

Abstract
Transforming growth factor (TGF)-β and fibroblast growth factor (FGF)-2 both promote repair in valve interstitial cell (VIC) injury models; however, the relationship between TGF-β and FGF-2 in wound repair are not well understood. VIC confluent monolayers were wounded by mechanical injury and incubated separately or in combination with FGF-2, neutralizing antibody to FGF-2, neutralizing antibody to TGF-β, and betaglycan antibody for 24 hours after wounding. Phosphorylated Smad2/3 (pSmad2/3) was localized at the wound edge (WE) and at the monolayer away from the WE. Down-regulation of pSmad2/3 protein expression via small-interfering RNA transfection was performed. The extent of wound closure was monitored for up to 96 hours. FGF-2 incubation resulted in a significant increase in nuclear pSmad2/3 staining at the WE. Neutralizing antibody to TGF-β alone or with FGF-2 present resulted in a similar significant decrease in pSmad2/3. Neutralizing antibody to FGF-2 alone or with FGF-2 present showed a similar significant decrease in pSmad2/3; however, significantly more staining was observed than treatment with neutralizing antibody to TGF-β. Incubation with betaglycan antibody inhibited FGF-2–mediated pSmad2/3 signaling. Wound closure corresponded with pSmad2/3 staining at the WE. Down-regulation of pSmad2/3 via small-interfering RNA transfection significantly reduced the extent to which FGF-2 promoted wound closure. Fibroblast growth factor-2 promotes in vitro VIC wound repair, at least in part, through the TGF-β/Smad2/3 signaling pathway.
Valve interstitial cells (VICs) are the most prevalent cells found in the three layers of the heart valve.1,2 In normal valves, VICs are in a quiescent phenotype, maintaining normal heart valve structure and function. However, in diseased valves, VICs become activated and regulate valve repair and remodeling.1–3
Transforming growth factor (TGF)-β1 is a homodimeric protein of the TGF-β family.1,4,5 In previous investigations, TGF-β1 was shown to regulate VIC wound repair, including VIC activation, proliferation, migration, and apoptosis.1,4 Each TGF-β protein signals by binding to specific type I and type II serine/threonine kinase receptors.4,6 Binding of the TGF-β protein results in the formation of a type I and type II receptor complex and phosphorylation of the type I receptor by the type II receptor.4,6,7 The phosphorylated type I receptor, in turn, phosphorylates specific receptor-regulated Smad2 and Smad3 proteins. Phosphorylation of Smad2/3 proteins results in their dissociation from the type I and type II receptor complex and their heteromerization with Smad4. The Smad2/3-Smad4 complex affects gene expression by translocating to the nucleus and interacting with transcriptional factors that are often associated with growth and remodeling.1,4 Betaglycan, also known as TGF-β type III receptor, is a transmembrane heparan and chondroitin sulfate proteoglycan, identified as the major binding molecule of TGF-β in many cell types, with multiple binding sites for TGF-β and a binding site at the heparan sulfate chains for fibroblast growth factor (FGF)-2.8–10 Betaglycan either promotes or reduces TGF-β signaling by either enhancing or interfering with TGF-β binding to its type I and type II receptors.9–11 In some cell types, on binding TGF-β, betaglycan presents TGF-β to the dimeric TGF-β type II receptor, which migrates toward the TGF-β type I receptor, forming the complex that promotes Smad2/3 signaling.12 In other cell types, betaglycan inhibits TGF-β signaling by preventing type I and type II receptor complex formation and thereby preventing Smad2/3 signaling.11 Certain cell types lacking endogenous betaglycan expression have also experienced increased affinity of TGF-β to the TGF-β type I and type II receptors.11
The effect of TGF-β on VIC wound repair was previously studied13; TGF-β and phosphorylated Smad2/3 (pSmad2/3) staining in the nucleus were increased in wounded monolayers at the wound edge (WE) compared with the monolayer away from the WE (AWM).13 The addition of exogenous TGF-β to the wounded cultures showed increased VIC activation, as characterized by increased α-smooth muscle actin (α-SMA) expression, VIC proliferation at the WE, and increased rate of wound closure.13–15 These effects were significantly reduced by incubation with neutralizing antibody to TGF-β, suggesting that TGF-β plays a role in regulating VIC wound repair. Coexpression of α-SMA and pSmad2/3 staining at the WE suggested an association between pSmad2/3 staining and VIC activation.13
Fibroblast growth factor-2 is a member of the FGF family of proteins that exhibit various tissue repair functions, including cell proliferation and migration.2,16–22 Various signaling pathways for FGFs have been identified, including binding to the FGF receptors with the aid of heparan sulfate proteoglycan coreceptors, inducing the dimerization and activation of FGF receptors.17 The activation of FGF receptors results in the activation of the mitogen-activated protein kinase signaling pathway.19,20,23,24 A previous investigation25 showed that FGF-2 promotes VIC repair (ie, VICs at the WE of a wounded monolayer experienced a significant up-regulation in FGF-2 compared with the nonwounded monolayer and treatment with neutralizing antibody to FGF-2 impeded wound closure in wounded VIC models).
The signaling pathway through which FGF-2 promotes wound repair in VIC models is not well understood. This investigation was undertaken to determine whether FGF-2 stimulates wound repair in in vitro wounded VIC models through the TGF-β/Smad signaling pathway.
Materials and Methods
Neutralizing Antibodies to TGF-β and FGF-2, Exogenous FGF-2, and Betaglycan Antibody
Pan-specific TGF-β neutralizing antibody (R&D Systems, Minneapolis, MN) was reconstituted in sterile PBS to make a 1-mg/ml concentration. At wounding, VIC monolayers were incubated in a 15-μg/ml concentration of neutralizing antibody to TGF-β. These concentrations were determined in a previous study13 to significantly reduce TGF-β activity in VICs incubated under the same conditions as in this study.
Human recombinant FGF-2 (Upstate Biotechnology, Lake Placid, NY) was reconstituted in 5-mmol Tris at pH 7.6 to make 100 μg/ml. Wounded monolayers and subconfluent and confluent monolayers were incubated in a 10-ng/ml concentration of FGF-2. These concentrations were determined in a previous study25 to significantly increase FGF-2 activity in VICs incubated under the same conditions as in this study.
