

Abstract
The genotype (M/M, M/V, or V/V) at polymorphic codon 129 of the human prion protein (PrP) gene and the type (1 or 2) of protease-resistant PrP (PrPres) in the brain are major determinants of the clinicopathological phenotypes of sporadic Creutzfeldt-Jakob disease (sCJD). According to this molecular typing system, sCJD has been classified into six subgroups (MM1, MM2, MV1, MV2, VV1, and VV2). Besides these pure subgroups, mixed cases presenting mixed neuropathological phenotypes and more than one PrPres type have been found in sCJD. To investigate the frequency of the co-occurrence of types 1 and 2 PrPres in sCJD patients classified as MM1, we produced type 2 PrPres-specific antibody Tohoku 2 (T2) that can specifically detect the N-terminal cleavage site of type 2 PrPres after protease treatment and examined brain samples from 23 patients with sCJD-MM1. Western blot analysis using the T2 antibody revealed that the minority type 2 PrPres could be detected in all sCJD-MM1 brain samples including those of the cerebellum where sCJD-MM2 prions rarely accumulate. These results show that the co-occurrence of types 1 and 2 PrPres within a single sCJD-MM1 patient is a universal phenomenon. The general co-occurrence of multiple PrPres fragments within a single prion strain questions the validity of the conventional molecular typing system.
Creutzfeldt-Jakob disease (CJD) is a lethal transmissible neurodegenerative disease caused by an abnormal isoform of prion protein (PrPSc), which is converted from the normal cellular isoform (PrPC).1 The genotype (M/M, M/V, or V/V) at polymorphic codon 129 of the human prion protein (PrP) gene and the type (type 1 or type 2) of PrPSc in the brain are major determinants of the clinicopathological phenotypes of sporadic CJD (sCJD).2–5 Type 1 and type 2 PrPSc are distinguishable according to the size of the proteinase K (PK)–resistant core of PrPSc (PrPres) (21 and 19 kDa, respectively), reflecting differences in the PK-cleavage site (at residues 82 and 97, respectively).2,5 According to this molecular typing system, sCJD has been classified into six subgroups (MM1, MM2, MV1, MV2, VV1, or VV2).
Besides these “pure” subgroups, “mixed” cases presenting mixed neuropathological phenotypes and more than one PrPres type have been reported.4,6–10 At first, the co-occurrence of types 1 and 2 PrPres within one individual was found in five of 14 patients with sCJD.6 Recently, a systematic regional study in a series of 225 patients revealed that 35% of the sCJD patients presented both PrPres types.9 In addition to these neuropathologically and biochemically mixed cases, monoclonal antibodies recognizing an epitope between residues 82 and 96 of human PrP (ie, specifically detecting type 1 PrPres after PK digestion), revealed that all CJD patients formerly classified as type 2 contained the minority type 1 PrPres despite the lack of mixed neuropathological phenotypes.11,12 The co-occurrence of multiple PrPres fragments without mixed neuropathological phenotypes has remained controversial. To investigate accurately the frequency of the co-occurrence of types 1 and 2 PrPres, we produced type 2 PrPres-specific polyclonal antibody Tohoku 2 (T2)13 and examined brain samples from 23 patients formerly classified as sCJD-MM1. Here we report that the minority type 2 PrPres could be detected with type 1 in all sCJD-MM1 patients examined.
Materials and Methods
Patients
All CJD cases included in this study were patients with clinically, genetically, and neuropathologically proven sCJD. The diagnosis of CJD and the type of PrPres were confirmed by neuropathological examination, PrP immunohistochemistry, and conventional Western blotting using monoclonal antibody 3F4 as described.14,15 The genotype and the absence of mutations in the open reading frame of the PrP gene were determined by sequence analysis.16 All subjects were homozygous for methionine at codon 129 of the PrP gene and were classified as follows: MM1, 23 cases; MM1+2 (MM1-dominant form), nine cases; MM2 (cortical form), one case. The clinical features of the patients are summarized in Table 1. Detailed information of this sCJD-MM2 patient has been reported previously.17 Four age-matched control subjects were included in this study and were also homozygous for methionine at codon 129 of the PrP gene.
Table 1.
Summary of Clinical Features
| Age at onset (years) | PSWC on EEG (months) | Myoclonus (months)⁎ | Akinetic mutism (months)⁎ | Duration of illness (months) | PrP deposition | |
|---|---|---|---|---|---|---|
| MM1 (n = 23) | 68.6 ± 7.8 | 2.0 ± 2.6 (23/23)† | 2.2 ± 2.5 (23/23)† | 3.0 ± 3.2 | 13.5 ± 7.8 | Synaptic |
| MM1+2 (n = 9) | 65.8 ± 10.2 | 5.3 ± 5.8 (8/9)† | 4.8 ± 5.4 (9/9)† | 5.7 ± 6.3 | 11.8 ± 10.9 | Synaptic + perivacuolar |
Values are mean ± SD.
Duration until appearance of PSWC, myoclonus, or akinetic mutism from onset.
Positive rate.
Sample Preparation and Western Blotting
Brain tissues were obtained at autopsy from the patients after receiving informed consent for research use. PrPres was extracted from brain tissues with collagenase treatment as described18 with some modifications. Samples were subjected to 13% SDS–polyacrylamide gel electrophoresis and Western blotting as described.19 The production of type 2 PrPres-specific polyclonal antibody T2 has been reported previously.13 The monoclonal antibody 3F420 and the T2 antibody were used as the primary antibodies. Goat–antimouse immunoglobulin polyclonal antibody labeled with the peroxidase-conjugated dextran polymer, EnVision+ (Dako, Carpinteria, CA) and antirabbit EnVision+ were used as the secondary antibodies. The signal intensities of the Western blots were quantified with Quantity One software using an imaging device, VersaDoc 5000 (Bio-Rad Laboratories, Hercules, CA).
Immunohistochemistry
Formalin-fixed brain tissues were treated with 99% formic acid for 1 hour to inactivate the infectivity and embedded in paraffin. Tissue sections were pretreated by hydrolytic autoclaving before PrP immunohistochemistry.14 The 3F4 antibody and the monoclonal antibody #7121,22 were used as the primary antibodies. Antimouse EnVision+ was used as the secondary antibody.
Results
T2-Reactive PrPres Fragments in the Cerebrums from sCJD-MM1 Patients
To determine whether type 2 PrPres could be detected in the sCJD patients formerly classified as type 1, we examined the cerebral samples from 23 patients with sCJD-MM1 by Western blotting using type 2 PrPres-specific polyclonal antibody T2 (Figure 1A). The T2 antibody is a proteolytic cleavage site–specific antibody and can specifically react with the N-terminally cleaved type 2 PrPres after PK digestion.13 Although the N-terminal cleavage of type 2 PrPres by PK treatment can occur at several sites,5 the T2 antibody detects the major cleavage products cleaved at residue 97.13 As the positive control for the T2 antibody, we also examined nine patients with sCJD-MM1+2 (MM1-dominant form) and one patient with sCJD-MM2 (cortical form). In the conventional Western blot analysis using the monoclonal antibody 3F4, which is the most popular anti-PrP antibody that reacts with all PrPres types, the sCJD-MM1 brains showed only type 1 PrPres (Figure 1B). In contrast, in the Western blot analysis using the T2 antibody, all sCJD-MM1 brains contained T2-reactive PrPres fragments (Figure 1C). The sizes of the T2-reactive fragments were identical to those of type 2 PrPres in the sCJD-MM2 brain. In the quantitative analysis, the mean signal intensities of PrPres in the sCJD-MM2 brain were assigned as 100/mm2 in each experiment using the 3F4 or T2 antibody (Figure 1D). To compare the amounts of the T2-reactive PrPres fragments among the patients, the mean signal intensities of the T2-reactive fragments were divided by those of the 3F4-reactive PrPres fragments, and the percentage of the T2-reactive fragments was calculated in each case (T2/3F4 ratio). The mean T2/3F4 ratio of the sCJD-MM1 patients was significantly lower than that of the sCJD-MM1+2 patients (Figure 1E). Thus, small but significant amounts of T2-reactive PrPres fragments could be detected in all sCJD-MM1 patients examined.
Figure 1.
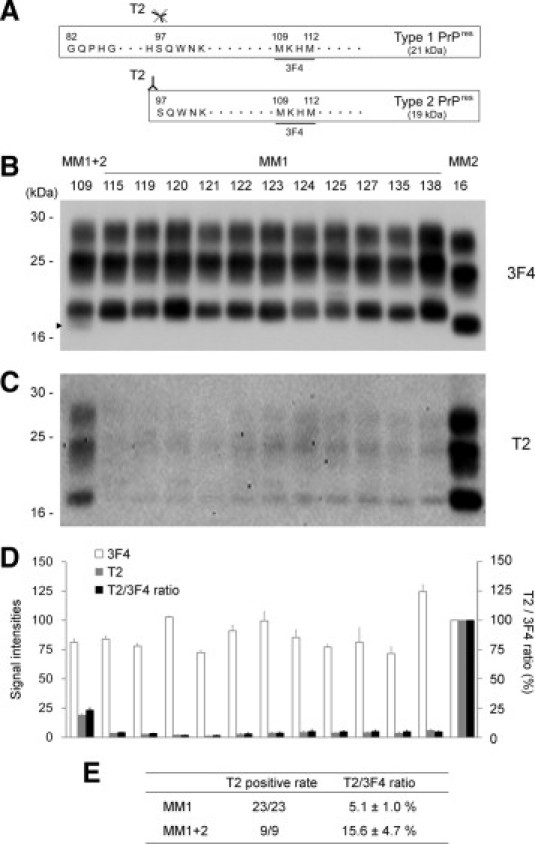
Tohoku 2 (T2)–reactive PrPres fragments in the cerebrums from sCJD-MM1 patients. A: The epitope for type 2 PrPres-specific antibody T2. The T2 antibody specifically detects the N-terminal cleavage site of type 2 PrPres.13 In contrast, 3F4 antibody detects both PrPres types. B: The conventional Western blot analysis using the 3F4 antibody was used for the classification of sCJD patients included in this study. The subgroup and case number of each patient are described above each lane. A faint type 2 PrPres band (arrowhead) was detected with the majority type 1 PrPres bands in the sCJD-MM1+2 brain (case 109). C: T2-reactive PrPres fragments could be detected in all sCJD-MM1 brains examined. D: The signal intensities of the 3F4-reactive PrPres fragments (white bars) or those of the T2-reactive PrPres fragments (gray bars). The mean signal intensities of PrPres in the sCJD-MM2 brain (case 16) were assigned as 100/mm2 in each experiment. To compare the amounts of the T2-reactive PrPres fragments among the patients, the mean signal intensities of the T2-reactive fragments were divided by those of the 3F4-reactive fragments, and the percentage of the T2-reactive fragments was calculated in each case (T2/3F4 ratio, black bars). The data are expressed as mean ± SEM (n = 3). E: Summary of the Western blot analysis using the T2 antibody. Values are means ± SEM.
T2-Reactive PrPres Fragments in the Cerebellums from sCJD-MM1 Patients
The consistent detection of the T2-reactive PrPres fragments in the cerebrums from sCJD-MM1 patients led us to suppose that co-occurrence of types 1 and 2 PrPres fragments might be a universal phenomenon. Indeed, the T2-reactive PrPres fragments were also detected in the cerebellum of a sCJD-MM1+2 patients in the present study. In most sCJD-MM1+2 patients, type 2 PrPres is rarely detected in the cerebellum by conventional Western blot analysis.9 In addition, the cerebellums from sCJD-MM1+2 patients lack the perivacuolar PrP deposition that is characteristic of sCJD-MM2 prions, whereas the cerebrums show a mixed PrP deposition pattern (ie, synaptic plus perivacuolar PrP deposition).9,10 In accordance with these reports, the cerebellums from the sCJD-MM1+2 patients in the present study lacked perivacuolar PrP deposition and showed only the synaptic-type deposition, whereas the cerebral cortices showed perivacuolar PrP deposition in addition to the synaptic-type deposition (Figure 2A). Furthermore, conventional Western blot analysis using the 3F4 antibody showed the lack of type 2 PrPres accumulation in the cerebellum of the sCJD-MM1+2 patient (case 45 CE) (Figure 2B). Nevertheless, Western blot analysis using the T2 antibody revealed that the cerebellum of the sCJD-MM1+2 patient (case 45 CE) contained T2-reactive PrPres fragments (Figure 2C). The sizes of the T2-reactive fragments were identical to those of type 2 PrPres in the sCJD-MM2 brain. In the quantitative analysis, the T2/3F4 ratio of the cerebellum (case 45 CE) was lower than that of the cerebrum (case 45) (Figure 2D). Thus, small amounts of T2-reactive PrPres fragments could be detected also in the cerebellum of the sCJD-MM1+2 patients.
Figure 2.
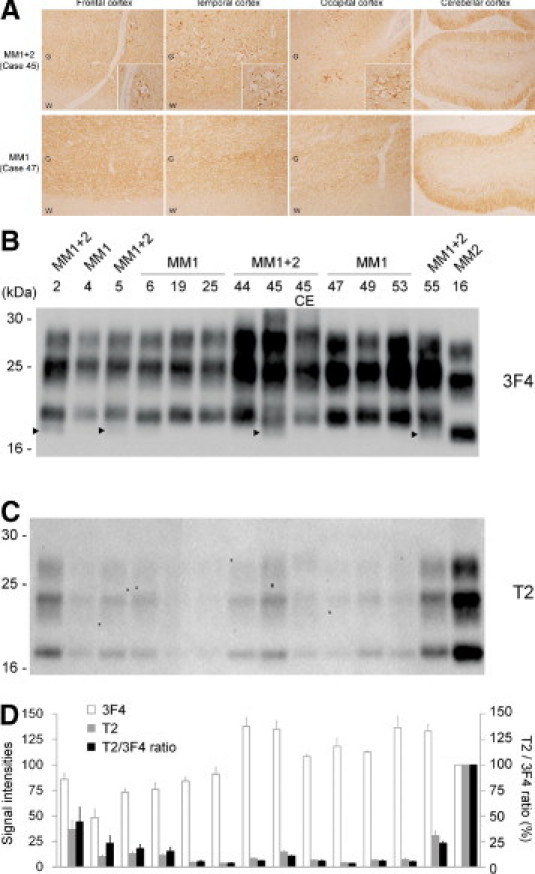
T2-reactive PrPres fragments in the cerebellum from the sCJD-MM1+2 patient. A: Imunohistochemistry using the anti-PrP antibody #71 showed that perivacuolar PrP deposition was restricted to within the cerebral cortices in the sCJD-MM1+2 patient (cases 45; ×40, insets × 200). The sCJD-MM1 patient showed only synaptic-type PrP deposition (case 47; ×40). G = gray matter; W = white matter. B: In the conventional Western blot analysis using the 3F4 antibody, faint type 2 PrPres bands (arrowheads) were detected in the cerebrums from the sCJD-MM1+2 patients (cases 2, 5, 45, and 55) but not in the cerebellum (case 45 CE). In case 44, although neuropathological examination showed focal perivacuolar PrP deposition, the conventional Western blot analysis failed to detect type 2 PrPres. C: T2-reactive PrPres fragments could be detected also in the cerebellum of the sCJD-MM1+2 patient (case 45 CE). D: The signal intensities of the 3F4-reactive PrPres fragments (white bars) or those of the T2-reactive PrPres fragments (gray bars), and the T2/3F4 ratios (black bars). The mean signal intensities of PrPres in the sCJD-MM2 brain (case 16) were assigned as 100/mm2 in each experiment. Data are expressed as mean ± SEM (n = 3).
For further analysis, we also examined the cerebellums from two sCJD-MM1 patients. In most sCJD-MM2 patients, the cerebellum lacks significant pathology, and PrP deposition is seldom or never observed.4 In line with this report, the cerebellum of the present sCJD-MM2 patient (case 16 CE) did not contain any PrPres signals when probed with the 3F4 or T2 antibody (Figure 3, A and B).17 In contrast, the cerebellums from the sCJD-MM1 patients contained the T2-reactive PrPres fragments (cases 47 CE and 49 CE). These T2-reactive PrPres fragments showed the exact type 2 PrPres migration pattern. In the quantitative analysis, the T2/3F4 ratios of the cerebellums (case 47 CE, 1.5% ± 0.8%; case 49 CE, 1.1% ± 0.5%) were lower than those of the cerebrums (case 47, 3.4% ± 0.5%; case 49, 5.9% ± 0.8%) (Figure 3C). Thus, trace amounts of T2-reactive PrPres fragments could be detected in all sCJD-MM1 brain samples including those of the cerebellum.
Figure 3.
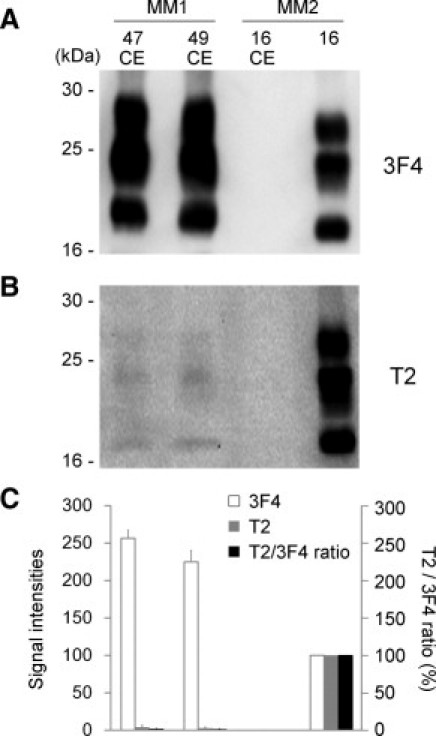
T2-reactive PrPres fragments in the cerebellums from the sCJD-MM1 patients. A: In the conventional Western blot analysis using the 3F4 antibody, PrPres was not detected in the cerebellum of the sCJD-MM2 patient (case 16 CE). B: The T2-reactive PrPres fragments could be detected in the cerebellums from the sCJD-MM1 patients (cases 47 CE and 49 CE), but not in the cerebellum from the sCJD-MM2 patient (case 16 CE). C: The signal intensities of the 3F4-reactive PrPres fragments (white bars) or those of the T2-reactive PrPres fragments (gray bars), and the T2/3F4 ratios (black bars). The mean signal intensities of PrPres in the sCJD-MM2 cerebrum (case 16) were assigned as 100/mm2 in each experiment. Data are expressed as mean ± SEM (n = 3).
T2-Reactive Truncated PrPC Fragments in Non-CJD Control Brains
The normal cellular isoform of PrP, denoted as PrPC, is cleaved by endogenous proteases and can generate various N-terminally truncated PrPC molecules.23,24 This raised the possibility that T2-reactive PrPC fragments (97-PrPC) might pre-exist in the brains and might be incorporated into MM1 PrPres aggregates in sCJD-MM1 patients. To determine whether the 97-PrPC fragments can be generated in normal brains, we examined brain samples from four age-matched control subjects without PK treatment. Western blot analysis using the T2 antibody showed that the cerebral cortices from the control subjects contained 97-PrPC (Figure 4, A and B). The glycosylation patterns of these 97-PrPC fragments were different from those of the T2-reactive PrPres fragments observed in the sCJD patients. After deglycosylation with PNGase F, these 97-PrPC fragments migrated at 19 kDa. Moreover, the cerebellums from the control subjects also contained the 97-PrPC (Figure 4, C and D). Thus, the T2-reactive truncated PrPC fragments could be detected in all control brain samples including those of the cerebellums.
Figure 4.
