

Abstract
Adhesion of circulating prostate cancer (PCa) cells to the microvascular endothelium is a critical step during cancer metastasis. To study PCa cell rolling and adhesion behavior, we developed a dynamic flow-based microtube system to mimic the microvascular environment. We found that PCa cell rolling capacity is mediated by E-selectin and can be enhanced by stromal cell-derived factor-1 under different wall shear stresses. Using this device, we tested if the chemopreventive agent, vitamin D, could interfere with PCa cell adhesion. We found that 1α,25-dihydroxyvitamin D3 (1,25-VD), the bioactive form of vitamin D, reduced PCa cell rolling numbers and increased rolling velocities resulting in a significant decreased number of PCa cells adhering to the microtube. The inhibitory effects of 1,25-VD on PCa cell heterotypic adhesion were further confirmed using microvascular endothelial cells in a static condition. Furthermore, we demonstrated that 1,25-VD can increase E-cadherin expression in PCa cells and promote the homotypic cell-cell aggregation, which can then hinder PCa cell adhesion to the endothelium. Blocking E-cadherin with a neutralizing antibody can reverse 1,25-VD-mediated suppression of PCa cell adhesion to the endothelium. Taken together, our data revealed that 1,25-VD promoted PCa cell aggregation by increasing E-cadherin expression, thus interfering with circulating PCa cell adhesion to microvascular endothelial cells and potentially reducing their metastatic potential.
Distal metastasis is the leading cause of cancer-associated mortality. Most cancers progress over time departing from primary carcinomas and metastasize to distal targeted organs. The progression from a locally growing tumor to lethal metastasis involves numerous cellular alterations that allow tumor cells to complete the complex series of events needed for metastatic spread. Following intravasation of PCa into the blood vessels, two major events — anoikis and cell adhesion to vessel endothelium — mediate hematogenous dissemination with blood circulation before cancer cells arrive at the distant organs. Anoikis is a programed cell death induced by detachment of anchorage-dependent cells from the surrounding extracellular matrix.1 Although most circulating tumor cells undergo anoikis following detachment to free flow, it has been shown that a subpopulation of malignant cells can develop an anoikis-resistant capability and survive circulatory travel.2 For circulating tumor cells that survive, heterotypic adhesion and firm arrest to the vessel endothelium within the microenvironment of a target organ is a required step for the progression to distal metastasis.3
Circulating PCa cells preferentially adhere to the vessel endothelia that express E-selectin4 via interactions between endothelial E-selectin and E-selectin ligands (such as PSGL-1, ESL-1, and CD44)5,6 or other adhesive molecules (such as α2 and β1 integrins)7 on the cancer cells. Weak cell-cell adhesion events initiate the tethering and rolling of cancer cells on the endothelium. This cell-cell adhesion is enhanced when chemokines, such as stromal cell-derived factor-1 (SDF-1), expressed by endothelial or stromal cells interact with their receptors (such as CXCR4) on the cancer cell surface.8 Following chemokine signaling, integrins play a further role in the development of stable attachments.9 The series of events of heterotypic cell-cell interactions that result in cancer cells arrest on the endothelium are requisite to the later stages of metastasis, including extravasation, migration, invasion, and proliferation of the metastatic tumor in the distant organs.
The role of vitamin D as an anti-prostate tumor agent was first proposed in 1990 by Schwartz et al,10 who described that the major PCa risk factors (such as age, race, and resident latitude) are correlated with the decreased vitamin D levels. Later studies further proved that vitamin D functions through multiple routes to inhibit tumor progression, including suppression of proliferation,11 migration,12 invasion,13,14 angiogenesis,15 and metastasis in vivo.16,17 Most of the studies of vitamin D effects on PCa are limited to the cell culture setting, hence the effects of vitamin D on PCa cells within the circulation system, where hematogenous dissemination of tumor metastasis occurs, are still largely unknown.
Cadherins are a family of cell surface glycoproteins that may play a role during cellular migration through their mediation of calcium-dependent homotypic cell adhesion.18 Specifically, E-cadherin, a member of the cadherin family, is expressed on the epithelial cells and forms the cell-cell tight junction complex.19 Many studies have demonstrated that E-cadherin function is suppressed during the development of different cancers, including breast, colon, prostate, stomach, liver, esophagus, skin, kidney, and lung.20 Decreased E-cadherin expression causes a decrease of homotypic cell adhesion and the increases of cell migration and invasion,21 suggesting a potential role of E-cadherin as a tumor suppressor that is defined by its cellular functions.20
To study the influence of 1,25-VD on PCa cell behavior within the circulatory system, we examined the effects of 1,25-VD on heterotypic PCa-endothelial cell adhesion, in both static and dynamic settings. We also examined the effects of 1,25-VD-induced homotypic PCa cell adhesion (cell aggregation) and subsequent effects of adhesion to the endothelium. This current study provides evidence of the functional mechanism by which 1,25-VD influences the adhesive behavior of PCa cells within the circulation system.
Materials and Methods
1,25-VD, Cell Lines, and Culture Conditions
1,25-VD was purchased from Sigma-Aldrich (St Louis, MO). Human dermal microvascular endothelial cell line (HMEC-1) was a generous gift from Dr. Jane Liesveld at the University of Rochester (Rochester, NY). Human PCa cells were obtained from the American Type Culture Collection. DU145 and PC-3 E-cadherin positive (+) and negative (−) PCa cell lines were a generous gift from Dr. Beatrice Knudsen at Fred Hutchinson Cancer Research Center (Seattle, WA). For HMEC-1 culture, the culture medium is MCDB 131 (Gibco, Carlsbad, CA), containing l-glutamine (10 mmol/L; Gibco), human epidermal growth factor (10 ng/ml; BD Biosciences, San Jose, CA), hydrocortisone (1 μg/ml; Sigma), and 10% heat-inactivated fetal bovine serum. For all PCa cell lines RPMI-1640 medium with 10% heat-inactivated fetal bovine serum and l-glutamine (10 mmol/L; Gibco) was used.
