

See related article on page 609
The glomerulus of the kidney is a complicated structure that consists of i) a capillary bed composed of specialized endothelial cells; ii) mesangial cells, modified mesenchymal cells that have a primary function of maintaining the three-dimensional structure of the capillary bed; iii) highly terminally differentiated visceral epithelial cells called podocytes; and iv) the glomerular basement membrane (GBM) found between the podocytes and the endothelial cells. The endothelial cells, GBM, and podocytes form the glomerular filtration barrier. The glomerulus is the primary target of most chronic renal diseases, and when severely injured it undergoes glomerulosclerosis, characterized by excessive extracellular matrix deposition and glomerular cell death. Numerous initiating events targeting any of the four major components of the glomerulus can result in glomerulosclerosis. Thus, it is imperative that we understand the molecular and cellular mechanisms that contribute to the homeostasis of the glomerular filtration barrier if we want to devise new tools to halt the progression of glomerulosclerosis and ideally prevent glomerular disease.
Growth factor–dependent signaling plays a critical role in the normal development and maintenance of the integrity of the glomerulus. This signaling can be autocrine and paracrine because it can occur between endothelial and mesangial cells, which are in direct contact with each other, and between podocytes and mesangial or endothelial cells, which are separated from each other by the GBM. The study by Khan and colleagues1 in this issue of the The American Journal of Pathology provides further evidence that paracrine signaling between mesangial and endothelial cells is important for the maintenance of normal glomerular integrity by providing novel proof that mesangial cell sequestration of latent transforming growth factor-β (TGF-β) by integrin αvβ8 modulates glomerular endothelial cell function.
Integrins, the transmembrane receptors for extracellular matrix, are composed of noncovalently linked α and β subunits. Twenty-four integrins with specific but overlapping functions are found in mammals.2 The best studied and characterized function of integrins is their ability to bind to the extracellular matrix, after which they mediate multiple cell functions, such as cell adhesion, migration, proliferation, and survival. These integrin-dependent functions are required for glomerular development and for maintenance of glomerular homeostasis in health and disease. The laminin binding receptor integrin α3β1 is essential for normal glomerular development, and mice in which the integrin α3 subunit is constitutively deleted have severe developmental abnormalities characterized by alterations in glomerular capillary loops, a disorganized GBM, and an inability of the foot processes of the podocytes to mature properly.3 Selective deletion of the integrin α3 subunit in the podocyte results in massive proteinuria and nephrotic syndrome, and the glomeruli have a disorganized GBM, podocyte foot process effacement, and, finally, glomerulosclerosis.4 A similar although more profound glomerular phenotype occurs when the integrin β1 subunit (which can bind 12 different α subunits, including α3) is selectively deleted in podocytes.5 The only other integrin known to regulate glomerular development by mediating an interaction with extracellular matrix is integrin α8β1. The α8 subunit is primarily expressed in mesangial cells, and mice lacking this integrin subunit have hypercellular glomeruli with an increased number of mesangial cells, increased mesangial matrix deposition, and minor abnormalities in the glomerular capillary networks. There is also evidence that this integrin-mediated cell-matrix interaction plays a role in maintaining the integrity of the glomerulus after injury as hypertensive integrin α8–deficient mice showed increased mesangial cell loss and mesangiolysis.6 Finally, although the collagen binding receptor integrins α1β1 and α2β1 do not play a role in normal glomerular development, integrin α1β1 plays a major role in regulating the degree of glomerulosclerosis in multiple injury models.7 Thus, it is clear that integrins play a critical role in glomerular development and homeostasis after injury by virtue of their ability to bind to their well-defined extracellular matrix ligands.
In addition to their role as matrix receptors, integrins bind various nonmatrix molecules, and these interactions play important roles in altering cell function. In this context, integrins can promote the invasion of infectious agents or toxins into cells; eg, integrin αvβ1 promotes infection by the human matapneumovirus, the collagen receptor integrins α1β1 and α2β1 act as receptors for the rotavirus enterotoxin, and most αv integrins, including αvβ8, function as receptors for the foot-and-mouth disease virus.8 Integrins also play a critical role in binding secreted and membrane-bound cell proteins to stimulate different cellular functions. For example, the axonal growth cone guidance molecule semaphorin 7A stimulates cytokine production in monocytes and macrophages by binding integrin α1β1; integrin αvβ3 binds matrix metalloproteinase-2 (MMP-2), thus directing endothelial cell migration and angiogenesis; and the serine protease urokinase plasminogen activator can promote cytoskeletal rearrangements and directional cell migration on binding to integrin αvβ5. Finally, integrins have been shown to bind growth factor family members, including connective tissue growth factor, neuregulin-1, vascular endothelial growth factor (VEGF), and TGF-β.
The TGF-β is probably the most studied growth factor with respect to the maintenance of glomerular homeostasis because its overexpression is associated with many glomerular diseases, especially diabetic nephropathy, the biggest cause of end-stage renal failure today. TGF-β is expressed by podocytes and mesangial cells, and it has multiple functions, including being a profibrotic cytokine. It has been implicated in increasing matrix production by mesangial cells, leading to mesangial expansion and, ultimately, glomerular fibrosis, as well as having effects on mesangial and podocyte cell survival and proliferation. Due to its pleiotropic effects, TGF-β requires tight local control of its activity. It is usually secreted and maintained in an inactive form via noncovalent interactions with the latency-associated peptide (LAP) of TGF-β. LAP cleavage and removal from latent TGF-β, or conformational changes in the LAP/TGF-β complex, are two essential steps in TGF-β activation. Enzymes such as membrane type-1 (MT1) MMP, aspartic and cysteine proteases, and plasmin have been shown to play a key role in LAP cleavage. In addition, thrombospondin-1, integrin αvβ6, and integrin αvβ8 are physiologically important TGF-β activators that act by inducing conformational changes in the LAP/TGF-β complex.9
Integrin αvβ6, a fibronectin and vitronectin receptor expressed principally by epithelial cells, was the first integrin shown to play a key role in TGF-β activation by inducing conformational changes in the LAP/TGF-β complex by binding to the arginine–glycine–aspartic acid sequence present in the LAP.10 Blocking this form of TGF-β activation was shown to decrease fibrosis in injury models because mice lacking the integrin β6 subunit were protected from bleomycin-mediated pulmonary fibrosis10 and in models of renal tubulointerstitial fibrosis.11 Because the regulation of TGF-β activation by this integrin requires the αv subunit to bind to an arginine–glycine–aspartic acid sequence, researchers explored the role of other αv-containing integrins, such as αvβ8, in TGF-β activation. The integrin β8 subunit is expressed in various epithelial cell types (squamous mucosa, airway epithelium, renal tubular epithelium, and neuroepithelium), mesenchymal cell types (fibroblasts, kidney mesangial cells, and astrocytes), and specific immune cell types (splenocytes, CD44+ T cells, and dendritic cells). The major, if not the only, ligand of integrin αvβ8 is latent TGF-β. Similar to integrin αvβ6, integrin αvβ8 binds to the integrin-recognition motif (arginine–glycine–aspartic acid) of the LAP of TGF-β and mediates its activation. However, unlike integrin αvβ6, which activates TGF-β via mechanical strain causing a conformational change in the LAP/TGF-β interaction, integrin αvβ8–bound latent TGF-β activation requires LAP cleavage by MT1-MMP.12 Interestingly, the β8 integrin subunit is unique in that it has a short cytoplasmic tail that bears no sequence homology to other β integrin subunits, does not bind to the actin cytoskeleton, and is not required for TGF-β activation.13
