, however, Ki-67 immunoreactivity became apparent. Hypoxia-exposed MKP-1+/+ lung had the highest number of Ki-67-positive cells, compared with those of MKP-1+/− and MKP-1−/− animals (Figure 5). These data suggest that, at 6 weeks of hypoxia exposure, cell replication might not be present in the remodeled vessels of MKP-1-deficient lung.
Figure 5.
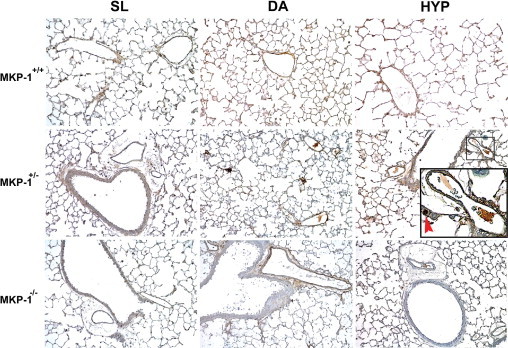
Ki-67-positive cells are most abundant in hypoxic lung of MKP-1+/+ mice. Representative images of lung from MKP-1+/+, MKP-1+/−, and MKP-1−/− mice exposed to SL, DA, and HYP for 6 weeks (n = 3 animals/group). In the MKP-1+/− lung section of HYP group, the small box marks an example of Ki-67-positive nuclei, magnified in the inset; the red arrow points to a Ki-67-positive nucleus. Original magnification ×400; ×1200 (inset).
Hypoxia-Induced Activation of MAP Kinases Is Highest in MKP-1−/− MEFs
To examine the mechanisms involved in hypoxia-induced modification of vasculature in mouse lung lacking MKP-1 expression, MEFs of all three genotypes were isolated and cultured. Hypoxia-stimulated activation of MAP kinases was evaluated in these MEFs using antibodies against phosphoMAP kinases. In MKP-1+/+ cells, ERK1/2 were maximally (twofold) phosphorylated at 30 minutes of hypoxia exposure. ERK1/2 dephosphorylation occurred by the 60 minutes time point and was maintained up to 120 minutes of hypoxia exposure in wild-type cells (Figure 6A). Basal levels of phosphorylated ERK1, as evidenced by detection of a strong upper band on an immunoblot, were higher in MKP-1+/− MEFs than in MKP-1+/+ and MKP-1−/− cells (Figure 6A). There was no statistically significant increase in ERK1/2 activation in hypoxia-exposed MKP-1+/− cells (Figure 6A). However, ERK1/2 phosphorylation was up-regulated fivefold in MKP-1−/− MEFs at 30 minutes of hypoxia exposure, which represents a time point of maximal ERK1/2 activation in response to hypoxic stimulation observed across the three genotypes (Figure 6A). Furthermore, ERK1/2 phosphorylation persisted in hypoxia-exposed MKP-1-null cells, albeit at lower levels, with threefold and twofold activation at 60 and 120 minutes, respectively (Figure 6A). Thus, MKP-1−/− MEFs acquire highest and sustained ERK1/2 activation in response to hypoxic stimulation.
Figure 6.
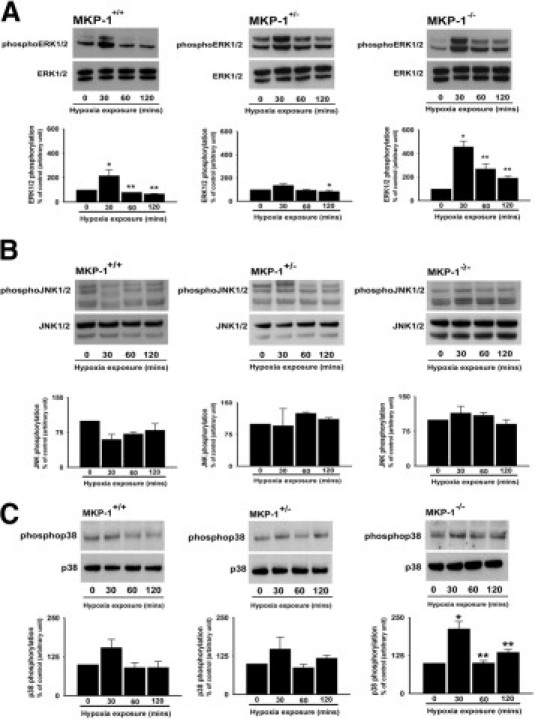
Hypoxic MKP-1−/− MEFs exhibit maximal phosphorylation of ERK1/2 and p38 MAP kinase. A: ERK1/2 phosphorylation is sustained in hypoxic MKP-1-null MEFs (n = 3 to 4 cell populations each isolated from individual embryo). MKP-1+/+: *P < 0.05 compared with 0 hours. **P < 0.05 compared with 30 minutes. MKP-1+/−: *P < 0.05 compared with 30 minutes. MKP-1−/−: *P < 0.01 compared with 0 hours. **P < 0.05 compared with 30 minutes. B: MEFs have high basal JNK1/2 phosphorylation levels (n = 3 to 4 populations, each cultured from a different embryo). C: Only MKP-1-deficient MEFs have significant p38 activation in response to hypoxia exposure (n = 3 to 4 cell populations, each isolated from single embryo). MKP-1−/−: *P < 0.05 compared with 0 hours. **P < 0.05 compared with 30 minutes.