Anti-FGF basic antibody (R&D Systems) was reconstituted in sterile PBS to make a 1-mg/ml concentration. Wounded monolayers and subconfluent and confluent monolayers were incubated in a 10-μg/ml concentration of neutralizing antibody to FGF-2. These concentrations were determined in a previous study25 to significantly reduce FGF-2 activity in VICs incubated under the same conditions as in this study.
Betaglycan antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was diluted 1:100 with standard medium, producing the 2 μg/ml used for incubation in cultures.
Cell Culture
The VIC cultures were harvested from explants from the distal third portion of the anterior leaflet of porcine mitral valves by methods previously described.26 The atrial and ventricular surfaces of the valve were scraped with a sterilized scalpel to remove endocardial cells. The valve was then rinsed with sterilized PBS to wash off the endothelial cells. The tissue was cut into 4 × 5-mm pieces; placed in 35-mm tissue culture dishes (Falcon; BD Biosciences, San Jose, CA); and grown in medium 199 with 10% fetal bovine serum (FBS), 1% penicillin, streptomycin, and amphotericin B (Fungizone; Life Technologies Inc, Rockville, MD) in an incubator at 37°C and in humidified 95% air and a 5% carbon dioxide atmosphere. The VICs that grew from the explants were subcultured in 10% FBS, 1% penicillin, streptomycin, and amphotericin B in medium 199 (standard medium), at 37°C, humidified 95% air, and a 5% carbon dioxide atmosphere. The VIC cultures used in the experiments were of passages 3 to 5 and were plated at 100,000 cells per well in experiments. In experiments in which subconfluent monolayers were required, 5000 cells per well were plated.
Wounding Experiments
The VIC monolayer was grown on an 18-mm diameter glass coverslip (Fisher Scientific, Pittsburgh, PA) in 12-well tissue culture dishes (Falcon) in 1 ml of standard medium per well. The cells were grown for 72 hours to allow for confluency. A flat cell scraper (width, 1 mm) was used to denude the middle of the monolayer in the wounded VIC monolayer, generating one wound with two WEs and two AWMs per coverslip. The wounded monolayer was then washed three times with standard medium to remove cell debris derived from the wounding process.
Wounded VIC monolayers were incubated for 24 hours after wounding with 1 ml of standard medium and treatments of exogenous FGF-2, neutralizing antibody to TGF-β, neutralizing antibody to TGF-β and exogenous FGF-2, neutralizing antibody to FGF-2, neutralizing antibody to FGF-2 and exogenous FGF-2, betaglycan antibody, or betaglycan antibody and exogenous FGF-2. There were also nontreated controls.
To show that, in subconfluent monolayer cultures under the conditions of our experiments, there is a difference in α-SMA staining compared with confluent monolayers, subconfluent and confluent VIC monolayers were incubated for 24 hours with the addition of exogenous FGF-2 or neutralizing antibody to FGF-2; there were also nontreated controls. At fixation, subconfluent monolayers covered approximately 20% of the coverslip surface.
In all conditions, three experiments were performed, with three sets of culture dishes for each treatment.
Immunofluorescent Staining
Wounded VIC monolayers and subconfluent and confluent monolayers were fixed with 4% formaldehyde for 15 minutes, washed three times with PBS, incubated in 0.1% Triton X-100 (The Dow Chemical Company, Midland, MI) in PBS for 3.5 minutes to permeabilize the VICs, and washed three times with PBS. The wounded monolayer coverslips were then incubated with anti-pSmad2/3 (serine 433/435) (1:50; Santa Cruz Biotechnology), and the subconfluent and confluent monolayer coverslips were incubated with mouse anti–α-SMA (1:500; Sigma, Oakville, ON, Canada) as primary antibodies for 1 hour. The coverslips were then washed three times in PBS. Secondary staining involved incubation of the coverslips with propidium iodide (1:5000; Sigma, St Louis, MO), goat anti-rabbit Alexa 488 (1:200; Molecular Probes, Invitrogen, Eugene, OR) for wounded monolayer coverslips, and goat anti-mouse Alexa 568 for subconfluent and confluent monolayer coverslips for 30 minutes. Negative control of wounded VIC monolayer coverslips incubated in rabbit IgG protein (Jackson ImmunoResearch, West Grove, PA) was also performed. The coverslips were then washed three times in PBS, dipped into distilled water, mounted onto slides with an antifade reagent (Prolong Gold; Molecular Probes, Invitrogen), and then cured at 4°C overnight. The coverslips were examined with a laser scanning confocal microscope (Olympus FluoView 1000; Olympus America Inc., Center Valley, PA) through an oil objective (×40 UplanFI N.A. 1.30), and data were quantified with a multilane argon laser (excitation wavelength, 488 nm) and a helium neon laser (excitation wavelength, 543 nm).
Quantification of pSmad2/3 and α-SMA
A total of 60 fields per coverslip were counted in the front five rows of the WE. The percentage of cells with nuclear pSmad2/3 staining/the total number of cells counted was compared for the different treatments. A strip of cells one field wide, parallel, and well away from the WE was used to count the staining at the AWM.
α-Smooth muscle actin staining was counted for the subconfluent and confluent monolayers in the same fashion as previously described, using 60 fields per coverslip.
Quantification of Wound Closure
The distance between the two WEs of each wound at five random points per wound was observed under a light microscope using phase contrast to outline the WE. Wound closure was measured at wounding and at 24, 48, 72, and 96 hours after wounding.
Small-Interfering RNA Transfection
Down-regulation of Smad2/3 gene expression was achieved through Smad2/3 small-interfering RNA (siRNA) (human sc-37238; Santa Cruz Biotechnology) transfection with a transfection reagent (INTERFERin siRNA; Polyplus transfection, New York, NY). Twenty-four hours before transfection, cells were plated into 12-well plates to reach 40% to 50% confluency at transfection. Each well was transfected with 6 μL of transfection reagent and 1.2 pmol of negative control siRNA or Smad2/3 siRNA (Santa Cruz Biotechnology) in media without FBS. Nontransfected cells were also prepared as the control. Six hours after transfection, the cultures were wounded and treated with or without FGF-2 in fresh media with FBS.
