

Abstract
Humoral immunity, which is the branch of the immune system governed by B cells, protects the body from extracellular pathogens through the secretion of immunoglobulins. Given the unpredictability of exogenous antigens, B cells must be accommodating to numerous genetic alterations to mold immunoglobulin specificity to recognize offending pathogens. Abnormalities in this process leave the host susceptible to permanent pathological modifications and in particular humoral autoimmunities in which secreted immunoglobulins mistake host proteins as pathogenic targets. Underlying the development of self-reactive immunoglobulins is activation-induced cytidine deaminase (AID), a mutagenic enzyme responsible for modifying the specificity of B cells by producing point mutations at the immunoglobulin gene locus. Ideally, these mutations result in an increased affinity for exogenous antigens. However, in pathological scenarios, these mutations produce or enhance a B cell's ability to target the host. AID-induced mutations occur in the germinal center microenvironment of peripheral lymphoid tissue, where pathogenic B-cell clones must evade overwhelming selection pressures to be released systemically. Recent research has revealed numerous genes and pathways responsible for eliminating self-reactive clones within the germinal center. On the basis of these studies, this review aims to clarify the link between AID and the generation of pathogenic immunoglobulins. Furthermore, it describes the selective pressures that pathogenic B cells must bypass within the germinal center to secrete immunoglobulins that ultimately result in disease.
When a foreign pathogen breaches the innate barriers of complex multicellular organisms, the host must use a calculated attack on this infection to reclaim sterility. Early vertebrates acquired what has evolved into a comprehensive system of immunological processes that allow for the theoretical targeting of any pathogen. This system necessitates an arsenal with a diverse enough repertoire to recognize any pathogen it is exposed to and with enough specificity to efficiently clear these threats from the system. This is an especially daunting responsibility given the limitless variety of pathogens that threaten the body and the amazing rate at which many of them adapt to evade detection. Central to this process is the humoral immune system, which uses secreted immunoglobulins produced by B cells to target pathogens with high affinity. Upon differentiation, these cells secrete immunoglobulins which bind extracellular pathogens and contribute to their destruction by i) direct lysis via complement factors, ii) neutralization of pathogen infectivity, and iii) enhancement of the opsonization process. Immunoglobulin production necessitates extensive genetic modification of its gene locus, a process that transpires during B-cell ontogeny. Although somatic restructuring of the immunoglobulin gene allows for flexibility in pathogen detection, the permanence of genetic manipulation leaves B cells susceptible to disease-causing alterations that include lymphomagenesis and autoimmunity.
The remodeling of the immunoglobulin gene begins early in B-cell development within the bone marrow and proceeds in various stages until terminal differentiation to an immunoglobulin-secreting plasma cell. Many of the steps that affect the immunoglobulin gene occur randomly and thus require selection-based checkpoints to eliminate clones deemed harmful to the system. These checkpoints, however, are not infallible, and aberrancies often lead to the release of pathogenic B-cell clones. The process of central tolerance in the bone marrow and the related mechanisms by which tolerance is bypassed are well reviewed elsewhere.1 Although circumvention of early tolerance checkpoints may result in anywhere from 50% to 75% of circulating B cells with some degree of self-recognition, these cells remain lowly reactive and, as a naive population, cannot, in and of themselves, secrete immunoglobulins.2,3 The development and secretion of highly active autoimmunoglobulins therefore requires further modification of the gene locus in the form of targeted point mutations. This modification is controlled by the B-cell–specific enzyme activation-induced cytidine deaminase (AID; reviewed below) and normally occurs in the germinal center (GC; reviewed below) microenvironment of peripheral lymphoid tissue. During a normal immune response, AID and the GC provide the biological tools necessary for the production of high-affinity, pathogen-specific immunoglobulins. However, the mechanistic details underlying this process are not well understood, and, indeed, anomalies can lead to severe clinical manifestations.
AID is a potent mutagen, and thus the production of high-affinity immunoglobulins leaves a B-cell vulnerable to off-target mutations and subsequent lymphomagenesis.4 This is illustrated by the fact that most B-cell lymphomas occur in either the GC or post-GC B-cell compartments with AID being linked to many of the etiological events.5 Abnormal GC behavior, however, is also associated with the onset of severe autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis, myasthenia gravis, and Sjögren's syndrome.6–9 Central to the pathology of these diseases is the presence of autoreactive immunoglobulins that contribute to inflammation via immune complex deposition and complement activation. Furthermore, the B cells carrying these autoreactive immunoglobulins may serve to activate cognate CD4+ T cells that exacerbate the disease phenotype.10,11 Most of these immunoglobulins are class switched and show signs of AID-mediated point mutations, prompting study into the relationship between this enzyme and autoimmunoglobulin generation.12 In reality, the interplay between AID and autoimmunity is rather complex and further convoluted by the as yet undefined selection checkpoints used by the GC. Although AID may play a role in generating highly reactive autoimmunoglobulins, breaches in GC selection are responsible for the spread of these B-cell clones to the afflicted tissues.13 This review aims to better define the connection among AID, GC selection, and the production of autoreactive immunoglobulins with a focus on recently published work in these fields.
Immunoglobulin Diversification
Genetic diversification of the immunoglobulin gene occurs at various stages of B-cell ontogeny and can be used as checkpoints of successful development. Primary diversification occurs in the bone marrow and constitutes somatic recombination of the immunoglobulin heavy chain locus. The antigen-binding domain of immunoglobulins is composed of 3 groups of repetitive gene segments known as the variable (V), diversity (D), and joining (J) segments. Somatic recombination of these segments produces a single exon composed of one of each of the V, D, and J segments that encode the antigen binding site of the immunoglobulin (Figure 1A). An analogous modification occurs at the light chain immunoglobulin locus, which differs only in its lack of D segments. The association of a randomly generated heavy chain with a randomly generated light chain allows for a considerable degree of plasticity for antigen specificity despite a limited encoding gene family.14 Along with being inherently random, V(D)J recombination occurs without impetus (ie, antigen independent) and consequently produces a primary B-cell repertoire that has little familiarity with exogenous antigenic epitopes. A naive B cell may therefore recognize a pathogen with only a weak affinity and must undergo further diversification to improve this interaction to the degree necessary for immunity.
Figure 1.