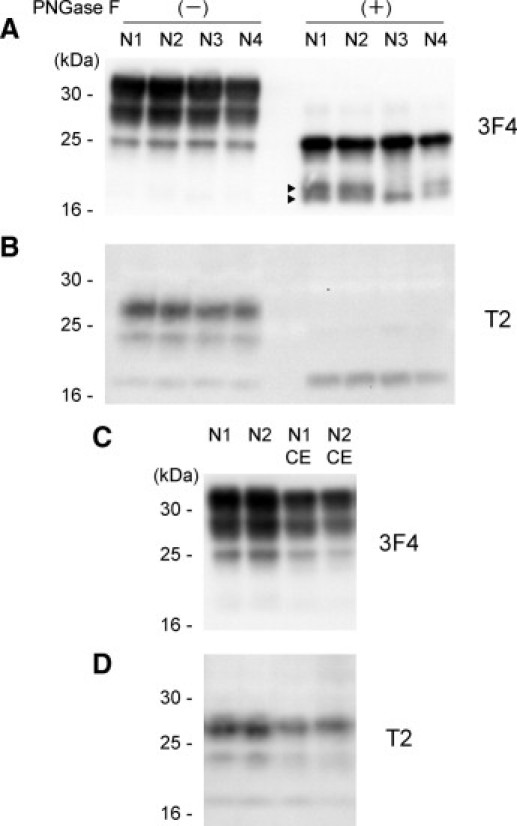
T2-reactive truncated PrPC fragments in non-CJD control brains. A: The cerebral cortices from age-matched control subjects (N1 to N4) were examined by conventional Western blot analysis using the 3F4 antibody without PK treatment. After deglycosylation with PNGase F, major PrPC fragments migrated at 25 kDa. In addition, various truncated PrPC fragments (arrowheads) were also observed. B: T2-reactive truncated PrPC fragments (97-PrPC) could be detected in all control brains. The 97-PrPC fragments migrated at 19 kDa after the deglycosylation. C and D: The 97-PrPC fragments could be detected also in the cerebellums from the control subjects (N1 CE and N2 CE).
Patchy Plaque-Type PrP Deposition in the sCJD-MM1 Patients with High T2/3F4 Ratios
In the 23 patients with sCJD-MM1 examined in the present study, two patients (cases 4 and 6) showed high T2/3F4 ratios similar to those of the sCJD-MM1+2 patients (Figure 2D). The clinical courses of cases 4 and 6 were not significantly different from those of the other sCJD-MM1 patients (Figure 5A). Indeed, the duration until the appearance of akinetic mutism and total duration of illness in case 6 were rather shorter than the mean values in the sCJD-MM1 patients. However, the high T2/3F4 ratios raised the possibility that these two sCJD patients might contain a focal deposition of MM2 prions overlooked in the previous examination. Therefore, we re-examined all brain sections from these patients by PrP immunohistochemistry. The cerebral and cerebellar cortices from the two patients lacked perivacuolar PrP deposition and showed diffuse synaptic-type PrP deposition. However, careful re-examination revealed a very focal accumulation of small PrP plaques in the cerebral cortices (Figure 5B). These patchy plaque-type PrP deposits were not accompanied by large vacuoles, in contrast to the perivacuolar PrP deposition (Figure 5, C and D). Meanwhile, besides perivacuolar and synaptic-type deposition, coarse PrP deposits similar to the patchy plaque-type PrP deposition were observed in the cerebral cortices from the sCJD-MM1+2 patients. These coarse PrP deposits in the sCJD-MM1+2 patients were also independent of vacuoles (Figure 5E). Thus, the sCJD-MM1 patients with the high T2/3F4 ratios showed the patchy plaque-type PrP deposition similar to the coarse PrP deposition in the sCJD-MM1+2 patients. Although these sCJD patients had been classified into MM1 by the conventional classification system because the lack of perivacuolar PrP deposition, the patchy plaque-type PrP deposition might indicate the existence of MM2 prions.
Figure 5.
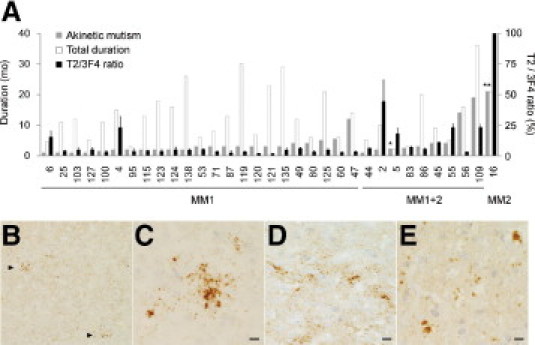
Patchy plaque-type PrP deposition in the sCJD-MM1 patients with high T2/3F4 ratios. A: The duration until the appearance of akinetic mutism (gray bars), total duration of illness (white bars), and the T2/3F4 ratio (black bars) of the sCJD patients. The sCJD-MM1 or sCJD-MM1+2 cases are arranged in order of the duration until the appearance of akinetic mutism. *The patient became unconsciousness and bedridden 1 month after the initial symptom because of respiratory failure. **The patient did not present akinetic mutism throughout the clinical course. B: PrP immunohistochemistry using the 3F4 antibody revealed patchy plaque-type PrP deposition (arrowheads) besides diffuse synaptic-type PrP deposition in the cerebral cortices from the sCJD-MM1 patient with the high T2/3F4 ratio (case 4; temporal cortex; ×40). C: The patchy plaque-type PrP deposition was independent of vacuoles (case 4; occipital cortex; ×400). D: The patchy plaque-type deposition in the other sCJD-MM1 patient with the high T2/3F4 ratio (case 6; frontal cortex; ×400). E: The cerebral cortices from the sCJD-MM1+2 patient showed coarse PrP deposition similar to the patchy plaque-type PrP deposition (case 5; parietal cortex; ×400). Scale bars = 10 μm.
Discussion
Our data comprised three major findings. First, using the novel antibody T2 that can specifically react with the N-terminal cleavage site of type 2 PrPres after PK digestion, type 2 PrPres could be detected in all sCJD-MM1 brain samples including those of the cerebellum where sCJD-MM2 prions rarely accumulate. Second, T2-reactive truncated PrPC fragments (97-PrPC) could be detected in age-matched control brains without PK treatment. Third, two sCJD-MM1 patients with relatively high type 2 PrPres contents showed unique PrP deposition, namely the patchy plaque-type deposition. Here we demonstrate that the co-occurrence of types 1 and 2 PrPres within a single sCJD-MM1 patient is a universal phenomenon.
The trace amounts of type 2 PrPres in the sCJD-MM1 brains might result from the involvement of 97-PrPC fragments into the MM1 PrPres aggregates. Despite the lack of perivacuolar PrP deposition that is characteristic of sCJD-MM2 prions, trace amounts of type 2 PrPres were detected in all sCJD-MM1 patients examined. In addition, type 2 PrPres could be detected not only in the cerebrums but also in the cerebellums where sCJD-MM2 prions rarely accumulate. These findings support the view of the co-occurrence of multiple PrPres fragments within MM1 prions rather than the co-occurrence of multiple prion strains within the same individual. One possible explanation for the co-occurrence of multiple PrPres fragments within a single prion strain is the involvement of multiple PrPC fragments into the PrPres aggregates. There is accumulating evidence that various N-terminally truncated PrPC molecules exist in human brains.23,24 Moreover, T2-reactive 97-PrPC fragments existed in normal human brains in the present study. These 97-PrPC fragments might be incorporated into the PrPres aggregates with full-length PrPSc and might acquire protease resistance as reported in deletion mutant PrPs or green fluorescent protein–tagged PrP.25,26 These findings lead us to speculate that the minority type 1 PrPres detected in the brains from CJD patients classified as type 211,12 might also be caused by the involvement of 82-PrPC fragments. Because the type 1 PrPres-specific antibodies used in these reports recognize an epitope between residues 82 and 96 of human PrP and can react with full-length PrPC, it remains to be determined whether 82-PrPC fragments can be generated in human brains. If an antibody that can specifically detect the N-terminal cleavage site at residue 82 is generated, 82-PrPC fragments might also be detected in human brains. To examine the possibility of the co-occurrence of multiple prion strains within one individual, whether transmissibilities and pathological phenotypes can be split into two groups in the transmission studies using the limiting dilution of these CJD brains should be addressed in the future.
The analysis of the type 2/total PrPres ratio (ie, the T2/3F4 ratio) using the T2 antibody may have significance in classifications of sCJD. In the present study, two of 23 sCJD-MM1 patients showed high T2/3F4 ratios and the unique PrP deposition designated as the patchy plaque-type. Because the patchy plaque-type deposition was similar to the coarse PrP deposition in the sCJD-MM1+2 patients, these deposits might be precursors of the perivacuolar PrP deposition or variant deposition patterns of MM2 prions. The finding of the patchy plaque-type deposition in the sCJD patients who have been neuropathologically and biochemically classified into MM1 raises the possibility that the co-occurrence of MM1 and MM2 prions within the same brain might be underestimated in the conventional classification. Hereafter, sCJD-MM1 patients need to be examined by the T2 antibody, and the focal accumulation of MM2 prions must be tested carefully in patients showing high T2/3F4 ratios compared with the mean value of the sCJD-MM1 patients (5.1%). On the other hand, although the mean T2/3F4 ratio of the sCJD-MM1+2 patients was significantly higher than that of the sCJD-MM1 patients, some of the sCJD-MM1+2 patients showed low T2/3F4 ratios indistinguishable from those of the sCJD-MM1 patients. These results suggest that examination using the T2 antibody is insufficient to discriminate sCJD-MM1+2 from sCJD-MM1 when the amount of type 2 PrPres is marginal and the examined brain region is limited. To avoid missing the focal accumulation of MM2 prions, multiple brain regions need to be examined by the T2 antibody, as suggested when using the conventional Western blot analysis.9
Unfortunately, immunohistochemical analysis of type 2 PrPres using the T2 antibody was unavailable in the present study. To eliminate the infectivity of sCJD prions, all brain sections had to be treated with formic acid, and infectious conformers were denatured. Therefore, the PrPres type-specific cleavage by PK treatment was impossible in tissue sections. We also attempted to detect the endogenous protease-cleaved type 2 PrPres without PK treatment. In this preliminary experiment, the patchy plaque-type PrP deposition in the sCJD-MM1 patients with the high T2/3F4 ratios showed the T2-immunoreactivities, whereas the synaptic-type deposition was not immunolabeled with the T2 antibody (A. Kobayahsi and T. Kitamoto, unpublished). However, as 97-PrPC fragments also exist in human brains, we cannot conclude whether the T2 immunoreactivities observed in the sCJD brains without PK treatment represent the existence of type 2 PrPres.
In conclusion, the present study, together with evidence from other groups,11,12 suggests that the co-occurrence of multiple PrPres fragments within a single sCJD patient is a universal phenomenon. These findings show that the conventional typing of PrPres merely represents the predominant PrPres subpopulation among multiple co-existing PrPres fragments. Besides the general co-occurrence of multiple PrPres fragments, the condition of PK digestion easily affects the size of PrPres.27,28 Indeed, insufficient PK digestion can generate type 1 PrPres-specific antibody–reactive fragments in the sCJD patients classified as type 2.29 Furthermore, it is possible that the conventional Western blot analysis fails to detect type 2 PrPres in sCJD-MM1+2 cases showing very focal perivacuolar PrP deposition in the brain.9 These confusing aspects of PrPres typing question the validity of the conventional molecular typing system. For a precise classification, it may be appropriate that the neuropathological phenotyping [synaptic (SY), perivacuolar (PV), plaque (PL), or patchy plaque (PP)] be combined with the molecular typing [eg, sCJD-MM1(+2)/SY+PV] the sCJD-MM patient showing synaptic + perivacuolar PrP deposition, but not type 2 PrPres in the conventional Western blot analysis.
Acknowledgments
We thank Hiroko Kudo for excellent technical assistance and Brent Bell for critical review of the manuscript.
Footnotes
Supported by Grants-in-Aid from the Research Committee of Prion disease and Slow Virus Infection, the Ministry of Health, Labour and Welfare of Japan (A.K., Y.I., and T.K.), and by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (A.K. and T.K.).
References
- 1.Prusiner S.B., Scott M.R., DeArmond J.P., Cohen F.E. Prion protein biology. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 2.Parchi P., Castellani R., Capellari S., Ghetti B., Young K., Chen S.G., Farlow M., Dickson D.W., Sima A.A., Trojanowski J.Q., Petersen R.B., Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 3.Parchi P., Capellari S., Chen S.G., Petersen R.B., Gambetti P., Kopp N., Brown P., Kitamoto T., Tateishi J., Giese A., Kretzschmar H. Typing prion isoforms. Nature. 1997;386:232–234. doi: 10.1038/386232a0. [DOI] [PubMed] [Google Scholar]
- 4.Parchi P., Giese A., Capellari S., Brown P., Schulz-Schaeffer W., Windl O., Zerr I., Budka H., Kopp N., Piccardo P., Poser S., Rojiani A., Streichemberger N., Julien J., Vital C., Ghetti B., Gambetti P., Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 5.Parchi P., Zou W., Wang W., Brown P., Capellari S., Ghetti B., Kopp N., Schulz-Schaeffer W.J., Kretzschmar H.A., Head M.W., Ironside J.W., Gambetti P., Chen S.G. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puoti G., Giaccone G., Rossi G., Canciani B., Bugiani O., Tagliavini F. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology. 1999;53:2173–2176. doi: 10.1212/wnl.53.9.2173. [DOI] [PubMed] [Google Scholar]
- 7.Schoch G., Seeger H., Bogousslavsky J., Tolnay M., Janzer R.C., Aguzzi A., Glatzel M. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS One. 2006;3:e14. doi: 10.1371/journal.pmed.0030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uro-Coste E., Cassard H., Simon S., Lugan S., Bilheude J.M., Perret-Liaudet A., Ironside J.W., Haik S., Basset-Leobon C., Lacroux C., Peoch' K., Streichenberger N., Langeveld J., Head M.W., Grassi J., Hauw J.J., Schelcher F., Delisle M.B., Andréoletti O. Beyond PrPres type 1 / type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 2008;4:e1000029. doi: 10.1371/journal.ppat.1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parchi P., Strammiello R., Notari S., Giese A., Langeveld J.P., Ladogana A., Zerr I., Roncaroli F., Cras P., Ghetti B., Pocchiari M., Kretzschmar H., Capellari S. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009;118:659–671. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cali I., Castellani R., Alshekhlee A., Cohen Y., Blevins J., Yuan J., Langeveld J.P., Parchi P., Safar J.G., Zou W.Q., Gambetti P. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132:2643–2658. doi: 10.1093/brain/awp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polymenidou M., Stoeck K., Glatzel M., Vey M., Bellon A., Aguzzi A. Coexistence of multiple PrPSc types in individuals with Creutzfeldt-Jakob disease. Lancet Neurol. 2005;4:805–814. doi: 10.1016/S1474-4422(05)70225-8. [DOI] [PubMed] [Google Scholar]
- 12.Yull H.M., Ritchie D.L., Langeveld J.P.M., van Zijderveld F.G., Bruce M.E., Ironside J.W., Head M.W. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:151–157. doi: 10.2353/ajpath.2006.050766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi A., Sakuma N., Matsuura Y., Mohri S., Aguzzi A., Kitamoto T. Experimental verification of a traceback phenomenon in prion infection. J Virol. 2010;84:3230–3238. doi: 10.1128/JVI.02387-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitamoto T., Shin R.-W., Doh-ura K., Tomokane N., Miyazono M., Muramoto T., Tateishi J. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol. 1992;140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- 15.Taguchi Y., Mohri S., Ironside J.W., Muramoto T., Kitamoto T. Humanized knock-in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am J Pathol. 2003;163:2585–2593. doi: 10.1016/S0002-9440(10)63613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitamoto T., Ohta M., Doh-ura K., Hitoshi S., Terao Y., Tateishi J. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun. 1993;191:709–714. doi: 10.1006/bbrc.1993.1275. [DOI] [PubMed] [Google Scholar]
- 17.Nozaki I., Hamaguchi T., Noguchi-Shinohara M., Ono K., Shirasaki H., Komai K., Kitamoto T., Yamada M. The MM2-cortical form of sporadic Creutzfeldt-Jakob disease presenting with visual disturbance. Neurology. 2006;67:531–533. doi: 10.1212/01.wnl.0000228224.35678.60. [DOI] [PubMed] [Google Scholar]
- 18.Grathwohl K.U., Horiuchi M., Ishiguro N., Shinagawa M. Improvement of PrPSc-detection in mouse spleen early at the preclinical stage of scrapie with collagenase-completed tissue homogenization and Sarkosyl-NaCl extraction of PrPSc. Arch Virol. 1996;141:1863–1874. doi: 10.1007/BF01718200. [DOI] [PubMed] [Google Scholar]
- 19.Asano M., Mohri S., Ironside J.W., Ito M., Tamaoki N., Kitamoto T. vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun. 2006;342:293–299. doi: 10.1016/j.bbrc.2006.01.149. [DOI] [PubMed] [Google Scholar]
- 20.Kascsak R.J., Rubenstein R., Merz P.A., Tonna-DeMasi M., Fersko R., Carp R.I., Wisniewski H.M., Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muramoto T., Tanaka T., Kitamoto N., Sano C., Hayashi Y., Kutomi T., Yutani C., Kitamoto T. Analyses of Gerstmann-Straussler syndrome with 102Leu219Lys using monoclonal antibodies that specifically detect human prion protein with 219Glu. Neurosci Lett. 2000;288:179–182. doi: 10.1016/s0304-3940(00)01232-5. [DOI] [PubMed] [Google Scholar]
- 22.Satoh K., Muramoto T., Tanaka T., Kitamoto N., Ironside J.W., Nagashima K., Yamada M., Sato T., Mohri S., Kitamoto T. Association of an 11–12 kDa protease-resistant prion protein fragment with subtypes of dura graft-associated Creutzfeldt-Jakob disease and other prion diseases. J Gen Virol. 2003;84:2885–2893. doi: 10.1099/vir.0.19236-0. [DOI] [PubMed] [Google Scholar]
- 23.Chen S.G., Teplow D.B., Parchi P., Teller J.K., Gambetti P., Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- 24.Jiménez-Huete A., Lievens P.M., Vidal R., Piccardo P., Ghetti B., Tagliavini F., Frangione B., Prelli F. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am J Pathol. 1998;153:1561–1572. doi: 10.1016/S0002-9440(10)65744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers M., Yehiely F., Scott M., Prusiner S.B. Conversion of truncated and elongated prion proteins into the scrapie isoform in cultured cells. 1993;90:3182–3186. doi: 10.1073/pnas.90.8.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bian J., Nazor K.E., Angers R., Jernigan M., Seward T., Centers A., Green M., Telling G.C. GFP-tagged PrP supports compromised prion replication in transgenic mice. Biochem Biophys Res Commun. 2006;340:894–900. doi: 10.1016/j.bbrc.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 27.Notari S., Capellari S., Giese A., Westner I., Baruzzi A., Ghetti B., Gambetti P., Kretzschmar H.A., Parchi P. Effects of different experimental conditions on the PrPSc core generated by protease digestion. J Biol Chem. 2004;279:16797–16804. doi: 10.1074/jbc.M313220200. [DOI] [PubMed] [Google Scholar]
- 28.Cali I., Castellani R., Yuan J., Al-Shekhlee A., Cohen M.L., Xiao X., Moleres F.J., Parchi P., Zou W.Q., Gambetti P. Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain. 2006;129:2266–2277. doi: 10.1093/brain/awl224. [DOI] [PubMed] [Google Scholar]
- 29.Notari S., Capellari S., Langeveld J., Giese A., Strammiello R., Gambetti P., Kretzschmar H.A., Parchi P. A refined method for molecular typing reveals that co-occurrence of PrPSc types in Creutzfeldt-Jakob disease is not the rule. Lab Invest. 2007;87:1103–1112. doi: 10.1038/labinvest.3700676. [DOI] [PubMed] [Google Scholar]