Western Blot Analysis
Cells were harvested 72 hours after transfection. Cytoplasmic and nuclear extractions were performed with extraction reagents (NE-PER; Thermo Scientific, Waltham, MA). Protein concentrations were determined through a bicinchoninic acid protein assay (Thermo Scientific). Cytoplasmic and nuclear extractions were combined and resolved via 12.5% SDS–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane via dry blotting (iBlot; Invitrogen) for 7 minutes. The membrane was then detected for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) loading control with GAPDH mouse antibody and goat anti-mouse secondary antibody (1:5000; Santa Cruz Biotechnology [for both]) and for pSmad2/3 with anti-pSmad antibody (1:2000; Santa Cruz Biotechnology) and goat anti-rabbit secondary antibody (1:5000, Santa Cruz Biotechnology) using a protein detection system (SNAP i.d.; Millipore, Billerica, MA). The blot was then subjected to substrate (Luminate forte Western HRP; Millipore) and exposed to film (CL-XPosure film; Thermo Scientific). Densitometry analysis was performed with computer software (ImageJ; National Institutes of Health, Bethesda, MA).
Statistical Analysis
Student's t test was performed to compare nontreated with treated experiments, and P < 0.05 was considered significant. Software (GraphPad Prism version 5; GraphPad Software Inc, San Diego, CA) was used for one-way analysis of variance, and a Bonferroni multiple-comparison posttest was used to analyze the statistical significance between various treatments.
Results
Exogenous FGF-2 Increases Nuclear pSmad2/3 Staining in VICs at the WE
At 24 hours after wounding, the VICs at the WE treated with exogenous FGF-2 (10 ng/ml) showed significantly increased nuclear pSmad2/3 staining of 90.96% ± 0.79% (stained cells/total cells) compared with nontreated wounded cultures with staining of 45.84% ± 1.16% (Figure 1, A, D, and G). The percentages of pSmad2/3 staining at AWM of the nontreated and treated VICs, all of which were less than 0.9%, did not exhibit statistically significant differences from each other (Figure 1, C and F).
Figure 1.
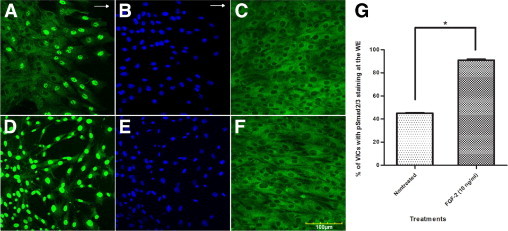
Representative immunofluorescent confocal photomicrographs of a wounded VIC monolayer 24 hours after wounding at the WE stained for pSmad2/3 (green) (A and D) and nuclear localization with propidium iodide (blue) (B and E), and AWM strip parallel to WE stained for pSmad2/3 (C and F) in nontreated cultures (A–C), and FGF-2 (10 ng/ml) treated cultures (D–F). The VICs treated with exogenous FGF-2 show increased nuclear pSmad2/3 staining at WE compared with nontreated WE. No difference was observed in pSmad2/3 staining at the AWM between treated and nontreated VICs. Arrows indicate direction of wound closure. G: Percentage of VICs showing nuclear pSmad2/3 staining at the WE with no treatment and with FGF-2 treatment. The VICs at the FGF-2–treated WE exhibit a statistically significant increase in the percentage of VICs showing nuclear pSmad2/3 staining compared with nontreated WE. Statistical significance between indicated groups at *P < 0.05, (n = 9). Original magnifications, ×400.
TGF-β Neutralizing Antibody and FGF-2 Neutralizing Antibody Decrease Nuclear pSmad2/3 Staining, with No Effects from Treatment with Exogenous FGF-2 in addition to Neutralizing Antibodies in VICs at the WE
At 24 hours after wounding and treatment, the VICs at the WE treated with TGF-β neutralizing antibody (15 μg/ml) showed nuclear pSmad2/3 staining of 11.53% ± 0.25% compared with VICs at the WE treated with FGF-2 (10 ng/ml) and TGF-β neutralizing antibody (15 μg/ml) with staining of 12.65% ± 0.57% showing no statistically significant difference (Figure 2, A, D, and M).
Figure 2.