Cell Adhesion Assay
HMEC-1 cells were grown as a monolayer in a 6-well plate. PCa cells were treated with ethanol (vehicle control) or 1,25-VD for 72 hours, labeled with green fluorescence by using calcein-AM solution (1 μmol/L; Sigma-Aldrich), following the manufacturer's instructions, detached by 5 mmol/L EDTA, washed 3 times with Hanks' balanced salt solution (HBSS) buffer (Gibco) with calcium, and then incubated for 1 hour. 106 PCa cells in 3 ml RPMI-1640 medium were directly seeded onto the monolayer of HMEC-1 cells. After 1-hour incubation, unbound PCa cells were gently washed away by PBS buffer and the attached PCa cell numbers were recorded by the fluorescent microscope (Nikon Eclipse E800; Nikon Instruments, Melville, NY). The cell numbers in each image were counted using ImageJ software (http://rsbweb.nih.gov/ij/; National Institutes of Health, Bethesda, MD). To generate clustered cells, 106 DU145 cells were centrifuged at 2300 × g for 5 minutes and then gently passed five times through a 1 ml standard pipette (Gilson, Middleton, WI) to form clustered cells. A mouse monoclonal E-cadherin blocking antibody (HECD-1, 2 μg/ml; Calbiochem, San Diego, CA) was applied in the assay to block E-cadherin mediated homotypic cell adhesive function.
Cell Rolling Assay
The cell capture microtube preparation (inside diameter = 300 μm, length 20 cm, MicroRenathane MRE-025; Braintree Scientific Inc., Braintree, MA) was previously described.22 The lumens of special polyurethane microtubes were coated with human recombinant IgG, E-selectin (chimera dual form, R&D Systems, Minneapolis, MN), and/or SDF-1 (R&D Systems) and incubated for 2 hours at room temperature. After PBS washing, the E-selectin or E-selectin/SDF-1 layer was maintained in calcium-enriched Hanks' balanced salt solution [HBSS (2 mmol/L Ca++), Gibco] until the rolling assay. The data readout procedure was modified and performed as previously described.15 A microscope-linked Hitachi CCD camera KP-M1AN (Hitachi, Tokyo, Japan) was used to monitor PCa cell rolling. Rolling of PCa cells on coated microtubes under different wall shear stress (WSS; 0.5, 1.0, 3.0, 5.0 dyne/cm2) was recorded by high-quality DVD for later analysis. Average rolling cell numbers were obtained using ImageJ software to retrieve 20 continuous frames. Rolling flux measurements and rolling velocities of PCa cells were acquired using ImageJ software. We defined rolling cells as cells that translated at an average velocity <50% of the calculated free stream velocity for more than 2 seconds while remaining in the field of view [FOV: 432 μm × 324 μm, using a 20x Plan Fluorite objective, NA 0.40 (Olympus America Inc., Center Valley, PA)]. Rolling cells that interacted with other cells were not included. The rolling velocity data were assayed under different WSS (0.5, 1.0, 3.0 dyne/cm2).
To determine docking cell number, the microtube was gently flushed by PBS buffer under 1 dyne/cm2 to remove unbound PCa cells. The remaining adherent PCa cells in the microtubes were considered “docked” cells. The average docking cell number was taken from an average of 20 continuous fields.
Western Blot Assay and Real-Time PCR Assay
The procedures were performed as previously described.14,23 Primer sequences are as follows: PSGL-1, sense-5′-CGGGGTACCGTACCATGTCCCCAAGCTTC-3′, antisense-5′-GCTCTAGAGTGGAGCTAGCAAAGGTCTC-3′; ESL-1, sense-5′-CAAGATGACGGCCATCATTTTCA-3′, antisense-5′-TTCCCCAAGACGAATGCTGC-3′; CD44, sense-5′-TTTGCATTGCAGTCAACAGTC-3′, antisense-5′-GTTACACCCCAATCTTCATGTCCAC-3′; integrin α2, sense-5′-GCAACTGGTTACTGGTTGGTT-3′, antisense-5′-GAGGCTCATGTTGGTTTTCATCT-3′; integrin β1, sense-5′-TTATTGGCCTTGCATTACTGCT-3′, antisense-5′-CCACAGTTGTTACGGCACTCT-3′; CXCR4, sense-5′-GGCAGCAGGTAGCAAAGTGA-3′, antisense-5′-TGATGACAAAGAGGAGGTCGG-3′; E-cadherin, sense-5′- CAGAAAGTTTTCCACCAAAG-3′, antisense-5′-AAATGTGAGCAATTCTGCTT-3′; β-catenin, sense-5′-AGGGATTTTCTCAGTCCTTC-3′, antisense-5′-CATGCCCTCATCTAATGTCT-3′.
Flow Cytometry Assay
After vehicle or 1,25-VD treatments, PCa cells were detached and washed twice in fluorescence activated cell sorter (FACS) staining washing buffer (1% heat-inactivated bovine serum and 0.1% sodium azide in PBS buffer). Then, 5 × 105 cells were incubated on ice for 30 minutes with saturating amounts of antibody against anti-CD44 antibody (eBioscience, San Diego, CA). Flow cytometric analyses were performed by using a dual-laser FACSCalibur flow cytometer (BD Biosciences).
Cell Aggregation Assay
The procedure was performed as previously described.24 Briefly, cells were detached, washed twice with PBS buffer, and suspended at 5 × 105 cells per ml in RPMI medium. Then, 1.5 × 104 cells were suspended as a hanging drop from the lid of the 24-well culture plate. After an 18-hour incubation period, the cells were subjected to the shear stress by passing them ten times through a standard 20-μl Gilson pipette tip. Cells were photographed under microscope to record aggregation behaviors within 30 minutes. The level of cell aggregation was determined by the cell aggregation index, 1-Na/N0, where Na and N0 are the number of particles after and before performing cell aggregation. Higher aggregation index value represents more cell aggregation.25
Statistical Analysis
The results were presented as mean ± SEM of values obtained from at least three repeated experiments. Data were assessed for statistical significance by using two-tailed Student's t-test comparisons (Microsoft Office Excel 2007 software; Microsoft Corporation, Redmond, WA). Statistically significant differences are presented as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Flow-Based Microtube System for the Capture and Characterization of Circulating PCa Cells
To study adhesive behavior of circulating PCa cells, we constructed a dynamic flow-based microtube system to mimic the bone microvascular environment. SDF-1 is a chemokine that is expressed in human endothelium and transduces signals via interaction with receptor CXCR4, abundantly expressed on the PCa cells.26 To mimic the PCa rolling behavior in a dynamic status, PCa cells were perfused at physiological WSS through microtube lumens coated with IgG control, E-selectin, SDF-1, or E-selectin and SDF-1; and their rolling behaviors were examined. As shown in Figure 1A, DU145 PCa cells exhibited adhesive rolling behavior on E-selectin-coated and E-selectin/SDF-1-coated microtubes, but not on IgG-coated microtubes or SDF-1-coated microtubes (data not shown). Though SDF-1 by itself was not sufficient to induce PCa rolling, significantly more PCa cells were observed to roll on E-selectin/SDF-1-coated microtubes than on E-selectin-coated microtubes (Figure 1A). These data suggest that E-selectin is required for PCa cell interaction with endothelium, an important step for PCa metastasis. SDF-1 by itself is not sufficient to induce PCa rolling; rather a cooperative effect with E-selectin is needed to enhance cell rolling. Moreover, we tested PCa rolling behavior under different WSS, from 1 to 5 dyne/cm2, the comparable WSS range found in microvesicles,27 E-selectin/SDF-1-coated tubes. As expected, with increasing WSS, the PCa cell rolling number was decreased (Figure 1B). The results demonstrate establishment of a controllable flow-based system that mimics the microvascular environment and allows us to study the rolling behavior of circulating PCa cells with the option of varying specific parameters for our experimental strategy.