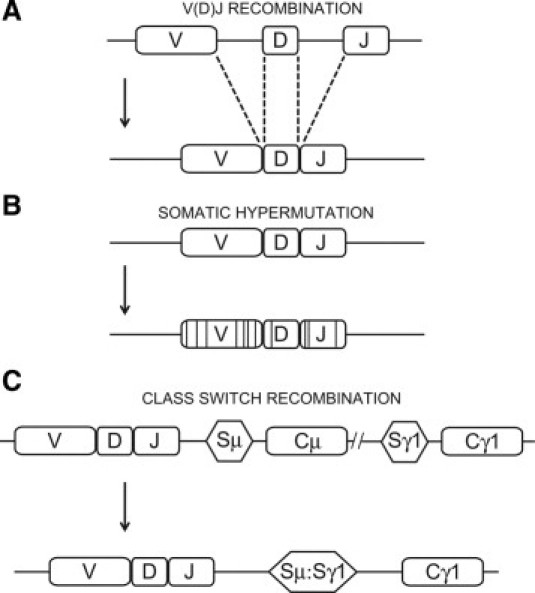
Mechanisms of immunoglobulin diversification. In humans and mice, the immunoglobulin locus of B cells undergoes three major modifications to ensure it has both the diversity and specificity needed to effectively clear pathogen. Primary diversification occurs in an antigen-independent manner through random recombination of the tandemly repeated V, D, and J gene segments at the immunoglobulin locus (A). On exposure to antigen, the immunoglobulin locus is further modified through the generation of random point mutations in the newly rearranged V(D)J region, a process known as somatic hypermutation (B). Constant regions downstream of the V(D)J dictate the immunoglobulin's effector function and can be irreversibly exchanged to alter this function while maintaining the same V(D)J specificity, a further level of diversification on antigen exposure. C: Class switch recombination from the default IgM switch region (Sμ) and associated constant regions (Cμ/Cδ) to the IgG1 switch region (Sγ1) and associated constant region (Cγ1).
Although secondary immunoglobulin diversification acts to complement V(D)J recombination, it is a mechanistically distinct and somewhat less characterized process. These modifications again work at the genetic level in the form of point mutations targeted to the now rearranged V(D)J region, a process dubbed somatic hypermutation (SHM; Figure 1B).15 Unlike primary diversification, which contains developmental checkpoints spurred by signals in the bone marrow, secondary diversification typically occurs in peripheral lymphoid tissues after a direct response to antigenic stimuli. Although SHM is also a random process, secondary diversification as a whole is fashioned to produce a discrete repertoire of B cells that can effectively respond to an offending pathogen. In humans and mice, SHM is allied with the generation of new immunoglobulin isotypes by promoting the exchange of the effector-encoding constant region of the default IgM with any IgG, IgA, or IgE (while maintaining the same antigen specificity). This process, known as class switch recombination (CSR; Figure 1C), allows for a varied immune attack using the unique effector functions of alternate immunoglobulin isotypes. The concerted efforts of immunoglobulin diversification allows for the initial generation of a large pool of naive B cells with a broad range of weak specificities (primary diversification), which is subsequently fine-tuned to respond in an antigen-specific manner with diverse effector capabilities (secondary diversification).
Secondary diversification occurs in the GC and is controlled at the molecular level by the enzyme AID.16,17 AID is a DNA-specific initiator of SHM and CSR that acts by deaminating cytidines to uridines specifically at the immunoglobulin locus.18–21 AID-generated uridines are engaged by various DNA repair proteins that, in the case of SHM, lead to the generation of affinity-altering point mutations and, in the case of CSR, spur recombinogenic events that substitute immunoglobulin isotypes. AID expression within the GC is a unique physiological scenario in which the cell actively mutates itself for the benefit of the body. Given the mutagenic potency of this enzyme, cells necessitate a multifaceted approach to controlling AID function, including limiting gene expression, restricting nuclear access and stability, using multiple regulatory phosphorylation sites, and associating with as yet unknown cofactors to regulate its activity.22–24 Others have even suggested that a strong negative regulation of the enzyme exists in B cells to prevent overmutation or off-target effects of AID.25 Despite these layers of protection, the body is still susceptible to adverse effects of secondary diversification, namely, the development of humoral autoimmunities as is the topic of this review.
The GC
The primary goal of B cells participating in the humoral response is to amass an army of plasma cells producing high-affinity, antigen-specific immunoglobulins that can help fight infection.26,27 In the process, the host must also build up a reserve of memory B cells to prevent persistent reinfection with the same pathogen (ie, to gain “immunity” to that pathogen).28 The source of both plasma and memory cells is the GC, a microenvironment that forms in peripheral lymphoid tissue 5 to 7 days after exposure to antigen and the site of secondary diversification (Figure 2). The GC serves as a niche for B-cell modification and selection, whereby affinity-altering SHMs produce a competitive milieu to allow only the most beneficial cells to exit as a plasma or memory cell.29 Although identified more than a century ago, no universally accepted model exists for GC behavior. Described below is a classic interpretation of this process based on well-established data.
Figure 2.
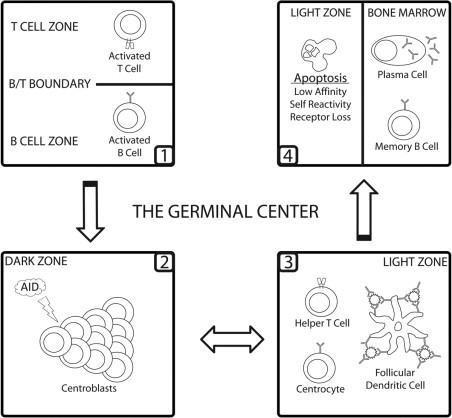
The germinal center reaction. After antigenic exposure, B and T cells are activated in their respective zones within peripheral lymphoid tissue. Activated cells then migrate to the B/T boundary where they interact with one another. T cells impart further activation signals onto cognate B cells, leading to the stimulation of pauciclonal GC founding populations. On GC seeding, founder cells are termed centroblasts and are characterized by the down-regulation of the immunoglobulin receptor from their cell surface and intense levels of proliferation and AID-mediated SHM. These cells then migrate to the light zone of the GC, where they reexpress the immunoglobulin receptor and are termed centrocytes. The light zone provides a competitive environment whereby centrocytes are positively selected based on their ability to bind antigen expressed on the surface of follicular dendritic cells (FDCs) in the form of immune complexes. FDCs and CD4+ T cells provide positively selected clones with the signals needed to exit the GC as a plasma or memory cell. Negatively selected cells die via apoptosis, where they are phagocytosed by local macrophages (not shown). A portion of cells may re-enter the dark zone to undergo further rounds of proliferation and SHM.
As antigen-loaded fluid percolates through the architecture of secondary lymphoid tissue, it encounters rare lymphocytes, both B and T cells, that have the capacity to recognize it.30,31 To successfully protect the body from infection, these rare cells must be selectively amplified and, in the case of B cells, further modified to provide an optimal immunological response. Antigen is recognized by B cells through surface-bound immunoglobulin receptors that provide a preliminary activatory signal. Costimulatory cues required for entry into the GC (namely, CD40-CD40L interactions) are subsequently provided by cognately activated CD4+ T cells.32,33 GCs are seeded by a pauciclonal population (ie, 3 or fewer) of activated B cells that migrate back to the B-cell zone of lymphoid tissue after interaction with cognate T cells.30,34
The efficiency of antigen-specific immunoglobulin development is heavily dependent on the architectural design of the GC. As the GC matures, it begins to form two phenotypically distinct zones with unique functions (Figure 2).26,35 These two regions, known as the dark and light zones, were originally characterized in 1930 based on cellular morphology in histological cross-sections of the lymph node.27 The dark zone, so-called because of the presence of large, densely packed, mitotically active B cells, is the first to arise, developing from the initial founding population. B cells in the dark zone, termed centroblasts, expand the meager founding population by proliferating every 6 to 10 hours.36,37 Centroblasts are further characterized by AID expression and a loss of surface immunoglobulin, presumably to allow uncomplicated modification of the immunoglobulin gene by SHM. The end result is an assortment of mutated B cells that have varying affinities for antigen. These cells must now undergo a selection process for high-affinity clones, resulting in the survival and expansion of only those cells deemed appropriate to fight off infection.
Because the process is random, SHMs acquired in the dark zone will be mostly deleterious to the cell's effectiveness in fighting infection, usually lowering its affinity toward the target antigen.38 Furthermore, mutation of the immunoglobulin locus risks the production of B cells that have acquired (or enhanced) the ability to target self-antigen, thereby making it dangerous to the host. Ineffectual clones must be targeted for removal from the system, whereas rare, high-affinity clones are preferentially selected for expansion. As a counterbalance to the chaotic proliferation and mutation observed in the dark zone, a discrete light zone forms apically to the T-cell regions of lymphoid tissue (Figure 2).35 The light zone is physically structured to facilitate selection using a competition-based system.26 Fundamental to this process is the follicular dendritic cell (FDC), a light zone–specific stromal cell with large processes that express complement and Fc receptors on their surface (Figure 2).39 This allows the FDC to construct a network of immune complexes that capture antigen as it enters lymphoid tissue. GC B cells, termed centrocytes on entry into the light zone, terminate SHM, exit cell cycle, and re-express surface immunoglobulin. Newly mutated cells test the affinity of their immunoglobulin receptors by competing for antigen bound to the surface of FDCs.40 Those that successfully bind antigen receive survival and maturation signals, of which little is known. Antigen-binding cells are positively selected to leave the GC and enter the bone marrow as long-term plasma cells or memory cells. Noncompetitive and autoreactive cells are removed from the system via apoptosis and are quickly phagocytosed by resident macrophages.26,38 Also present in the light zone is a small population of CD4+ T cells that are generally believed to serve as a further checkpoint for mutated centrocytes.26,41 It has been hypothesized that these cells have the task of ensuring that centrocytes do not possess any self-reactive specificities and further provide co-stimulatory/differentiation signals that allow them to exit the GC based on OX40-OX40L and CD40-CD40L interactions.42 Light zone T cells may also provide the signaling necessary to induce CSR in centrocytes.5 Others have proffered that the presence of soluble antigen plays the major role in deleting self-reactive clones through the competitive inhibition of their interactions with FDCs.43,44 In either case, given the rarity of high-affinity antigen-specific B cells, the light zone becomes a virtual graveyard for apoptotic cells and an important checkpoint for the removal of self-reactive clones.
Although the simplicity of the GC model previously described is appealing, its basic tenets have been questioned by recent intravital two-photon microscopic experiments, which have allowed for the live imaging of murine GC cells in vivo.27 Some of the observations made using this technique are inconsistent with the accepted model, particularly calling into question the phenotypic segregation of centroblasts and centrocytes as unique entities. Importantly, the authors of these reports found that centrocytes located in the light zone of the GC are, in fact, mitotically active albeit at a much lower rate than centroblasts. Furthermore, both centroblasts and centrocytes were shown to undergo apoptosis in vivo.45 These observations conflict with the classic view of the GC, which restricts proliferation to the dark zone–resident centroblasts and apoptosis-mediated selection to the light zone–resident centrocytes. It is unclear, however, whether the observed proliferation and apoptosis serve the same purpose in the dark zone as they do in the light zone. These studies also emphasized similarities in appearance and motility of centroblasts and centrocytes and noted frequent migration of these cells between the two zones, implying they may be more related than previously thought.45,46 However, caution must be taken in the analyses of these results because of the technical limitations of two-photon microscopy. Many of the conclusions, including those on proliferation, apoptosis, and migration, were based on rare observations and limited tracking: a major shortcoming of this technology.35 Despite this, it is clear whether the current model of the GC must be refined as future findings begin to reconcile the observations made in these studies with what is already established.
AID and the Production of Autoreactive Immunoglobulins
Given the mutagenic capacity of AIDs, it is somewhat expected that aberrancies in its expression or even its normal physiological function may lead to disease.47 A role in autoimmunity can be predicted based on the fact that autoimmunoglobulins are often heavily mutated and class switched. Furthermore, sites of autoantibody production, such as ectopic GCs and tertiary lymphoid organs, strongly express AID and exhibit hallmarks of SHM and CSR.6 Indeed, using the BXD2 autoimmune mouse model, which develops high titers of autoimmunoglobulins and associated disease, Hsu et al48 described an association between the levels of AID expression and autoimmunoglobulin production. BXD2 mice produce spontaneous GCs with overexpressed levels of AID compared with wild-type (WT) cells. In addition, pathogenic autoimmunoglobulins were heavily mutated and switched to the IgG isotype. By blocking cognate T-cell interactions, the authors were able to decrease AID levels in B cells and showed concomitant decreases in autoimmunoglobulin production, confirming a role for AID. Similarly, in rheumatoid arthritis patients, both circulating B cells and those found in ectopic lymphoid follicles expressed significantly higher levels of AID when compared with controls.49 Furthermore, AID expression showed a strong correlation with serum presence of rheumatoid factor, anticyclic citrullinated peptide, interferon-γ, and interleukin-7, all of which contribute to the development of disease. These data provide both direct and circumstantial evidence of a role for AID in producing the autoreactive immunoglobulins that may lead to disease.
The most convincing evidence supporting the link between AID and autoimmunoglobulin production was illustrated by a series of experiments using the autoimmune prone MRL/lpr mouse, a standard model for disease that mimics human SLE. In the complete absence of AID, MRL/lpr mice showed a reduction in glomerulonephritis and a significant increase in overall survival.50 Although these studies were complicated by the inability of these mice to produce class-switched IgG antibodies, it emphasized the importance autoimmunoglobulins play in the onset of disease. Also noted in the study was a striking increase in autoreactive IgM antibodies in the AID-deficient MRL/lpr mice (despite the absence of SHM) could not compensate for a lack of autoreactive IgG. In an independent study by the same group, the authors remedied their previous model's inability to switch to IgG by using AID heterozygous mice on the same MRL/lpr background.51 Although serum levels of switched immunoglobulins in vivo were comparable between WT and AID heterozygous mice, the authors showed a gene dosage effect on CSR in vitro, a phenomenon that is now an established characteristic of AID heterozygosity.52,53 Interestingly, AID heterozygous mice on the MRL/lpr background had a marked reduction in levels of SHM in vivo owing to the aforementioned gene dosage effect. Lowered SHM profoundly reduced the affinity of autoreactive immunoglobulins to self-targets and had an associated delay in the onset of disease, including kidney destruction.51 These findings were significant in pinpointing SHM as the major contributor to autoimmunoglobulin production, with CSR being necessary but not sufficient for disease onset. More recently, a report by Zan et al54 studied the MRL/lpr mouse in greater detail and noted enhanced expression of not only AID but the low-fidelity repair factors associated with SHM, including polymerase θ, η, and ζ. The up-regulation of these proteins was associated with increased SHM and CSR levels, and associated abnormalities resulted in an expanded spectrum of AID-targeted mutation hotspots in the MRL/lpr mouse immunoglobulin gene. The authors argue that the overactive secondary diversification process is ultimately responsible for producing heavily mutated class-switched immunoglobulins with autoreactive specificities. Taken together, these data conclusively link AID with production of heavily mutated clones that, on breaching GC selection, lead to the development of autoimmune disorders.
AID Deficiency and Autoimmunity
Although the link between AID and the generation of autoimmunoglobulins is somewhat expected, there is a paradoxical counterbalance to this association that has been revealed in recent reports. Patients with inactivating mutations in the AID gene present autoimmune phenotypes in 21% of cases, including arthritis, autoimmune hepatitis, and Crohn's disease.55 Indeed, hyper-IgM syndrome patients (a pathological manifestation of impaired B/T interaction, GC production, or AID function) have been shown to be susceptible to a gamut of autoimmune phenotypes.56 In the mouse model, aged AID−/− mice develop ectopic lymphoid neogenesis (a hallmark of numerous autoimmune diseases), which ultimately leads to aberrant B-cell expansion and severe autoimmune gastritis.57 In this study, disease was mediated by gastric mucosa–specific autoimmunoglobulins and autoreactive CD4+ T cells. In contrast to the aforementioned results from MRL/lpr mice, Chen et al58 used AID-deficient lpr mice on the C57BL/6 background to show a profound increase in spleen size, overactive GC responses, elevated levels of IgM autoimmunoglobulins, and advanced glomerulonephritis, suggesting that the absence of AID aggravates the autoimmune phenotype associated with the lpr mouse. Although the previously discussed report by Jiang et al50 noted the ability of AID−/− mice to spontaneously produce anti-dsDNA IgM, which was exacerbated in the context of MRL/lpr mice, the differences in these studies may be attributed to variation in the genetic background of the MRL and C57BL/6 strains.
As mentioned before, mildly autoreactive B cells are known to evade central tolerance and enter the primary repertoire after successful V(D)J recombination. In the GC, iterative rounds of SHM and selection aim to guide these clones away from their autoreactive specificities in favor of antigenic epitopes.50 AID−/− mice that have lost the ability to refine their immunoglobulins through SHM may accumulate these autoreactive clones in the GC. We recently showed that AID−/− B cells accumulate within the GC due to an inability to undergo normal apoptosis.59 This finding provided an explanation for the large GC structures observed in lymphoid tissues of AID−/− mice and humans and also provides an explanation for the inability of AID−/− mice to clear self-reactive cells from the GC, thereby contributing to the autoimmune phenotype observed by others.16,17 Furthermore, GCs in the AID−/− mouse arise spontaneously, a common feature of humoral autoimmunities where B cells react in response to self-antigen. As others have suggested, unregulated B-cell activation on its own is sufficient to produce autoimmune phenotypes, as seen with mice deficient in FcγRIIB, Shp1, or protein kinase cδ.57,60–62 To this effect, we showed enhanced proliferation of AID−/− cells in vitro after stimulation through the B-cell receptor and CD40, which mimics in vivo T-cell activation. The finding that an absence of AID may also affect the onset of autoimmune disease underscores the importance of AID regulation during secondary diversification. Indeed both the overabundance and underabundance of AID may contribute to autoimmune disease via vastly different pathways, which will be better understood with further study.
It has been suggested that the inability of AID−/− mice to produce class-switched IgG antibodies may contribute to GC deregulation, B-cell expansion, and autoimmunoglobulin production due to an inability to crosslink the inhibitory FcγRIIB receptor on the surface of B cells.57 Indeed, mice lacking the FcγRIIB receptor are prone to humoral autoimmune disease.60,63 However, using mixed bone marrow chimeras with WT and AID−/− donors, we still observed an apoptosis defect and subsequent accumulation of AID−/− B cells in splenic GCs. In this model, secreted IgG is provided by WT donors and would therefore compensate for an FcγRIIB defect by binding this receptor on the surface of AID−/− cells. We therefore concluded that, at least in the AID−/− model, the FcγRIIB receptor is not a contributing factor in GC selection, with the defect likely being B-cell intrinsic. Although loss of the FcγRIIB receptor is clearly associated with the progression of autoimmune disorders, our work and the work of others suggest that these defects do not arise during the removal of self-reactive clones from the GC.59,64
The GC: Apoptosis and Autoimmunity
Abnormal GC behavior, such as spontaneous or ectopic formation, is often linked to the production of pathogenic, self-reactive immunoglobulins.6,65 For example, chronic B-cell activation in these lymphoid microenvironments has been shown to lead to the development of humoral autoimmune diseases, such as SLE.66,67 In severe cases of autoimmunity, including myasthenia gravis and Sjögren's syndrome, ectopic GC formation is found in the afflicted organs producing highly specific autoimmunoglobulins.7–9,68 Defective cellular mechanisms, such as the delayed clearance of apoptotic cells within the GC, provide a suitable source of intracellular components to which autoimmune B cells can react.64,69 Reports have also revealed a role for GC-specific components, such as FDCs, in disease progression.70 Given the various possibilities, the exact role of the GC in mediating humoral autoimmunity is not fully known and likely depends on the nature of the disease. Ultimately, however, the root of pathology must be in the evasion of GC B-cell selection, which normally eliminates these cells before they are released systemically.13,71 Although little is known of the mechanisms mediating GC selection, it is widely accepted that the primary means by which this process occurs is through the apoptosis of ineffective B-cell clones. In mouse models, defects in apoptotic signaling have a profound effect on GC selection and the production of autoimmunoglobulins (reviewed in Table 1).
Table 1.
B-Cell–Associated Humoral Autoimmunity Models and Proposed Mechanisms of Disease
| Proposed mechanism | Mouse genotype | Autoimmune phenotype | References |
|---|---|---|---|
| Promote survival of autoreactive GC B cells through the extrinsic apoptotic pathway | lpr and/or gld | Lymphoproliferation, autoimmunoglobulin production, multiorgan lymphocyte infiltration and disease, glomerulonephritis | 72–74 |
| CD19-conditional Fas−/− | Lymphoproliferation, abnormal GC activity, lymphadenopathy, autoimmunoglobulin-mediated destruction of kidney, liver, and lungs | 75 | |
| Promote survival of autoreactive GC B cells through the intrinsic apoptotic pathway | Bcl-2 Tg | Abnormal GC activity, enhanced cell survival, spontaneous production of autoimmunoglobulins, lupus-like glomerulonephritis. | 76–78 |
| bim−/− | Abnormal GC B cell survival and selection, expanded plasma cell compartment, autoimmunoglobulin production, fatal glomerulonephritis | 79,80 | |
| Abnormal B-cell signaling and proliferation | CD22−/− | Anti-dsDNA, cardiolipin, myeloperoxidase IgG | 81 |
| PD-1−/− | IgG3-mediated arthritis, glomerulonephritis, exacerbation of lpr phenotype | 82 | |
| FcγRIIB−/− | Antinuclear immunoglobulins, fatal autoimmune glomerulonephritis, multiorgan inflammation | 60 | |
| CD19-conditional Ptpn6 (Shp1 deficient) | Unregulated B-cell proliferation, altered immunoglobulin responses, anti-DNA immunoglobulins, glomerulonephritis | 61 | |
| PKCδ−/− | Spontaneous GCs, unregulated proliferation, autoimmunoglobulin-mediated glomerulonephritis, multiorgan lymphocyte infiltration | 62 | |
| CD19 Tg | Breakdown of peripheral tolerance and production of anti-HEL autoimmunoglobulins | 83 | |
| BAFF Tg | Abnormal GCs, high levels of rheumatoid factors, circulating immune complexes, anti-DNA autoimmunoglobulins, deposition in the kidneys | 84 | |
| Lyn−/− | Impaired B-cell signaling and proliferation, increased serum immunoglobulin, antinuclear IgG, glomerulonephritis | 85 | |
| AID−/− | Autoimmune gastritis, exacerbation of BL/6/lpr phenotype, anti-dsDNA IgM, abnormal GC function, variable disease outcomes | 50,51,57,59,96 |
Autoreactive B-cell clones must bypass dominant apoptotic signals to escape the various layers of GC selection, although the signaling cascades responsible for GC death remain to be fully characterized. Of the two major apoptotic pathways, the so-called extrinsic pathway is regulated by death receptors of the tumor necrosis factor family, which play the initiating role in the cell's death on ligand engagement.86–88 The Fas receptor (also known as Apo-1 and CD95) is the most extensively studied member of the this family, and its role in lymphocyte apoptosis is well established.89 Death through the Fas receptor is paramount to lymphocyte homeostasis because mutations in this receptor in humans and mice (the lpr mouse model) or its ligand (the gld mouse model) lead to autoimmune lymphoproliferative syndrome: an excessive accumulation of abnormal lymphocytes due to a break in selection.75,90,91 The extrinsic apoptotic pathway was initially assumed to be the major contributor to GC selection due to the observed overexpression of the Fas receptor on the surface of GC B cells.92 In vivo analyses of the contribution of the Fas receptor to GC function and selection, however, have been somewhat contradictory. The first experiments were performed using the murine lpr model, which, surprisingly, concluded that immunized lpr mice produced normally functioning GCs that generated high-affinity antibodies and a normal memory cell compartment.92 Conversely, recent experiments using the same experimental procedure and more detailed flow cytometric characterization showed defects in clonal selection within the GCs of immunized lpr mice, leading to a significant defect in the generation of the antigen-specific memory cell compartment and survival of heavily mutated B-cell clones.72 Furthermore, study of the contribution of Fas to the elimination of autoreactive GC B cells has produced equally conflicting results, with some experiments showing the buildup of autoreactive GC B cells in immunized lpr mice and others showing that Fas plays no role in the elimination of these cells.73,93 A fundamental limitation of the lpr system, however, is a Fas deficiency in all immune cells throughout their development, thereby causing the abnormal expansion of other hematopoietic cells whose effects on GC function remain unknown. Important experiments using conditional deletion of the Fas gene in the B-cell lineage have conclusively shown that Fas is indeed responsible for the selection of cells in the GC, with these mice showing an expanded memory cell compartment and, importantly, accumulation of autoreactive B cells.75
In the absence of external cues for apoptosis, death signals can be triggered from stimuli within the cell. The second major apoptotic pathway is known as the intrinsic pathway and responds to internal cellular stress cues, such as DNA damage or viral infection. Intrinsic apoptosis is regulated by the Bcl-2 family of pro-apoptotic and anti-apoptotic factors.86–88 When the balance between these proteins is disturbed, mitochondrial membrane integrity is destabilized, causing the release of death-signaling factors. The intrinsic apoptotic pathway is also important for lymphocyte development as evidenced by experiments using transgenic expression of prosurvival or deletion of proapoptotic members of the Bcl-2 family in mice.74,79,94,95 These genetic alterations lead to an accumulation of lymphocytes in the system and associated disease, similar to what is seen in Fas-deficient mice. Importantly, enforced expression of the prosurvival protein Bcl-2 or deletion of the proapoptotic protein Bim affects GC selection in a manner that leads to the development of humoral autoimmunity with both mice developing lupus-like disease.76–80 We and others have shown that both intrinsic and extrinsic apoptotic pathways act simultaneously within the GC, and a study performed recently by Aït-Azzouzene et al96 showed that members of both pathways make significant contributions to the deletion of self-reactive clones. Further research is required to fully understand the context in which either pathway is effective and how that affects the onset of disease and potential therapeutic targets.
Conclusions
The lesson learned from the studies reviewed in this report is the importance of fine-tuning AID levels during the secondary diversification process in the GC. Although overexpression of the gene clearly entails the threat of producing harmful self-reactive immunoglobulins, the complete absence of this gene has a similar effect, albeit through a distinct pathway. The numerous layers regulating the expression and function of this important enzyme are therefore understandable. Less obvious are the mechanisms by which inappropriately mutated B cells are able to bypass normal GC selection. As previously explained, evasion of the apoptotic pressures in the GC are the route by which these cells can enter systemic circulation. A better understanding of these pathways would provide potential clues for therapeutic treatment of GC-derived humoral autoimmunities.
As the origin of humoral autoimmunity is better understood, the importance of B cells in the generation of autoimmunoglobulins is being appreciated as a major contributor to pathology. Consequently, novel therapies have targeted B cells to alleviate the symptoms associated with these diseases. Importantly, the use of the B-cell–depleting monoclonal antibody rituximab for multiple autoimmune disorders has been promising, especially in the treatment of rheumatoid arthritis.97 Other therapies have focused on stunting GC production by blockading B/T interactions, notably through the obstruction of CD40 or CTLA4 signaling in SLE. The modulation of B-cell–specific survival factors, such as BAFF and APRIL, have shown varying efficiencies in inhibiting B-cell function and bypassing disease phenotypes.98 A number of therapeutic strategies still in the rudimentary stages of development focus on other aspects of B-cell signaling and activation in an effort to inhibit the production of pathogenic immunoglobulins.99,100 Future research into the mechanisms controlling GC selection and the regulation of AID will allow for a more specific targeting of disease-causing aberrations and the techniques necessary to prevent or treat them.
Acknowledgments
We are grateful to the Martin Laboratory for helpful discussions regarding this manuscript.
Footnotes
Supported by a grant from the Canadian Institutes of Health Research (MOP89783) and a Canada Research Chair award (A.M.).
CME Disclosure: The authors did not disclose any relevant financial relationships.
References
- 1.Goodnow C.C. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 2.Meffre E., Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. 2008;20:632–638. doi: 10.1016/j.coi.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Wardemann H., Yurasov S., Schaefer A., Young J.W., Meffre E., Nussenzweig M.C. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 4.Ramiro A., San-Martin B.R., McBride K., Jankovic M., Barreto V., Nussenzweig A., Nussenzweig M.C. The role of activation-induced deaminase in antibody diversification and chromosome translocations. Adv Immunol. 2007;94:75–107. doi: 10.1016/S0065-2776(06)94003-6. [DOI] [PubMed] [Google Scholar]
- 5.Klein U., Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8:22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]
- 6.Nacionales D.C., Weinstein J.S., Yan X.J., Albesiano E., Lee P.Y., Kelly-Scumpia K.M., Lyons R., Satoh M., Chiorazzi N., Reeves W.H. B cell proliferation, somatic hypermutation, class switch recombination, and autoantibody production in ectopic lymphoid tissue in murine lupus. J Immunol. 2009;182:4226–4236. doi: 10.4049/jimmunol.0800771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim H.J., Berek C. B cells in rheumatoid arthritis. Arthritis Res. 2000;2:126–131. doi: 10.1186/ar77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuckerman N.S., Howard W.A., Bismuth J., Gibson K., Edelman H., Berrih-Aknin S., Dunn-Walters D., Mehr R. Ectopic GC in the thymus of myasthenia gravis patients show characteristics of normal GC. Eur J Immunol. 2010;40:1150–1161. doi: 10.1002/eji.200939914. [DOI] [PubMed] [Google Scholar]
- 9.Stott D.I., Hiepe F., Hummel M., Steinhauser G., Berek C. Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease: the salivary glands of patients with Sjögren's syndrome. J Clin Invest. 1998;102:938–946. doi: 10.1172/JCI3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin R.H., Mamula M.J., Hardin J.A., Janeway C.A., Jr Induction of autoreactive B cells allows priming of autoreactive T cells. J Exp Med. 1991;173:1433–1439. doi: 10.1084/jem.173.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takemura S., Klimiuk P.A., Braun A., Goronzy J.J., Weyand C.M. T cell activation in rheumatoid synovium is B cell dependent. J Immunol. 2001;167:4710–4718. doi: 10.4049/jimmunol.167.8.4710. [DOI] [PubMed] [Google Scholar]
- 12.Shlomchik M., Mascelli M., Shan H., Radic M.Z., Pisetsky D., Marshak-Rothstein A., Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han S., Zheng B., Dal Porto J., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl, IV: affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J Exp Med. 1995;182:1635–1644. doi: 10.1084/jem.182.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fanning L.J., Connor A.M., Wu G.E. Development of the immunoglobulin repertoire. Clin Immunol Immunopathol. 1996;79:1–14. doi: 10.1006/clin.1996.0044. [DOI] [PubMed] [Google Scholar]
- 15.Neuberger M.S. Antibody diversification by somatic mutation: from Burnet onwards. Immunol Cell Biol. 2008;86:124–132. doi: 10.1038/sj.icb.7100160. [DOI] [PubMed] [Google Scholar]
- 16.Revy P., Muto T., Levy Y., Geissmann F., Plebani A., Sanal O., Catalan N., Forveille M., Dufourcq-Labelouse R., Gennery A., Tezcan I., Ersoy F., Kayserili H., Ugazio A.G., Brousse N., Muramatsu M., Notarangelo L.D., Kinoshita K., Honjo T., Fischer A., Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 17.Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 18.Petersen-Mahrt S.K., Harris R.S., Neuberger M.S. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–104. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 19.Di Noia J., Neuberger M.S. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 20.Bransteitter R., Pham P., Scharff M.D., Goodman M.F. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin A., Bardwell P.D., Woo C.J., Fan M., Shulman M.J., Scharff M.D. Activation-induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature. 2002;415:802–806. doi: 10.1038/nature714. [DOI] [PubMed] [Google Scholar]
- 22.Aoufouchi S., Faili A., Zober C., D'Orlando O., Weller S., Weill J.C., Reynaud C.A. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008;205:1357–1368. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basu U., Wang Y., Alt F.W. Evolution of phosphorylation-dependent regulation of activation-induced cytidine deaminase. Mol Cell. 2008;32:285–291. doi: 10.1016/j.molcel.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Z., Pone E.J., Al-Qahtani A., Park S.R., Zan H., Casali P. Regulation of aicda expression and AID activity: relevance to somatic hypermutation and class switch DNA recombination. Crit Rev Immunol. 2007;27:367–397. doi: 10.1615/critrevimmunol.v27.i4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muto T., Okazaki I.M., Yamada S., Tanaka Y., Kinoshita K., Muramatsu M., Nagaoka H., Honjo T. Negative regulation of activation-induced cytidine deaminase in B cells. Proc Natl Acad Sci U S A. 2006;103:2752–2757. doi: 10.1073/pnas.0510970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolniak K.L., Shinall S.M., Waldschmidt T.J. The germinal center response. Crit Rev Immunol. 2004;24:39–65. doi: 10.1615/critrevimmunol.v24.i1.20. [DOI] [PubMed] [Google Scholar]
- 27.Allen C.D., Okada T., Cyster J.G. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McHeyzer-Williams L.J., McHeyzer-Williams M.G. Antigen-specific memory B cell development. Annu Rev Immunol. 2005;23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- 29.Manser T. Textbook germinal centers. J Immunol. 2004;172:3369–3375. doi: 10.4049/jimmunol.172.6.3369. [DOI] [PubMed] [Google Scholar]
- 30.Jacob J., Kassir R., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl, I: the architecture and dynamics of responding cell populations. J Exp Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garside P., Ingulli E., Merica R.R., Johnson J.G., Noelle R.J., Jenkins M.K. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 32.Clark L.B., Foy T.M., Noelle R.J. CD40 and its ligand. Adv Immunol. 1996;63:43–78. doi: 10.1016/s0065-2776(08)60854-8. [DOI] [PubMed] [Google Scholar]
- 33.Kawabe T., Naka T., Yoshida K., Tanaka T., Fujiwara H., Suematsu S., Yoshida N., Kishimoto T., Kikutani H. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 34.Jacob J., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl, II: a common clonal origin for periarteriolar lymphoid sheath-associated foci and germinal centers. J Exp Med. 1992;176:679–687. doi: 10.1084/jem.176.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen C.D., Ansel K.M., Low C., Lesley R., Tamamura H., Fujii N., Cyster J.G. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y.J., Zhang J., Lane P.J., Chan E.Y., MacLennan I.C. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. Eur J Immunol. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- 37.Cozine C.L., Wolniak K.L., Waldschmidt T.J. The primary germinal center response in mice. Curr Opin Immunol. 2005;17:298–302. doi: 10.1016/j.coi.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 38.Tarlinton D.M., Smith K.G. Apoptosis and the B cell response to antigen. Int Rev Immunol. 1997;15:53–71. doi: 10.3109/08830189709068171. [DOI] [PubMed] [Google Scholar]
- 39.Allen C.D., Cyster J.G. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. 2008;20:14–25. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park C.S., Choi Y.S. How do follicular dendritic cells interact intimately with B cells in the germinal centre. Immunology. 2005;114:2–10. doi: 10.1111/j.1365-2567.2004.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker L.S., Gulbranson-Judge A., Flynn S., Brocker T., Lane P.J. Co-stimulation and selection for T-cell help for germinal centres: the role of CD28 and OX40. Immunol Today. 2000;21:333–337. doi: 10.1016/s0167-5699(00)01636-4. [DOI] [PubMed] [Google Scholar]
- 42.Cornall R.J., Goodnow C.C., Cyster J.G. The regulation of self-reactive B cells. Curr Opin Immunol. 1995;7:804–811. doi: 10.1016/0952-7915(95)80052-2. [DOI] [PubMed] [Google Scholar]
- 43.Pulendran B., Smith K.G., Nossal G.J. Soluble antigen can impede affinity maturation and the germinal center reaction but enhance extrafollicular immunoglobulin production. J Immunol. 1995;155:1141–1150. [PubMed] [Google Scholar]
- 44.Shokat K.M., Goodnow C.C. Antigen-induced B-cell death and elimination during germinal-centre immune responses. Nature. 1995;375:334–338. doi: 10.1038/375334a0. [DOI] [PubMed] [Google Scholar]
- 45.Allen C.D., Okada T., Tang H.L., Cyster J.G. Imaging of germinal center selection events during affinity maturation. Science. 2007;315:528–531. doi: 10.1126/science.1136736. [DOI] [PubMed] [Google Scholar]
- 46.Schwickert T.A., Lindquist R.L., Shakhar G., Livshits G., Skokos D., Kosco-Vilbois M.H., Dustin M.L., Nussenzweig M.C. In vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]
- 47.Diamond B., Katz J.B., Paul E., Aranow C., Lustgarten D., Scharff M.D. The role of somatic mutation in the pathogenic anti-DNA response. Annu Rev Immunol. 1992;10:731–757. doi: 10.1146/annurev.iy.10.040192.003503. [DOI] [PubMed] [Google Scholar]
- 48.Hsu H.C., Wu Y., Yang P., Wu Q., Job G., Chen J., Wang J., Accavitti-Loper M.A., Grizzle W.E., Carter R.H., Mountz J.D. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J Immunol. 2007;178:5357–5365. doi: 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- 49.Xu X., Hsu H.C., Chen J., Grizzle W.E., Chatham W.W., Stockard C.R., Wu Q., Yang P.A., Holers V.M., Mountz J.D. Increased expression of activation-induced cytidine deaminase is associated with anti-CCP and rheumatoid factor in rheumatoid arthritis. Scand J Immunol. 2009;70:309–316. doi: 10.1111/j.1365-3083.2009.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang C., Foley J., Clayton N., Kissling G., Jokinen M., Herbert R., Diaz M. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol. 2007;178:7422–7431. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang C., Zhao M.L., Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126:102–113. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takizawa M., Tolarova H., Li Z., Dubois W., Lim S., Callen E., Franco S., Mosaico M., Feigenbaum L., Alt F.W., Nussenzweig A., Potter M., Casellas R. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med. 2008;205:1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sernandez I.V., de Yebenes V.G., Dorsett Y., Ramiro A.R. Haploinsufficiency of activation-induced deaminase for antibody diversification and chromosome translocations both in vitro and in vivo. PLoS One. 2008;3:e3927. doi: 10.1371/journal.pone.0003927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zan H., Zhang J., Ardeshna S., Xu Z., Park S.R., Casali P. Lupus-prone MRL/faslpr/lpr mice display increased AID expression and extensive DNA lesions, comprising deletions and insertions, in the immunoglobulin locus: concurrent upregulation of somatic hypermutation and class switch DNA recombination. Autoimmunity. 2009;42:89–103. doi: 10.1080/08916930802629554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quartier P., Bustamante J., Sanal O., Plebani A., Debre M., Deville A., Litzman J., Levy J., Fermand J.P., Lane P., Horneff G., Aksu G., Yalcin I., Davies G., Tezcan I., Ersoy F., Catalan N., Imai K., Fischer A., Durandy A. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to activation-induced cytidine deaminase deficiency. Clin Immunol. 2004;110:22–29. doi: 10.1016/j.clim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 56.Jesus A.A., Duarte A.J., Oliveira J.B. Autoimmunity in hyper-IgM syndrome. J Clin Immunol. 2008;28(Suppl 1):S62–S66. doi: 10.1007/s10875-008-9171-x. [DOI] [PubMed] [Google Scholar]
- 57.Hase K., Takahashi D., Ebisawa M., Kawano S., Itoh K., Ohno H. Activation-induced cytidine deaminase deficiency causes organ-specific autoimmune disease. PLoS ONE. 2008;3:e3033. doi: 10.1371/journal.pone.0003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen L., Guo L., Tian J., Zheng B., Han S. Deficiency in activationinduced cytidine deaminase promotes systemic autoimmunity in lpr mice on a C57BL/6 background. Clin Exp Immunol. 2010;159:169–175. doi: 10.1111/j.1365-2249.2009.04058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zaheen A., Boulianne B., Parsa J.Y., Ramachandran S., Gommerman J.L., Martin A. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood. 2009;114:547–554. doi: 10.1182/blood-2009-03-211763. [DOI] [PubMed] [Google Scholar]
- 60.Bolland S., Ravetch J.V. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–285. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 61.Pao L.I., Lam K.P., Henderson J.M., Kutok J.L., Alimzhanov M., Nitschke L., Thomas M.L., Neel B.G., Rajewsky K. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 62.Miyamoto A., Nakayama K., Imaki H., Hirose S., Jiang Y., Abe M., Tsukiyama T., Nagahama H., Ohno S., Hatakeyama S., Nakayama K.I. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature. 2002;416:865–869. doi: 10.1038/416865a. [DOI] [PubMed] [Google Scholar]
- 63.Rahman Z.S., Niu H., Perry D., Wakeland E., Manser T., Morel L. Expression of the autoimmune Fcgr2b NZW allele fails to be upregulated in germinal center B cells and is associated with increased IgG production. Genes Immun. 2007;8:604–612. doi: 10.1038/sj.gene.6364423. [DOI] [PubMed] [Google Scholar]
- 64.Rahman Z.S., Alabyev B., Manser T. FcgammaRIIB regulates autoreactive primary antibody-forming cell, but not germinal center B cell, activity. J Immunol. 2007;178:897–907. doi: 10.4049/jimmunol.178.2.897. [DOI] [PubMed] [Google Scholar]
- 65.Bombardieri M., Barone F., Humby F., Kelly S., McGurk M., Morgan P., Challacombe S., De Vita S., Valesini G., Spencer J., Pitzalis C. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjögren's syndrome. J Immunol. 2007;179:4929–4938. doi: 10.4049/jimmunol.179.7.4929. [DOI] [PubMed] [Google Scholar]
- 66.Tsubata T. B cell abnormality and autoimmune disorders. Autoimmunity. 2005;38:331–337. doi: 10.1080/08916930500123959. [DOI] [PubMed] [Google Scholar]
- 67.Kozono Y., Kotzin B.L., Holers V.M. Resting B cells from New Zealand Black mice demonstrate a defect in apoptosis induction following surface IgM ligation. J Immunol. 1996;156:4498–4503. [PubMed] [Google Scholar]
- 68.Sims G.P., Shiono H., Willcox N., Stott D.I. Somatic hypermutation and selection of B cells in thymic germinal centers responding to acetylcholine receptor in myasthenia gravis. J Immunol. 2001;167:1935–1944. doi: 10.4049/jimmunol.167.4.1935. [DOI] [PubMed] [Google Scholar]
- 69.Luzina I.G., Atamas S.P., Storrer C.E., daSilva L.C., Kelsoe G., Papadimitriou J.C., Handwerger B.S. Spontaneous formation of germinal centers in autoimmune mice. J Leukoc Biol. 2001;70:578–584. [PubMed] [Google Scholar]
- 70.Victoratos P., Kollias G. Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity. 2009;30:130–142. doi: 10.1016/j.immuni.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 71.Vuyyuru R., Mohan C., Manser T., Rahman Z.S. The lupus susceptibility locus Sle1 breaches peripheral B cell tolerance at the antibody-forming cell and germinal center checkpoints. J Immunol. 2009;183:5716–5727. doi: 10.4049/jimmunol.0804215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takahashi Y., Ohta H., Takemori T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity. 2001;14:181–192. doi: 10.1016/s1074-7613(01)00100-5. [DOI] [PubMed] [Google Scholar]
- 73.Hoch S., Boyd M., Malone B., Gonye G., Schwaber J., Schwaber J. Fas-mediated apoptosis eliminates B cells that acquire self-reactivity during the germinal center response to NP. Cell Immunol. 2000;203:103–110. doi: 10.1006/cimm.2000.1681. [DOI] [PubMed] [Google Scholar]
- 74.Moulian N., Berrih-Aknin S. Fas/APO-1/CD95 in health and autoimmune disease: thymic and peripheral aspects. Semin Immunol. 1998;10:449–456. doi: 10.1006/smim.1998.0155. [DOI] [PubMed] [Google Scholar]
- 75.Hao Z., Duncan G.S., Seagal J., Su Y.W., Hong C., Haight J., Chen N.J., Elia A., Wakeham A., Li W.Y., Liepa J., Wood G.A., Casola S., Rajewsky K., Mak T.W. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity. 2008;29:615–627. doi: 10.1016/j.immuni.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hande S., Notidis E., Manser T. Bcl-2 obstructs negative selection of autoreactive, hypermutated antibody V regions during memory B cell development. Immunity. 1998;8:189–198. doi: 10.1016/s1074-7613(00)80471-9. [DOI] [PubMed] [Google Scholar]
- 77.Notidis E., Hande S., Manser T. Enforced expression of Bcl-2 selectively perturbs negative selection of dual reactive antibodies. Dev Immunol. 2001;8:223–234. doi: 10.1155/2001/83595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Strasser A., Whittingham S., Vaux D.L., Bath M.L., Adams J.M., Cory S., Harris A.W. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci U S A. 1991;88:8661–8665. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fischer S.F., Bouillet P., O'Donnell K., Light A., Tarlinton D.M., Strasser A. Proapoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood. 2007;110:3978–3984. doi: 10.1182/blood-2007-05-091306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bouillet P., Metcalf D., Huang D.C., Tarlinton D.M., Kay T.W., Kontgen F., Adams J.M., Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 81.O'Keefe T.L., Williams G.T., Batista F.D., Neuberger M.S. Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med. 1999;189:1307–1313. doi: 10.1084/jem.189.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nishimura H., Nose M., Hiai H., Minato N., Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 83.Inaoki M., Sato S., Weintraub B.C., Goodnow C.C., Tedder T.F. CD19-regulated signaling thresholds control peripheral tolerance and autoantibody production in B lymphocytes. J Exp Med. 1997;186:1923–1931. doi: 10.1084/jem.186.11.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mackay F., Woodcock S.A., Lawton P., Ambrose C., Baetscher M., Schneider P., Tschopp J., Browning J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gutierrez T., Halcomb K.E., Coughran A.J., Li Q.Z., Satterthwaite A.B. Separate checkpoints regulate splenic plasma cell accumulation and IgG autoantibody production in Lyn-deficient mice. Eur J Immunol. 2010;40:1897–1905. doi: 10.1002/eji.200940043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zimmermann K.C., Bonzon C., Green D.R. The machinery of programmed cell death. Pharmacol Ther. 2001;92:57–70. doi: 10.1016/s0163-7258(01)00159-0. [DOI] [PubMed] [Google Scholar]
- 87.Movassagh M., Foo R.S. Simplified apoptotic cascades. Heart Fail Rev. 2008;13:111–119. doi: 10.1007/s10741-007-9070-x. [DOI] [PubMed] [Google Scholar]
- 88.Schultz D.R., Harrington W.J., Jr Apoptosis: programmed cell death at a molecular level. Semin Arthritis Rheum. 2003;32:345–369. doi: 10.1053/sarh.2003.50005. [DOI] [PubMed] [Google Scholar]
- 89.Strasser A., Bouillet P. The control of apoptosis in lymphocyte selection. Immunol Rev. 2003;193:82–92. doi: 10.1034/j.1600-065x.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- 90.Sobel E.S. Cellular interactions in the lpr and gld models of systemic autoimmunity. Adv Dent Res. 1996;10:76–80. doi: 10.1177/08959374960100011601. [DOI] [PubMed] [Google Scholar]
- 91.Vaishnaw A.K., Toubi E., Ohsako S., Drappa J., Buys S., Estrada J., Sitarz A., Zemel L., Chu J.L., Elkon K.B. The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum. 1999;42:1833–1842. doi: 10.1002/1529-0131(199909)42:9<1833::AID-ANR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 92.Smith K.G., Nossal G.J., Tarlinton D.M. FAS is highly expressed in the germinal center but is not required for regulation of the B-cell response to antigen. Proc Natl Acad Sci U S A. 1995;92:11628–11632. doi: 10.1073/pnas.92.25.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alabyev B., Vuyyuru R., Manser T. Influence of Fas on the regulation of the response of an anti-nuclear antigen B cell clonotype to foreign antigen. Int Immunol. 2008;20:1279–1287. doi: 10.1093/intimm/dxn087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smith K.G., Light A., O'Reilly L.A., Ang S.M., Strasser A., Tarlinton D. bcl-2 transgene expression inhibits apoptosis in the germinal center and reveals differences in the selection of memory B cells and bone marrow antibody-forming cells. J Exp Med. 2000;191:475–484. doi: 10.1084/jem.191.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takahashi Y., Cerasoli D.M., Dal Porto J.M., Shimoda M., Freund R., Fang W., Telander D.G., Malvey E.N., Mueller D.L., Behrens T.W., Kelsoe G. Relaxed negative selection in germinal centers and impaired affinity maturation in bcl-xL transgenic mice. J Exp Med. 1999;190:399–410. doi: 10.1084/jem.190.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aït-Azzouzene D, Kono DH, Gonzalez-Quintial R, McHeyzer-Williams LJ, Lim M, Wickramarachchi D, Gerdes T, Gavin AL, Skog P, McHeyzer-Williams MG, Nemazee D, Theofilopoulos AN: Deletion of IgG-switched autoreactive B cells and defects in Fas(lpr) lupus mice. J Immunol 185:1015–1027 [DOI] [PMC free article] [PubMed]
- 97.Edwards J.C., Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- 98.Browning J.L. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006;5:564–576. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 99.Mok M.Y. The immunological basis of B-cell therapy in systemic lupus erythematosus. Int J Rheum Dis. 2010;13:3–11. doi: 10.1111/j.1756-185X.2009.01458.x. [DOI] [PubMed] [Google Scholar]
- 100.Blank M., Shoenfeld Y. B cell targeted therapy in autoimmunity. J Autoimmun. 2007;28:62–68. doi: 10.1016/j.jaut.2007.02.001. [DOI] [PubMed] [Google Scholar]