To develop a MAP kinase activation profile of hypoxic MEFs, we next evaluated JNK1/2 and p38 MAP kinase activation. Basal levels of phosphorylated JNK1/2 were high and not affected by hypoxia across all three cell types (Figure 6B). In the case of p38 MAP kinase, the basal phosphorylation levels were again considerably high in cells of all three genotypes (Figure 6C). There was an increase in p38 MAP kinase phosphorylation in MKP-1+/+ and MKP-1+/− cells after 30 minutes of hypoxia exposure; however, the up-regulation of phosphorylation levels was not statistically significant (Figure 6C). By contrast, p38 MAP kinase phosphorylation was augmented more than twofold in MKP-1−/− MEFs at 30 minutes of hypoxia exposure (Figure 6C). Taken together, these data suggest that response of MKP-1-deficient MEFs to hypoxia relies on phosphorylation of ERK1/2 and p38 MAP kinase, but not on JNK1/2 activation.
VEGF Expression Is Blunted in Hypoxic MKP-1-Deficient MEFs
To evaluate the levels of αSMA and VEGF, quiescent MEFs of all genotypes were exposed to either normoxia or hypoxia. The basal αSMA expression was noticeably higher in MKP-1-null cells than in the wild-type MEFs (Figure 7A). To evaluate the role of MAP kinases in αSMA expression, cells were stimulated with hypoxia either in the absence or in the presence of various pharmacological antagonists of MAP kinase pathways. Only up on blockade of JNK1/2 with SP600125, hypoxia significantly up-regulated αSMA levels in MKP-1+/+ MEFs suggesting that JNK1/2 might be a negative regulator of αSMA expression in these cells (Figure 7A). Neither hypoxia exposure nor blockade of MAP kinases with various inhibitors had any effect on αSMA levels in MKP-1-deficient cells (Figure 7A). In MKP-1+/− MEFs, the findings regarding αSMA expression resembled those from the null cells. αSMA levels were unaffected by different inhibitors of MAP kinase pathways (data not shown). Therefore, αSMA expression in MEFs might be regulated by MKP-1.
Figure 7.
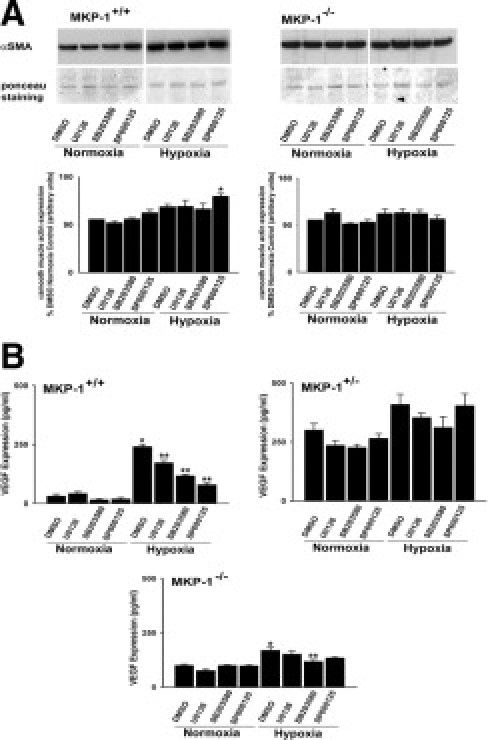
MKP-1 depletion affects αSMA and VEGF expression in MEFs. A: Blockade of JNK pathways with SP600125 up-regulates αSMA expression in hypoxic MKP-1+/+ MEFs (n = 3 to 4 cell populations, each isolated from an individual embryo). *P < 0.05 compared with the results of SP600125 treated cells under normoxic conditions. B: Hypoxia selectively up-regulates VEGF expression in MKP-1+/+ MEFs (n = 3 to 4 populations, each cultured from an individual embryo). MKP-1+/+: *P < 0.001 compared with normoxic dimethyl sulfoxide (DMSO). **P < 0.001 compared with hypoxic DMSO. MKP-1−/−: *P < 0.001 compared with normoxic DMSO and **P < 0.05 compared with hypoxic DMSO.