Figure 1.
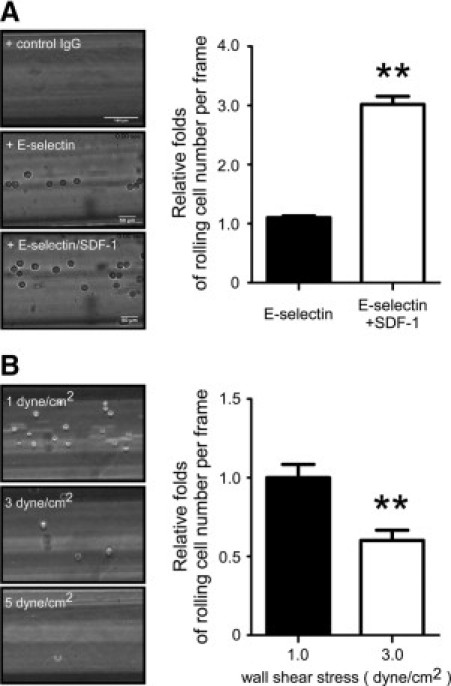
Establishment of a dynamic microvessel system mimicking the bone microvascular environment. A: PCa cell rolling behavior is mediated by E-selectin and SDF-1. DU145 PCa cells were infused into the microtubes coated with human control IgG (40 μg/ml), E-selectin (40 μg/ml), or E-selection (40 μg/ml) plus SDF-1 (10 μg/ml) under wall shear stress at 1 dyne/cm2. PCa cell rolling behaviors were recorded and rolling cell numbers were calculated. Results are the mean ± SEM of three experiments; **P < 0.01. B: PCa cell rolling capacity under different wall shear stress. DU145 cells rolling on the E-selectin/SDF-1 coated microtubes under different wall shear stress, at 1, 3, and 5 dyne/cm2. PCa cell rolling behaviors were recorded and rolling numbers were calculated. The result was presented as the relative fold of rolling cell number per frame. Results are the mean ± SEM of three experiments; **P < 0.01.
1,25-VD Suppresses PCa Rolling and Docking Capacity in the E-Selectin/SDF-1-Coated Microtube Under Flow
Numerous preclinical and epidemiological data support that vitamin D signals play important roles in the development, progression, and therapy for PCa. We sought to determine whether vitamin D could influence the circulating PCa cell adhesion to endothelium, and thus potentially mediate hematogenous dissemination during metastasis. Two PCa cell lines that express vitamin D receptor (VDR)28,29 (DU145 and PC-3 cells) were treated with 1,25-VD or vehicle control and then were injected into the E-selectin/SDF-1-coated microtubes under the flow condition. The rolling behaviors of cells were recorded and analyzed. Treatment with 1,25-VD suppressed PCa cell rolling as a function of total number of cells captured by microtubes, when compared to vehicle-treated cells as a control, under WSS ranging from 0.5 to 3 dyne/cm2 (Figure 2A). Rolling cell results as a function of WSS were consistent with our previous data demonstrating that rolling numbers are inversely correlated with WSS for both control and 1,25-VD-treated DU145 cells. Another key parameter to quantify cell adhesion is cell rolling velocity. We found that 1,25-VD increased cell rolling velocities of DU145 cells under different WSS (Figure 2B). We noted that under flow, most of the rolling PC-3 cells form static adhesion to the E-selectin/SDF-1-coated microtubes, and the PC-3 rolling velocity cannot be determined under this experimental condition. This increased rolling velocity, following 1,25-VD treatment could also contribute to a decrease in the PCa cell ability to adhere to endothelium.
Figure 2.