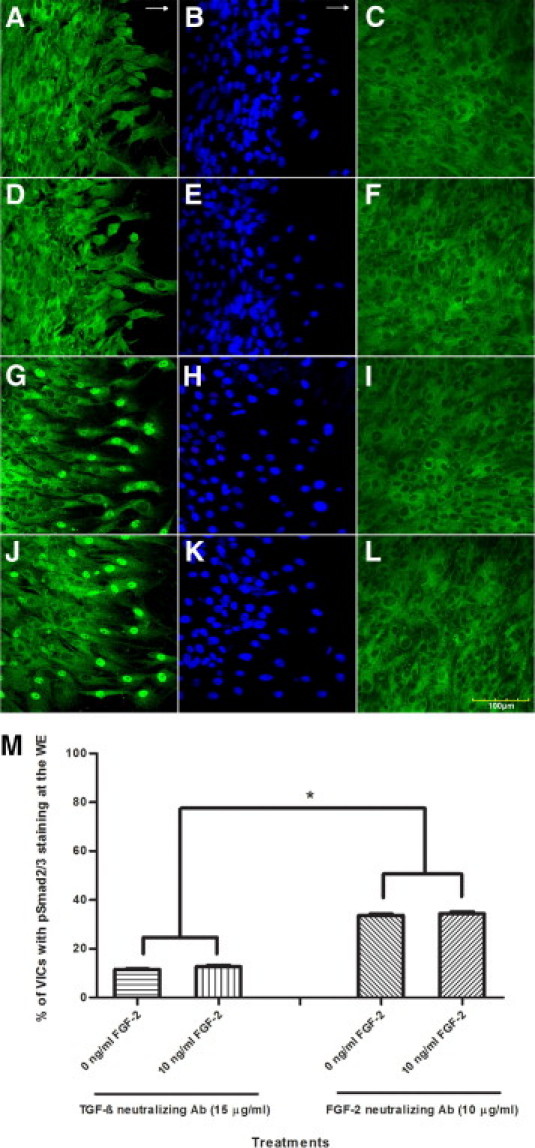
Representative immunofluorescent confocal photomicrographs of a wounded VIC monolayer 24 hours after wounding at the WE stained for pSmad2/3 (green) (A, D, G, and J) and nuclear localization with propidium iodide (blue) (B, E, H, and K), and AWM strip parallel to WE stained for pSmad2/3 (C, F, I, and L) in wounded cultures treated with neutralizing antibody to TGF-β (15 μg/ml) (A–C), with neutralizing antibody to TGF-β (15 μg/ml) and exogenous FGF-2 (10 ng/ml) (D–F), with neutralizing antibody to FGF-2 (10 μg/ml) (G–I) and with neutralizing antibody to FGF-2 (10 μg/ml) and exogenous FGF-2 (10 ng/ml) (J–L). Same nuclear pSmad2/3 staining at the WE is observed for VICs treated with neutralizing antibody to TGF-β only and VICs treated with neutralizing antibody and exogenous FGF-2. The same nuclear pSmad2/3 staining at the WE is observed for VICs treated with neutralizing antibody to FGF-2 only and VICs treated with neutralizing antibody to FGF-2 and exogenous FGF-2. Treatment with neutralizing antibody to FGF-2 shows greater nuclear pSmad2/3 staining at the WE than VICs treated with neutralizing antibody to TGF-β at the WE. No difference was observed in pSmad2/3 staining at the AWM among the treatments. Arrows indicate direction of wound closure. M: Percentage of VICs showing nuclear pSmad2/3 staining at the WE with treatments of neutralizing antibody to TGF-β, exogenous FGF-2, and neutralizing antibody to TGF-β, neutralizing antibody to FGF-2, and exogenous FGF-2 and neutralizing antibody to FGF-2. Treatment of exogenous FGF-2 in combination with neutralizing antibody to TGF-β does not yield a statistically significant difference of pSmad2/3 staining at the WE from when treated with neutralizing antibody to TGF-β alone. Treatment of exogenous FGF-2 in combination with neutralizing antibody to FGF-2 does not yield a statistically significant difference of pSmad2/3 staining at the WE from when treated with neutralizing antibody to FGF-2 alone. A statistically significant increase is observed in percentage of VICs showing nuclear pSmad2/3 staining at the WE treated with neutralizing antibody to FGF-2 compared with WE treated with neutralizing antibody to TGF-β. Statistical significance between indicated groups at *P < 0.05, (n = 9). Original magnifications, ×400.
At 24 hours after wounding and treatment, the VICs at the WE treated with FGF-2 neutralizing antibody (10 μg/ml) showed nuclear pSmad2/3 staining of 33.64% ± 0.56% compared with VICs at the WE treated with exogenous FGF-2 (10 ng/ml) and neutralizing antibody to FGF-2 (10 μg/ml) with staining of 34.40% ± 0.73% showing no statistically significant difference (Figure 2, G, J, and M).
The VICs that were treated with neutralizing antibody to TGF-β exhibited a statistically significant decrease in nuclear pSmad2/3 staining at the WE compared with the treatment of neutralizing antibody to FGF-2 (Figure 2, M). Both treatment with neutralizing antibody to TGF-β and neutralizing antibody to FGF-2 exhibited a statistically significant decrease in nuclear pSmad2/3 staining at the WE compared with the nontreated control.
The percentages of pSmad2/3 staining at AWM of the nontreated and treated VICs, all of which were less than 0.9%, did not exhibit statistically significant differences from each other (Figure 2, C, F, I, and L).
Betaglycan Antibody Exhibits Statistically Same Nuclear pSmad2/3 Staining at WE and AWM as Nontreated Monolayers, with No Effects from Treatment with Exogenous FGF-2 in addition to Betaglycan Antibody in VICs at the WE
Immunofluorescent staining showed betaglycan presence in VICs in all conditions studied. At 24 hours after wounding and treatment, the VICs at the WE treated with betaglycan antibody (2 μg/ml) showed nuclear pSmad2/3 staining of 50.79% ± 1.14% compared with VICs treated with FGF-2 (10 ng/ml) in addition to betaglycan antibody with staining of 51.19% ± 1.93% (Figure 3, E, G, and I). Both treatments with betaglycan antibody showed a statistically significant decrease in nuclear pSmad2/3 staining compared with wounded monolayers treated with exogenous FGF-2 alone at the WE, which showed staining of 90.44% ± 1.52% (Figure 3, C and I). The increase in nuclear pSmad2/3 staining observed when wounded cultures are treated with betaglycan antibody compared with nontreated wounded monolayers at the WE (45.57% ± 1.83%) is not statistically significant (Figure 3, A and I).
Figure 3.
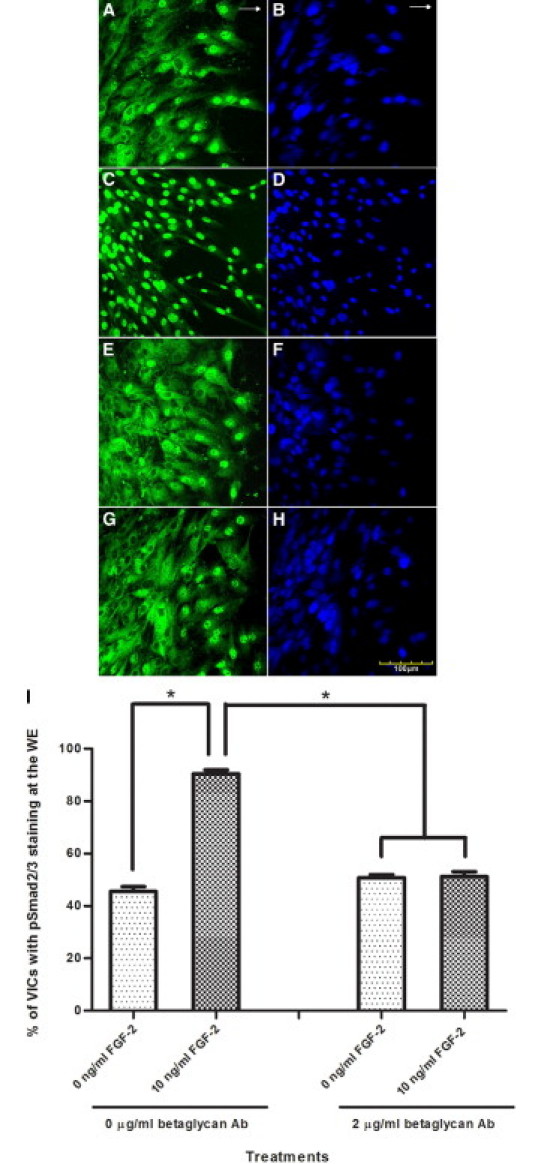
Representative immunofluorescent confocal photomicrographs of the WE of wounded VIC monolayers 24 hours after wounding stained for pSmad2/3 (green) (A, C, E, and G) and for nuclear localization with propidium iodide (blue) (B, D, F, and H) in wounded cultures with no treatment (A and B), with exogenous FGF-2 (10 ng/ml) (C and D), with betaglycan antibody (2 μg/ml) (E and F), and with betaglycan antibody (2 μg/ml) and exogenous FGF-2 (10 ng/ml) (G and H). Increased nuclear pSmad2/3 staining at the WE is observed for VICs treated with exogenous FGF-2 only compared with nontreated cultures. Same nuclear pSmad2/3 staining at the WE is observed for VICs treated with betaglycan antibody and VICs treated with betaglycan antibody and exogenous FGF-2. Decreased pSmad2/3 staining at the WE is observed in cultures treated with betaglycan antibody compared with cultures treated with exogenous FGF-2 only. Arrows indicate direction of wound closure. I: Percentage of VICs showing nuclear pSmad2/3 staining at the WE with no treatment, exogenous FGF-2, betaglycan antibody, and betaglycan antibody and FGF-2. A statistically significant increase is observed in percentage of VICs showing nuclear pSmad2/3 staining at the WE treated with exogenous FGF-2 compared with nontreated WE. Treatment of exogenous FGF-2 in conjunction with betaglycan antibody does not yield a statistically significant difference in nuclear pSmad2/3 staining at the WE from VICs treated with betaglycan antibody alone. Statistical significance between indicated groups at *P < 0.05 (n = 3 for nontreated and only exogenous FGF-2–treated cultures, and n = 9 for betaglycan antibody–treated cultures). Original magnifications, ×400.
The percentages of pSmad2/3 staining at AWM of the nontreated and treated VICs, all of which were less than 0.9%, did not exhibit statistically significant differences from each other.
Subconfluent VIC Monolayer Exhibits More Active VICs with α-SMA Staining Compared with the Confluent VIC Monolayer
At 24 hours after treatment, α-SMA staining of VICs for each treatment of the subconfluent monolayer was significantly greater than α-SMA staining of VICs of their respective treatments in the confluent monolayer (Figure 4). In the subconfluent monolayer, α-SMA staining for nontreated (97.01% ± 0.44%) and exogenous FGF-2–treated (96.90% ± 0.19%) VICs did not exhibit a statistically different percentage staining from each other (P < 0.05; Figure 4, A–D and G). Monolayers treated with neutralizing antibody to FGF-2 exhibited a statistically significant decrease in α-SMA staining (90.58% ± 0.50%) compared with nontreated and FGF-2–treated monolayers (Figure 4G).
Figure 4.
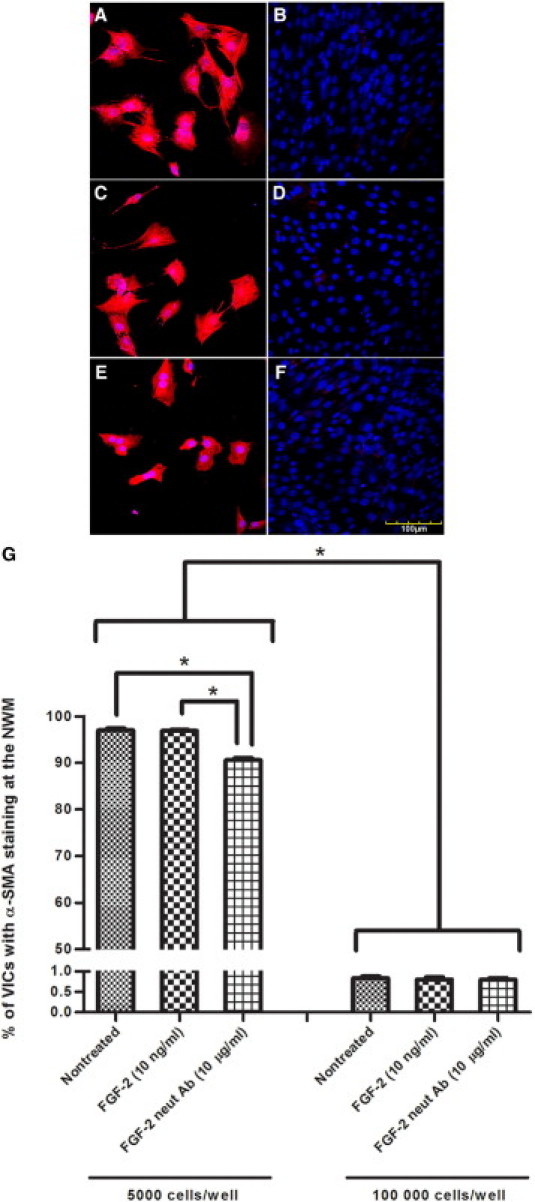
Representative immunofluorescent confocal photomicrographs of subconfluent and confluent VIC monolayers, stained for α-SMA (red) and nuclear localization with propidium iodide (blue), 24 hours after no treatment (A and B), treatment with exogenous FGF-2 (C and D), and treatment with neutralizing antibody to FGF-2 (E and F), in subconfluent monolayers of 5000 cells/well inoculation (A, C, and E) and confluent monolayer of 100,000 cells/well inoculation (B, D, and F). α-SMA staining for all treatments is greater in subconfluent monolayer than their respective treatments in confluent monolayers. G: Percentage of VICs showing α-SMA staining in the monolayers with treatments of exogenous FGF-2 and neutralizing antibody to FGF-2. A statistically significant increase in α-SMA staining is observed in the subconfluent monolayer for nontreated and treated monolayers compared with their respective treatments in the confluent monolayer. Treatment with neutralizing antibody to FGF-2 resulted in a statistically significant decrease in α-SMA staining compared with the nontreated monolayers and monolayers treated with exogenous FGF-2. Nontreated and exogenous FGF-2–treated VIC subconfluent monolayers did not exhibit a statistically significant difference in α-SMA staining. Confluent monolayers did not yield statistically significant differences in α-SMA staining for different treatments. Significance between indicated groups at *P < 0.05, (n = 4). Original magnification, ×400.
The percentages of α-SMA staining of the confluent monolayer of the nontreated and treated VICs, all of which were less than 0.9%, did not exhibit statistically significant differences from each other (Figure 4, B, D, F, and G).
Extent of Wound Closure Corresponds with pSmad2/3 Staining in VICs
Wounds closed progressively over 96 hours, with most treatments closing by 72 and 96 hours (Figure 5). The FGF-2–treated wounds showed a significantly greater extent of wound closure for all points compared with nontreated wounds. Cell cultures treated with neutralizing antibody to TGF-β, regardless of the addition of FGF-2, resulted in a statistically reduced extent of wound closure compared with nontreated cultures and cultures treated with FGF-2 alone. The addition of FGF-2 to cultures treated with neutralizing antibody to TGF-β did not show a statistically significant difference in the extent of wound closure when compared with treatment of neutralizing antibody to TGF-β alone. Cell cultures treated with betaglycan antibody, regardless of the addition of FGF-2, resulted in a statistically reduced extent of wound closure compared with cultures treated with FGF-2 alone. No significant difference was observed for cell cultures treated with betaglycan antibody compared with nontreated cultures (Figure 5B).
Figure 5.
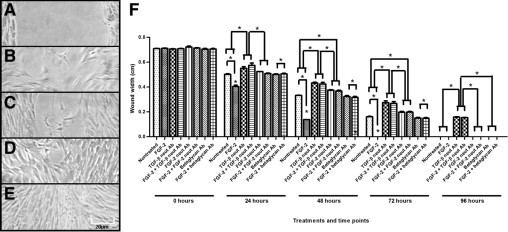
Wound closure of a wounded VIC monolayer, up to 96 hours. Representative phase-contrast micrographs of wounded VICs at 0 (A), 24 (B), 48 (C), 72 (D), and 96 (E) hours. F: Measure of width of the wound, between the two WEs at points of 0, 24, 48, 72, and 96 hours, for various treatments of VICs. Treatment with exogenous FGF-2 resulted in a significantly smaller wound width; therefore, increased wound closure, compared with nontreated VICs, for all points up to complete closure of wounds. Treatment with neutralizing antibody to TGF-β resulted in significantly greater wound width and decreased wound closure compared with nontreated VICs for all points; further addition of FGF-2 in the presence of neutralizing antibody to TGF-β did not yield significantly different results from neutralizing antibody to TGF-β alone for all points. Treatment with neutralizing antibody to FGF-2 resulted in significantly greater wound width and decreased wound closure compared with nontreated VICs for all points up to wound closure, except for 24 hours after wounding; further addition of FGF-2 in the presence of neutralizing antibody to FGF-2 did not yield significantly different results from neutralizing antibody to FGF-2 alone for all points. Treatment with neutralizing antibody to FGF-2 yielded significantly greater wound closure than treatment with neutralizing antibody to TGF-β for all points. Treatment with betaglycan antibody did not result in significantly different wound width compared with nontreated VICs for all points; further addition of FGF-2 in the presence of betaglycan antibody did not yield significantly different results from betaglycan antibody alone for all points. Treatment of FGF-2 in the presence of, individually, neutralizing antibody to TGF-β, neutralizing antibody to FGF-2, or betaglycan antibody resulted in significantly greater wound width and decreased wound closure compared with treatment with FGF-2 alone for all points up to complete closure of wounds. Significance between indicated groups at *P < 0.05, (n = 3).
Down-Regulation of pSmad2/3 Gene Expression Decreases Effect of FGF-2 on Wound Closure in VICs
Down-regulation of pSmad2/3 protein expression by siRNA transfection was confirmed by Western blotting (Figure 6, A and B). Cultures transfected with control siRNA showed a similar extent of wound closure when compared with nontransfected cultures. Transfection with Smad2/3 siRNA resulted in significantly reduced wound closure of FGF-2–treated and nontreated cultures when compared with control siRNA-transfected cultures (Figure 6C).
Figure 6.
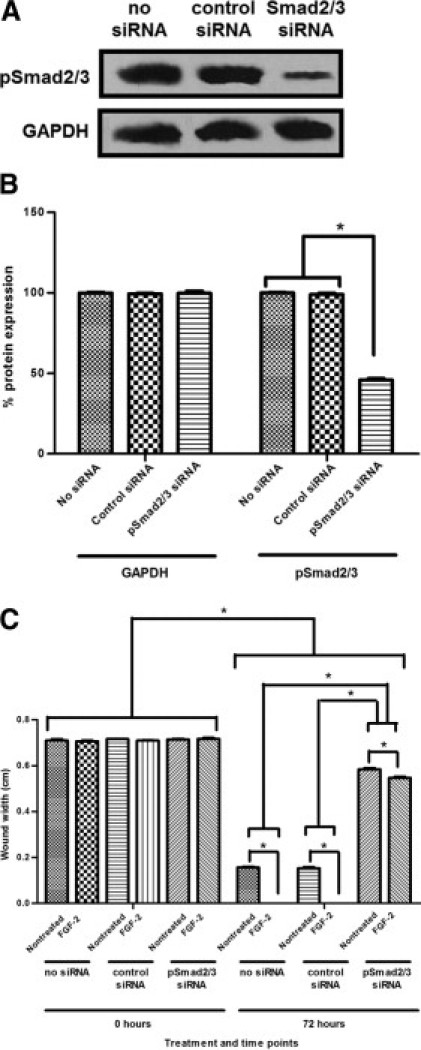
Down-regulation of pSmad2/3 in wounded VIC monolayers by Smad2/3 siRNA transfection. A: Representative pSmad2/3 protein expression of wounded VIC monolayer 72 hours after siRNA transfection detected by Western blot analysis with anti-pSmad2/3 antibody. The membrane was also probed with anti-GAPDH antibody as an internal control for equal loading and transfer. Down-regulated pSmad2/3 protein expression for Smad2/3 siRNA transfected VICs is observed compared with VICs with no siRNA transfection and control siRNA transfection. B: pSmad2/3 protein expression as detected by Western blot analysis and quantified by densitometry (ImageJ). Protein expression of pSmad2/3 in Smad2/3 siRNA–transfected VIC monolayers is significantly less than nontransfected and control siRNA-transfected VIC monolayer. Protein expression of pSmad2/3 in nontransfected and control siRNA-transfected VIC monolayers do not differ significantly. Protein expression of GAPDH for all transfections does not differ significantly from each another. Significance between indicated groups at *P < 0.05, (n = 3). C: Wounded VICs transfected with Smad2/3 siRNA showed significantly greater wound width and less wound closure at 72 hours with no treatment compared with nontransfected and control siRNA-treated VIC monolayers. For all transfections, FGF-2 treatment resulted in a statistically significant decrease of wound width and increased wound closure. Nontransfected and control siRNA-transfected VICs did not differ significantly in wound closure. Significance between indicated groups at *P < 0.05, (n = 3).
Discussion
Separate previous investigations13,25 of VIC wound repair have shown that the TGF-β/Smad2/3 signaling pathway is associated with wound repair and that FGF-2 is up-regulated in VICs at the WE, suggesting that FGF-2 plays a role in wound repair. Our current study shows that treatment with FGF-2 resulted in increased activation of pSmad2/3 in the nucleus of VICs at the WE, an effect that is inhibited by treatment with neutralizing antibody to FGF-2 and treatment with neutralizing antibody to TGF-β. The latter suggests that FGF-2 promotes in vitro VIC wound repair, at least in part, through the TGF-β/Smad2/3 signaling pathway. The finding that the inhibition by neutralizing antibody to TGF-β is significantly more pronounced than the inhibition by neutralizing antibody to FGF-2 suggests that there are other factors that also regulate repair. Wound closure analyses supported results obtained from pSmad2/3 expression analyses. Similarly, down-regulation of pSmad2/3 via siRNA also confirmed the upstream effects of FGF-2 on the TGF-β/Smad signaling pathway and the possibility that FGF-2 interacts with other factors that also regulate wound repair. These factors have yet to be identified.
In contrast to our findings, FGF-2 has been reported to prevent TGF-β–mediated Smad2/3 activation by blocking the TGF-β signaling pathway.27 An important difference in that study compared with ours is that they studied subconfluent cultures, whereas we studied the effect of FGF-2 on confluent monolayers that show activation of VICs at the WE. Our study showed that FGF-2 promotes wound repair through the TGF-β/Smad2/3 signaling pathway; previous studies have shown that the treatment of FGF-2 promotes cell proliferation and migration.25 It is possible that the addition of FGF-2 to subconfluent monolayers would promote cell growth, which results in increased cell-cell contact and a less activated monolayer. We see the same effect of reduced activation of Smad2/3 by FGF-2 treatment at the AWM, where the monolayer is confluent with prominent cell-cell contact; we also showed that subconfluent VICs are activated, with significantly increased α-SMA staining, compared with confluent monolayers. Thus, in cell wounding, FGF-2 promotes repair by activation of the TGF-β/Smad2/3 signaling pathway. The FGF-2–mediated TGF-β/Smad2/3 signaling pathway that we report is consistent with the role of FGF-2 in the wound repair of many tissues.19,21,22 Another difference between our studies is that, while we looked at mitral VICs, the other study27 investigated aortic VICs.
Our study suggests that FGF-2 promotes TGF-β/Smad2/3 signaling via betaglycan, a membrane heparan and chondroitin sulfate proteoglycan.8 Betaglycan binds TGF-β with high affinity, possibly decreasing the amount of TGF-β available for binding to the TGF-β type I and type II receptors in certain cell types.11 This decreased binding of TGF-β to type I and type II receptors results in down-regulation of the Smad2/3 signaling pathway, thereby producing less nuclear pSmad2/3. In the presence of betaglycan antibody, the FGF-2–mediated twofold increase in nuclear Smad2/3 at the WE was eliminated. Supporting results were also obtained from wound closure analyses. We suggest the following (Figure 7): FGF-2 is known to selectively modify the glycosaminoglycan chains of betaglycan on binding to it, altering betaglycan function,11 which likely affects the binding site or binding affinity of betaglycan for TGF-β. In VICs, FGF-2 likely reduces TGF-β binding to betaglycan, where treatment with FGF-2 would result in more TGF-β being available to bind to the type I and type II receptor complex, leading to Smad2/3 signaling (Figure 7). The exact molecular mechanisms by which betaglycan, TGF-β, and FGF-2 interact in VICs are yet to be determined.
Figure 7.
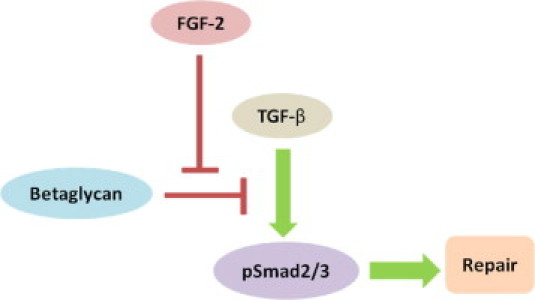
Summary of a putative pathway that regulates FGF-2–mediated wound repair through the TGF-β/Smad signaling pathway. The TGF-β promotes the phosphorylation of Smad2/3, resulting in VIC wound repair. Betaglycan binds endogenous TGF-β, resulting in less available TGF-β to bind to its signaling receptors. The FGF-2 also binds betaglycan, which makes more TGF-β available for receptor signaling.
Our conclusion is that FGF-2 promotes in vitro VIC wound repair, at least in part, through the TGF-β/Smad2/3 signaling pathway. The FGF-2 regulates the TGF-β/Smad pathway by binding to betaglycan, which itself binds to TGF-β and reduces TGF-β/Smad signaling. Thus, FGF-2 enhances VIC wound repair by reducing the inhibitory effect of betaglycan on the TGF-β/Smad signaling pathway.
Footnotes
Supported by the Heart and Stroke Foundation of Ontario (grant NA6204) and the Canadian Institutes for Health Research (grant 84228).
References
- 1.Liu A.C., Joag V.R., Gotlieb A.I. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Durbin A.D., Gotlieb A.I. Advances towards understanding valve response to injury. Cardiovasc Pathol. 2002;11:69–77. doi: 10.1016/s1054-8807(01)00109-0. [DOI] [PubMed] [Google Scholar]
- 3.Durbin A., Nadir N., Rosenthal A., Gotlieb A.I. Nitric oxide promotes in vitro interstitial cell heart valve repair. Cardiovasc Pathol. 2005;14:12–18. doi: 10.1016/j.carpath.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 4.ten Dijke P., Hill C.S. New insights into TGF-β-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Blobe G.C., Schiemann W.P., Lodish H.F. Role of transforming growth factor β in human diseases. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 6.Nakao A., Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hanai J., Heldin C.H., Miyazono K., ten Dijke P. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y., Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 8.Andres J.L., DeFalcis D., Noda M., Massagué J. Binding of two growth factor families to separate domains of the proteoglycan betaglycan. J Biol Chem. 1992;267:5927–5930. [PubMed] [Google Scholar]
- 9.Kaname S., Ruoslahti E. Betaglycan has multiple binding sites for transforming growth factor-β1. Biochem J. 1996;315:815–820. doi: 10.1042/bj3150815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendoza V., Vilchis-Landeros M.M., Mendoza-Hernández G., Huang T., Villarreal M.M., Hinck A.P., López-Casillas F., Montiel J.L. Betaglycan has two independent domains required for high affinity TGF-β binding: proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor. Biochemistry. 2009;48:11755–11765. doi: 10.1021/bi901528w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eickelberg O., Centrella M., Reiss M., Kashgarian M., Wells R.G. Betaglycan inhibits TGF-β signaling by preventing type I-type II receptor complex formation. J Biol Chem. 2002;277:823–829. doi: 10.1074/jbc.M105110200. [DOI] [PubMed] [Google Scholar]
- 12.Lee J.D., Hempel N., Lee N.Y., Blobe G.C. The type II TGF-β receptor suppresses breast cancer progression through GIPC-mediated inhibition of TGF-β signaling. Carcinogen. 2010;31:175–183. doi: 10.1093/carcin/bgp271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu A.C., Gotlieb A.I. Transforming growth factor-β regulates in vitro heart valve repair by activated valve interstitial cells. Am J Pathol. 2008;173:1275–1285. doi: 10.2353/ajpath.2008.080365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cushing M.C., Liao J., Anseth K.S. Activation of valvular interstitial cells is mediated by transforming growth factor-β1 interactions with matrix molecules. Matrix Biol. 2005;24:428–437. doi: 10.1016/j.matbio.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Darby I., Skalli O., Gabbiani G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. 1990;63:21–29. [PubMed] [Google Scholar]
- 16.Bikfalvi A., Klein S., Pintucci G., Rifkin D.B. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- 17.Javerzat S., Auguste P., Bikfalvi A. The role of fibroblast growth factors in vascular development. Trends Mol Med. 2002;8:483–489. doi: 10.1016/s1471-4914(02)02394-8. [DOI] [PubMed] [Google Scholar]
- 18.Lemiere S., Azar R., Belloc F., Gürsel D., Pyronnet S., Bikfalvi A., Auguste P. Overexpression of high molecular weight FGF-2 forms inhibits glioma growth factor by acting on cell-cycle progression and protein translation. Exp Cell Res. 2008;314:3701–3711. doi: 10.1016/j.yexcr.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 19.Virag J.A., Rolle M.L., Reece J., Hardouin S., Feigl E.O., Murry C.E. Fibroblast growth factor-2 regulates myocardial infarct repair. Am J Pathol. 2007;171:1431–1440. doi: 10.2353/ajpath.2007.070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eswarakumar V.P., Lax I., Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Ortega S., Ittmann M., Tsang S., Ehrlich M., Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribatti D., Nico B., Vacca A., Roncali L., Presta M. Endogenous and exogenous fibroblast growth factor-2 modulate wound healing in the chick embryo chorioallantoic membrane. Angiogenesis. 1999;3:89–95. doi: 10.1023/a:1009049932252. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z., Coomans C., David G. Membrane heparan sulfate proteoglycan-supported FGF-2-FGFR1 signaling. J Biol Chem. 2001;276:41921–41929. doi: 10.1074/jbc.M106608200. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto T., Turesson I., Book M., Gerwins P., Claesson-Welsh L. p38 MAP kinase negatively regulates endothelial cell survival, proliferation, and differentiation in FGF-2-stimulated angiogenesis. J Cell Biol. 2002;156:149–160. doi: 10.1083/jcb.200103096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gotlieb A.I., Rosenthal A., Kazemian P. Fibroblast growth factor 2 regulation of mitral valve interstitial cell repair in vitro. J Thorac Cardiovasc Surg. 2002;124:591–597. doi: 10.1067/mtc.2002.123812. [DOI] [PubMed] [Google Scholar]
- 26.Lester W., Rosenthal A., Granton B., Gotlieb A.I. Porcine mitral valve interstitial cells in culture. Lab Invest. 1988;59:710–719. [PubMed] [Google Scholar]
- 27.Cushing M.C., Mariner P.D., Liao J.T., Sims E.A., Anseth K.S. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J. 2008;22:1769–1777. doi: 10.1096/fj.07-087627. [DOI] [PMC free article] [PubMed] [Google Scholar]