Integrin αvβ8 can regulate cell functions in a TGF-β–independent and a TGF-β–dependent manner. In lung epithelial cells, engagement of integrin αvβ8 by matrix inhibits cell proliferation via activation of the cyclin-dependent kinase inhibitor p21Cip1. Integrin αvβ8 also regulates brain development, where it was shown to alter endothelial cell proliferation, migration, and differentiation due to activation of TGF-β by astrocytes.14 In contrast to the idea that integrin αvβ8 is an activator of TGF-β, the study by Khan and colleagues1 shows that mesangial cells in the glomerulus sequester latent TGF-β and prevent its activation in an integrin αvβ8–dependent manner. They then propose that this event, in turn, reduces the release of bioactive TGF-β, thus leading to increased glomerular endothelial cell survival. The authors came to this conclusion by analyzing integrin β8–null mice that were bred onto outbred and C57BL/6 congenic backgrounds, which permitted embryonic survival and evaluation of renal phenotypes. The mice developed azotemia and albuminuria, indicating glomerular injury, at 4 weeks of age, which was more pronounced at 12 weeks. However, other than focal podocyte foot process effacement, there was no morphologic evidence of injury in any of the cell types (podocytes, mesangial cells, or endothelial cells) in the glomerulus. The authors were unable to define a mechanism for the subtle morphologic changes in vivo; however, they isolated mesangial cells from integrin β8–null mice and showed that these cells had decreased levels of membrane-bound latent TGF-β and increased secreted bioactive TGF-β compared with wild-type mesangial cells. Based on this observation, they argue that mesangial cells are different from other cells in that they do not activate the latent LAP/TFG-β complex. Their explanation for the difference between mesangial cell function and the other cells is that mesangial cells do not secrete MT1-MMP, a key factor in promoting integrin αvβ8–mediated TGF-β activation. In this context, MT1-MMP is expressed at extremely low levels in mesangial cells,15 and cultured mesangial cells rendered quiescent by prolonged incubation in serum-free media do not express MT1-MMP, making them unable to cleave the LAP of TGF-β. This explanation is unlikely to be the mechanism resulting in the in vitro or in vivo phenotype because it is known that MT1-MMP activation requires signaling pathways such as protein kinase C, which does not function when cells are quiescent. Furthermore, MT1-MMP is detected in the mesangium of the glomerulus in vivo (R. Zent, unpublished observation), and MT1-MMP–null mice do not have proteinuria at the time of their death (R. Zent, unpublished observation). In addition, other MMP-null mice, such as MMP-9–, MMP-2–, and MMP-9/MMP-2 double–null mice are also all normal with no evidence of proteinuria. There are multiple other possible explanations for the observed phenotype that the authors did not consider. For example, it is possible that in integrin β8–null mice, TGF-β binds to other cell surface receptors on mesangial cells, which have a lower affinity for TGF-β than does integrin αvβ8. However, these receptors, including, eg, integrin αvβ6, could potentially be more potent activators of TGF-β than is integrin αvβ8, thus resulting in increased TGF-β activation in integrin β8–null mesangial cells.
Another interesting observation made by Khan et al1 is that the integrin β8–null mice displayed increased expression of platelet endothelial cell adhesion molecule-1 (PECAM-1) in glomerular endothelial cells, which helps protect them from apoptosis. The authors suggest that the mechanism whereby this effect occurs is due to induction of PECAM-1 by TGF-β as previously described in endothelial cells16 and macrophages.17 However, in this study, the authors were unable to induce PECAM-1, even with exogenously added TGF-β. These results suggest that PECAM-1 induction in glomerular endothelial cells is not directly dependent on TGF-β or that immortalized glomerular endothelial cells used in this study do not induce PECAM-1 expression in response to TGF-β. In addition to TGF-β, other cytokines, such as tumor necrosis factor-α and interferon-γ, have been shown to regulate PECAM-1 expression. These mechanisms of PECAM-1 expression regulation might have been acting in a TGF-β–independent manner.
Despite these deficiencies in the study by Khan and colleagues,1 this work does have novelty in at least two aspects: i) it provides evidence that in glomeruli, integrin αvβ8 expressed on mesangial cells is important in binding latent TGF-β, thus delaying its activation, and ii) it provides the first evidence that mesangial cells can affect glomerular endothelial cell stability, unless PECAM-1 is up-regulated. These observations show, for the first time, that mesangial cells can regulate endothelial cell function and raise some new and interesting insights into the mechanism whereby homeostasis of the glomerulus is maintained. This is an important addition to the already described cross-talk between other cell types in the glomerulus. However, it is unclear how such profound differences in integrin αvβ8–dependent TGF-β activation occur in glomerular and nonglomerular cells. In nonglomerular cells, there is evidence that MT1-MMP mediates cleavage and activation of latent TGF-β bound to integrin αvβ8 (Figure 1A). In contrast, Khan and colleagues1 suggest that because mesangial cells do not express MT1-MMP, latent TGF-β bound to integrin αvβ8 is not cleaved and, thus, remains inactive (Figure 1B). Therefore, when mesangial cells do not express integrin αvβ8, latent TGF-β does not bind to this integrin and is available to bind to other receptors, where it is made available to other proteases for cleavage or conformational changes to the LAP/TGF-β complex (Figure 1C). The increased active TGF-β in the glomeruli of integrin β8–null mice is proposed to lead to endothelial cell apoptosis, unless PECAM-1 is up-regulated.
Figure 1.
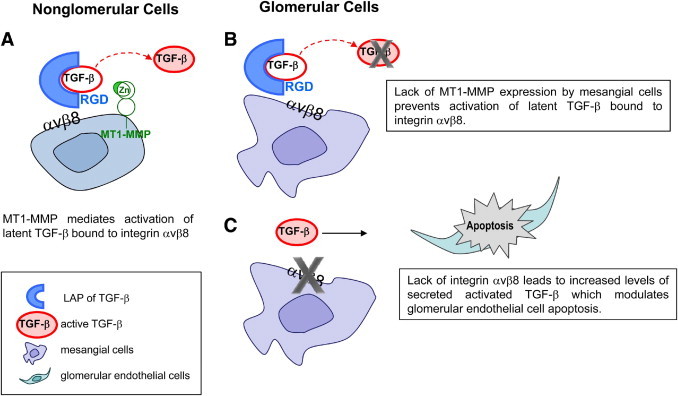
Schematic representation of integrin αvβ8–mediated TGF-β activation in nonglomerular (A) and glomerular (B and C) cells. A: In nonglomerular cells, integrin αvβ8–bound latent TGF-β is normally activated by MT1-MMP. B: In contrast, in the glomerulus of the kidney, lack of MT1-MMP expression by mesangial cells prevents integrin αvβ8–mediated TGF-β activation. C: Deletion of the integrin β8 subunits results in increased release of activated TGF-β by mesangial cells, which modulates glomerular endothelial cell apoptosis. RGD, arginine–glycine–aspartic acid.
The best example that demonstrates the complex role of growth factors in glomerular cell homeostasis and the importance of cross-talk between different cell types in the glomerulus is VEGF. Glomerular endothelial cells have been demonstrated to require podocyte-expressed VEGF for normal development and glomerular maintenance. Selective deletion of VEGF in podocytes leads to abnormalities that range from no glomerular capillary formation when no VEGF is produced to less severe developmental phenotypes when low amounts of VEGF are expressed.18 Production of VEGF by the podocyte is also necessary to maintain the mature glomerular filtration barrier integrity, and selective down-regulation of VEGF in mature podocytes results in glomerular endothelial cell damage and thrombotic microangiopathy.19 Moreover, VEGF produced by podocytes is important in promoting and supporting glomerular endothelial cell migration, differentiation, and survival. Interestingly, in mice where VEGF expression by podocytes was decreased, there was also mesangial cell loss, suggesting that this growth factor is a key regulator of glomerular homeostasis by controlling endothelial and mesangial cell stability. Podocytes are not the only glomerular cells that produce VEGF. Up-regulation of VEGF has been detected in mesangial cells exposed to high glucose or in response to stretch.20 However, whether mesangial cell–provided VEGF can directly affect glomerular endothelial cell functions in vivo has not been investigated because no selective mesangial cell markers are available, and there are no mesangial cell–specific cre mice. The study by Khan et al1 is unique because it shows a mesangial cell–specific function for integrin αvβ8, as this receptor is expressed only by mesangial cells in the glomerulus.
It is likely that there are many parallels between the mechanisms whereby VEGF and TGF-β secretion and local activation regulate glomerular development and homeostasis. Similar to VEGF, TGF-β is required for normal development of the metanephric mesenchyme, yet excessive TGF-β signaling is implicated in glomerular diseases, suggesting that the levels of TGF-β need to be regulated in an extremely precise manner in specific parts of the glomerulus. Mesangial cells are the principal producers of TGF-β, thus they need to precisely regulate the activation of this growth factor. Sequestration of latent TGF-β by integrin αvβ8 as suggested by Khan et al1 is novel and significantly adds to the already described mechanisms of TGF-β activation, such as regulated proteolytic cleavage of the LAP and modulation by activators such as thrombospondin-1 and integrin αvβ6. Up-regulation of thrombospondin-1 and integrin αvβ6 has been observed in diabetic nephropathy and in focal and segmental glomerulosclerosis, implying a role in altering disease progression. It is also likely that the protective role of integrin αvβ8–dependent sequestration of TGF-β will be altered in disease states because it has been reported that a high glucose level activates nuclear factor of activated T cells, a transcription factor shown to up-regulate the TGF-β activator MT1-MMP in mesangial cells.
In conclusion, although the study by Khan and colleagues1 demonstrates that integrin αvβ8 plays a role in the regulation of glomerular homeostasis, it does not define the mechanism whereby this integrin regulates TGF-β activation in vivo. More definitive studies need to be performed to show that lack of MT1-MMP expression or activation in the mesangium is the reason for decreased TGF-β cleavage from the LAP in vivo. It is also an open question as to how integrin αvβ8 regulates PECAM-1 expression. Although it is possibly due to the ability of integrin αvβ8 to regulate TGF-β activation, it might be due to other yet to be defined functions of the integrin that are independent of TGF-β. No matter what the mechanism is, it will be important to define whether enhanced PECAM-1 expression affects glomerular endothelial cell functions, such as permselectivity. Finally, it will be interesting to define the role of integrin αvβ8 in disease states of the kidney as this is an intriguing potential mechanism for regulating TGF-β activation. Ultimately, if we plan to target TGF-β as a strategy to ameliorate glomerulosclerosis in human disease, understanding the ever-increasing complex mechanisms of how TGF-β activity is regulated in the glomerulus is critically important.
Footnotes
Supported in part by a Merit Review from the Department of Veterans Affairs (A.P. and R.Z.); O'Brien Center grant P30DK79341-01 (A.P. and R.Z.); NIH grants 2P01DK065123 (A.P. and R.Z.) and DK075594, DK65123, and DK083187 (R.Z.); and an American Heart Association established investigator award (R.Z.).
CME Disclosure: The authors did not disclose any relevant financial relationships.
Contributor Information
Ambra Pozzi, Email: ambra.pozzi@vanderbilt.edu.
Roy Zent, Email: roy.zent@vanderbilt.edu.
References
- 1.Khan S L.-R.S., McCarty J.H., Sorenson C.M., Sheibani N., Reichardt L.R., Kim J.H., Wang B., Sedor J.R., Shelling J.R. Mesangial cell integrin αvβ8 provides glomerular endothelial cell cytoprotection by sequestering TGF-β and regulating PECAM-1. Am J Pathol. 2011;178:609–620. doi: 10.1016/j.ajpath.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Askari J.A., Buckley P.A., Mould A.P., Humphries M.J. Linking integrin conformation to function. J Cell Sci. 2009;122:165–170. doi: 10.1242/jcs.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kreidberg J., Donovan M., Goldstein S., Rennke H., Shepherd K., Jones R., Jaenisch R. α 3 β 1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- 4.Sachs N., Kreft M., van den Bergh Weerman M.A., Beynon A.J., Peters T.A., Weening J.J., Sonnenberg A. Kidney failure in mice lacking the tetraspanin CD151. J Cell Biol. 2006;175:33–39. doi: 10.1083/jcb.200603073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pozzi A., Jarad G., Moeckel G.W., Coffa S., Zhang X., Gewin L., Eremina V., Hudson B.G., Borza D.B., Harris R.C., Holzman L.B., Phillips C.L., Fassler R., Quaggin S.E., Miner J.H., Zent R. β1 integrin expression by podocytes is required to maintain glomerular structural integrity. Dev Biol. 2008;316:288–301. doi: 10.1016/j.ydbio.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartner A., Cordasic N., Klanke B., Muller U., Sterzel R.B., Hilgers K.F. The α8 integrin chain affords mechanical stability to the glomerular capillary tuft in hypertensive glomerular disease. Am J Pathol. 2002;160:861–867. doi: 10.1016/s0002-9440(10)64909-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zent R., Yan X., Su Y., Hudson B.G., Borza D.B., Moeckel G.W., Qi Z., Sado Y., Breyer M.D., Voziyan P., Pozzi A. Glomerular injury is exacerbated in diabetic integrin α1-null mice. Kidney Int. 2006;70:460–470. doi: 10.1038/sj.ki.5000359. [DOI] [PubMed] [Google Scholar]
- 8.Jackson T., Clark S., Berryman S., Burman A., Cambier S., Mu D., Nishimura S., King A.M. Integrin αvβ8 functions as a receptor for foot-and-mouth disease virus: role of the β-chain cytodomain in integrin-mediated infection. J Virol. 2004;78:4533–4540. doi: 10.1128/JVI.78.9.4533-4540.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimura S.L. Integrin-mediated transforming growth factor-β activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol. 2009;175:1362–1370. doi: 10.2353/ajpath.2009.090393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A., Rifkin D.B., Sheppard D. The integrin α v β 6 binds and activates latent TGF β 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 11.Ma L.J., Yang H., Gaspert A., Carlesso G., Barty M.M., Davidson J.M., Sheppard D., Fogo A.B. Transforming growth factor-β–dependent and –independent pathways of induction of tubulointerstitial fibrosis in β6(−/−) mice. Am J Pathol. 2003;163:1261–1273. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mu D., Cambier S., Fjellbirkeland L., Baron J.L., Munger J.S., Kawakatsu H., Sheppard D., Broaddus V.C., Nishimura S.L. The integrin α(v)β8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishimura S.L., Sheppard D., Pytela R. Integrin αvβ8: interaction with vitronectin and functional divergence of the β8 cytoplasmic domain. J Biol Chem. 1994;269:28708–28715. [PubMed] [Google Scholar]
- 14.Cambier S., Gline S., Mu D., Collins R., Araya J., Dolganov G., Einheber S., Boudreau N., Nishimura S.L. Integrin α(v)β8–mediated activation of transforming growth factor-β by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boucher E., Mayer G., Londono I., Bendayan M. Expression and localization of MT1-MMP and furin in the glomerular wall of short- and long-term diabetic rats. Kidney Int. 2006;69:1570–1577. doi: 10.1038/sj.ki.5000316. [DOI] [PubMed] [Google Scholar]
- 16.Neubauer K., Lindhorst A., Tron K., Ramadori G., Saile B. Decrease of PECAM-1-gene-expression induced by proinflammatory cytokines IFN-γ and IFN-α is reversed by TGF-β in sinusoidal endothelial cells and hepatic mononuclear phagocytes. BMC Physiol. 2008;8:9. doi: 10.1186/1472-6793-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lastres P., Almendro N., Bellon T., Lopez-Guerrero J.A., Eritja R., Bernabeu C. Functional regulation of platelet/endothelial cell adhesion molecule-1 by TGF-β 1 in promonocytic U-937 cells. J Immunol. 1994;153:4206–4218. [PubMed] [Google Scholar]
- 18.Eremina V., Baelde H.J., Quaggin S.E. Role of the VEGF: a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 2007;106:32–37. doi: 10.1159/000101798. [DOI] [PubMed] [Google Scholar]
- 19.Eremina V., Jefferson J.A., Kowalewska J., Hochster H., Haas M., Weisstuch J., Richardson C., Kopp J.B., Kabir M.G., Backx P.H., Gerber H.P., Ferrara N., Barisoni L., Alpers C.E., Quaggin S.E. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gruden G., Araf S., Zonca S., Burt D., Thomas S., Gnudi L., Viberti G. IGF-I induces vascular endothelial growth factor in human mesangial cells via a Src-dependent mechanism. Kidney Int. 2003;63:1249–1255. doi: 10.1046/j.1523-1755.2003.00857.x. [DOI] [PubMed] [Google Scholar]