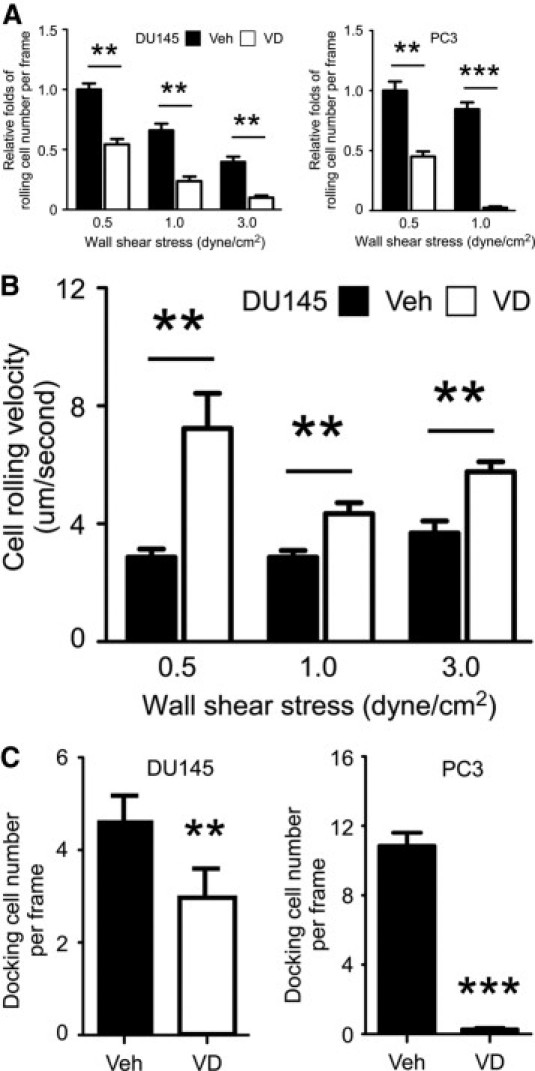
The effects of 1,25-VD on the PCa cell rolling behaviors. A: 1,25-VD reduced PCa cell rolling number on the E-selectin/SDF-1-coated surface under different wall shear stress. DU145 or PC-3 cells were treated with vehicle (Veh) or 100 nmol/L 1,25-VD for 72 hours, then were perfused into microtubes. Rolling cell number was recorded under 0.5, 1, and 3 dyne/cm2 wall shear stress. B: 1,25-VD elevates DU145 PCa cell rolling velocity under different wall shear stress. The rolling velocity was determined by the rolling distance per second and calculated by the formula as described in Materials and Methods. C: 1,25-VD reduces docking PCa cells on the E-selectin/SDF-1 coated microtubes. After rolling, we injected PBS buffer with calcium to flush away the unbound PCa cells. The adhered PCa cell numbers in the microtubes representing the docking cell number were calculated. The result was presented as the docking cell number per frame. Results are the mean ± SEM of three experiments; **P < 0.01, ***P < 0.001, vehicle vs. 1,25-VD treatment.
After rolling, only the cells that form a firm adhesion and dock to endothelium have the opportunity to continue further metastatic events. We, therefore, observed effects of 1,25-VD on PCa cells docking within E-Selectin/SDF-1-coated microtubes. As expected, 1,25-VD-treated PCa cells exhibited statistically less cellular docking than vehicle control (Figure 2C). Taken together, we demonstrated for the first time that 1,25-VD treatment suppressed PCa cell rolling capacity by decreasing rolling numbers and increasing cell rolling velocities, thereby reducing the number of PCa cells docking on the E-selectin/SDF-1-coated microtubes under flow conditions. This result provides a novel functional mechanism by which 1,25-VD exerts its anti-tumor effects by interfering with circulating PCa cells adhering to endothelium and, thus, decreases the further invasion and metastases in distal organs.
1,25-VD Suppresses PCa Cell Adhesion to Human Microvascular Endothelial Layer
To further confirm 1,25-VD effects on PCa heterotypic adhesion, a static cell-cell adhesion assay was applied by using human microvascular endothelial cells (HMEC-1). The expression of E-selectin and SDF-1 in HMEC-1 cells were first examined by immunofluorescent staining. HMEC-1 cells do express E-selectin, as well as SDF-1, but to a lesser extent (data not shown). 1,25-VD effects on PCa cell attachment to HMEC cells were then examined, and we found that treatment of PCa cells with 1,25-VD reduced cell adhesion to the HMEC-1 cell monolayer when compared with vehicle-treated cells in a static condition (Figure 3). In conjunction with the rolling data, we conclude that 1,25-VD can suppress PCa cell adhesion to endothelium in both dynamic flow and static conditions.
Figure 3.
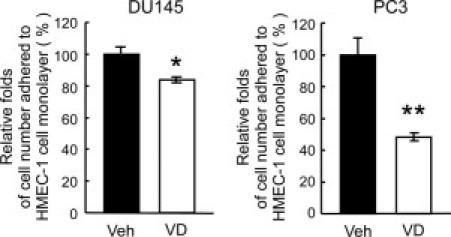
The effect of 1,25-VD on PCa cell adhesion to endothelial cells. 1,25-VD suppressed PCa cell adhesion to endothelial cells. A monolayer of HMEC-1 was cultured in the 6-well plates. DU145 and PC-3 cells were treated with ethanol or 100 nmol/L 1,25VD for 72 hours, then labeled with calcium-AM, and seeded into the culture plates. The unbound PCa cells were gently washed away. The adhered PCa cell numbers representing the cell-cell adhesion intensity were calculated. Results are the mean ± SEM of three experiments; *P < 0.05, **P < 0.01, vehicle vs. 1,25-VD treatment.
1,25-VD Has No Effect on E-Selectin and SDF-1 but Induces E-Cadherin Expression on PCa Cells
To study the mechanism by which 1,25-VD regulates PCa cell heterotypic adhesion, we first examined the effects of 1,25-VD on E-selectin and SDF-1 adhesive ligands expressions in the PCa cells using quantitative PCR assay. As shown in Figure 4A, there was no significant alteration of mRNA expression for E-selectin or SDF-1 ligand following 1,25-VD treatments. The expression of CD44, a well-known regulator of PCa cell and immune cell adhesion to vessel endothelium,6,30 was examined by flow cytometry. As shown in Figure 4B, 1,25-VD had no effect on surface CD44 expression in DU145 cells. These results suggest that demonstrated effects on heterotypic adhesion by 1,25-VD are not mediated via the regulation of E-selectin- or SDF-1-associated adhesion molecule expression.
Figure 4.
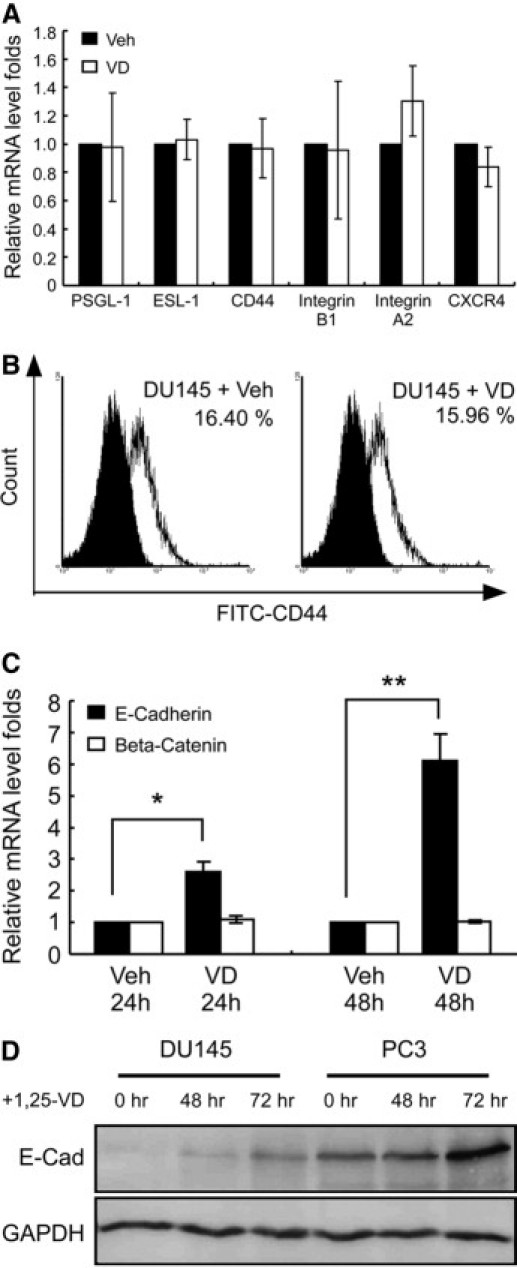
1,25-VD up-regulates E-cadherin. A: 1,25-VD had no effect on the E-selectin ligands or SDF-1 receptor CXCR4 mRNA expression level in DU145 cells. DU145 cells were treated with either vehicle (Veh) or 100 nmol/L 1,25-VD for 24 hours, then the mRNA levels were examined by real-time PCR assay. Results are the mean ± SEM of three experiments. B: 1,25-VD had no effect on CD44 expression level on the surface. DU145 cells were treated with either vehicle or 100 nmol/L 1,25-VD for 48 hours, then CD44 expression was examined by flow cytometry. The black area represents the CD44-negative cells and the white area represents the CD44-positive cells. C, D: 1,25-VD increased E-cadherin expression. DU145 or PC-3 cells were treated with either vehicle or 100 nmol/L 1,25-VD for 24, 48, and 72 hours, and then cells were harvested to determine E-cadherin and β-catenin mRNA levels by quantitative PCR (C) and E-cadherin protein levels by western blotting (D). Results are the mean ± SEM of three experiments; *P < 0.05, **P < 0.01, vehicle vs. 1,25-VD treatment.
E-cadherin has been shown to be regulated by vitamin D. Decreased E-cadherin expression causes the loss of homotypic cell adhesion and, subsequently, an increase in potential for heterotypic cell adhesion events, including migration and invasion.21 Consistent with a previous report,31 we found that 1,25-VD promoted E-cadherin expression in PCa cells at both mRNA and protein levels (Figure 4, C and D). This induction of E-cadherin expression might account for the observed 1,25-VD-induced effects on PCa adhesion, including homotypic aggregation.
1,25-VD Promotes PCa Cell Aggregation through E-Cadherin
E-cadherin is known to promote formation of homotypic cell-cell tight junctions. To test if increasing E-cadherin expression via 1,25-VD treatment can alter PCa homotypic behaviors and cell-cell aggregation, and influence PCa heterotypic adhesion, we tested E-cadherin effects on PCa cell aggregation using E-cadherin negative (−) and positive (+) clones of DU145 and PC-3. We showed that both DU145 and PC3 E-cadherin (+) cells exhibited more cell clusters than E-cadherin (−) cells in suspension status, suggesting that E-cadherin can promote PCa cell aggregation (Figure 5A). Next we tested whether the elevated E-cadherin expression induced by 1,25-VD could promote PCa cell aggregation and influence subsequent PCa cell adhesive behaviors. As shown in Figure 5B, we found that 1,25-VD promoted both DU145 and PC-3 cell aggregation in which more and larger cell clusters were found in 1,25-VD-treated cells. More importantly, this 1,25-VD-induced cell aggregation was blocked by applying E-cadherin blocking antibody (2 μg/ml HECD-1). These data suggests that 1,25-VD promoted PCa cell aggregation is mediated through E-cadherin.
Figure 5.
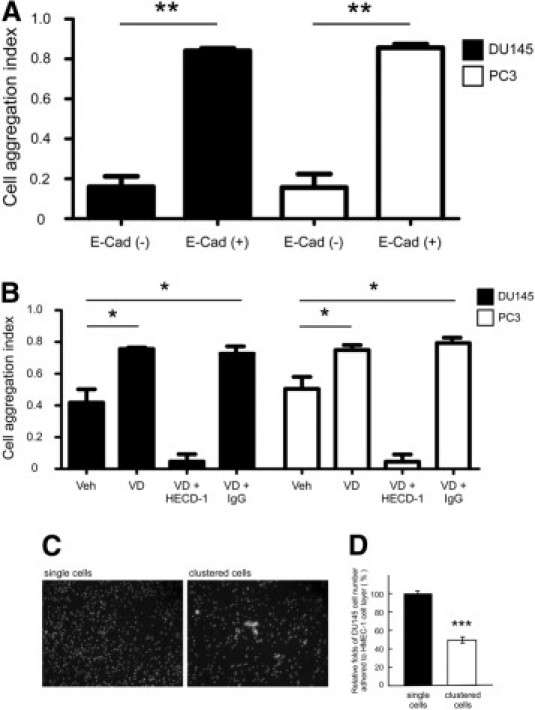
1,25-VD promotes PCa cell aggregation by enhancing E-cadherin expression, and PCa cell aggregation suppresses heterotypic adhesion. A: E-cadherin promoted cell aggregation. DU145 or PC-3 E-cadherin (−) and (+) cells were used for the cell aggregation. Cells were detached and washed twice with PBS buffer, and suspended at the 5 × 105 cells per ml in RPMI medium. Then, 1.5 × 104 cells were suspended as a hanging drop from the lid of the 24-well culture plate and incubated at 5% CO2 and 37°C overnight. After an 18-hour incubation, the cells were subjected to the shear stress by passing through standard 20-μl Gilson pipette tips ten times. Cells were photographed under microscope with 10× phase-contrast objective to record aggregation behaviors within 30 minutes. Results are the mean ± SEM of three experiments; **P < 0.01, E-cadherin (−) versus E-cadherin (+) PCa cells. B: 1,25-VD regulates DU145 and PC-3 cell aggregation through E-cadherin expression. DU145 or PC-3 cells were treated with ethanol as vehicle or 1,25-VD 100 nmol/L for 72 hours, then incubated with E-cadherin blocking antibody (HECD-1, 2 μg/ml; Calbiochem) to inactivate E-cadherin adhesive function and the cell aggregation assay was performed. Results are the mean ± SEM of three experiments; *P < 0.05, vehicle versus 1,25-VD or 1,25-VD/IgG treatment. C: The images of DU145 single cells and clustered cells (original magnification, ×40). To generate clustered cells, 106 DU145 cells in 1 ml of culture medium were centrifuged at 2300 × g for 5 minutes and then the medium was gently passed five times through a 1 ml standard Gilson pipette. D: Clustered cells exhibited fewer cells adhered to the endothelial cell (HMEC-1) layer. To assay the effects of cell aggregation on PCa cell heterotypic adhesion, the cell numbers adhered to HMEC-1 cell layer from single cells and clustered cells were compared. After clustered cells were generated, the same number of single cells and clustered cells were incubated with Calcein-AM to express green fluorescence and seeded on the HMEC-1 cell monolayer. Results are the mean ± SEM of three experiments; ***P < 0.001, single cells group versus clustered cells group.
To further test if homotypic aggregation of PCa cells might interfere with subsequent heterotypic adhesion to other cells, we artificially forced DU145 PCa cells to aggregate by centrifugation and compared their adhesion. As shown in Figure 5C, after centrifugation, DU145 cells aggregated to form clusters (left panel) when compared with control DU145 cells (right panel). Aggregated DU145 cells adhered less to endothelium when compared to DU145 single cells (Figure 5D), supporting the hypothesis that cell aggregation interferes with PCa cell adhesion to endothelium.
1,25-VD Suppresses PCa Adhesion to Endothelium through E-Cadherin
To further support the premise that 1,25-VD-induced E-cadherin upregulation is responsible for influencing cell adhesion, we compared the adhesion behavior between the DU145 and PC-3 E-cadherin (−) and (+) cells. As shown in Figure 6A, perfused E-cadherin (+) cells exhibited fewer rolling cell numbers than E-cadherin (−) cells. Similarly, less E-cadherin (+) cells adhering to HMEC-1 cells than E-cadherin (−) cells using the static heterotypic cell-cell adhesion assay (data not shown). Again, our data confirmed that E-cadherin can suppress PCa cell adhesion to endothelium. Finally, to demonstrate that 1,25-VD-induced E-cadherin expression is responsible for ultimate reduction of PCa heterotypic cell adhesion, we applied E-cadherin antibody HECD-1 to neutralize the E-cadherin epitope. Indeed, HECD-1 treatment reversed 1,25-VD reduced cell adhesion to endothelium (Figure 6B). Together, we identified a novel mechanism of 1,25-VD-inhibitory effects on circulating PCa cell adhesion to endothelium through the regulation of E-cadherin-mediated cell aggregation, consequently suppressing PCa cell adhesion to endothelium (Figure 6C).
Figure 6.
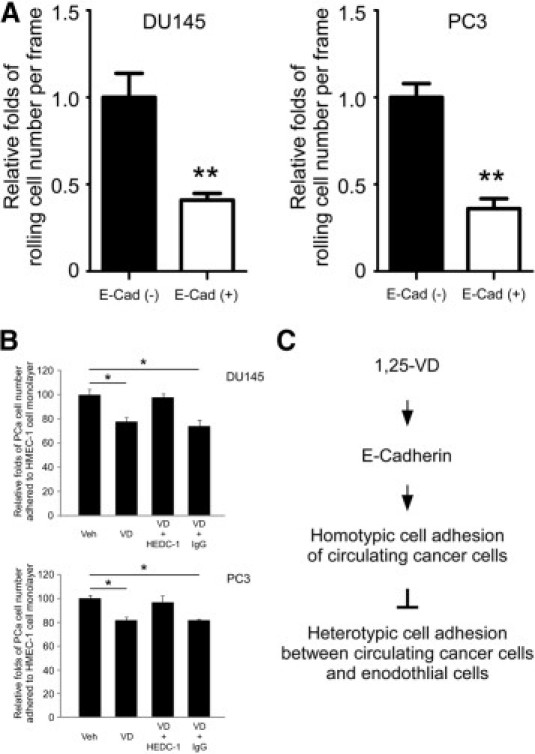
1,25-VD suppressed PCa cell adhesion to endothelial cells through E-cadherin. A: E-cadherin reduced PCa cell rolling numbers on the E-selectin/SDF-1 coated surface. DU145 or PC-3 E-cadherin (−) and (+) cells were applied in the rolling assay. Results are the mean ± SEM of three experiments; **P < 0.01, E-cadherin (−) cells vs. (+) cells. B: E-cadherin neutralizing antibody blocks the 1,25-VD effects on PCa cell adhesion to endothelial cells. DU145 or PC-3 cells were treated with vehicle or 1,25-VD for 72 hours, incubated with E-cadherin blocking antibody (HECD-1; 2 μg/ml) and then tested for cell adhesion assay. The relative fold of PCa cell number adhered to HMEC-1 cell layer represents the cell-cell adhesion ability. The result exhibited no statistical significance between vehicle and 1,25-VD-treated groups. Results are the mean ± SEM of three experiments; *P < 0.05, vehicle versus 1,25-VD or 1,25-VD/IgG treatment. C: The proposed mechanism of 1,25-VD suppressing circulating PCa cell adhesive behaviors is illustrated.
Discussion
The current paradigm for cancer metastasis is that malignant cells escape from the primary organ and circulate in the blood until they find an environment in which to reside in a dormant condition for later proliferation. Thus, detection of circulating cancer cells represents an early indication of metastatic potential. Disruption of circulating cancer cells arrival and arrest on endothelium of targeted sites would prevent cancer cell metastasis. To study dynamic interactions between circulating PCa cells and microvessels, we developed a flow-based microtube system as a mimic to the microvasculature. E-selectin is constitutively expressed on the bone marrow microvessels,32 and initiates the adhesive rolling of immune cells30 and circulating cancer cells33 on the endothelial layer. SDF-1 is a chemokine secreted by endothelial cells or stromal cells, and interacts with its receptor CXCR4 on the PCa cell surface. The interaction between SDF-1 and CXCR4 has been suggested to be responsible for PCa homing to bone,26 which eventually leads to bone metastasis. In addition to PCa cells, the combined effects of SDF-1 and E-selectin has been shown to increase the adhesion of cells across a number of flow environment26 and SDF-1-stimulated migration of CD34+ hematopoietic cells that was dependent on E-selectin.34 In another study, SDF-1 was shown to result in the creation of actin-based protrusions with increased expression of CD44, a ligand to E-selectin, on the surface of blood-born cells.35 As expected, more PCa cells roll and dock on the microtubes coated with E-selectin and SDF-1 than on the microtubes coated with E-selectin only. This observed SDF-1-induced enhancement of PCa cell capture suggests that SDF-1 in the bone endothelium has the potential to attract more circulating PCa cells and thus contributes to PCa homing and metastasis to the bone.
Identification of signals that control PCa cells behavior in the circulation can potentially provide an approach for the disruption of PCa cell adhesion on the endothelium of target organs, which may be a potential therapy for blocking distal organ metastasis. Vitamin D deficiency has been linked to increased PCa risk and disease progression and has led to a hypothesis of an anti-tumor role for vitamin D/VDR. In this study, we provide first-time evidences of vitamin D-inhibitory effects on circulating PCa cell adhesion to the E-selectin/SDF-1 hybrid surface under constant WSS. Three output metrics are involved in this inhibitory effect — total cell adhesion, cell rolling number (flux), and rolling velocity — all suggesting that 1,25-VD treatment can suppress circulating PCa cell arrest and recruitment to the vessel endothelium. Previous in vivo studies have shown that elevated rolling velocities reduce neutrophil recruitment.36,37 Similarly, it has been shown that a reduction in rolling velocities increased neutrophil adhesion.38 Our data demonstrated that 1,25-VD-treated DU145 cells exhibit elevated rolling velocities and fewer cells arrest on E-selectin/SDF-1-coated surfaces, similar to previous neutrophil studies,36,37 resulting in a decrease in cellular arrest. A number of mechanisms have been proposed by which elevated rolling velocity reduces cellular arrest, including a decrease in contact time for potential arrest of the rolling cell on the endothelium.39 More specific to PCa, CD44, an E-selectin ligand, has been reported to reduce neutrophil rolling velocity,30 and thus has been suggested to play a role in PCa metastasis to bone marrow endothelium.6 In our studies, we did not find significantly different CD44 expression between ethanol-treated (control vehicle) and 1,25-VD-treated DU145 cells (Figure 4, A and B), nor did we observe differences in expression of other E-selectin ligands expression. Therefore, vitamin D effect on the cell adhesion to endothelium is not likely to be mediated through the 1,25-VD/VDR transcriptional regulation of those E-selectin ligands.
Previous studies have attempted to relate the effects of cancer cell homotypic adhesion (cell aggregation) phenotypes on cancer progression. Earlier studies have suggested that cancer cells with higher malignancies have higher aggregation capabilities.40 However, loss of E-cadherin-dependent cell homotypic adhesion has been inversely correlated to cancer progression.41 Here, our data showed that 1,25-VD promotes E-cadherin-mediated homotypic cell adhesion that results in a greater number of circulating cell clusters. Due to the increased flow stress over the larger cross-sectional area of the cell aggregates, one possible result is a disruption of “normal” (though pathological) docking of circulating tumor cells to the vessel's endothelial surface under flow. This increased homotypic aggregation and decreased heterotypic adhesion would result in a decrease in the subsequent steps in the metastatic cascade, including extravasation, migration, and invasion to target organs. Thus, our results provide a novel hypothesis that homotypic adhesion of circulating cancer cells could interfere with later heterotypic cell adhesion.
Additionally, loss of E-cadherin has been reported to induce epithelial-mesenchymal transition (EMT) via a disruption in cell adhesion. EMT is thought to be a critical step during cancer metastasis because decreased cell adhesion results in the detachment of cancer cells at the primary site.42 Identification of the mediators/pathways that control the EMT process could lead to a strategy to inhibit cancer metastasis. Hence, our finding that vitamin D induces E-cadherin expression in PCa cells may have a bearing on the suppression of PCa metastasis due to its potential for mediation of the EMT process. To investigate this possibility, more studies on vitamin D's effect on EMT need to be performed.
The regulation of E-cadherin by 1,25-VD has been reported in colon cancer,31 yet the molecular mechanism by which 1,25-VD upregulates E-cadherin is still unknown. In our study, we showed that 1,25-VD-induced E-cadherin mRNA expression occurs at 24 hours and with much higher induction at 48 hours. Additionally, there is no potential VDR-responsive element in the E-cadherin promoter region, suggesting this regulation is via an indirect mechanism, rather than direct binding to E-cadherin promoter—for instance, activation of EGF/EGFR suppression of E-cadherin via activation of E-cadherin transcriptional repressors of Snail and Slug.43 Interestingly, 1,25-VD can suppress EGFR expression,44 therefore, it is possible that 1,25-VD promotes E-cadherin expression through the inhibition of EGFR/Snail/Slug signaling. Further experiments are required to determine the putative interplay between those molecules on the 1,25-VD treatment in context of the E-cadherin promoter.
Vitamin D–based therapy represents an emerging class of potentially promising drugs for treatment of PCa. Our report on the 1,25-VD inhibitory effects on PCa cell adhesion to the endothelium adds one more anti-tumor mechanism possessed by 1,25-VD. However, when a high concentration of 1,25-VD is used as a therapeutic agent, it has the potential to result in hypercalcemia and significant weight loss17 and these adverse side effects have hampered the clinical application of 1,25-VD. The development of low calcemic vitamin D-based therapy is highly desirable. A few vitamin D analogues that possess more potent anti-tumor activities with low calcemic side effects have been developed. For instance, seocalcitol (EB1089), a potent vitamin D analog that induces differentiation and inhibits growth of various cancers with less calcemic levels,17 was tested in Phase I/II clinical trials of breast cancer,45 pancreatic cancer,46 and hepatoma.47 Also, the intermittent treatment of high doses of 1,25-VD that exhibited anti-tumor effects with a transient hypercalcemia was reported in the treatment of prostate cancer.48,49 In addition, the results from the laboratory experiments showing 1,25-VD can suppress the expression of cyclooxygenase-2 (COX-2), the key prostaglandins (PG) synthesis enzyme, provide a functional mechanism of vitamin D–based combination therapy. Indeed, the combination of 1,25-VD with a COX-2 inhibitor, such as non-steroidal anti-inflammatory drug (NSAID), synergistically suppressed PCa cancer progression in vitro and in vivo.50 Even though the results of 1,25-VD application in cancer patient studies are still inconclusive, more well controlled clinical trials are needed to confirm the benefits of 1,25-VD in cancer therapy. Our current study demonstrated one 1,25-VD anti-PCa metastasis characteristics occurred by reducing circulating PCa cell adhesion to the endothelium, therefore it would be of interest to test if those low-calcemic vitamin D–based drugs can suppress PCa metastasis.
In summary, our data has provided evidence that 1,25-VD can suppress data has provided evidencesheterotypic adhesion of circulating PCa cells in a microfluidic system that mimics the endothelial microvasculature of bone marrow by promoting E–cadherin–mediated homotypic cell adhesion. The identification of the vitamin D signal as a mediator of circulating PCa cell behavior provides a novel strategy for the disruption of cancer cell adhesion to the blood vessels during heamtogenous dissemination, and a possible strategy decrease in metastatic potential.
Acknowledgments
We thank Dr. Beatrice Knudsen (Fred Hutchinson Cancer Research Center, Seattle, WA) for providing E-cadherin (+) and (−) DU145 and PC-3 cells. We also thank Dr. Jane Liesveld (University of Rochester, Rochester, NY) for providing human endothelial cell line, HMEC-1.
Footnotes
Supported by the NIH (CA 143876 to M.K.) and by the Department of Urology, University of Rochester Medical Center, Rochester, NY.
References
- 1.Frisch S.M., Screaton R.A. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- 2.Kim H.R., Lin H.M., Biliran H., Raz A. Cell cycle arrest and inhibition of anoikis by galectin-3 in human breast epithelial cells. Cancer Res. 1999;59:4148–4154. [PubMed] [Google Scholar]
- 3.Orr F.W., Wang H.H. Tumor cell interactions with the microvasculature: a rate-limiting step in metastasis. Surg Oncol Clin N Am. 2001;10:357–381. ix–x. [PubMed] [Google Scholar]
- 4.Dimitroff C.J., Lechpammer M., Long-Woodward D., Kutok J.L. Rolling of human bone-metastatic prostate tumor cells on human bone marrow endothelium under shear flow is mediated by E-selectin. Cancer Res. 2004;64:5261–5269. doi: 10.1158/0008-5472.CAN-04-0691. [DOI] [PubMed] [Google Scholar]
- 5.Dimitroff C.J., Descheny L., Trujillo N., Kim R., Nguyen V., Huang W., Pienta K.J., Kutok J.L., Rubin M.A. Identification of leukocyte E-selectin ligands: P-selectin glycoprotein ligand-1 and E-selectin ligand-1, on human metastatic prostate tumor cells. Cancer Res. 2005;65:5750–5760. doi: 10.1158/0008-5472.CAN-04-4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Draffin J.E., McFarlane S., Hill A., Johnston P.G., Waugh D.J. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res. 2004;64:5702–5711. doi: 10.1158/0008-5472.CAN-04-0389. [DOI] [PubMed] [Google Scholar]
- 7.Fornaro M., Manes T., Languino L.R. Integrins and prostate cancer metastases. Cancer Metastasis Rev. 2001;20:321–331. doi: 10.1023/a:1015547830323. [DOI] [PubMed] [Google Scholar]
- 8.Sun Y.X., Fang M., Wang J., Cooper C.R., Pienta K.J., Taichman R.S. Expression and activation of alpha v beta 3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate. 2007;67:61–73. doi: 10.1002/pros.20500. [DOI] [PubMed] [Google Scholar]
- 9.Engl T., Relja B., Marian D., Blumenberg C., Muller I., Beecken W.D., Jones J., Ringel E.M., Bereiter-Hahn J., Jonas D., Blaheta R.A. CXCR4 chemokine receptor mediates prostate tumor cell adhesion through alpha5 and beta3 integrins. Neoplasia. 2006;8:290–301. doi: 10.1593/neo.05694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz G.G., Hulka B.S. Is vitamin D deficiency a risk factor for prostate cancer?: (Hypothesis) Anticancer Res. 1990;10:1307–1311. [PubMed] [Google Scholar]
- 11.Moreno J., Krishnan A.V., Feldman D. Molecular mechanisms mediating the anti-proliferative effects of Vitamin D in prostate cancer. J Steroid Biochem Mol Biol. 2005;97:31–36. doi: 10.1016/j.jsbmb.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Sung V., Feldman D. 1,25-Dihydroxyvitamin D3 decreases human prostate cancer cell adhesion and migration. Mol Cell Endocrinol. 2000;164:133–143. doi: 10.1016/s0303-7207(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz G.G., Wang M.H., Zang M., Singh R.K., Siegal G.P. 1 alpha,25-Dihydroxyvitamin D (calcitriol) inhibits the invasiveness of human prostate cancer cells. Cancer Epidemiol Biomarkers Prev. 1997;6:727–732. [PubMed] [Google Scholar]
- 14.Bao B.Y., Yeh S.D., Lee Y.F. 1alpha,25-dihydroxyvitamin D3 inhibits prostate cancer cell invasion via modulation of selective proteases. Carcinogenesis. 2006;27:32–42. doi: 10.1093/carcin/bgi170. [DOI] [PubMed] [Google Scholar]
- 15.Bao B.Y., Yao J., Lee Y.F. 1alpha, 25-dihydroxyvitamin D3 suppresses interleukin-8-mediated prostate cancer cell angiogenesis. Carcinogenesis. 2006;27:1883–1893. doi: 10.1093/carcin/bgl041. [DOI] [PubMed] [Google Scholar]
- 16.Getzenberg R.H., Light B.W., Lapco P.E., Konety B.R., Nangia A.K., Acierno J.S., Dhir R., Shurin Z., Day R.S., Trump D.L., Johnson C.S. Vitamin D inhibition of prostate adenocarcinoma growth and metastasis in the Dunning rat prostate model system. Urology. 1997;50:999–1006. doi: 10.1016/S0090-4295(97)00408-1. [DOI] [PubMed] [Google Scholar]
- 17.Lokeshwar B.L., Schwartz G.G., Selzer M.G., Burnstein K.L., