

Abstract
Systemic exposure to cell-free hemoglobin (Hb) or its breakdown products after hemolysis or with the use of Hb-based oxygen therapeutics may alter the function and integrity of the blood-brain barrier. Using a guinea pig exchange transfusion model, we investigated the effect of a polymerized cell-free Hb (HbG) on the expression of endothelial tight junction proteins (zonula occludens 1, claudin-5, and occludin), astrocyte activation, IgG extravasation, heme oxygenase (HO), iron deposition, oxidative end products (4-hydroxynonenal adducts and 8-hydroxydeoxyguanosine), and apoptosis (cleaved caspase 3). Reduced zonula occludens 1 expression was observed after HbG transfusion as evidenced by Western blot and confocal microscopy. Claudin-5 distribution was altered in small- to medium-sized vessels. However, total expression of claudin-5 and occludin remained unchanged except for a notable increase in occludin 72 hours after HbG transfusion. HbG-transfused animals also showed increased astrocytic glial fibrillary acidic protein expression and IgG extravasation after 72 hours. Increased HO activity and HO-1 expression with prominent enhancement of HO-1 immunoreactivity in CD163-expressing perivascular cells and infiltrating monocytes/macrophages were also observed. Consistent with oxidative stress, HbG increased iron deposition, 4-hydroxynonenal and 8-hydroxydeoxyguanosine immunoreactivity, and cleaved caspase-3 expression. Systemic exposure to an extracellular Hb triggers blood-brain barrier disruption and oxidative stress, which may have important implications for the use of Hb-based therapeutics and may provide indirect insight on the central nervous system vasculopathies associated with excessive hemolysis.
Hemoglobin (Hb)–induced neurotoxicity is a major pathophysiological feature of intracerebral (ICH) and subarachnoid hemorrhage.1–3 The lysis of extravasated red blood cells exposes the central nervous system (CNS) to significant quantities of Hb or its breakdown products, triggering harmful oxidative and inflammatory events.1–3 Commonly studied experimental models of ICH and subarachnoid hemorrhage include cerebral vessel perforation, collagenase digestion, or the direct injection of Hb or blood into brain compartments outside the confines of the blood-brain barrier (BBB).3–7 Given that an intact BBB blocks CNS entry of extracellular Hb, considerably less attention has focused on studying the potential CNS effects of plasma cell-free Hb. For example, relatively little is known about how systemic exposure to Hb may affect the function and integrity of the BBB. This may be particularly important given the well-documented susceptibility of vascular endothelium to Hb or heme-mediated oxidative injury and the reported vasculopathies associated with chronic or intermittent hemolysis.8–12
The development of oxygen-carrying solutions that use cell-free Hb as a basis for chemical modification has been linked to serious adverse events, including stroke and myocardial infarction, in late-phase clinical trials.13 The mechanisms underlying these events are not fully understood but could be related, in part, to the uncontrolled pro-oxidative activity of cell-free Hb or heme.8,9 Similarly, neurologic complications (eg, stroke) are commonly observed in disease states associated with chronic or intermittent intravascular hemolysis, including genetic and drug-induced hemolytic anemias, microbial infections, and trauma.11,12,14–18 However, the mechanisms are multifaceted and not fully defined.12,17,18
Extracellular Hb can oxidize to methemoglobin (ferric, Fe3+), ferryl heme intermediate (Fe4+), hemichromes, and free heme or iron, which can initiate or propagate oxidative damage to lipids, nucleic acids, and proteins.8,9 Cell-free Hb is also an effective scavenger of nitric oxide, a key mediator of vascular homeostasis.11,14 Heme catabolism and iron sequestration systems play a key role in mediating tissue responses to cell-free Hb.8,9,19 Heme oxygenase (HO), the rate-limiting enzyme of heme catabolism, exists as two main isoforms, inducible HO-1 and constitutive HO-2 that catalyze the degradation of heme to biliverdin, free iron, and carbon monoxide.20 HO-1 is up-regulated by heme, heavy metals, lipopolysaccharide, and several other stimuli. In the brain, HO-1 induction has been reported in ICH, subarachnoid hemorrhage, and other neurodegenerative conditions.7,21–26 HO-1 up-regulation is viewed as an adaptive response that confers protection through anti-inflammatory, anti-apoptotic, and antioxidant mechanisms.20,24,25 However, under settings of excess heme exposure, HO-1 over-activation may lead to iron or other by-product toxicities and can serve as a marker of oxidative stress.7,23,26,27
The BBB is composed of endothelial cells, basal lamina, astrocytic end-foot processes, pericytes, perivascular macrophages, and neurons.28,29 Tight junctions (TJs) are specialized interendothelial structural complexes that restrict paracellular permeability. TJs are composed of transmembrane proteins, including occludin, claudins (primarily claudin-3 and -5), junctional adhesion molecules (JAMs), and cytoplasmic zonula occludens (ZO) family members (ZO-1, -2, -3). ZO proteins are accessory proteins that bind the other proteins to the actin cytoskeleton.28,29 Astrocytes, pericytes, and perivascular macrophages play a central role in BBB regulation. Activated astrocytes express increased levels of glial fibrillary acidic protein (GFAP), which serves as a reliable marker of BBB dysfunction or injury.30,31 Pericytes are enveloped within the basal lamina and provide direct structural support to the cerebral endothelium as well as metabolic support through the release of growth factors.32,33 Perivascular macrophages are located abluminally and serve as phagocytic regulators of the perivascular environment.32
In this study, we examined the effects of a polymerized cell-free Hb on BBB integrity and oxidative stress in a guinea pig exchange transfusion model. This model was selected on the basis of previous studies that identified the guinea pig as a potentially useful species for studying the oxidative toxicity of cell-free Hb.9,19,34 The guinea pig, similar to humans, lacks the ability to produce ascorbate, which is a powerful reductant that minimizes the oxidation of cell-free Hb in the circulation.9,34 Here, we provide evidence that systemic Hb exposure triggers BBB disruption and oxidative stress in this model.
Materials and Methods
Materials
Polymerized bovine hemoglobin (Oxyglobin; HbG), a product approved for veterinary use, was a kind gift from Biopure Corporation (Cambridge, MA). This solution consists of a heterogeneous mixture of glutaraldehyde-polymerized bovine hemoglobin at a concentration of 13 g/dl in modified Lactated Ringer's solution. The solution contains unstabilized tetramers (<5%), stabilized 64-kDa tetramers (∼35%), 65- to 130-kDa oligomers (∼50%), and 500-kDa polymers (<10%). Detailed physicochemical characterization has been previously reported.35 The initial content of HbG in the ferric form was 5.7%. All other chemicals were obtained from Sigma (St. Louis, MO) unless indicated otherwise.
Animal Experiments
Male Hartley guinea pigs (Charles Rivers Laboratories, Wilmington, MA) were acclimated for 1 week on arrival to the Food and Drug Administration/Center for Biologics Evaluation and Research animal care facility. All animals weighed 350 to 400 g before surgery. Animal protocols were approved by the Food and Drug Administration/Center for Biologics Evaluation and Research Institutional Animal Care and Use Committee, with all experimental procedures performed in adherence to the National Institutes of Health guidelines on the use of experimental animals. For surgery, guinea pigs were anesthetized i.p. with a ketamine HCl (100 mg/kg) and xylazine HCl (5 mg/kg) cocktail (Phoenix Scientific Inc., St. Joseph, MO). All procedures were performed under aseptic conditions. A midline incision was made around the neck region, allowing for blunt dissection and exposure of the right common carotid artery and the left external jugular vein. Saline-filled catheters containing 50 IU heparin/ml prepared from sterile PE50 tubing (Clay Adams of Becton-Dickenson, Sparks, MD) were placed in each vessel and tunneled under the skin to the back of the neck. Animals were treated for pain immediately after surgery with 5 mg/kg ketoprofen, given subcutaneously, and topical application of lidocaine 0.1%. All animals were allowed 24 hours of recovery before experimentation. Animals then underwent a 50% exchange transfusion with HbG by connecting the arterial and venous catheters to separate syringe pumps (Model 11; Harvard Apparatus, Holliston, MA) set on withdrawal (1 ml/min) and infuse (1 ml/min), respectively. Fifty percent transfusion volume in the guinea pig was calculated as 50% transfusion (ml) = [0.06 (ml/g) × body weight (g) + 0.77]/2. The mean doses of HbG administered and the resulting end-transfusion maximum plasma HbG concentrations were 3272 ± 106 mg/kg and 4.0 ± 0.2 g/dl, respectively.34 The half-life of HbG was calculated as 15.7 hours.34 Two additional control groups included animals subjected to a 50% exchange transfusion with 5% human albumin (n = 9; Baxter Healthcare Corp., Round Lake, IL) or shed guinea pig whole blood (n = 9). At the end of the desired time period, animals were humanely euthanized and perfused via the arterial catheter with 60 ml of cold saline. Brains were excised, and right and left hemispheres were separated. One-half was stored in 10% formalin, and the other half was frozen at −80°C. Alternatively, animals were perfused with cold saline followed by 10% formalin before brain collection.
Antibodies
Mouse monoclonal and rabbit polyclonal antibodies to ZO-1 (catalog no. 33–9100, 61–7300) and mouse monoclonal antibodies to claudin-5 (catalog no. 35–2500) and occludin (catalog no. 33–1500) were purchased from Invitrogen (Carlsbad, CA). Mouse anti-GFAP (clone SMI-22) was purchased from Calbiochem (San Diego, CA). Rabbit polyclonal anti–HO-1 (catalog no. SPA-894) and anti–HO-2 (catalog no. OSA-200) were purchased from Assay Designs (Ann Arbor, MI). Mouse monoclonal anti-CD163 (clone EDHu-1) was obtained from AbD Serotec (Raleigh, NC). Mouse monoclonal antibodies to 8-hydroxydeoxyguanosine (8-OHdG; N45.1) and 4-hydroxynonenal protein adducts (HNEJ-2) were purchased from Oxis International (Foster City, CA). Rabbit polyclonal anti-cleaved caspase-3 antibody (catalog no. 9661) was purchased from Cell Signaling Technologies (Danvers, MA). Donkey anti-guinea pig IgG–horseradish peroxidase was purchased from Millipore (Billerica, MA). Rabbit polyclonal anti-actin was obtained from Santa Cruz Biotechnologies (Santa Cruz, CA).
Preparation of Whole Brain Lysates
Frozen brains were sectioned coronally, and samples (250 mg) consisting primarily of frontal cortex were dounce-homogenized in the presence of ice-cold modified radioimmunoprecipitation assay buffer (50 mmol/L Tris, 150 mmol/L NaCl, 1% IgePal-630, 0.5% deoxycholate, 1 mmol/L EDTA) containing protease inhibitors (Cocktail Set III; Calbiochem). Lysates were centrifuged at 10,000 rpm at 4°C for 30 minutes, and supernatants were collected, divided into aliquots, and stored at −80°C. Protein concentrations were measured with the use of the BCA protein assay (Thermo Scientific, Rockford, IL).
IgG Extravasation
BBB integrity was assessed with the use of a one-step immunoblot assay for guinea pig IgG.36 Briefly, brain lysates (10 μg) were resolved on 4% to 12% Bis-Tris gels, transferred to polyvinylidene fluoride membranes, and blocked for 1 hour in Tris-buffered saline containing 0.1% Tween 20 with 5% nonfat dry milk. Membranes were incubated with donkey anti-guinea pig IgG-horseradish peroxidase–conjugated antibody (dilution 1:5000) in 1% nonfat dry milk for 2 hours at room temperature. Signal was developed with the ECL Plus kit (GE Healthcare, Piscataway, NJ) and detected on HyperECL film. Blots were stripped and reprobed for actin to control for protein loading. The protein band representing the light chain of IgG (∼25 kDa) was analyzed by densitometry with the use of the ImageJ software (National Institutes of Health, Bethesda, MD) and normalized to total actin levels. Diluted guinea pig plasma (1:400) was analyzed as a positive control sample.
Western Blot Analyses
Brain lysates were resolved on 4% to 12% Bis-Tris or 3% to 8% Tris-Acetate (for ZO-1) gels, transferred to polyvinylidene fluoride membranes, and blocked for 1 hour in Tris-buffered saline containing 0.1% Tween 20 with 5% nonfat dry milk. Membranes were incubated overnight at 4°C with antibodies to ZO-1 (1:1000), occludin (1:500), claudin-5 (1:1000), GFAP (1:5000), HO-1 (1:2000), HO-2 (1:5000), and cleaved caspase-3 (1:2000) in Tris-buffered saline containing 0.1% Tween 20 with 1% nonfat dry milk or 1% bovine serum albumin (for occludin), washed, and then incubated with a relevant horseradish peroxidase–conjugated secondary antibody for 1 hour. Signal was developed with the use of the ECL Plus kit and detected on HyperECL film. Membranes were stripped and reprobed for actin. Densitometry analysis was performed with the use of the ImageJ software with normalization to actin.
Heme Oxygenase Activity
Brain microsomes were prepared by ultracentrifugation as previously described.37 Microsomal membranes (0.5 to 1 mg/ml) were added to reaction mixtures containing glucose 6-phosphate (2 mmol/L), glucose-6-phosphate dehydrogenase (1.6 U/ml), hemin (15 μmol/L), NADPH p450 reductase (0.1 U/ml), 0.5 mg/ml of rat liver cytosol prepared from a 105,000 × g supernatant fraction as a source of biliverdin reductase, in potassium phosphate buffer (100 mmol/L, pH 7.4) containing 2 mmol/L MgCl2. After adding NADP (0.8 mmol/L) to the experimental tube and assay buffer to the reference tube, the mixtures were incubated in a shaking water bath for 30 minutes at 37°C in the dark. The tubes were then placed on ice for 2 minutes and subsequently analyzed on a dual-beam scanning spectrophotometer (Perkin Elmer Lambda 18, Shelton, CT). Bilirubin was calculated by the difference in absorbance between 464 and 530 nm (40 mmol/L cm−1). Heme oxygenase activity was expressed as nanomoles of bilirubin formed per hour per milligram of cell protein. Protein content was determined with the use of the Bio-Rad assay (Bio-Rad, Hercules, CA).
Immunohistochemistry
Brains were fixed in 10% formalin and embedded in paraffin, and 5-μm sections were prepared. Sections were de-waxed in xylene and rehydrated in graded ethanol and deionized water. Antigen retrieval was performed by heat treatment in a microwave oven for 15 minutes in 10 mmol/L sodium citrate buffer (pH 6.0), cooled for 30 minutes at room temperature, and rinsed with deionized water and PBS containing 0.05% Tween-20 (PBS-T). Sections were incubated with 3% H2O2 in PBS-T for 15 minutes at room temperature. For 4-HNE and 8-OHdG, this peroxidase inhibition step was carried out after the primary antibody incubation to avoid detecting any potential oxidative damage caused by H2O2. Sections were blocked with PBS-T containing 10% normal horse or goat serum and 0.25% Triton-X for 1 hour at room temperature followed by incubation with primary antibodies to GFAP (1:500), HO-1 (1:400), 4-HNE (1 μg/ml), and 8-OHdG (1 μg/ml) overnight at 4°C. Signal was developed by peroxidase-conjugated avidin-biotin complexes (Vectastain Elite ABC; Vector Laboratories, Burlingame, CA) and diaminobenzidine (SigmaFast DAB; Sigma). Slides were washed, counterstained with hematoxylin unless otherwise indicated, washed, and dehydrated in graded ethanol and xylene. For negative controls, sections were incubated without the primary antibody.
Slides were imaged with the use of an Olympus IX71 inverted microscope equipped with an Olympus DP70 digital camera (Olympus America, Melville, NY). Images were obtained from the cerebral cortex or midbrain regions unless otherwise noted. Standardized microscope and software settings were applied during image capture and processing. Semiquantitative analysis of 8-OHdG staining was performed with the use of the Photoshop software (Adobe Systems, Mountain View, CA). A total of five random cortical fields were imaged at ×200 magnification and evaluated with the optical density function. Staining intensity was reported as mean of arbitrary units (AU) ± SEM. Sections incubated with anti-8-OHdG antibody in the presence of 500 μmol/L 8-OHdG as an absorption control showed minimal reactivity.
Immunofluorescence
Paraffin-embedded slides were dewaxed and rehydrated. Slides were heat treated in a microwave oven for 15 minutes in 10 mmol/L Tris, 1 mmol/L EDTA buffer, pH 9.0 (claudin-5) or 10 mmol/L sodium citrate buffer, pH 6.0 (HO-1, CD163, 4-HNE, 8-OHdG); cooled for 30 minutes at room temperature; and rinsed with deionized water and PBS-T. For ZO-1, sections were incubated with pepsin (Invitrogen) for 10 minutes at 37°C and rinsed with PBS-T. Sections were then blocked in PBS-T with 5% goat serum and 0.25% Triton-X for 1 hour at room temperature followed by overnight incubation with mouse monoclonal anti–claudin-5 (1:100), rabbit polyclonal anti–ZO-1 (1:50), rabbit polyclonal anti–HO-1 (1:400), mouse anti-CD163 (1:100), mouse anti–8-OHdG (1 μg/ml), or mouse anti–4-HNE (1 μg/ml) at 4°C. Sections were rinsed and incubated with relevant Alexa Fluor 488– and Alexa Fluor 555–conjugated secondary antibodies for 1 hour at room temperature. Nuclei were counterstained with Hoechst 33342. For double-labeling experiments, primary antibodies were mixed together and incubated overnight at 4°C. Images were acquired with a Zeiss LSM 710 confocal laser scanning microscope (Carl Zeiss MicroImaging Inc., Jena, Germany). Alexa Fluor 488, Alexa Fluor 555, and Hoechst 33342 were excited with 488-, 543-, and 750-nm wavelengths, respectively. Images of 1024 × 1024 pixels were acquired with a 100× oil objective. Image analysis was performed with the Zeiss Zen software. Alternatively, images were processed with an Olympus IX71 inverted microscope equipped with an Olympus DP70 digital camera.
Brain Iron Measurement
Brain cortical tissue (50 mg) was homogenized in ddH20 at 1:10 w/v. Homogenates were mixed with 500 μl of an acid mixture containing 1 mmol/L HCl and 10% trichloro acetic acid, and incubated at 50°C for 1 hour with intermittent shaking.38 The samples were then centrifuged at 15,000 × g for 15 minutes at room temperature. The clear supernatant (90 μl) was mixed with 30 μl of 20 mg/ml ascorbic acid followed by 20 μl ferrozine (0.85% w/v in hydroxylamine hydrochloride). The samples were allowed to completely develop for 30 minutes. Absorbance was measured at 560 nm with the use of the Synergy 4 Hybrid Multi-Mode microplate reader (Biotek, Vinooski, VT). A standard curve was generated with the use of an iron standard (500 μg/dl).
Non-Heme Iron Histochemistry with DAB Intensification
Non-heme ferric iron deposition was detected with the use of Perls method with DAB intensification.39 Sections were incubated with Perls iron reagent containing 5% potassium ferrocyanide and 2% hydrochloric acid for 45 minutes at room temperature and rinsed in deionized water. Sections were then incubated with 0.3% hydrogen peroxide and 0.01 mol/L sodium azide in methanol for 30 minutes at room temperature. All sections were then rinsed in 0.1 mol/L phosphate buffer, pH 7.4, incubated with DAB for 5 minutes, washed in deionized water, and lightly counterstained with Gill's II hematoxylin. Images of the cerebral cortex were obtained with the use of the Olympus IX71 inverted microscope.
Data Presentation and Statistics
Data are represented as means ± SEMs for replicate experiments. A value of P < 0.05 was considered statistically significant. Statistical significance for all comparisons was determined by analysis of variance using the JMP Discovery Statistical Software v5.1 (Version 3.2; SAS Institute, Cary, NC).
Results
Altered TJ Protein Expression and Organization after HbG Transfusion
To determine the effect of HbG on cerebral TJs, we examined the expression of ZO-1, occludin, and claudin-5 by Western blot. In HbG-transfused animals, ZO-1 expression was significantly reduced after 4, 12, and 24 hours compared with nontreated sham controls (Figure 1A). At 72 hours, no significant difference in ZO-1 was detected between both groups. For occludin, no change in expression was observed at 4, 12, or 24 hours. However, a notable threefold increase in occludin was measured after 72 hours (Figure 1B). Total expression of claudin-5 was not altered at any time point after HbG transfusion (Figure 1C). In shed blood or albumin-transfused animals, there was no significant change in the expression of ZO-1, occludin, and claudin-5 compared with nontreated sham controls at 12 and 72 hours (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Figure 1.

Tight junction protein expression after HbG transfusion. Brains were harvested from sham-operated controls (NT) and HbG-transfused animals after the indicated times. Lysates were prepared and analyzed for ZO-1 (A), occludin (B), and claudin-5 (C) by Western blot as described in Materials and Methods. Representative immunoblots for ZO-1 (4 hours), occludin (72 hours), and claudin-5 (12 hours) are shown with each lane representing a separate animal. Bands for ZO-1, occludin, and claudin-5 migrated at approximately 200, 65, and 22 kDa, respectively. Densitometry values for protein expression as a ratio to actin were normalized to NT and presented as the means ± SEMs. *P < 0.05 vs NT (4 to 5 animals per time point).
To further examine the distribution of TJ proteins, ZO-1 and claudin-5 were analyzed by confocal microscopy. Blood vessels from nontreated sham controls showed intense and continuous reactivity for ZO-1 in large and medium- to small-sized vessels (Figure 2, A–C). Conversely, in HbG-transfused animals, ZO-1 immunoreactivity was comparatively lower and appeared more punctuate and diffuse (Figure 2, D–F). Claudin-5 immunoreactivity in sham controls showed intense and continuous interendothelial borders (Figure 2, G–I). In large vessels, both groups generally showed a similar claudin-5 staining pattern. However, in HbG-transfused animals, a high number of medium- to small-sized vessels exhibited a discontinuous and more diffuse claudin-5 staining pattern compared with similar-sized vessels in sham controls (Figure 2, J–L).
Figure 2.
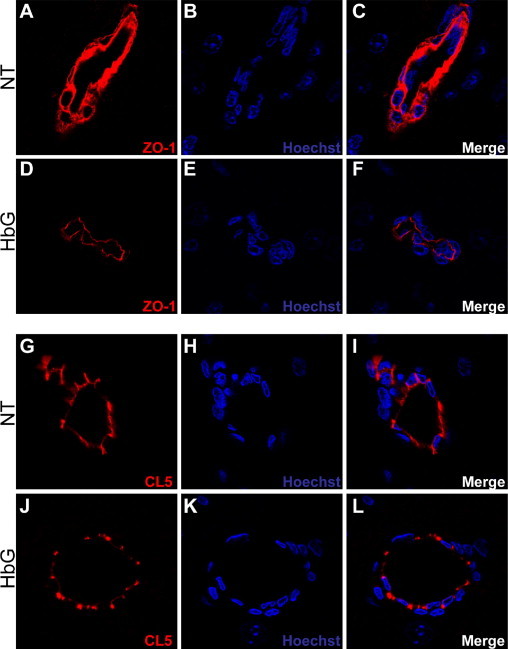
Altered tight junction organization after HbG transfusion. Brain sections from sham controls (A–C, G–I) and HbG 24 hours after transfusion (D–F, J–L) were analyzed for ZO-1 (A–F) or claudin-5 (CL5) (G–L) by laser scanning confocal microscopy as described in Materials and Methods. Reduced ZO-1 intensity is observed after HbG transfusion (D and F) compared with sham controls (A and C). Intense and continuous endothelial CL5 immunoreactivity is observed in medium-sized vessels of sham controls (G and I), whereas CL5 staining was more diffuse in similar-sized vessels of HbG-transfused animals (J and L). Nuclei were counterstained with Hoechst 33342 (blue). Original magnifications, ×1000.
Astrocyte Activation and IgG Extravasation after HbG Transfusion
Up-regulation of GFAP in astrocytes is considered a reliable marker of BBB disruption in models of CNS disease and toxicity.30,31 GFAP expression was analyzed over the course of 72 hours after HbG transfusion by Western blot. GFAP was detectable at 4, 12, and 24 hours but showed high variability, whereas a significant expression was observed at 72 hours (P < 0.01) (Figure 3A). Consistent with these data, increased GFAP immunoreactivity was observed in astrocyte processes surrounding many medium- to small-sized blood vessels in the brain parenchyma (Figure 3B). The glial limitans of the brain surface also showed enhanced GFAP immunoreactivity compared with sham control animals. GFAP was not up-regulated after exchange transfusion with shed blood or albumin (data not shown).
Figure 3.

GFAP induction and IgG extravasation in HbG-transfused animals. A: Western blot analysis of GFAP expression in brains harvested from sham controls (NT) and at the indicated times after HbG transfusion. A representative immunoblot for GFAP (51 kDa) at 72 hours is shown. Densitometry values expressed as a ratio to actin were normalized to NT and presented as the means ± SEMs. *P < 0.01 vs NT (3 to 6 animals per time point). B: GFAP immunohistochemistry was performed as described in Materials and Methods. Minimal GFAP immunoreactivity was observed in brain parenchyma of sham controls (left panel), whereas GFAP was detectable around blood vessels 24 hours after HbG transfusion (right panel). Original magnifications, ×600 (B) and ×200 (insets). C: Western blot analysis of guinea pig IgG. Brain lysates were analyzed with a donkey anti-guinea pig IgG antibody conjugated to horseradish peroxidase. Guinea plasma sample (GPL) served as a positive control. A representative immunoblot for IgG (72 hours) is shown. Densitometry was performed on a 25-kDa protein band representing the light chain of IgG. Data from at least four to five animals per group were normalized to NT and presented as the means ± SEMs. *P < 0.001 vs NT.
As a measure of BBB permeability, we analyzed plasma IgG extravasation via Western blot. In HbG-transfused animals, there were minor changes in IgG extravasation at 4 hours (P = 0.17), 12 hours (P = 0.26), and 24 hours (P = 0.07), but a significant increase at 72 hours (P < 0.001) compared with sham controls (Figure 3C). These data suggest that, within the first 24 hours, there were no major changes in permeability to large-sized molecules (IgG; mol. wt., 150 kDa), but a leakier barrier develops by 72 hours. In contrast, there was no significant increase in IgG extravasation in the shed blood or albumin groups after 72 hours (see Supplemental Figure S1 at http://ajp.amjpathol.org). In separate analyses, we used a histochemical stain for Hb40 to monitor for possible HbG extravasation during the first 24 hours when significant quantities of circulating HbG were still present. Consistent with the IgG observations, we did not observe widespread HbG extravasation into the brain parenchyma during the first 24 hours.
Heme Oxygenase Induction after HbG Transfusion
As an index of HbG or heme exposure, we analyzed total HO activity and the expression of HO-1 and HO-2. In HbG-transfused animals, we observed progressive increases in HO activity that peaked at 12 and 24 hours after transfusion and then declined by 72 hours (Figure 4A). Total HO activity was unchanged in animals transfused with shed blood (P = 0.26, n = 4) or albumin (P = 0.92, n = 3) compared with sham controls, suggesting HO induction was not caused by the surgery or the transfusion procedure itself. Western blot analyses of whole brain lysates showed increased HO-1 expression in HbG-transfused animals, whereas HO-2 expression remained the same compared with sham controls (Figure 4B).
Figure 4.
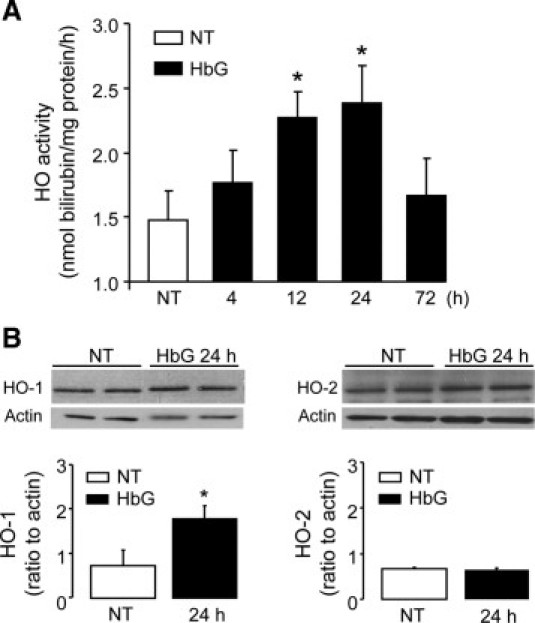
Brain HO induction in HbG-transfused guinea pigs. A: Brain microsomes were harvested from sham controls and at the indicated times after HbG transfusion. HO activity was measured by the bilirubin spectrophotometric assay and was reported as nanomole bilirubin formed per hour per milligram of protein. Values are shown as the means ± SEMs for at least four animals per group for each time point. *P < 0.05 vs NT. B: Western blot analysis of HO-1 (left panel) and HO-2 (right panel) in brain lysates from sham controls (NT) and 24 hours after HbG transfusion with the use of rabbit polyclonal antibodies as described in Materials and Methods. Representative immunoblots for HO-1 and HO-2 are shown for two different animals per group. Densitometry analysis was performed on at least four animals per group and was expressed relative to actin as the means ± SEMs. *P < 0.05 vs NT.
To localize HO-1–expressing cell types, brains sections were analyzed by immunohistochemistry. In HbG-transfused animals, intense HO-1 reactivity was observed in cells forming the walls of small- to medium-sized vessels (capillaries, arterioles) and postcapillary venules (Figure 5, D–F), whereas HO-1 was not detectable in similar-sized vessels in sham controls (Figure 5A). Most of the vessel-associated HO-1–positive cells possessed nuclei positioned abluminally, suggesting they represent pericytes or perivascular macrophages or both. In some cases, highly HO-1–reactive cells, which morphologically resembled monocytes/macrophages, accumulated in the lumen of postcapillary venules (Figure 5F). HO-1 immunoreactivity was variably observed in endothelial cells of medium-sized vessels, although this was not nearly as prominent as the perivascular HO-1 staining. Large vessels surrounding the brains of HbG-transfused animals showed extensive perivascular HO-1 reactivity compared with sham controls (see Supplemental Figure S2 at http://ajp.amjpathol.org). In albumin- or shed blood–transfused controls, HO-1 immunoreactivity was not enhanced in the parenchyma or in large vessels surrounding the brain (Figure 5, B and C). HO-1 staining was detected in scattered neuron populations, but there were no apparent differences in staining intensity between HbG-transfused animals and sham controls.
Figure 5.

Brain HO-1 expression in HbG-transfused guinea pigs. A–F: For HO-1 immunohistochemistry, brain sections were stained with a rabbit polyclonal HO-1 antibody and detected by Vectastain Elite with DAB as a substrate and counterstained with hematoxylin. A: Sham-operated controls (NT). Minimal HO-1 immunoreactivity in small- and medium-sized vessels in brain parenchyma. B and C: Exchange transfusion with shed blood (Sb) and albumin (Alb) produced minimal blood vessel HO-1 expression after 24 hours. D–F: HbG-transfused guinea pigs after 24 hours. High-intensity HO-1 immunoreactivity in walls of small- and medium-sized vessels (D and E) and in cells accumulating within postcapillary venules (F). G–L: Co-localization of HO-1 and CD163. Brain sections were stained with antibodies to HO-1 and CD163 and detected with the use of relevant secondary antibodies conjugated with Alex Fluor 488 (HO-1, green) and Alex Fluor 555 (CD163, red). Nuclei were counterstained with Hoechst 33342 (blue). Sham-operated controls (G–I) and HbG-transfused guinea pigs after 24 hours (J–L). CD163-positive and HO-1–negative staining observed in a medium-sized vessel of a sham control animal (G, arrowheads). Colocalized HO-1 and CD163 expression in cells trapped in the lumen of a medium-sized vessel of an HbG-transfused animal (L). Brightfield; original magnifications, ×600 (A–F). Double-label epifluorescence microscopy; original magnification, ×600 (G–L).
To further identify the HO-1–expressing cells, sections were double labeled for HO-1 and CD163, a marker of pericytes/perivascular macrophages.41,42 In sham controls, CD163-positive perivascular cells with minimal HO-1 reactivity were detectable as flattened and elongated cells around small- to medium-sized blood vessels (Figure 5, G–I). In HbG-transfused animals, HO-1–positive cells that lined the vessel walls and accumulated luminally in venules were also found to be CD163 positive (Figure 5, J–L). Enhanced HO-1 immunoreactivity was not observed in astrocytes or microglia on the basis of staining for GFAP and ionized calcium binding adaptor molecule 1, respectively (data not shown).
To assess whether enhanced HO-1 expression co-localized with sites of BBB disruption, sections were double labeled for HO-1 and claudin-5 or GFAP. Sham control vessels with minimal HO-1 reactivity showed intact claudin-5 distribution (Figure 6, A–C) and minimal GFAP staining (Figure 6, G–I). In HbG-transfused animals, vessels with intense perivascular or luminal HO-1 reactivity consistently showed irregular or discontinuous claudin-5 immunoreactivity, suggesting a close correlation between vessel exposure to HbG/heme and sites of TJ alterations (Figure 6, D–F). Similarly, vessels with enhanced GFAP immunoreactivity frequently showed increased HO-1 expression (Figure 6, J–L).
Figure 6.

Relationship between HO-1, claudin-5 (CL5), and GFAP expression. Brain sections were stained with antibodies to HO-1, claudin-5, and GFAP and detected with relevant secondary antibodies conjugated with Alex Fluor 488 (HO-1, green) and Alex Fluor 555 (CL5 and GFAP, red). Nuclei were counterstained with Hoechst 33342 (blue). A–C and G–I: Sham-operated controls. D–F and J–L: HbG-transfused guinea pigs after 24 hours. HO-1–negative staining in vessels showing intact CL5 immunoreactivity (C) and minimal GFAP staining (I). In HbG-transfused animals, HO-1–positive pericytes/perivascular macrophages (arrowheads) lined the walls of vessels with diffuse CL5 staining and enhanced GFAP reactivity. Laser scanning confocal microscopy; original magnification, ×700 (A–F). Double-label epifluorescence microscopy; original magnification, ×400 (G–L).
Non-Heme Iron Deposition after HbG Transfusion
Because breakdown of Hb/heme produces iron, we examined brain iron status after HbG transfusion. To quantitatively assess iron accumulation, brain homogenates were analyzed with the use of a colorimetric ferrozine-based assay. Importantly, this assay was performed with deproteinization conditions that cause minimal release of iron from heme proteins.38 In HbG animals, a significant increase in iron was measured 24 hours after transfusion compared with sham controls (Figure 7A). Non-heme iron accumulation was also visualized with the use of the Perls DAB intensification method. Iron deposition was not observed in brain sections from sham controls or from shed blood– or albumin-transfused animals (Figure 7, B–D). In HbG-transfused animals, iron deposits were detectable in brain parenchyma at 12 and 24 hours and variably observed at 4 and 72 hours. Iron deposits were primarily detected in neurons, and, in some cases, within cells forming the blood vessels (Figure 7, E and F).
Figure 7.
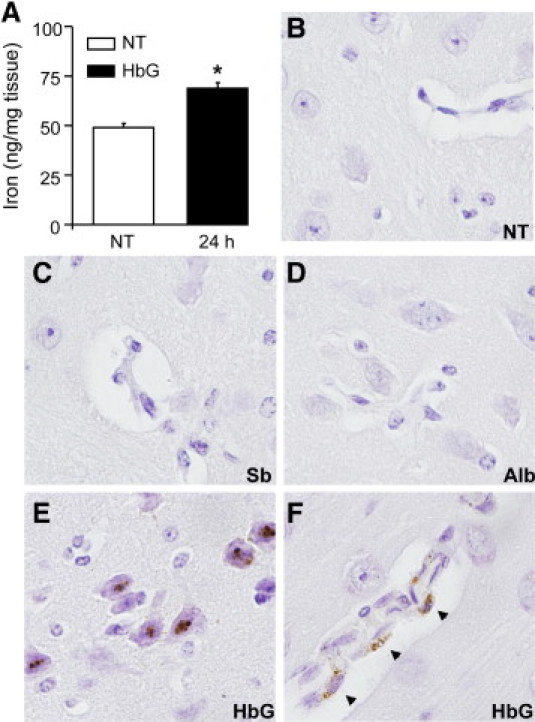
Iron deposition after HbG transfusion. A: Spectrophotometric determination of iron in brain homogenates prepared from sham controls (NT) and HbG-transfused animals after 24 hours as described in Materials and Methods. Data were obtained from at least four animals per group. Values are expressed as nanogram of iron per milligram of tissue and presented as means ± SEMs. *P < 0.05 vs NT. B–F: Perls non-heme iron histochemistry with DAB intensification in sham controls (B) and 24 hours after transfusion with shed blood (C), albumin (D), and HbG (E and F). In HbG animals, non-heme iron deposits were observed primarily in neurons (E) and were variably detected in walls of blood vessels (F, arrowheads). Sections were counterstained with hematoxylin. Original magnification, ×600 (B–F).
HbG Induces Oxidative Stress and Apoptosis
To further investigate whether systemic HbG exposure produces oxidative stress at the level of the BBB, we analyzed 4-HNE–modified adducts, a marker of lipid peroxidation, and 8-OHdG, a marker of oxidative DNA damage. In HbG-transfused animals, the endothelial lining of many medium- to small-sized vessels displayed increased 4-HNE immunoreactivity (Figure 8, A–C). Double-labeling analyses showed strong co-localization of 4-HNE and HO-1 in cellular infiltrates. Consistent with these 4-HNE observations, increased 8-OHdG immunoreactivity was detected in vessels, although endothelial 8-OHdG staining was generally not as intense as that observed for 4-HNE (Figure 8, D–F). Co-localization of 8-OHdG and HO-1 was also prominent among cellular infiltrates. Minimal 4-HNE and 8-OHdG staining was observed in sham controls (see Supplemental Figure S2 at http://ajp.amjpathol.org). Interestingly, in the brain parenchyma, increased 4-HNE and 8-OHdG reactivity was detected in neurons of the cerebral cortex (Figure 8, G and H). This was observed as early as 4 hours but was most prominent at 24 hours. In brain cortical regions, a fourfold increase in the staining intensity of 8-OHdG was observed in the HbG group compared with sham controls (Figure 8I). Consistent with an increase in oxidative toxicity, HbG-transfused animals showed a 2.5-fold increase in cleaved caspase 3 expression, a specific marker of apoptosis, by Western blot (Figure 8J).
Figure 8.
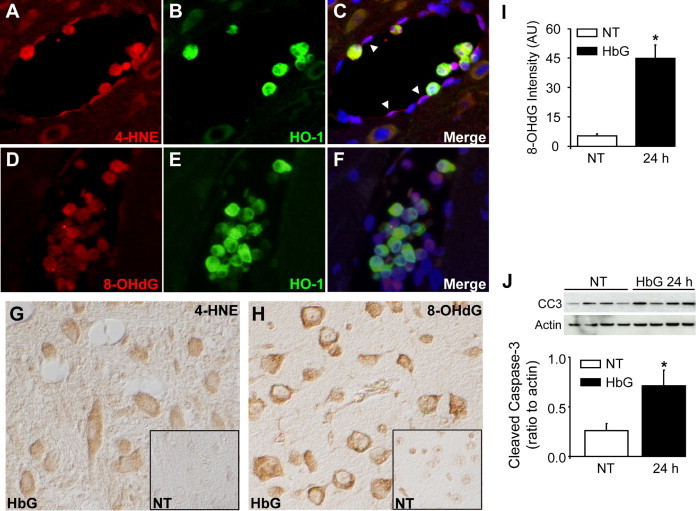
Oxidative markers, HO-1, and cleaved caspase 3 expression after HbG transfusion. Brain sections were stained with antibodies to HO-1, 4-HNE–modified adducts, and 8-OHdG and detected with relevant secondary antibodies conjugated with Alex Fluor 488 (HO-1, green) and Alex Fluor 555 (4-HNE and 8-OHdG, red) and counterstained with Hoechst 33342 (blue). HbG-transfused guinea pigs after 24 hours showing increased immunoreactivity for 4-HNE (A–C) and 8-OHdG (D–F) in endothelial cells (arrowheads) and co-localization with HO-1–positive cellular infiltrates. G and H: Immunohistochemical staining for 4-HNE–modified adducts (G) and 8-OHdG (H) in brain parenchyma of HbG-transfused guinea pigs after 24 hours. Inset images represent respective sham controls. In HbG-transfused animals, increased neuronal staining for 4-HNE and 8-OHdG was observed in cortical brain regions. Nuclear counterstain was omitted. I: Semiquantitative image analysis of 8-OHdG staining intensity as described in Materials and Methods. Data were obtained from seven animals per group and presented as the means ± SEMs. J: Western blot analysis of cleaved caspase 3 in sham controls and HbG-transfused guinea pigs. A representative Western blot is shown with each lane representing a separate animal. Data are expressed as a ratio to actin (mean ± SEM; n = 4 animals per group). *P < 0.05 vs NT. Double-label epifluorescence microscopy; original magnification, ×400 (A–F). Differential contrast microscopy; original magnification, ×400 (G and H).
Discussion
An intact BBB plays a critical role in blocking the entry of potentially neurotoxic cell-free Hb into the CNS. Yet, there is a paucity of information on how plasma cell-free Hb may alter the function and integrity of the BBB in vivo. Here, we provide evidence that systemic exposure to a long-circulating polymerized cell-free Hb solution induces BBB disruption as evidenced by TJ alterations, GFAP up-regulation, and IgG extravasation. This was accompanied by increased HO-1 expression in pericytes/perivascular macrophages and infiltrating monocyte/macrophages but not in microglia, astrocytes, or neurons. Notably, enhanced HO-1 immunoreactivity was observed in vessels that showed altered claudin-5 immunostaining and increased GFAP reactivity. Importantly, shed blood– or albumin-transfused control animals did not show tight junction protein alterations or enhanced HO-1 or GFAP in blood vessels. Together, these data suggest that HbG or its breakdown products or both may preferentially target the endothelial-perivascular cell interface in this setting. This idea is further supported by the prominent oxidative end product accumulation (4-HNE–modified protein adducts and 8-OHdG) along the endothelial lining and immediate perivascular compartments in vessels that co-expressed HO-1.
BBB disruption is typically characterized by changes in the expression, distribution, or phosphorylation of TJ proteins.28,29 ZO-1 is a key cytoplasmic TJ accessory protein that links the transmembrane proteins, occludin and the claudins, to the actin cytoskeleton. In this study, HbG produced a significant reduction in ZO-1 expression as early as 4 hours after transfusion, reached maximal decline by 12 hours, and partially recovered by 72 hours. Previous studies have shown that oxidative stress can alter ZO-1 expression and the molecular association between ZO-1 and the transmembrane proteins.43–49 For example, oxidative stress induced by hypoxia-reoxygenation disrupted ZO-1–occludin interactions in vivo.49 Recent findings suggest that oxidative stress induced by intracerebral injection of Hb triggers matrix metalloproteinase–mediated proteolysis of TJ proteins.5 In our study, we detected an accumulation of oxidative end products within the endothelial and perivascular compartments after HbG transfusion. These data support a possible link between oxidative stress and the reduction in ZO-1 expression, claudin-5 disorganization, and delayed occludin up-regulation.
Unlike ZO-1, the total expression levels of claudin-5 and occludin as determined by Western blot were unchanged after HbG transfusion. Nevertheless, we did observe altered claudin-5 immunoreactivity in many medium- to small-sized vessels after HbG transfusion. This could be related to weakened interactions between ZO-1 and claudin-5. Alternatively, others have reported that oxidative stress may modulate the function and localization of TJ proteins through non-degrading changes in their phosphorylation.47 Further investigations on the phosphorylation status of TJ proteins are warranted because this may help shed further light on the molecular mechanisms underlying HbG-mediated TJ disruption. With regard to the occludin data, it is conceivable that the observed up-regulation at 72 hours represents an adaptive response designed to reinforce TJ integrity. In support of this view, others have shown that heat shock triggers occludin synthesis as a protective response to heat-induced barrier disruption.50 The occludin results are also in line with the astrocytic GFAP induction at 72 hours, which is also known to contribute to barrier repair processes. Others have also shown that occludin alterations during BBB disruption are characterized by the breakdown of functional occludin oligomers in different cellular compartments.49,51 Notably, the latter studies used a polyclonal antibody capable of detecting occludin oligomers, whereas we used a monoclonal antibody that mainly recognizes the monomer 60-kDa form of occludin. Therefore, it is possible that the increased expression of occludin monomer at 72 hours could reflect changes in occludin oligomerization that we were unable to detect with the monoclonal antibody.
In this study, HbG increased total HO activity levels by 1.6-fold after 24 hours, which compared favorably with the magnitude of HO-1 protein expression by Western blot. Several studies have provided evidence for HO-mediated CNS protection.20,24,25 HO activity has been implicated in the protection of astrocytes and cerebral endothelial cells but may promote neuronal cytotoxicity.23,52,53 Others have shown that HO-1 may exacerbate early brain injury after ICH, and administration of HO inhibitors reduces Hb- and ICH-induced brain edema in vivo.7,23,26 In our study, enhanced HO-1 expression served as a useful marker of HbG/heme exposure and oxidative stress that correlated well with sites of altered TJ organization, enhanced GFAP reactivity, and oxidative end products. The present study was not specifically designed to evaluate whether the HO-1 response was protective or deleterious in this setting. However, recent experiments in our laboratory have shown that pretreatment with zinc protoporphyrin IX, a nonselective HO inhibitor, can attenuate the increase in brain HO activity and the reduction in ZO-1 expression after HbG transfusion (see Supplemental Figure S3 at http://ajp.amjpathol.org). These data imply a potential relationship between increased HO activity and the observed BBB alterations; however, further work will be required to fully understand the role of HO in this system.
The present study provided evidence for enhanced HO-1 immunoreactivity primarily in pericytes/perivascular macrophages, infiltrating monocytes, and variably in endothelial cells. This is in striking contrast to previous ICH and subarachnoid hemorrhage studies that have reported up-regulation of HO-1 in microglia, astrocytes, and neurons.22,54,55 These differences in the cellular expression may be explained, in part, by the absence of widespread leakage of HbG into the brain parenchyma.
We previously showed that plasma HbG undergoes rapid oxidation to methemoglobin, with levels reaching 10% to 15% as early as 4 hours after transfusion and up to 30% by 24 hours.34 Heme is known to dissociate more readily from Hb as ferric (Fe3+) heme relative to ferrous (Fe2+) heme, implying the vasculature could be subjected to greater amounts of free (Fe3+) heme.8,56 Our previous findings also showed that plasma HbG oxidation correlated with globin chain destabilization and presumably increased heme release.34 As an extension of these earlier findings, we have found that free heme is detectable by mass spectrometry in 24-hour plasma samples from HbG-transfused animals (see Supplemental Figure S4 at http://ajp.amjpathol.org). Several possible mechanisms could account for HbG-mediated oxidative stress and BBB disruption, including: i) The release of heme from oxidized HbG in amounts sufficient to exceed plasma-scavenging protein capacity. (This may be especially relevant in this exchange transfusion setting, where animals experience a significant loss of plasma Hb/heme-binding proteins. Highly lipophilic heme could then be taken up by endothelial cells or pericytes/perivascular macrophages and cause oxidative stress.) ii) Small amounts of HbG may traverse endothelial cells and be scavenged by pericytes/perivascular macrophages that may either be an indicator of, or in some circumstances, a trigger for localized oxidative stress. iii) Redox reactions driven by intact ferric Hb (eg, ferryl species) could trigger a localized oxidative stress at endothelial-perivascular interface. iv) Free iron-driven oxidation reactions may be involved. In support of the latter mechanism, we have found that administration of dexrazoxane (Zinecard), a commercially available iron chelator, immediately before HbG transfusion prevented the HbG-induced decrease in ZO-1 and attenuated the brain iron accumulation (see Supplemental Figure S3 at http://ajp.amjpathol.org). The protective effects of dexrazoxane in different models have generally been attributed to the compound's iron-chelating properties; however, several studies have reported an additional protective mechanism by dexrazoxane associated with a non-chelating antioxidant function that may be more potent than typical small molecule reducing agents (eg, ascorbate, urate, and tocopherol) found circulating in plasma.57,58 Therefore, additional studies will be required to determine the precise mechanism of protection by dexrazoxane in the present setting.
BBB dysfunction associated with the “loosening” of TJs facilitates the entry of leukocytes into the brain parenchyma. For example, diminished ZO-1 expression has been shown to correlate with monocyte infiltration.59 In the present study, HO-1 and CD163-positive infiltrates were identified in HbG-transfused animals. CD163, the putative monocyte/macrophage membrane receptor for Hb-haptoglobin complexes, is mainly expressed on resident macrophages in the liver, spleen, lung, as well as perivascular/meningeal macrophages and pericytes in the CNS, and additionally peripheral blood monocytes but not on microglia.41,42 Our findings may be consistent with the previously reported findings that Hb and heme activate proinflammatory pathways leading to up-regulation of endothelial adhesion molecules, vascular cell adhesion molecule 1 and intercellular adhesion molecule 1, which promote leukocyte adhesion and transmigration.60,61 Activated monocyte/macrophages accumulating at the BBB interface could act as an important source of reactive oxygen/nitrogen species that could exacerbate the oxidative reactions of HbG, heme, or iron.
BBB dysfunction and oxidative stress are common features of neurologic disorders, including cerebral ischemia, traumatic brain injury, and stroke.44,45 Besides the enhanced 4-HNE and 8-OHdG immunoreactivity associated with blood vessels in HbG-transfused animals, both markers were elevated in cortical neurons after 24 hours. This could be related, in part, to the greater sensitivity of neurons to heme- and iron-mediated toxicity compared with glial cells.62,63 Our observations that non-heme iron and oxidative markers accumulate in neurons provide preliminary evidence that systemic HbG exposure may affect other sensitive cells of the CNS. The precise temporal relationship between the HbG-mediated BBB events and the development of other CNS events will be important to delineate.
In summary, we report that systemic HbG exposure produces BBB disruption and oxidative stress in a guinea pig exchange transfusion model. These findings have potential implications in the area of Hb-based oxygen therapeutics and may offer indirect insight into the mechanisms underlying the CNS vasculopathies associated with chronic or excessive hemolysis.11,12 Interestingly, the longer circulation half-life of HbG compared with native Hb (15 hours versus ∼1 hour) does offer an experimental opportunity to evaluate how the BBB responds to a more sustained Hb exposure. Hb neurotoxicity with concomitant traumatic brain injury remains a concern in the Hb therapeutic field.13 However, our present findings suggest that even in normal animals the interaction between systemic extracellular Hb and the BBB may have important acute consequences. These findings also suggest the need for a better understanding of Hb exposure and BBB function in disease state models of endothelial dysfunction whereby acute insults may lead to long-term pathologic events.
Acknowledgments
We thank Dr. Abdu Alayash for reviewing the manuscript and Dr. Eugene Herman (CDER/FDA) for critical discussions about dexrazoxane. We are thankful to Drs. Jaroslav Vostal and Xuan Chi (Laboratory of Cellular Hematology, CBER/FDA) for the use of the laser scanning confocal microscope.
Footnotes
Supported by a Critical Path Initiative award from CBER/FDA (F.D. and P.W.B.).
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any agency determination or policy.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2010.12.006.
Supplementary data
Supplement Figure S1.

Tight junction protein expression and IgG extravasation 12 hours and 72 hours after transfusion with shed blood or colloidally matched albumin. Brain lysates were prepared and analyzed for ZO-1 (A), occludin (B), claudin-5 (C), and IgG (D) by Western blot as described in Materials and Methods. Data corresponding to 12 hours and 72 hours HbG-transfused guinea pigs are included for comparison purposes (from Figures 1 and 3). Densitometry values for protein expression as a ratio to actin were normalized to sham controls (NT) and presented as the means ± SEMs. *P < 0.05 vs NT. For NT and HbG, n = 4 to 5 animals per time point; for shed blood and albumin, n = 3 to 4 animals per time point.
Supplement Figure S2.
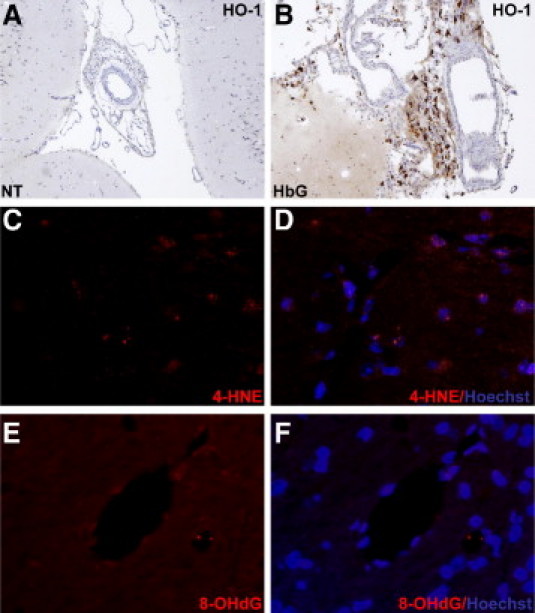
Brain HO-1 expression in sham controls (A) and 24 hours after HbG transfusion (B). HO-1 was detected with the use of Vectastain Elite with DAB as a substrate and counterstained with hematoxylin. HbG-transfused animals showed extensive perivascular HO-1 immunoreactivity around large vessels compared with sham controls. C–F: Representative images of 4-HNE–modified adducts and 8-OHdG staining in sham controls. 4-HNE and 8-OHdG were detected with an Alex Fluor 555 (red)–conjugated secondary antibody and counterstained with Hoechst 33342 (blue). Minimal immunoreactivity was observed in the sham controls for 4-HNE (C and D) and 8-OHdG (E and F) compared with HbG (see Figure 8). The same image capture and postprocessing settings were used for the corresponding images in Figure 8, A–F. Brightfield; original magnification, ×200 (A and B). Epifluorescence microscopy; original magnification, ×400 (C–F).
Supplemental Figure S3.

Effect of zinc protoporphyrin IX (ZnPP) and dexrazoxane (Dex) in HbG-transfused animals. Guinea pigs were injected with ZnPP (50 mg/kg i.p., n = 5) 2 hours before HbG transfusion or infused with Dex (150 mg/kg i.v., n = 5) immediately before HbG transfusion. A: Western blot analysis of ZO-1 expression in brains harvested from sham controls (NT) and 24 hours after HbG transfusion with ZnPP or Dex treatment. Representative immunoblots are shown for ZO-1 and actin with each lane representing a separate animal. Densitometry values for ZO-1 as a ratio to actin were normalized to NT and presented as the means ± SEMs. For comparison purposes, ZO-1 data corresponding to 24-hour HbG-transfused guinea pigs are included (from Figure 1). *P < 0.05 vs NT. B: ZnPP inhibits HbG-induced HO activity. Brain microsomes were harvested from NT and 24 hours after transfusion with HbG alone or HbG + ZnPP pretreatment. HO activity was measured as described in Materials and Methods and expressed as fold relative to NT animals. Values are shown as the means ± SEMs for at least four animals per group for each group. *P < 0.05 vs NT. C: Dex reduces brain iron accumulation in HbG-transfused animals. Iron was measured with the ferrozine assay as described in Materials and Methods. Brain homogenates were prepared from sham controls and 24 hours after transfusion with HbG alone or HbG + Dex co-treatment. For direct comparison purposes, fresh samples from the 24-hour HbG alone group were prepared and analyzed concurrently with the other two groups. Data were obtained from four animals per group. Values are expressed as nanogram of iron per milligram of tissue and presented as the means ± SEMs. *P < 0.01 vs NT.
Supplemental Figure S4.
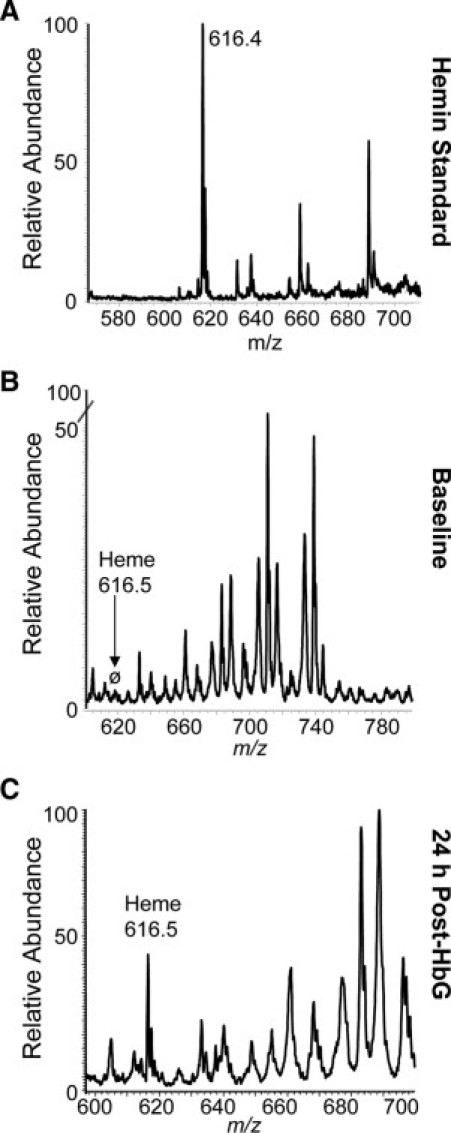
Free heme identification by mass spectrometry. Guinea pig plasma samples were collected at baseline (before transfusion) and 24 hours after HbG transfusion and processed through a 10-kDa Centricon filter (plasma filtrate). For heme identification, filtrates were run on a Thermofinnigan LTQ electrospray ionization mass spectrometry. Representative mass spectra are shown for standard hemin (A), baseline plasma filtrate (B), and 24 hours after HbG plasma filtrate (C). The hemin standard showed an ion at m/z 616.4 (theoretical = 616.47). The characteristic heme ion was absent in baseline samples, whereas the spectra of a sample 24 hours after HbG (n = 4) showed an ion consistent with heme at 616.5 m/z, suggesting the presence of free heme in plasma. Importantly, spectra from matrix-associated laser desorption and ionization mass spectrometry showed no Hb, albumin, or hemopexin protein mass ions in filtrate, suggesting that the heme ion detected in 24-hour samples is not associated with heme-containing protein traversing the filter.
References
- 1.Wagner K.R., Dwyer B.E. Hematoma removal, heme, and heme oxygenase following hemorrhagic stroke. Ann N Y Acad Sci. 2004;1012:237–251. doi: 10.1196/annals.1306.020. [DOI] [PubMed] [Google Scholar]
- 2.Robinson S.R., Dang T.N., Dringen R., Bishop G.M. Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009;14:228–235. doi: 10.1179/135100009X12525712409931. [DOI] [PubMed] [Google Scholar]
- 3.Keep R.F., Xiang J., Ennis S.R., Andjelkovic A., Hua Y., Xi G., Hoff J.T. Blood-brain barrier function in intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:73–77. doi: 10.1007/978-3-211-09469-3_15. [DOI] [PubMed] [Google Scholar]
- 4.Prunell G.F., Mathiesen T., Svendgaard N.A. Experimental subarachnoid hemorrhage: cerebral blood flow and brain metabolism during the acute phase in three different models in the rat. Neurosurgery. 2004;54:426–436. doi: 10.1227/01.neu.0000103670.09687.7a. [DOI] [PubMed] [Google Scholar]
- 5.Katsu M., Niizuma K., Yoshioka H., Okami N., Sakata H., Chan P.H. Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood-brain barrier dysfunction in vivo. J Cereb Blood Flow Metab. 2010;30:1939–1950. doi: 10.1038/jcbfm.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang F.P., Xi G., Keep R.F., Hua Y., Nemoianu A., Hoff J.T. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 7.Wang J., Doré S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–1652. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belcher J.D., Beckman J.D., Balla G., Balla J., Vercellotti G. Heme degradation and vascular injury. Antioxid Redox Signal. 2010;12:233–248. doi: 10.1089/ars.2009.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buehler P.W., D'Agnillo F. Toxicological consequences of extracellular hemoglobin: biochemical and physiological perspectives. Antioxid Redox Signal. 2010;12:275–291. doi: 10.1089/ars.2009.2799. [DOI] [PubMed] [Google Scholar]
- 10.D'Agnillo F., Alayash A.I. Redox cycling of diaspirin cross-linked hemoglobin induces G2/M arrest and apoptosis in cultured endothelial cells. Blood. 2001;98:3315–3323. doi: 10.1182/blood.v98.12.3315. [DOI] [PubMed] [Google Scholar]
- 11.Rother R.P., Bell L., Hillmen P., Gladwin M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 12.Kato G.J., Hebbel R.P., Steinberg M.H., Gladwin M.T. Vasculopathy in sickle cell disease: biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol. 2009;84:618–625. doi: 10.1002/ajh.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silverman T.A., Weiskopf R.B. Hemoglobin-based oxygen carriers: current status and future directions. Transfusion. 2009;49:2495–2515. doi: 10.1111/j.1537-2995.2009.02356.x. [DOI] [PubMed] [Google Scholar]
- 14.Pamplona A., Hanscheid T., Epiphanio S., Mota M.M., Vigário A.M. Cerebral malaria and the hemolysis/methemoglobin/heme hypothesis: shedding new light on an old disease. Int J Biochem Cell Biol. 2009;41:711–716. doi: 10.1016/j.biocel.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 15.Bunn H.F., Nathan D.G., Dover G.J., Hebbel R.P., Platt O.S., Rosse W.F., Ware R.E. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood. 2010;116:687–692. doi: 10.1182/blood-2010-02-268193. [DOI] [PubMed] [Google Scholar]
- 16.Elsayed N.M., Gorbunov N.V., Kagan V.E. A proposed biochemical mechanism involving hemoglobin for blast overpressure-induced injury. Toxicology. 1997;121:81–90. doi: 10.1016/s0300-483x(97)03657-3. [DOI] [PubMed] [Google Scholar]
- 17.Samadder N.J., Casaubon L., Silver F., Cavalcanti R. Neurological complications of paroxysmal nocturnal hemoglobinuria. Can J Neurol Sci. 2007;34:368–371. doi: 10.1017/s0317167100006855. [DOI] [PubMed] [Google Scholar]
- 18.Vichinsky E.P., Neumayr L.D., Gold J.I., Weiner M.W., Rule R.R., Truran D., Kasten J., Eggleston B., Kesler K., McMahon L., Orringer E.P., Harrington T., Kalinyak K., De Castro L.M., Kutlar A., Rutherford C.J., Johnson C., Bessman J.D., Jordan L.B., Armstrong F.D., Neuropsychological Dysfunction and Neuroimaging Adult Sickle Cell Anemia Study Group Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. 2010;303:1823–1831. doi: 10.1001/jama.2010.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butt O.I., Buehler P.W., D'Agnillo F. Differential induction of renal heme oxygenase and ferritin in ascorbate and nonascorbate producing species transfused with modified cell-free hemoglobin. Antioxid Redox Signal. 2010;12:199–208. doi: 10.1089/ars.2009.2798. [DOI] [PubMed] [Google Scholar]
- 20.Abraham N.G., Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 21.Schipper H.M., Song W., Zukor H., Hascalovici J.R., Zeligman D. Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. Neurochem. 2009;110:469–485. doi: 10.1111/j.1471-4159.2009.06160.x. [DOI] [PubMed] [Google Scholar]
- 22.Turner C.P., Bergeron M., Matz P., Zegna A., Noble L.J., Panter S.S., Sharp F.R. Heme oxygenase-1 is induced in glia throughout brain by subarachnoid hemoglobin. J Cereb Blood Flow Metab. 1998;18:257–273. doi: 10.1097/00004647-199803000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Koeppen A.H., Dickson A.C., Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63:587–597. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- 24.Panahian N., Yoshiura M., Maines M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem. 1999;72:1187–1203. doi: 10.1111/j.1471-4159.1999.721187.x. [DOI] [PubMed] [Google Scholar]
- 25.Guan L., Wen T., Zhang Y., Wang X., Zhao J. Induction of heme oxygenase-1 with hemin attenuates hippocampal injury in rats after acute carbon monoxide poisoning. Toxicology. 2009;262:146–152. doi: 10.1016/j.tox.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Gong Y., Tian H., Xi G., Keep R.F., Hoff J.T., Hua Y. Systemic zinc protoporphyrin administration reduces intracerebral hemorrhage-induced brain injury. Acta Neurochir Suppl. 2006;96:232–236. doi: 10.1007/3-211-30714-1_50. [DOI] [PubMed] [Google Scholar]
- 27.Chen M., Regan R.F. Time course of increased heme oxygenase activity and expression after experimental intracerebral hemorrhage: correlation with oxidative injury. J Neurochem. 2007;103:2015–2021. doi: 10.1111/j.1471-4159.2007.04885.x. [DOI] [PubMed] [Google Scholar]
- 28.Hawkins B.T., Davis T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 29.Persidsky Y., Ramirez S.H., Haorah J., Kanmogne G.D. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 30.Kaur C., Sivakumar V., Zhang Y., Ling E.A. Hypoxia-induced astrocytic reaction and increased vascular permeability in the rat cerebellum. Glia. 2006;54:826–839. doi: 10.1002/glia.20420. [DOI] [PubMed] [Google Scholar]
- 31.Jensen M.B., Finsen B., Zimmer J. Morphological and immunophenotypic microglial changes in the denervated fascia dentata of adult rats: correlation with blood-brain barrier damage and astroglial reactions. Exp Neurol. 1997;143:103–116. doi: 10.1006/exnr.1996.6337. [DOI] [PubMed] [Google Scholar]
- 32.Guillemin G.J., Brew B.J. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- 33.Krueger M., Bechmann I. CNS pericytes: concepts, misconceptions, and a way out. Glia. 2010;58:1–10. doi: 10.1002/glia.20898. [DOI] [PubMed] [Google Scholar]
- 34.Buehler P.W., D'Agnillo F., Hoffman V., Alayash A.I. Effects of endogenous ascorbate on oxidation, oxygenation, and toxicokinetics of cell-free modified hemoglobin after exchange transfusion in rat and guinea pig. J Pharmacol Exp Ther. 2007;323:49–60. doi: 10.1124/jpet.107.126409. [DOI] [PubMed] [Google Scholar]
- 35.Buehler P.W., Boykins R.A., Jia Y., Norris S., Freedberg D.I., Alayash A.I. Structural and functional characterization of glutaraldehyde-polymerized bovine hemoglobin and its isolated fractions. Anal Chem. 2005;77:3466–3478. doi: 10.1021/ac050064y. [DOI] [PubMed] [Google Scholar]
- 36.Kuang F., Wang B.R., Zhang P., Fei L.L., Jia Y., Duan X.L., Wang X., Xu Z., Li G.L., Jiao X.Y., Ju G. Extravasation of blood-borne immunoglobulin G through blood-brain barrier during adrenaline-induced transient hypertension in the rat. Int J Neurosci. 2004;114:575–591. doi: 10.1080/00207450490422731. [DOI] [PubMed] [Google Scholar]
- 37.Maines M. Carbon monoxide and nitric oxide homology: differential modulation of heme oxygenases in brain and detection of protein and activity. Methods Enzymol. 1996;268:473–488. doi: 10.1016/s0076-6879(96)68049-5. [DOI] [PubMed] [Google Scholar]
- 38.Carter P. Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (ferrozine) Anal Biochem. 1971;40:450–458. doi: 10.1016/0003-2697(71)90405-2. [DOI] [PubMed] [Google Scholar]
- 39.Meguro R., Asano Y., Odagiri S., Li C., Iwatsuki H., Shoumura K. Nonheme-iron histochemistry for light and electron microscopy: a historical, theoretical and technical review. Arch Histol Cytol. 2007;70:1–19. doi: 10.1679/aohc.70.1. [DOI] [PubMed] [Google Scholar]
- 40.Okajima K. On the selective staining of the erythrocyte. Anat Rec. 1916;11:295–296. [Google Scholar]
- 41.Balabanov R., Washington R., Wagnerova J., Dore-Duffy P. CNS microvascular pericytes express macrophage-like function, cell surface integrin alpha M, and macrophage marker ED-2. Microvasc Res. 1996;52:127–142. doi: 10.1006/mvre.1996.0049. [DOI] [PubMed] [Google Scholar]
- 42.Van Gorp H., Delputte P.L., Nauwynck H.J. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol Immunol. 2010;47:1650–1660. doi: 10.1016/j.molimm.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 43.Walter J.K., Rueckert C., Voss M., Mueller S.L., Piontek J., Gast K., Blasig I.E. The oligomerization of the coiled coil-domain of occludin is redox sensitive. Ann N Y Acad Sci. 2009;1165:19–27. doi: 10.1111/j.1749-6632.2009.04058.x. [DOI] [PubMed] [Google Scholar]
- 44.Allen C.L., Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 45.Pun P.B., Lu J., Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–364. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- 46.Brooks T.A., Hawkins B.T., Huber J.D., Egleton R.D., Davis T.P. Chronic inflammatory pain leads to increased blood-brain barrier permeability and tight junction protein alterations. Am J Physiol Heart Circ Physiol. 2005;289:H738–H743. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haorah J., Knipe B., Gorantla S., Zheng J., Persidsky Y. Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J Neurochem. 2007;100:324–336. doi: 10.1111/j.1471-4159.2006.04245.x. [DOI] [PubMed] [Google Scholar]
- 48.Rao R.K., Basuroy S., Rao V.U., Karnaky K.J., Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002;368:471–481. doi: 10.1042/BJ20011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lochhead J.J., McCaffrey G., Quigley C.E., Finch J., Demarco K.M., Nametz N., Davis T.P. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dokladny K., Ye D., Kennedy J.C., Moseley P.L., Ma T.Y. Cellular and molecular mechanisms of heat stress-induced up-regulation of occludin protein expression regulatory role of heat shock factor-1. Am J Pathol. 2008;172:659–670. doi: 10.2353/ajpath.2008.070522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCaffrey G., Staatz W.D., Quigley C.A., Nametz N., Seelbach M.J., Campos C.R., Brooks T.A., Egleton R.D., Davis T.P. Tight junctions contain oligomeric protein assembly critical for maintaining blood-brain barrier integrity in vivo. J Neurochem. 2007;103:2540–2555. doi: 10.1111/j.1471-4159.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 52.Chen-Roetling J., Benvenisti-Zarom L., Regan R.F. Cultured astrocytes from heme oxygenase-1 knockout mice are more vulnerable to heme-mediated oxidative injury. J Neurosci Res. 2005;82:802–810. doi: 10.1002/jnr.20681. [DOI] [PubMed] [Google Scholar]
- 53.Regan R.F., Guo Y., Kumar N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/s0304-3940(00)00817-x. [DOI] [PubMed] [Google Scholar]
- 54.Matz P., Turner C., Weinstein P.R., Massa S.M., Panter S.S., Sharp F.R. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996;713:211–222. doi: 10.1016/0006-8993(95)01511-6. [DOI] [PubMed] [Google Scholar]
- 55.Beschorner R., Adjodah D., Schwab J.M., Mittelbronn M., Pedal I., Mattern R., Schluesener H.J., Meyermann R. Long-term expression of heme oxygenase-1 (HO-1. HSP-32) following focal cerebral infarctions and traumatic brain injury in humans. Acta Neuropathol. 2000;100:377–384. doi: 10.1007/s004010000202. [DOI] [PubMed] [Google Scholar]
- 56.Bunn H.F., Jandl J.H. Exchange of heme among hemoglobins and between hemoglobin and albumin. J Biol Chem. 1968;243:465–475. [PubMed] [Google Scholar]
- 57.Hasinoff B.B., Hellmann K., Herman E.H., Ferrans V.J. Chemical, biological and clinical aspects of dexrazoxane and other bisdioxopiperazines. Curr Med Chem. 1998;5:1–28. [PubMed] [Google Scholar]
- 58.Galetta F., Franzoni F., Cervetti G., Regoli F., Fallahi P., Tocchini L., Carpi A., Antonelli A., Petrini M., Santoro G. In vitro and in vivo study on the antioxidant activity of dexrazoxane. Biomed Pharmacother. 2010;64:259–263. doi: 10.1016/j.biopha.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 59.Boven L.A., Middel J., Verhoef J., De Groot C.J., Nottet H.S. Monocyte infiltration is highly associated with loss of the tight junction protein zonula occludens in HIV-1-associated dementia. Neuropathol Appl Neurobiol. 2000;26:356–360. doi: 10.1046/j.1365-2990.2000.00255.x. [DOI] [PubMed] [Google Scholar]
- 60.Silva G., Jeney V., Chora A., Larsen R., Balla J., Soares M.P. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J Biol Chem. 2009;284:29582–29595. doi: 10.1074/jbc.M109.045344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wagener F.A., Feldman E., de Witte T., Abraham N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc Soc Exp Biol Med. 1997;216:456–463. doi: 10.3181/00379727-216-44197. [DOI] [PubMed] [Google Scholar]
- 62.Gaasch J.A., Lockman P.R., Geldenhuys W.J., Allen D.D., Van der Schyf C.J. Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem Res. 2007;32:1196–1208. doi: 10.1007/s11064-007-9290-4. [DOI] [PubMed] [Google Scholar]
- 63.Goldstein L., Teng Z.P., Zeserson E., Patel M., Regan R.F. Hemin induces an iron-dependent, oxidative injury to human neuron-like cells. J Neurosci Res. 2003;73:113–121. doi: 10.1002/jnr.10633. [DOI] [PubMed] [Google Scholar]