Because we hypothesized an effect of MKP-1 on VEGF expression, we next examined whether hypoxia stimulates increase in VEGF levels in the MEFs. Notably, eightfold up-regulation of VEGF levels was observed in hypoxia-exposed wild-type cells (Figure 7B). Blockade of ERK1/2 and p38 MAP kinase with U0126 and SB203580, respectively, markedly blunted the increase in VEGF levels (Figure 7B). However, the greatest inhibitory effects on VEGF were found in hypoxic wild-type MEFs in the presence of JNK1/2 attenuation by SP600125 (Figure 7B). In the case of MKP-1+/− MEFs, the basal levels were considerably greater (300 pg/ml) compared with that in wild-type cells (31 pg/ml) (Figure 7B). Hypoxia did not have any effect on VEGF expression in these cells, nor did different inhibitors of MAP kinase pathways (Figure 7B). MKP-1-null cells also possessed higher basal VEGF levels (100 pg/ml), compared with MKP-1+/+ MEFs (31 pg/ml) (Figure 7B). There was only a 70% increase in VEGF levels in hypoxic MKP-1−/− MEFs, a value that is markedly lower than for wild-type cells (Figure 7B). Furthermore, VEGF expression in hypoxia-exposed MKP-1−/− MEFs was reduced only in the presence of p38 inhibitor with SB203580 (Figure 7B).
MKP-1−/− MEFs Lack Hypoxia-Stimulated Replication Responses
To examine the role of MKP-1 in hypoxia-induced proliferative responses of the cells, growth-arrested MEFs were exposed to either normoxia or hypoxia for 72 hours. Increase in proliferation was observed in hypoxic MKP-1+/+ and MKP-1+/− MEFs, but not in MKP-1−/− MEFs (Figure 8). We then evaluated the effects of different MAP kinase inhibitors on proliferative responses of the cells. Hypoxia-induced replication in wild-type cells was reduced in the presence of ERK1/2 inhibition by U0126, JNK1/2 attenuation by SP600125, and p38 MAP kinase blockade by SB203580 (Figure 8). In the case of MKP-1+/− and MKP-1−/− MEFs, replication rate was reduced only in the presence of JNK1/2 inhibition under hypoxic conditions (Figure 8). Our findings suggest that MEFs in the absence of MKP-1 expression might acquire dysregulated cell proliferation in response to hypoxic stimulation.
Figure 8.
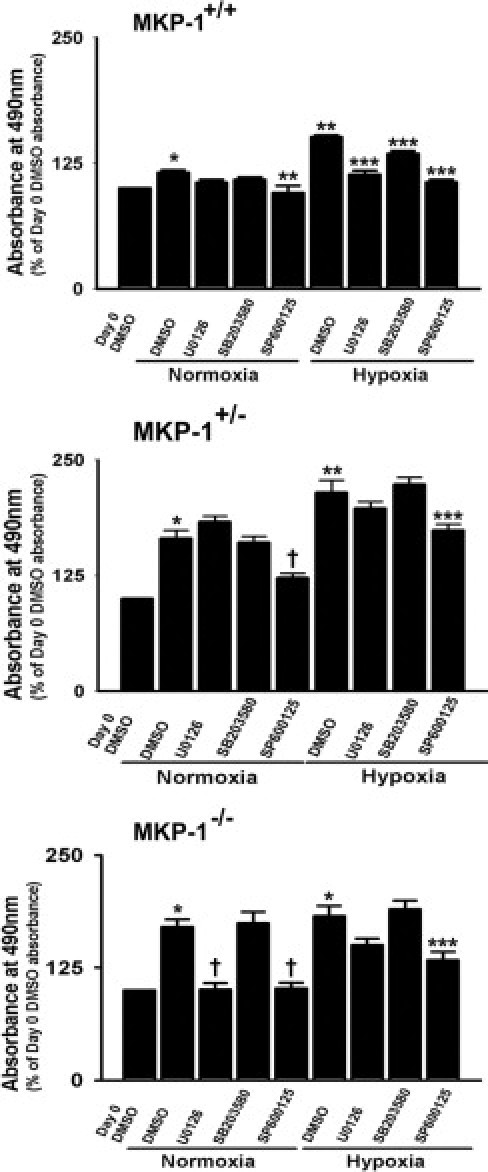
Hypoxia stimulates proliferation in MKP-1+/+ and MKP-1+/− but not in MKP-1−/− MEFs. In MKP-1+/+ MEFs, hypoxia-induced replication was attenuated by inhibitors of MAP kinase pathway (n = 8 replicate wells with 3 to 4 populations of MEFs, each isolated and cultured from individual embryo). *P < 0.05 compared with absorbance value of day 0 DMSO for cells from all three genotypes. **P < 0.001 compared with day 3 DMSO under normoxic condition for MKP-1+/+ and MKP-1+/− MEFs. ***P < 0.001 compared with day 3 DMSO under hypoxic conditions for all three genotypes. †P < 0.001 compared with day 3 DMSO under normoxic conditions for MKP-1+/− and MKP-1−/− MEFs. Data reported as mean ± SEM.
Discussion
In the present study, we found that MKP-1 functions as a key phosphatase in hypoxic lung through the regulation of VEGF levels and subsequent vessel density. We found that p38 MAP kinase, a substrate of MKP-1, is strongly activated in lung of MKP-1-deficient mice in response to chronic hypoxia exposure. Hypoxia also dramatically up-regulated αSMA expression in MKP-1-deficient lung. Most importantly, we demonstrated that chronic hypoxia exposure induces reduction and structural remodeling of αSMA-positive vessels (<50 μm OD) in lung lacking the MKP-1 gene. Using isolated MKP-1-deficient MEFs, we also found that these cells have maximal and sustained hypoxia-induced activation of ERK1/2 and p38 MAP kinase. However, the cells have faulty hypoxia-stimulated VEGF expression, which might contribute to decrease in lung vasculature density of MKP-1-null mice in response to chronic hypoxia exposure. Our data strongly suggest that MKP-1 acts as an important phosphatase for the maintenance of vessels through the regulation of VEGF levels in hypoxic lung and thus represents a promising pharmacological target for the treatment of numerous vascular disorders.
MKP-1 belongs to a family of dual-specific phosphatases, able to dephosphorylate both threonine and tyrosine residues. Expression of MKP-1 is increased by cellular stressors such as hypoxia, UV light, oxidative stress, glucocorticoid stimulation, and heat shock.32–36 MKP-1-null mice exhibit enhanced ERK1/2, JNK1/2, and p38 MAP kinase activities and are resistant to diet-induced obesity, suggesting that this phosphatase might be a crucial regulator of body weight.14 The concept of MKP-1 as a weight-controlling phosphatase is in accord with our present findings, that chronic exposure to hypoxia interfered with weight gain in MKP-1-null mice (Figure 1B). Recently, Jin et al16 reported similar findings, that chronic hypoxia exposure (4 weeks) attenuated gain in body weight of adult MKP-1-null mice. It will be of broad interest to dissect the mechanisms involved in the regulatory role of MKP-1 in total body weight gain on challenge with chronic hypoxia exposure.
Studies using MKP-1-deficient mice have indicated a role for MKP-1 as a negative regulator of Toll-like receptors, which are important in innate immunity.37 Although recent studies have demonstrated that MKP-1 plays an important role in ex vivo aortic angiogenesis9 and is a hypoxia-inducible phosphatase,4,32 the role of MKP-1 in the maintenance of vascular density in the lung, especially on hypoxic challenge, remains unknown. Our data regarding the loss and structural remodeling of vessels (<50 μm OD) in hypoxic MKP-1-null lung suggest that MKP-1 might act as a regulator of progression of various vascular diseases.
Hypoxia has been shown to promote angiogenesis via increase in MKP-1 expression.38 Treating sections of descending aorta from wild-type and MKP-1-null mice with VEGF or thrombin has revealed that endothelial cell sprouting is greatly reduced in MKP-1-null aortic sections, compared with control mice, confirming proangiogenic activity of MKP-1.9 In the present study, marked reduction in vessel densities in the hypoxic lung of MKP-1-deficient animals also strongly supports a critical regulatory role of this phosphatase as an angiogenic signaling mediator.
The classical paradigm in vessel formation implicates endothelial proliferation, migration, tube assembly, and remodeling as sequential steps.5 The loss of vessels could be due to alterations in endothelial cells and/or pericytes resulting in apoptosis. Indeed, vascular endothelial cells in lung of MKP-1-deficient mice have heightened p38 MAP kinase activation after 6 weeks of hypoxia exposure (Figure 2B). Given that p38 MAP kinase plays an important role in endothelial cell apoptosis,39 exaggerated p38 MAP kinase phosphorylation in endothelial cells might be one of the factors involved in vessel loss in MKP-1-deficient mice, a hypothesis that needs more rigorous examination in lung of MKP-1-null animals.
A role for phosphatases in either vessel formation or maintenance is also supported by other studies demonstrating that the phenotype of vascular endothelial protein tyrosine phosphatase knockout mice is embryonic lethal, with impaired vessel remodeling and maintenance.40,41 Although MKP-1-deficient mice appear to be normal under control conditions, our data strongly suggest that lungs of these mice have blunted response to hypoxia, due to the defective expression of the major angiogenic factor VEGF, as well as flaws in vessel formation or maintenance.
Decrease in vascularity has been shown to be involved in vascular remodeling.42 Endothelial cell apoptosis might contribute to vascular remodeling via release of secondary mediators leading to smooth muscle cell proliferation.43 In the present study, endothelial cell apoptosis might also be an important mechanism by which vascular remodeling develops in hypoxic MKP-1+/− and MKP-1−/− lung. However, proliferative index of vascular cells as evaluated by Ki-67 staining (Figure 5) was not higher in hypoxic MKP-1-deficient lung. In fact, Ki-67-positive nuclei were most numerous in hypoxic lung of wild-type mice. Greater proliferating cell nuclear antigen (PCNA) levels have been recently detected in lung of hypoxic (4 weeks exposure) MKP-1−/− mice than that in hypoxic wild-type mice.16 Because these PCNA levels have been evaluated by Western blot analysis in total lung homogenate, it is very difficult to localize hypoxia-stimulated enhanced proliferative responses in lung of MKP-1-deficient mice.
Duration of hypoxia exposure might also be an important determinant for the detection of hypoxia-induced replication rate in lung of MKP-1-deficient mice. We have future plans to examine the proliferative responses of vascular wall cells in MKP-1 null lungs more completely, by performing time-course studies of hypoxia-induced BrdU incorporation in the lungs of these mice. Our experiments with isolated MEFs demonstrated that cells lacking MKP-1 expression lose the ability to divide under hypoxic conditions. In contrast, hypoxia stimulates an increase in cell numbers in wild-type MEFs (Figure 8). Therefore, cell proliferation after 6 weeks of hypoxia exposure might not be a major determining factor for vascular remodeling in MKP-1-deficient lung. Marked up-regulation of αSMA expression in MKP-1-deficient lung under hypoxic conditions (Figure 3A) suggests that smooth muscle cell hypertrophy could be involved in remodeling process of the vascular wall in the lung lacking MKP-1 expression.
VEGF is an important survival factor for endothelial cells and hypoxia up-regulates its expression.5,44 Although MKP-1 has been identified as a novel VEGF-inducible gene,9,10 regulation of hypoxia-induced VEGF expression by MKP-1 remains unexplored. The present findings demonstrate that VEGF expression is tightly regulated by MKP-1 in hypoxic MEFs. Hypoxia induces marked up-regulation of VEGF expression in wild-type cells. However, MKP-1-null cells lack this response, suggesting that this phosphatase is an important mediator of hypoxia-induced VEGF expression. Another important point is that the increase in VEGF levels in hypoxic MKP-1+/+ MEFs is highly dependent on JNK1/2 activation; attenuation of JNK1/2 with SP600125 led to greatest reduction in VEGF expression in these cells, compared with ERK and p38 MAP kinase inhibitors (Figure 7B). In contrast, MKP-1-deficient cells do not require JNK1/2 activation for VEGF expression. Therefore, defective VEGF expression and the subsequent rarefaction of vascular structures, together with remodeling, suggest that lack of MKP-1 expression in endothelial cells might induce impaired responses in MKP-1-deficient mice challenged by hypoxia.
In the present study, hypoxia-induced sustained activation of ERK1/2 in isolated MKP-1−/− MEFs (Figure 6A) was not recapitulated in the in vivo phosphorylation pattern of this kinase (Figure 2A). Only MKP-1-deficient lung from the SL group had increased ERK1/2 phosphorylation. Discrepancies in the activation patterns might be due either to the extended in vivo experimental period or to over-expression of other members of MKP family. Six weeks of hypoxia exposure might be an exceedingly long time for the detection of ERK1/2 phosphorylation in the lung. However, in the case of p38 MAP kinase, we found hypoxia-stimulated sustained phosphorylation of the kinase, both in vitro and in vivo, suggesting that p38 MAP kinase activation might be a major signaling kinase in lung of MKP-1-deficient mice. The role of p38 MAP kinase-responsive genes in the physiological responses of MKP-1-null lung to hypoxia needs further evaluation.
MKP-1 likely acts as a signaling switch that attenuates the phosphorylation of p38 MAP kinase, JNK, and ERK, thus allowing the expression of genes necessary for VEGF production and vessel formation in the lung. Consequently, strategies specifically targeting MKP-1 may be of benefit in potentiating hypoxic signaling and enhancing the formation and maturation of neovessels in the context of therapeutic angiogenesis.
Acknowledgments
We thank Lexicon Pharmaceuticals, Inc., for providing MKP-1-null mice. We also thank Dr. Kurt Stenmark, Dr. Carlyne Cool (University of Colorado Denver), and Dr. James West (Vanderbilt University) for critical comments on the manuscript.
Footnotes
Supported by NIH grant HL64917 (M.D.).
References
- 1.Camps M., Nichols A., Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- 2.Misra-Press A., Rim C.S., Yao H., Roberson M.S., Stork P.J.S. A novel mitogen-activated protein kinase phosphatase: Structure, expression and regulation. J Biol Chem. 1995;270:14587–14596. doi: 10.1074/jbc.270.24.14587. [DOI] [PubMed] [Google Scholar]
- 3.Zakkar M., Chaudhury H., Sandvik G., Enesa K., Luong L.A., Cuhlmann S., Mason J.C., Krams R., Clark A.R., Haskard D.O., Evans P.C. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ Res. 2008;103:726–732. doi: 10.1161/CIRCRESAHA.108.183913. [DOI] [PubMed] [Google Scholar]
- 4.Seta K.A., Kim R., Kim H.W., Millhorn D.E., Beitner-Johnson D. Hypoxia-induced regulation of MAPK phosphatase-1 as identified by subtractive suppression hybridization and cDNA microarray analysis. J Biol Chem. 2001;276:44405–44412. doi: 10.1074/jbc.M103346200. [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 6.Tuder R.M., Flook B.E., Voelkel N.F. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia: Modulation of gene expression by nitric oxide. J Clin Invest. 1995;95:1798–1807. doi: 10.1172/JCI117858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christou H., Yoshida A., Arthur V., Morita T., Kourembanas S. Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol. 1998;18:768–776. doi: 10.1165/ajrcmb.18.6.2980. [DOI] [PubMed] [Google Scholar]
- 8.Marti H.H., Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci USA. 1998;95:15809–15814. doi: 10.1073/pnas.95.26.15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinney C.M., Chandrasekharan U.M., Mavrakis L., DiCorleto P.E. VEGF and thrombin induce MKP-1 through distinct signaling pathways: role for MKP-1 in endothelial cell migration. Am J Physiol Cell Physiol. 2008;294:C241–C250. doi: 10.1152/ajpcell.00187.2007. [DOI] [PubMed] [Google Scholar]
- 10.Bellou S., Hink M.A., Bagli E., Panopoulou E., Bastiaend P.I.H., Murphy C., Fotsis T. VEGF auto-regulates its proliferative and migratory ERK1/2 and p38 cascades by enhancing the expression of DUSP1 and DUSP5 phosphatases in endothelial cells. Am J Physiol Cell Physiol. 2009;297:C1477–C1489. doi: 10.1152/ajpcell.00058.2009. [DOI] [PubMed] [Google Scholar]
- 11.Dorfman K., Carrasco D., Gruda M., Ryan C., Lira S.A., Bravo R. Disruption of the erp/MKP-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene. 1996;13:925–931. [PubMed] [Google Scholar]
- 12.Salojin K.V., Owusu I.B., Millerchip K.A., Potter M., Platt K.A., Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol. 2006;176:1899–1907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Q., Wang X., Nelin L.D., Yao Y., Matta R., Manson M.E., Baliga R.S., Meng X., Smith C.V., Bauer J.A., Chang C.H., Liu Y. MAP kinase phosphatase-1 controls innate immune response and suppresses endotoxic shock. J Exp Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J.J., Roth R.J., Anderson E.J., Hong E.G., Lee M.K., Choi C.S., Neufer P.D., Shulman G.I., Kim J.K., Bennett A.M. Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to diet-induced obesity. Cell Metab. 2006;4:61–73. doi: 10.1016/j.cmet.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Carison J., Cui W., Zhang Q., Xu X., Mercan F., Bennett A.M., Vignery A. Role of MKP-1 in osteoclasts and bone homeostasis. Am J Pathol. 2009;175:1564–1573. doi: 10.2353/ajpath.2009.090035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin Y., Calvert T.J., Chen B., Chicoine L.G., Joshi M., Bauer J.A., Liu Y., Nelin L.D. Mice deficient in MKP-1 develop more severe pulmonary hypertension and greater lung protein levels of arginase in response to chronic hypoxia. Am J Physiol Heart Circ Physiol. 2010;298:H1518–H1528. doi: 10.1152/ajpheart.00813.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaoka T., Gebb S.A., Karoor V., Homma N., Morris K.G., McMurtry I.F., Oka M. Involvement of RhoA/Rho kinase signaling in pulmonary hypertension of the fawn-hooded rat. J Appl Physiol. 2006;100:996–1002. doi: 10.1152/japplphysiol.01028.2005. [DOI] [PubMed] [Google Scholar]
- 18.Homma N., Nagaoka T., Morio Y., Ota H., Gebb S.A., Karoor V., McMurtry I.F., Oka M. Endothelin-1 and serotonin are involved in activation of RhoA/Rho kinase signaling in the chronically hypoxic hypertensive rat pulmonary circulation. J Cardiovasc Pharmacol. 2007;50:697–702. doi: 10.1097/FJC.0b013e3181593774. [DOI] [PubMed] [Google Scholar]
- 19.Thomas M., Docx C., Holmes A.M., Beach S., Duggan N., England K., Leblanc C., Lebret C., Schindler F., Raza F., Walker C., Crosby A., Davies R.J., Morrell N.W., Budd D.C. Activin-Like Kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am J Pathol. 2009;174:380–389. doi: 10.2353/ajpath.2009.080565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dempsey E.C., Wick M.J., Karoor V., Barr E.J., Tallman D.W., Wehling C.A., Walchak S.J., Laudi S., Le M., Oka M., Majka S., Cool C.D., Fagan K.A., Klemm D.J., Hersh L.B., Gerard N.P., Gerard C., Miller Y.E. Neprilysin null mice develop exaggerated pulmonary vascular remodeling in response to chronic hypoxia. Am J Pathol. 2009;174:782–796. doi: 10.2353/ajpath.2009.080345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W., Liang Q., Balzar S., Wenzel S., Gorska M., Alam R. Cell-specific activation profile of extracellular signal-regulated kinase 1/2, Jun N-terminal kinase, and p38 mitogen-activated protein kinases in asthmatic airways. J Allergy Clin Immunol. 2008;121:893–902.e2. doi: 10.1016/j.jaci.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Gesslein B., Håkansson G., Carpio R., Gustafsson L., Perez M.T., Malmsjö M. Mitogen-activated protein kinases in the porcine retinal arteries and neuroretina following retinal ischemia-reperfusion. Mol Vis. 2010;16:392–407. [PMC free article] [PubMed] [Google Scholar]
- 23.Li J., Kuzin I., Moshkani S., Proulx S.T., Xing L., Skrombolas D., Dunn R., Sanz I., Schwarz E.M., Bottaro A. Expanded CD23+/CD21hi B cells in inflamed lymph nodes are associated with the onset of inflammatory-erosive arthritis in TNF-transgenic mice and are targets of anti-CD20 therapy. J Immunol. 2010;184:6142–6150. doi: 10.4049/jimmunol.0903489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan S.J., Pham S., Wei Y., Mateo D., St-Pierre M., Fletcher T.M., Vazquez-Padron R.I. Stress-induced senescence exaggerates postinjury neointimal formation in the old vasculature. Am J Physiol Heart Circ Physiol. 2010;298:H66–H74. doi: 10.1152/ajpheart.00501.2009. [DOI] [PubMed] [Google Scholar]
- 25.Giatromanolaki A., Koukourakis M.I., Sivridis E., Turley H., Wykoff C.C., Gatter K.C., Harris A.L. DEC1 (STRA13) protein expression relates to hypoxia-inducible factor 1-alpha and carbonic anhydrase-9 overexpression in non-small cell lung cancer. J Pathol. 2003;200:222–228. doi: 10.1002/path.1330. [DOI] [PubMed] [Google Scholar]
- 26.Das M., Burns N., Wilson S.J., Zawada W.M., Stenmark K.R. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of PKCzeta in the pulmonary artery adventitia. Cardiovasc Res. 2008;78:440–448. doi: 10.1093/cvr/cvn014. [DOI] [PubMed] [Google Scholar]
- 27.Morrell N.W., Atochina E.N., Morris K.G., Danilov S.M., Stenmark K.R. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J Clin Invest. 1995;96:1823–1833. doi: 10.1172/JCI118228. [Erratum appeared in J Clin Invest 1996, 97:271] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long L., MacLean M.R., Jeffery T.K., Morecroft I., Yang X., Rudarakanchana N., Southwood M., James V., Trembath R.C., Morrell N.W. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res. 2006;98:818–827. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 29.Crosby A., Jones F.M., Southwood M., Stewart S., Schermuly R., Butrous G., Dunne D.W., Morrell N.W. Pulmonary vascular remodeling correlates with lung eggs and cytokines in murine schistosomiasis. Am J Respir Crit Care Med. 2010;181:279–288. doi: 10.1164/rccm.200903-0355OC. [DOI] [PubMed] [Google Scholar]
- 30.Das M., Bouchey D.M., Moore M.J., Hopkins D.C., Nemenoff R.A., Stenmark K.R. Hypoxia-induced proliferative response of vascular adventitial fibroblasts is dependent on G-protein-mediated activation of mitogen-activated protein kinases. J Biol Chem. 2001;276:15631–15640. doi: 10.1074/jbc.M010690200. [DOI] [PubMed] [Google Scholar]
- 31.Short M.D., Fox S.M., Lam C.F., Stenmark K.R., Das M. Protein kinase Czeta attenuates hypoxia-induced proliferation of fibroblasts by regulating MAP kinase phosphatase-1 expression. Mol Biol Cell. 2006;17:1995–2008. doi: 10.1091/mbc.E05-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laderoute K.R., Mendonca H.L., Calaoagan J.M., Knapp A.M., Giaccia A.J., Stork P.J. Mitogen-activated protein kinase phosphatase-1 (MKP-1) expression is induced by low oxygen conditions found in solid tumor microenvironments: A candidate MKP for the inactivation of hypoxia-inducible stress-activated protein kinase/c-Jun N-terminal protein kinase activity. J Biol Chem. 1999;274:12890–12897. doi: 10.1074/jbc.274.18.12890. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y., Gorospe M., Yang C., Holbrook N.J. Role of mitogen-activated protein kinase phosphatase during the cellular response to genotoxic stress: Inhibition of c-Jun N-terminal kinase activity and AP-1 dependent gene activation. J Biol Chem. 1995;270:8377–8380. doi: 10.1074/jbc.270.15.8377. [DOI] [PubMed] [Google Scholar]
- 34.Keyse S.M., Emslie E.A. Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature. 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- 35.Kassel O., Sancono A., Krätzschmar J., Kreft B., Stassen M., Cato A.C. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001;20:7108–7116. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong H.R., Dunsmore K.E., Page K., Shanley T.P. Heat shock-mediated regulation of MKP-1. Am J Physiol Cell Physiol. 2005;289:C1152–C1158. doi: 10.1152/ajpcell.00138.2005. [DOI] [PubMed] [Google Scholar]
- 37.Chi H., Flavell R.A. Acetylation of MKP-1 and the control of inflammation. Sci Signal. 2008;41:pe44. doi: 10.1126/scisignal.141pe44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boutros T., Chevet E., Metrakos P. Mitogen-activated protein (MAP) kinases/MAP kinase phosphatase regulation: roles in cell growth, death and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- 39.McMullen M.E., Bryant P.W., Glembotski C.C., Vincent P.A., Pumiglia K.M. Activation of p38 has opposing effects on the proliferation and migration of endothelial cells. J Biol Chem. 2005;280:20995–21003. doi: 10.1074/jbc.M407060200. [DOI] [PubMed] [Google Scholar]
- 40.Baumer S., Keller L., Holtman A., Funke R., August B., Gamp A., Wolburg H., Wolburg-Buchholz K., Deutsch U., Vestweber D. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood. 2006;107:4754–4762. doi: 10.1182/blood-2006-01-0141. [DOI] [PubMed] [Google Scholar]
- 41.Dominguez M.G., Hughes V.C., Pan L., Simmons M., Daly C., Anderson K., Noguera-Troise I., Murphy A.J., Valenzuela D.M., Davis S., Thurston G., Yancopoulos G.D., Gale N.W. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci USA. 2007;104:3243–3248. doi: 10.1073/pnas.0611510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Howell K., Ooi H., Preston R., McLoughlin P. Structural basis of hypoxic pulmonary hypertension: the modifying effect of chronic hypercapnia. Exp Physiol. 2003;89:66–72. doi: 10.1113/expphysiol.2003.026765. [DOI] [PubMed] [Google Scholar]
- 43.Sakao S., Taraseviciene-Stewart L., Wood K., Cool C.D., Voelkel N.F. Apoptosis of pulmonary microvascular endothelial cells stimulates vascular smooth muscle cell growth. Am J Physiol Lung Cell Mol Physiol. 2006;291:L362–L368. doi: 10.1152/ajplung.00111.2005. [DOI] [PubMed] [Google Scholar]
- 44.Voelkel N.F., Hoeper M., Maloney J., Tuder R.M. Vascular endothelial growth factor in pulmonary hypertension. Ann N Y Acad Sci. 1996;796:186–193. doi: 10.1111/j.1749-6632.1996.tb32580.x. [DOI] [PubMed] [Google Scholar]