

Abstract
Mucosal inflammation in the gut is characterized by infiltration of innate and adaptive immune cells and by an alteration in serotonin–producing enterochromaffin cells. We investigated the role of serotonin in the function of dendritic cells (DCs) and sequential T-cell activation in relation to generation of gut inflammation. DCs isolated from tryptophan hydroxylase-1–deficient (TPH1−/−) mice, which have reduced serotonin in the gut, and wild-type (TPH1+/+) mice with or without dextran sulfate sodium (DSS)–induced colitis were stimulated with lipopolysaccharide to assess interleukin-12 (IL-12) production. Isolated DCs from TPH1+/+ and TPH1−/− mice were also cocultured with CD4+ T cells of naive TPH1+/+ mice to assess the role of serotonin in priming T cells. In addition, serotonin-pulsed DCs were transferred to TPH1−/− mice to assess the effect on DSS-induced colitis. Consistent with a reduced severity of colitis, DCs from DSS-induced TPH1−/− mice produced less IL-12 compared with the TPH1+/+ mice. In vitro serotonin stimulation restored the cytokine production from TPH1−/− DCs and adoptive transfer of serotonin-pulsed DCs into TPH1−/− up-regulated colitis. Furthermore, CD4+ T cells primed by TPH1−/− DCs produce reduced the levels of IL-17 and interferon-γ. This study provides novel information on serotonin-mediated immune signaling and promotion of interactions between innate and adaptive immune responses in the context of gut inflammation, which may ultimately lead to improved strategies to combat gut inflammatory disorders.
Inflammation in the gastrointestinal (GI) tract is characterized by mucosal recruitment of activated cells from both the innate and adaptive immune systems. In addition to immune cells, inflammation in the gut is associated with an alteration in serotonin producing enterochromaffin (EC) cells. EC cells are the most well-characterized subset of GI endocrine cells, which are dispersed throughout the GI mucosa and are the main source of serotonin in the gut.1–4 Serotonin is an important enteric mucosal signaling molecule that influences gut physiology (motor and secretory function) after inflammation and is considered important in gut function. Changes in intestinal EC cell numbers and serotonin levels are observed in patients with inflammatory bowel disease (IBD)5–7 and also in experimental colitis.8,9 In addition, an alteration in the number of EC cells and in serotonin level is observed in enteric infection–induced inflammation in the gut10–13 and functional disorders such as irritable bowel syndrome (IBS).7,14 The association between alteration in EC cell numbers and serotonin production and various GI diseases highly emphasizes the significance of serotonin in intestinal homeostasis. Because of the strategic location of EC cells in gut mucosa, it is likely that serotonin plays an important role in immune activation in relation to gut pathology and pathophysiology in various GI disorders, including IBD.
EC cells synthesize serotonin from its precursor l-tryptophan. Tryptophan hydroxylase (TPH) catalyzes the rate-limiting step in the synthesis of serotonin from tryptophan and has been detected prominently in EC cells.15 Recent studies show that there are two isoforms of TPH enzymes that regulate the serotonin system. TPH1 is mainly present in peripheral organs, such as the EC cells and spleen, whereas TPH2 predominates in the brainstem and myenteric plexus neurons.16,17
Recently, using tryptophan hydroxylase-1–deficient (TPH1−/−) mice, which have significantly reduced amounts of serotonin in the gut, and mice treated with the serotonin synthesis inhibitor para-chloro-d, l-phenylalanine (PCPA), we demonstrated a critical role of serotonin in the generation of colitis in two different models of colitis [dextran sulfate sodium (DSS) and dinitrobenzene sulfonic acid (DNBS)].8 Decreased severity of colitis and down-regulation of proinflammatory cytokine production were observed in TPH1−/− mice compared with wild-type mice and in PCPA-treated mice after induction of colitis.
Dendritic cells (DCs) are important innate immune cells and perform a key role in activation of immune response. DCs are the major source of proinflammatory mediators, including cytokines, reactive oxygen, and nitrogen intermediates, and play a critical role in the generation of inflammation in the intestinal tract. The character of a T-cell (key player in adaptive immune system) response, whether effector T cells (TH1, TH2, TH17) or regulatory T cells (Tr, TH3), reflects instruction by the initiating DCs. Studies on human DCs revealed the presence of different serotoninergic receptors on DCs, and functional studies indicated that activation of serotonin 4 and serotonin 7 receptor enhanced the release of the cytokines interleukin-1β (IL-1β) and IL-8.18 Although DCs expressing serotonin receptors are situated close to EC cells, and are considered to play an important role in immune activation and generation of intestinal inflammation, the role of serotonin in modulating DC function in the context of intestinal inflammation remains to be determined. In this study we investigated the role of serotonin in the function of DCs and sequential T-cell activation in relation to generation of gut inflammation in a well-defined experimental model of colitis.
Materials and Methods
Animals
TPH1−/− mice on C57BL/6 background were originally produced by gene mutation as described by Côté et al.19 Briefly, exon 2 of the TPH1 locus has been substituted by the nlslacZneopolyA cassette. These mice are viable, express normal amounts of serotonin in the brain, and show no observed food intake differences or body weight difference from wild-type mice. Breeding pairs of TPH1−/− mice and their wild-type (TPH1+/+) littermates were obtained from Université Pierre et Marie Curie, CNRS, Paris, France, and were kept and bred under specific pathogen-free conditions. C57BL/6 were purchased from Taconic (Albany, NY). All experiments were approved by the McMaster University Animal Ethics Committee and conducted under the Canadian Guidelines for Animal Research.
Induction of DSS Colitis
DSS (mol. wt., 40 kDa; ICN Biomedicals Inc, Soho, Ohio) was added to the drinking water in a final concentration of 5% (wt/vol) for 5 days.8,20 Controls were all time matched and consisted of mice that received normal drinking water only. Mean DSS consumption was noted per cage each day.
Assessment of the Severity of Colitis: Disease Activity Index
Disease activity index (DAI) scores have historically correlated well with the pathological findings in a DSS-induced model of ulcerative colitis.21 DAI is the combined score of weight loss, stool consistency, and bleeding. Scores were defined as follows: weight: 0, no loss; 1, 5% to 10%; 2, 10% to 15%; 3, 15% to 20%; and 4, 20% weight loss; stool: 0, normal; 2, loose stool; and 4, diarrhea; and bleeding: 0, no blood; 2, presence (Hemoccult II positive result; Beckman Coulter, Fullerton, CA); and 4, gross blood. The DAI was scored from day 0 to day 5 during DSS treatment.
Macroscopic Scores
Five days after the beginning of the DSS treatment, the mice were sacrificed and the abdominal cavity was opened, the colon was located, and observations on distension, fluid content, hyperemia, and erythema were recorded. The colon was removed and opened longitudinally, and macroscopic damage was immediately assessed. Macroscopic scores were performed using a previously described scoring system for DSS colitis.21
Colonic Histology and Myeloperoxidase Activity
Formalin-fixed colon segments were paraffin embedded and 3-μm sections were stained with H&E. Colonic damage was scored based on a published scoring system that considers architectural derangements, goblet cell depletion, edema or ulceration, and degree of inflammatory cell infiltrate.21 Myeloperoxidase (MPO) activity was determined after an established protocol.22 Briefly, MPO activity, used as a marker of neutrophilic infiltration, was extracted and the activity was measured using a modified version of the method described by Bradley et al.23 Tissue samples were homogenized (50 mg/ml) in ice-cold 50 mmol/L potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyl trimethyl ammonium bromide (Sigma, Mississauga, Ontario, Canada). The homogenate was freeze thawed three times, briefly sonicated, and then centrifuged at 12 000 × g for 12 minutes at 4°C. The supernatant was then added to a solution of O-dianisidine (Sigma) and hydrogen peroxide. The absorbance of the colorimetric reaction was measured by a spectrophotometer. MPO is expressed in units per milligram of wet tissue, with 1 unit being the quantity of enzyme able to convert 1 μmol of hydrogen peroxide to water in 1 minute at room temperature.
DC and CD4+ T-Cell Isolation
Spleens were cut into small pieces and digested for 30 minutes at 37°C with spleen dissociation medium (Stem Cell Technology, Vancouver, British Columbia, Canada). After incubation dissociation and centrifugation, the resulting cell suspension was treated with supplemented medium [PBS: 2% fetal bovine serum (FBS) and 1 mmol/L EDTA]. Spleen CD11c+ cells were isolated by positive selection using EasySep mouse CD11c+ selection kit (Stem Cell Technology, Vancouver, BC, Canada) following manufacturer’s instruction. CD4+ T cells were isolated from the mixed splenocytes by negative selection using an EasySep mouse CD4+ T cell enrichment cocktail with magnetic nanoparticles (Stem Cell Technologies, Vancouver, BC, Canada). Cell purity was 90% as determined by flow cytometry, using anti‐mouse CD4 (LT34) monoclonal antibody (BD Pharmingen, San Diego, CA).
Immunophenotyping of Isolated CD11c+ Cells
After MACS isolation, CD11c+ preparations were surface stained using various anti-mouse monoclonal antibodies. The following antibodies were used: anti-major histocompatibility complex II (MHCII)–phycoerythrin (PE), anti–CD40–fluorescein isothiocyanate, anti–CD80–peridinin chlorophyll protein–Cy5.5, and anti–CD86-allophycocyanin. All antibodies were purchased from BD Biosciences except anti–MHCII-PE (eBioscience, San Diego, CA). Specific phenotyping procedures used four-color analysis to determine various DC subsets. Data were collected on a BD LSR II cytometer using FACSDiva software (BD Biosciences, Mississawga, ON, Canada) and analyzed using FlowJo software (Treestar Inc, Ashland, OR).
Bone Marrow Isolation
Bone marrow cells were harvested from the femur and tibiae of naive mice as previously described24 and cultured in RPMI 1640 medium containing 10% FBS, 100 U/ml of penicillin, 100 μg/ml of streptomycin, 2 mmol/L l-glutamine, 0.1% 2-mercaptoethanol, 0.1 mmol/L nonessential amino acids, 1 mmol/L sodium pyruvate, and 20 ng/ml of recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF; PeproTech, Rocky Hill, NJ). Three and six days after initial culture, cells were replenished with fresh medium supplemented with GM-CSF. On day 7, DC culture was completed.
Adoptive Transfer of Spleen CD11c+ Cells
Spleen CD11c+ cells from TPH1−/− mice were isolated as described above. A total of 2.106 cells per mouse were injected i.p. into TPH1−/− host mice. After 24 hours, 5% DSS-induced colitis was induced for 5 days.
Cytokine Assessment from Bone Marrow–Derived DCs, DCs, and T Cells Cocultured with DCs
Myeloid DCs (106/ml) were cultured with increasing doses (10−7 to 10−3M) of serotonin for 48 hours. Spleen CD11c+ cells from colitic and noncolitic TPH1−/− mice were isolated as described above. CD11c+ DCs were cultured and exposed to lipopolysaccharide (LPS) (100 ng/ml; Sigma) for 24 hours. The level of IL-12p40 was determined in the presence or absence of serotonin (10−7 and 10−5M) and/or nuclear factor κB (NF-κB) inhibitor, pyrrolidine dithiocarbamate (PDTC; 10.10−6M; Sigma). For coculture, CD11c+ DCs from mice treated with DSS were cultured and exposed to LPS (100 ng/ml) for 24 hours before being cocultured with CD4+ T cells isolated from naive mice at a ratio of 1:3 (DC:T)25 in plate coated with 10 μg/ml of anti-CD3 and 2 μg/ml of anti-CD28. Cell culture supernatants were collected at days 3, 5, and 7 for cytokine analysis [interferon-γ (IFN-γ) and IL-17]. Cytokine levels (IL-1β, IL-12p40, IL-17, and IFN-γ) were determined using enzyme-linked immunosorbent assay (ELISA) commercial kit (R&D Systems, Minneapolis, MN).
Colonic Cytokine Production
A colonic sample was homogenized in 700 μL of Tris HCl buffer containing protease inhibitors (Sigma). Samples were centrifuged for 30 minutes, and the supernatant was frozen at −80°C until assay. Cytokine levels [IL-1β, IL-6, and tumor necrosis factor-α (TNF-α)] were determined using a commercial ELISA kit (R&D Systems). Cytokine (IL-1β, IL-6, IL-4, IL-12, and IL-17) and prostaglandin E2 (PGE2) levels were determined using a commercial ELISA kit (R&D Systems).
Histamine Level
Histamine was extracted from each sample by adding 10 μL of 0.2N HClO4 per milligram of tissue. Samples were homogenized and supernatant was collected and neutralized to pH 6.8 by addition of an equal volume of 1M potassium borate. Histamine levels were determined using a commercial ELISA kit (Beckman Coulter).
Statistical Analysis
Results are presented as mean ± SEM. Statistical analysis was performed using one- or two-way analysis of variance followed by the Student-Newman-Keuls multiple comparisons post hoc analysis and P < 0.05 was considered significant.
Results
Down-Regulation of Cytokine Production from DCs and Attenuation of DSS-Induced Colitis in Mice with Reduced Levels of Serotonin in the Gut
We confirmed our previous study8 showing that in TPH1−/− mice that have reduced serotonin in the gut, acute colitis is significantly decreased after 5 days of DSS treatment. Macroscopic and MPO activity decreased significantly from 3.7 ± 0.25 to 2.2 ± 0.3 and 2.1 ± 0.4 to 0.6 ± 02, respectively. This reduction in the severity of DSS-induced colonic inflammation in TPH1−/− mice was associated with down-regulation of IL-17, IFN-γ, and IL-12p40 in colonic tissue (Figure 1, A–C). However, histamine and PGE2 levels were not significantly modified (Figure 1, D and E).
Figure 1.

Effects of the lack of serotonin in the gut in the development of DSS-induced colitis. Effects of the lack of serotonin on proinflammatory cytokines [IL-17 (A), IFN-γ (B), and IL-12p40 (C)]. Histamine (D) and PGE2 (E) levels in DSS-induced colitis. TPH1+/+ and TPH1−/− mice were given a 5% DSS solution in the drinking water to induce colitis and were sacrificed on day 5 after DSS. Each value represents mean ± SEM from eight mice. Significantly lower (*P < 0.05) in DSS-treated TPH1−/− mice compared with DSS-treated TPH1+/+ mice. F: IL-12p40 production from DC (2.106 cells/ml) isolated from colitic (TPH1−/−) mice and cultured with medium or with the presence of LPS (100 ng/ml) *P < 0.05 compared with medium. **P < 0.05 compared with TPH1+/+. Data are representative of two independent experiments with quadruplicated cultures using seven mice, mean ± SEM.
DCs are critical in the production of IL-12p40 in many immune responses, including those associated with IBD. To elucidate the mechanism by which serotonin is influencing the development of colitis, we next assessed the role of serotonin in DC function in relation to gut inflammation. DCs isolated from TPH1−/− mice with DSS-induced colitis released significantly less IL-12p40 compared with DCs isolated from TPH1+/+ mice when stimulated with LPS for 24 hours (Figure 1F), implying a role of serotonin-mediated activation of DCs in the pathogenesis of colitis in this model. Interestingly, in control conditions we also observed a significant decrease in the amount of IL-12p40 released in the culture supernatant of nonstimulated DCs (Figure 1A).
Analysis of cell surface molecules by flow cytometry revealed the expression of MHCII, CD40, CD80, and CD86 on the isolated CD11c+ cells from both the TPH1+/+ and TPH1−/− mice (Figure 2). CD11c+ cells isolated from TPH1+/+ mice expressed MHCII (12%), CD40 (27.5%), CD80 (1.7%), and CD86 (22.5%). CD11c+ cells isolated from TPH1−/− mice expressed MHCII (9.2%), CD40 (26.8%), CD80 (1.5%), and CD86 (21.2%).
Figure 2.

Expression of MHCII, CD40, CD80, and CD86 by CD11c+ cells isolated by positive selection using EasySep mouse CD11c+ selection kit. CD40 and MHCII costaining of CD11c+ isolated DC from TPH1+/+ mouse (A), CD80 and CD86 costaining of CD11c+ isolated DC from TPH1+/+ mouse (B), CD40 and MHCII costaining of CD11c+ isolated DC from TPH1−/− mouse (C), and CD80 and CD86 costaining of CD11c+ isolated DCs from TPH1−/− mouse (D). Data are representative of six samples.
Serotonin Stimulates Cytokine Production from DCs
The role of serotonin in DC function was further studied by examining the ability of serotonin to stimulate naive DCs. A significantly lower amount of IL-12p40 in the culture supernatant from naive TPH1−/− mice was observed compared with TPH1+/+ mice (Figure 3A). However, addition of serotonin in culture media of DCs from TPH1−/− mice significantly up-regulated IL-12p40 production compared with DCs from TPH1−/− cultured without the addition of serotonin (Figure 3A). Addition of NF-κB inhibitor PDTC in the culture media significantly decreased the production of serotonin-stimulated IL-12p40 cytokines by DCs in response to LPS. Serotonin alone significantly increased the levels of IL-12p40, and this effect was inhibited by the addition of NF-κB inhibitor (Figure 3B).
Figure 3.
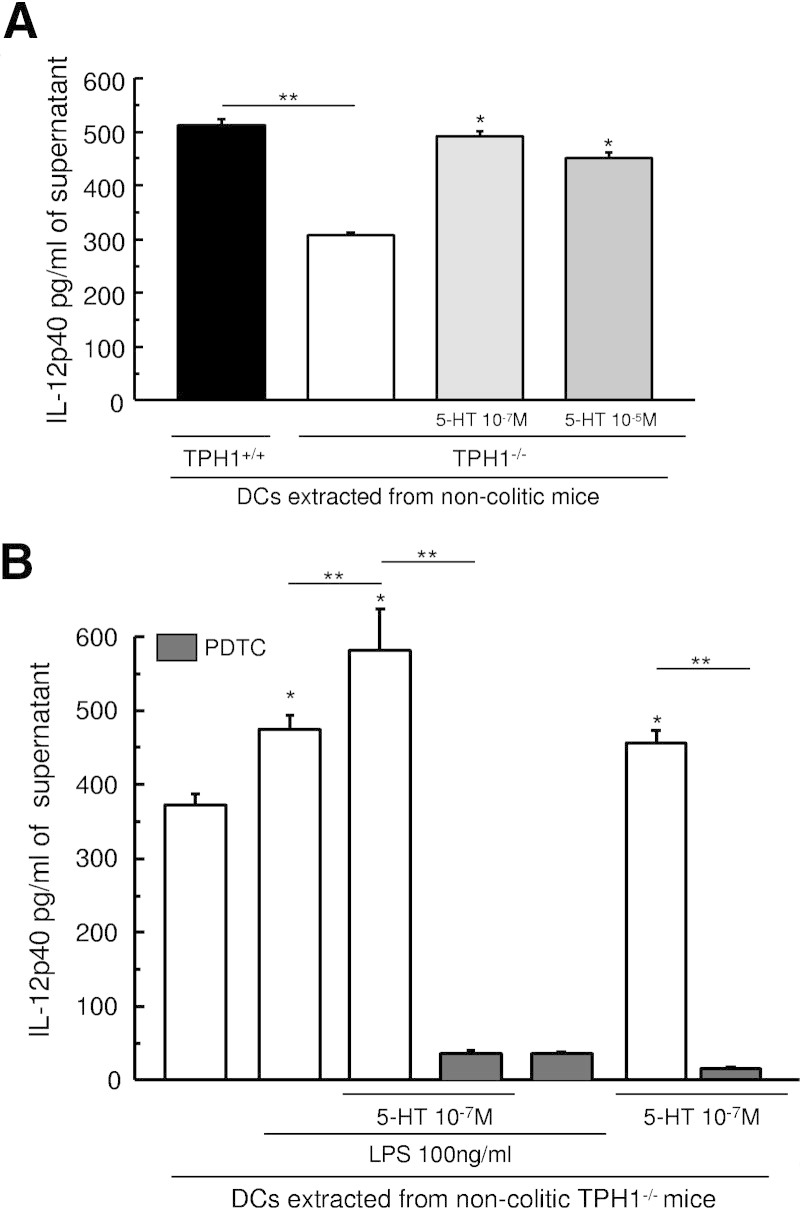
Effect of the lack of serotonin on IL-12p40 produced by DCs. A: IL-12p40 production from DCs (2.106 cells/ml) isolated from noncolitic (TPH1−/−) mice and cultured with medium or with serotonin at two different doses (10−7 and 10−5M). *P < 0.05 compared with untreated TPH1−/−. **P < 0.05 compared with untreated TPH1+/+. B: IL-12p40 production from DCs (2.106 cells/ml) isolated from noncolitic (TPH1−/−) mice and cultured with medium or with serotonin (10−7M) with or without LPS (100 ng/ml) and with or without NF-κB inhibitor (PDTC; 10 μmol/L). The levels of IL-12p40 present in the culture supernatant were investigated by ELISA. *P < 0.05 compared with control. **P < 0.05. Data are representative of two independent experiments with quadruplicated cultures, mean ± SEM.
We also investigated the effect of serotonin on bone marrow–derived dendritic cells (BMDCs) from naive mice. Bone marrow progenitors were cultured in the presence of GM-CSF to generate myeloid DCs.24 Serotonin dose dependently increased significantly the release of IL-12p40 and IL-1β in cell culture supernatant compared with untreated BMDCs (Figure 4, A and B).
Figure 4.
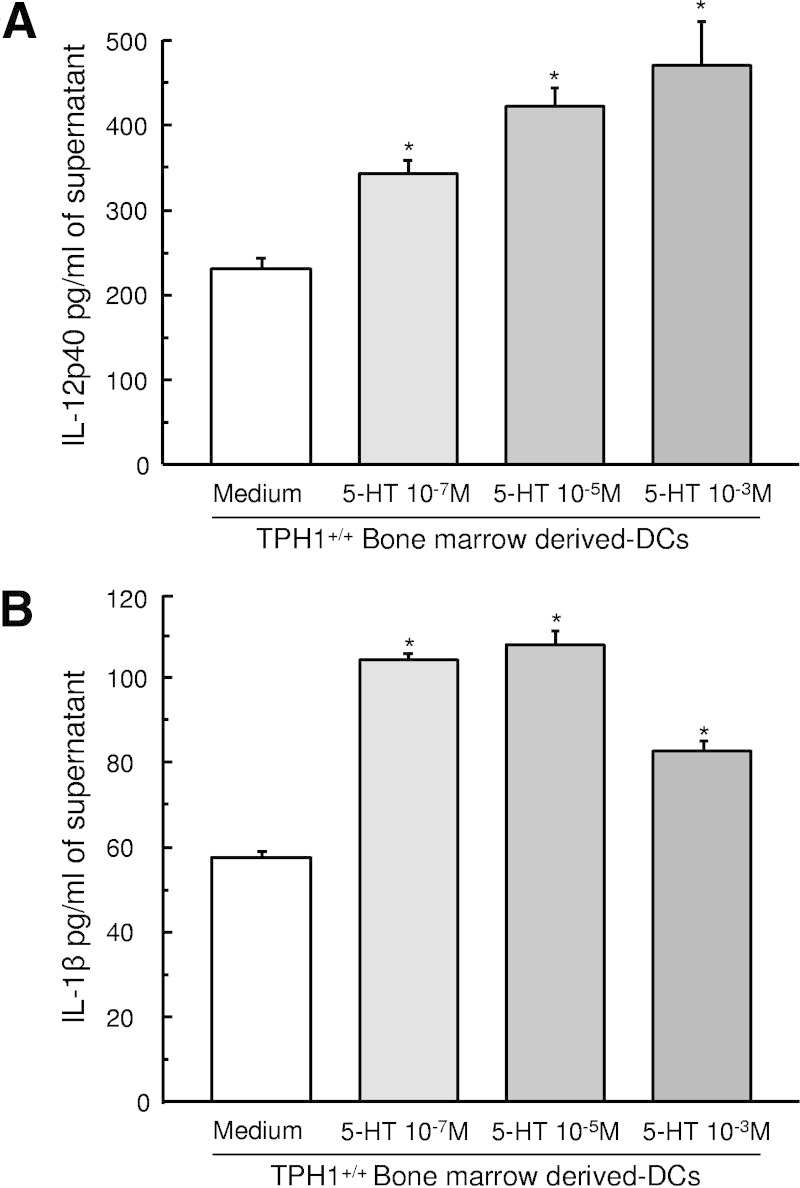
Effect of serotonin on IL-12p40 and IL-1β production from BMDCs. Bone marrow progenitors were cultured in the presence of GM-CSF (20 ng/ml) for 7 days to generate myeloid DCs. BMDCs were treated with serotonin dose dependently (10−7, 10−5, and 10−3M) for 48 hours. The levels of IL-12p40 (A) and IL-1β (B) present in the culture supernatant were investigated by ELISA. *P < 0.05 compared with medium. Data are representative of two independent experiments with quadruplicated cultures using seven mice, mean ± SEM.
Serotonin Regulates the Ability of DC Priming of CD4 T Cells
We next assessed whether the lack of serotonin in the gut in TPH1−/− mice can affect the ability of DCs to prime CD4+ T cells. CD4+ T cells isolated from naive mice and cocultured with DCs isolated from colitic TPH1−/− mice released significantly lower amounts of IL-17 and IFN-γ compared with CD4+ T cells cocultured with DCs isolated from colitic TPH1+/+ mice when stimulated with LPS. Interestingly, in control conditions in the presence of medium only, we also observed a significant decrease in the amount of IL-17 and IFN-γ released in the culture supernatant of nonstimulated DCs (Figure 5). We observed this immunosuppressive function of DCs isolated from TPH1−/− mice on days 3, 5, and 7 of culture.
Figure 5.

Role of serotonin in function of DCs and sequential CD4+ T-cell activation. DCs from DSS-treated TPH1−/− and TPH1+/+ were cultured in the presence or absence of LPS for 24 hours before being cocultured with CD4+ T lymphocytes isolated from naive TPH1+/+ mice. Supernatant were collected at day 3 (A), day 5 (B), and day 7 (C). The levels of IL-17 and IFN-γ present in the culture supernatant were investigated by ELISA. *P < 0.05 compared with medium. **P < 0.05 compared with TPH1+/+. Data are representative of two independent experiments with quadruplicated cultures using seven mice, mean ± SEM.
Adoptive Transfer of Serotonin-Pulsed DCs Modulates DSS-Induced Colitis
We then investigated whether the transfer of serotonin-pulsed DCs from noncolitic TPH1−/− mice could influence the development of an acute colitis in TPH1−/− mice. As illustrated in Figure 6, the deterioration of colitis in mice receiving serotonin-pulsed DCs, compared with untreated DCs, was evident in the 1.9-fold increase in the macroscopic damage score (Figure 6A) and in the 2.9-fold increase in MPO activity (Figure 5B). The increased severity of DSS colitis in mice receiving serotonin-pulsed DCs was also evident histologically (Figure 6, C and D). The histological damage score increased from 1.5 ± 0.6 in mice receiving nonpulsed DCs to 3.3 ± 1.3 in mice transferred with serotonin-pulsed DCs. Similarly, cytokine concentrations in the colon were also increased in mice receiving serotonin-pulsed DCs. The fold increase in IL-1β was 3.3 times higher in mice receiving serotonin-pulsed DCs compared with untreated mice with DSS-induced colitis, and respective values for IL-6 and IL-17 were 1.7- and 1.3-fold increases. Levels of IL-1β, IL-6, and IL-17 in colonic tissue were significantly increased when mice were transferred with nonpulsed DCs (Figure 7, A–D). The IL-4 level was not significantly modified.
Figure 6.

Influence of adoptive transfer of DCs in the development of DSS-induced colitis in TPH1−/− mice Macroscopic score (A), MPO activity (B), and histological score (C–D) after 5 days of DSS-induced colitis. DCs (2.106 cells per mouse, i.p.) pulsed with serotonin (10−7M) for 24 hours increased all of the different markers. *P < 0.05 compared with control TPH1−/−. **P < 0.05 compared with naive DCs. Each value represents mean ± SEM from eight mice.
Figure 7.
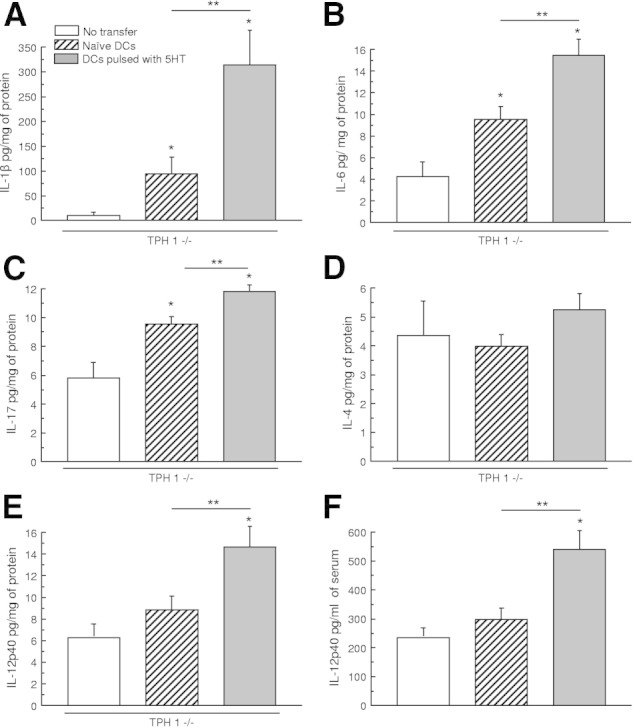
Influence of adoptive transfer of DCs in TPH1−/− mice cytokine profile after induction of colitis. IL-1β (A), IL-6 (B), IL-17 (C), IL-4 (D), and IL-12p40 (E) level in colonic tissue after 5 days of DSS-induced colitis. DCs (2.106 cells per mouse, i.p.) pulsed with serotonin (10−7M) for 24 hours increased all of the different markers but not IL-14 when transferred to a TPH1−/− mouse. F: IL-12p40 in serum was increased. *P < 0.05 compared with control TPH1−/−. **P < 0.05 compared with naive DCs. Each value represents mean ± SEM from eight mice.
Confirmation of the adoptive transfer was assessed by the level of IL-12p40 in colonic tissue and serum. The fold increase in IL-12 was significantly higher in 1.7 in colonic tissues and 1.8 in serum in mice receiving serotonin-pulsed DCs compared with untreated TPH1−/− DCs (Figure 7, E and F).
Discussion
Mucosal inflammation in conditions ranging from infective acute enteritis or colitis to IBD is accompanied by alteration in EC cell numbers and serotonin content in the gut. It is now well recognized that this altered serotonin response plays an important role in inflammation-induced gut physiological changes, such as motility and fluid and mucin secretion. There is also emerging evidence that the immune system regulates EC cell biology and serotonin production26 and serotonin plays an important role in the generation of intestinal inflammation and immune regulatory effects.3,8,27 Nevertheless, the precise mechanism by which serotonin regulates intestinal inflammation and immune cell function remains to be determined. DCs are key cells of an innate immune system and play a crucial role in the generation of adaptive immune response. Because of the strategic location in the intestinal mucosa, DCs may interact with EC cells and perform a key role in the activation of the immune response and the generation of gut inflammation. In this study, for the first time to our knowledge, we provide evidence of an essential role of serotonin in DC activation and generation of inflammation in the model of experimental colitis.
IBD, including mainly Crohn's disease and ulcerative colitis, is the most common and serious chronic inflammatory condition of the human bowel.28,29 Inflammation in the gut is characterized by mucosal recruitment of a variety of inflammatory cells, including T lymphocytes, macrophages, DCs, neutrophils, and plasma cells. The persistent release of inflammatory cytokines promotes adhesion, migration, and activation of immune and inflammatory cells and causes tissue damage. Given the importance of the complex network of cytokine profiles and its current applications as targeted biological agents, precise analysis on the source and mechanism of inflammatory cytokine production is critical in developing effective therapeutic strategy in IBD. DCs are powerful professional antigen-presenting cells and are the orchestrators of the immune response. DCs are not only responsible for linking innate and adaptive immune response, they also direct the type and quality of subsequent adaptive immune responses in the context of the situation. DCs have been implicated in Crohn's disease pathogenesis because defective migration and function of DCs and aberrant immunogenic response to pathogens have been suggested to contribute to the generation of intestinal inflammation.30 The importance of DCs in the pathogenesis of colitis has been highlighted by the findings that an increased number of DCs was found in the mesenteric lymph nodes in the T-cell transfer model of colitis31 and amelioration of DSS-induced colitis by depletion of DCs after the administration of diphtheria toxin in CD11c-DT receptor transgenic mice.32 In IBD, CD11c+ DCs are activated, their expression of microbial recognition receptors is up-regulated, and more DCs produce inflammatory cytokines, implying an important role of DCs in the pathogenesis of IBD.33 CD is a TH1/Th17 cell–driven intestinal inflammatory disease, and once initiated, the inflamed tissue is characterized by the expression of innate (IL-12p40, IL-6, TNF-α) and adoptive derived (IFN-γ and IL-17) proinflammatory cytokines.34 TH1/TH17 differentiation is controlled primarily by the IL-12 family of cytokines produced by the activated DCs.35 DSS-induced colitis is induced by the addition of DSS polymers to the drinking water and results in a reproducible acute colitis characterized by colonic mucosal inflammation with ulcerations, body weight loss, and bloody diarrhea. Both innate and adaptive (TH1, TH17) immunological mechanisms are shown to be important in the pathogenesis of colitis in this model.36,37 Recent studies from our laboratory demonstrated a key role of serotonin in the development of DSS-induced colitis in TPH1−/− mice, which have reduced levels of serotonin in the gut.8 This was associated with the down-regulation of the production of proinflammatory cytokines (IL-1β, IL-6, and TNF-α). We also observed that restoration of serotonin levels in TPH1−/− mice by the serotonin precursor 5-hydroxytryptophan increased the severity of DSS-induced colitis and the production of proinflammatory mediators. In this study, we have confirmed our previous findings of attenuated DSS-induced colitis in TPH1−/− mice compared with the TPH1+/+ mice. We also observed that this reduced severity of colitis in TPH1−/− mice is accompanied with down-regulation of IL-12p40, IL-17, and IFN-γ levels in colonic tissues. Given that there are other mediators in the in vivo setting that may also contribute to the immune activation and inflammatory process after DSS exposure, it was important to confirm that the reduced inflammation seen in TPH1−/− mice is not due to factors other than serotonin deficiency. We investigated the colonic levels of PGE2 and histamine as potential mediators in the process and observed absence of a significant difference in the levels of colonic PGE2 and histamine between TPH1−/− and TPH1+/+ mice in DSS-induced colitis, implying that serotonin is the critical factor for the attenuation of inflammation in TPH1−/− mice after exposure to DSS. Our data on the proinflammatory role of serotonin corroborate recent studies that demonstrated that colitis induced by 2,4,6-trinitrobenzene sulfonic acid or associated with IL-10 deficiency is increased in severity when coupled with the serotonin–enhancing effects of the knockout of serotonin reuptake transporter.27,38
The attenuated inflammation in DSS-induced colitis was accompanied by reduced levels of IL-12p40 production from DCs isolated from TPH1−/− mice compared with the DCs from TPH1+/+ mice. We also observed that DCs isolated from noncolitic TPH1−/− mice produced significantly less IL-12p40 in response to LPS compared with the TPH1+/+ mice. However, in vitro stimulation with serotonin restored IL-12p40 production from DCs isolated from naive TPH1−/− mice. Serotonin also stimulated IL-12p40 and IL-1β production from BMDCs, which further imply an important role of serotonin in immunomodulation. Serotoninergic receptors have been identified on DCs,18 and a recent study demonstrated that by binding to serotonin 3, serotonin 4, and serotonin 7 receptors, serotonin up-regulated production of proinflammatory cytokines and chemokines from DCs.39 It has also been suggested that by up-taking serotonin at sites of inflammation, DCs may shuttle serotonin to naive T cells and thereby modulate T-cell proliferation and differentiation.40 NF-κB–mediated overproduction of key proinflammatory mediators (IL-1β, IL-6, TNF-α, IL-12, and nitric oxide) is attributed to the initiation and progression of colonic inflammation in both human and animal models.41,42 Serotonin can influence regulation of gut inflammation by modulating NF-κB activation and production of proinflammatory cytokines from innate immune cells. In this study, we observed that serotonin-stimulated production of proinflammatory cytokines by DCs was significantly inhibited by the addition of NF-κB inhibitor. Taken together, these observations suggest that serotonin plays an important role in activation of DC function via the NF-κB signaling pathway and that the serotonin-mediated modulation of DC function is critical in the pathogenesis of colitis in this model.
Because TH1- and TH17-type immune responses have been characterized in DSS-induced colitis36 and DCs play a crucial role in priming the immune response to TH1 and TH17 cells, we next examined whether reduced levels of serotonin in TPH1−/− mice have any influence on T-cell priming and production of TH1 and TH17 cytokines. Our results demonstrated that DCs isolated from DSS-treated TPH1−/− mice had a significantly reduced ability to stimulate naive CD4+ T cells in vitro to produce IFN-γ and IL-17 compared with the DCs isolated from DSS-treated TPH1+/+ mice. In addition, in studies on the adoptive transfer of serotonin-pulsed DCs, we observed a significant increase in macroscopic and histological scores in TPH1−/− mice that received serotonin-pulsed DCs along with DSS administration compared with the TPH1−/− mice that received DCs without serotonin pulsing. This was associated with significant up-regulation of MPO activity and inflammatory cytokines (IL-1β, IL-6, IL-12p40, and IL-17). These observations further provide evidence in favor of a crucial role of serotonin in DC function in relation to generation of gut inflammation.
Taken together, these studies show an important role of serotonin in the pathogenesis of inflammation in the gut by influencing proinflammatory cytokine production from DCs via activation of the NF-κB pathway and sequential T-cell instigation. These observations provide new insight on the mechanism of gut inflammation. In addition to enhancing our understanding of the pathogenesis of colitis, this study provides novel information on serotonin in the context of immune cell signaling. Serotonin deficiency is not only shown to be important in modulating intestinal inflammation, recent studies have shown that serotonin deficiency causes slower growth of colon cancer allografts in vivo43 and reduces development of osteoporosis by promoting bone formation.44 Thus, the data obtained from the present study are important not only in understanding the mechanism of GI inflammation but also in gaining insight into the pathogenesis of non-GI disorders where there is alteration in serotonin signaling. In a wider perspective, these data may have an implication in understanding the role of this gut hormone in the pathogenesis of both GI and non-GI inflammation, which may ultimately lead to improved therapeutic strategies in inflammatory disorders.
Acknowledgments
We thank Yonghong Wan (McMaster University) and Yasuaki Motomura (Kyushu University) for their valuable input and support.
Footnotes
Supported by grants from the Crohn's and Colitis Foundation of Canada and by the Canadian Institutes of Health Research to W.I.K.
N.L. and J.-E.G. contributed equally to this work.
References
- 1.Gershon M.D. Serotonin and its implication for the management of irritable bowel syndrome. Rev Gastroenterol Disord. 2003;3(Suppl 2):S25–S34. [PubMed] [Google Scholar]
- 2.Ham T.S. Regional distribution and relative frequency of gastrointestinal endocrine cells in large intestines of C57BL/6 mice. J Vet Sci. 2002;3:233–238. [PubMed] [Google Scholar]
- 3.Khan W.I., Ghia J.E. Gut hormones: emerging role in immune activation and inflammation. Clin Exp Immunol. 2010;161:19–27. doi: 10.1111/j.1365-2249.2010.04150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim D.Y., Camilleri M. Serotonin: a mediator of the brain-gut connection. Am J Gastroenterol. 2000;95:2698–2709. doi: 10.1111/j.1572-0241.2000.03177.x. [DOI] [PubMed] [Google Scholar]
- 5.Ahonen A., Kyosola K., Penttila O. Enterochromaffin cells in macrophages in ulcerative colitis and irritable colon. Ann Clin Res. 1976;8:1–7. [PubMed] [Google Scholar]
- 6.Belai A., Boulos P.B., Robson T., Burnstock G. Neurochemical coding in the small intestine of patients with Crohn's disease. Gut. 1997;40:767–774. doi: 10.1136/gut.40.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coates M.D., Mahoney C.R., Linden D.R., Sampson J.E., Chen J., Blaszyk H., Crowell M.D., Sharkey K.A., Gershon M.D., Mawe G.M., Moses P.L. Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and irritable bowel syndrome. Gastroenterology. 2004;126:1657–1664. doi: 10.1053/j.gastro.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 8.Ghia J.E., Li N., Wang H., Collins M., Deng Y., El-Sharkawy R.T., Cote F., Mallet J., Khan W.I. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology. 2009;137:1649–1660. doi: 10.1053/j.gastro.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 9.Linden D.R., Chen J.X., Gershon M.D., Sharkey K.A., Mawe G.M. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2003;285:G207–G216. doi: 10.1152/ajpgi.00488.2002. [DOI] [PubMed] [Google Scholar]
- 10.Grondahl M.L., Jensen G.M., Nielsen C.G., Skadhauge E., Olsen J.E., Hansen M.B. Secretory pathways in Salmonella typhimurium–induced fluid accumulation in the porcine small intestine. J Med Microbiol. 1998;47:151–157. doi: 10.1099/00222615-47-2-151. [DOI] [PubMed] [Google Scholar]
- 11.Kordasti S., Sjovall H., Lundgren O., Svensson L. Serotonin and vasoactive intestinal peptide antagonists attenuate rotavirus diarrhoea. Gut. 2004;53:952–957. doi: 10.1136/gut.2003.033563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Hara J.R., Skinn A.C., MacNaughton W.K., Sherman P.M., Sharkey K.A. Consequences of Citrobacter rodentium infection on enteroendocrine cells and the enteric nervous system in the mouse colon. Cell Microbiol. 2006;8:646–660. doi: 10.1111/j.1462-5822.2005.00657.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang H., Steeds J., Motomura Y., Deng Y., Verma-Gandhu M., El-Sharkawy R.T., McLaughlin J.T., Grencis R.K., Khan W.I. CD4+ T cell-mediated immunological control of enterochromaffin cell hyperplasia and 5-hydroxytryptamine production in enteric infection. Gut. 2007;56:949–957. doi: 10.1136/gut.2006.103226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camilleri M., Northcutt A.R., Kong S., Dukes G.E., McSorley D., Mangel A.W. Efficacy and safety of alosetron in women with irritable bowel syndrome: a randomised, placebo-controlled trial. Lancet. 2000;355:1035–1040. doi: 10.1016/S0140-6736(00)02033-X. [DOI] [PubMed] [Google Scholar]
- 15.Fitzpatrick P.F. Tetrahydropterin-dependent amino acid hydroxylases. Annu Rev Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 16.Walther D.J., Bader M. A unique central tryptophan hydroxylase isoform. Biochem Pharmacol. 2003;66:1673–1680. doi: 10.1016/s0006-2952(03)00556-2. [DOI] [PubMed] [Google Scholar]
- 17.Walther D.J., Peter J.U., Bashammakh S., Hortnagl H., Voits M., Fink H., Bader M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299:76. doi: 10.1126/science.1078197. [DOI] [PubMed] [Google Scholar]
- 18.Idzko M., Panther E., Stratz C., Muller T., Bayer H., Zissel G., Durk T., Sorichter S., Di Virgilio F., Geissler M., Fiebich B., Herouy Y., Elsner P., Norgauer J., Ferrari D. The serotoninergic receptors of human dendritic cells: identification and coupling to cytokine release. J Immunol. 2004;172:6011–6019. doi: 10.4049/jimmunol.172.10.6011. [DOI] [PubMed] [Google Scholar]
- 19.Côt́ F., Thevenot E., Fligny C., Fromes Y., Darmon M., Ripoche M.A., Bayard E., Hanoun N., Saurini F., Lechat P., Dandolo L., Hamon M., Mallet J., Vodjdani G. Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc Natl Acad Sci U S A. 2003;100:13525–13530. doi: 10.1073/pnas.2233056100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghia J.E., Blennerhassett P., Collins S.M. Impaired parasympathetic function increases susceptibility to inflammatory bowel disease in a mouse model of depression. J Clin Invest. 2008;118:2209–2218. doi: 10.1172/JCI32849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper H.S., Murthy S.N., Shah R.S., Sedergran D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 22.Boughton-Smith N.K., Wallace J.L., Whittle B.J. Relationship between arachidonic acid metabolism, myeloperoxidase activity and leukocyte infiltration in a rat model of inflammatory bowel disease. Agents Actions. 1988;25:115–123. doi: 10.1007/BF01969102. [DOI] [PubMed] [Google Scholar]
- 23.Bradley P.P., Priebat D.A., Christensen R.D., Rothstein G. Measurement of cutaneous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 24.Lutz M.B., Kukutsch N., Ogilvie A.L., Rossner S., Koch F., Romani N., Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 25.Murugaiyan G., Mittal A., Weiner H.L. Identification of an IL-27/osteopontin axis in dendritic cells and its modulation by IFN-gamma limits IL-17-mediated autoimmune inflammation. Proc Natl Acad Sci U S A. 2010;107:11495–11500. doi: 10.1073/pnas.1002099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Motomura Y., Ghia J.E., Wang H., Akiho H., El-Sharkawy R.T., Collins M., Wan Y., McLaughlin J.T., Khan W.I. Enterochromaffin cell and 5-hydroxytryptamine responses to the same infectious agent differ in Th1 and Th2 dominant environments. Gut. 2008;57:475–481. doi: 10.1136/gut.2007.129296. [DOI] [PubMed] [Google Scholar]
- 27.Bischoff S.C., Mailer R., Pabst O., Weier G., Sedlik W., Li Z., Chen J.J., Murphy D.L., Gershon M.D. Role of serotonin in intestinal inflammation: knockout of serotonin reuptake transporter exacerbates 2,4,6-trinitrobenzene sulfonic acid colitis in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G685–G695. doi: 10.1152/ajpgi.90685.2008. [DOI] [PubMed] [Google Scholar]
- 28.Podolsky D.K. Inflammatory bowel disease (2) N Engl J Med. 1991;325:1008–1016. doi: 10.1056/NEJM199110033251406. [DOI] [PubMed] [Google Scholar]
- 29.Qiu B.S., Vallance B.A., Blennerhassett P.A., Collins S.M. The role of CD4+ lymphocytes in the susceptibility of mice to stress-induced reactivation of experimental colitis. Nat Med. 1999;5:1178–1182. doi: 10.1038/13503. [DOI] [PubMed] [Google Scholar]
- 30.Kelsall B.L. A focus on dendritic cells and macrophages as key regulators of mucosal immunity. Mucosal Immunol. 2008;1:423–424. doi: 10.1038/mi.2008.66. [DOI] [PubMed] [Google Scholar]
- 31.Malmstrom V., Shipton D., Singh B., Al-Shamkhani A., Puklavec M.J., Barclay A.N., Powrie F. CD134L expression on dendritic cells in the mesenteric lymph nodes drives colitis in T cell-restored SCID mice. J Immunol. 2001;166:6972–6981. doi: 10.4049/jimmunol.166.11.6972. [DOI] [PubMed] [Google Scholar]
- 32.Berndt B.E., Zhang M., Chen G.H., Huffnagle G.B., Kao J.Y. The role of dendritic cells in the development of acute dextran sulfate sodium colitis. J Immunol. 2007;179:6255–6262. doi: 10.4049/jimmunol.179.9.6255. [DOI] [PubMed] [Google Scholar]
- 33.Hart A.L., AL-Hassi H.O., Rigby R.J., Bell S.J., Emmanuel A.V., Knight S.C., Kamm M.A., Stagg A.J. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology. 2005;129:50–65. doi: 10.1053/j.gastro.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez-Munoz F., Dominguez-Lopez A., Yamamoto-Furusho J.K. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–4288. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoeve M.A., Savage N.D., de Boer T., Langenberg D.M., de Waal Malefyt R., Ottenhoff T.H., Verreck F.A. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol. 2006;36:661–670. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 36.Alex P., Zachos N.C., Nguyen T., Gonzales L., Chen T.E., Conklin L.S., Centola M., Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dieleman L.A., Ridwan B.U., Tennyson G.S., Beagley K.W., Bucy R.P., Elson C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 38.Haub S., Ritze Y., Bergheim I., Pabst O., Gershon M.D., Bischoff S.C. Enhancement of intestinal inflammation in mice lacking interleukin 10 by deletion of the serotonin reuptake transporter. Neurogastroenterol Motil. 2010;22:826–834. doi: 10.1111/j.1365-2982.2010.01479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller T., Durk T., Blumenthal B., Grimm M., Cicko S., Panther E., Sorichter S., Herouy Y., Di Virgilio F., Ferrari D., Norgauer J., Idzko M. 5-hydroxytryptamine modulates migration, cytokine and chemokine release and T-cell priming capacity of dendritic cells in vitro and in vivo. PLoS One. 2009;4:e6453. doi: 10.1371/journal.pone.0006453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Connell P.J., Wang X., Leon-Ponte M., Griffiths C., Pingle S.C., Ahern G.P. A novel form of immune signaling revealed by transmission of the inflammatory mediator serotonin between dendritic cells and T cells. Blood. 2006;107:1010–1017. doi: 10.1182/blood-2005-07-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacDermott R.P. Alterations in the mucosal immune system in ulcerative colitis and Crohn's disease. Med Clin North Am. 1994;78:1207–1231. doi: 10.1016/s0025-7125(16)30096-7. [DOI] [PubMed] [Google Scholar]
- 42.Neurath M.F., Becker C., Barbulescu K. Role of NF-kappaB in immune and inflammatory responses in the gut. Gut. 1998;43:856–860. doi: 10.1136/gut.43.6.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nocito A., Dahm F., Jochum W., Jang J.H., Georgiev P., Bader M., Graf R., Clavien P.A. Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 2008;68:5152–5158. doi: 10.1158/0008-5472.CAN-08-0202. [DOI] [PubMed] [Google Scholar]
- 44.Yadav V.K., Balaji S., Suresh P.S., Liu X.S., Lu X., Li Z., Guo X.E., Mann J.J., Balapure A.K., Gershon M.D., Medhamurthy R., Vidal M., Karsenty G., Ducy P. Pharmacological inhibition of gut-derived serotonin synthesis is a potential bone anabolic treatment for osteoporosis. Nat Med. 2010;16:308–312. doi: 10.1038/nm.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]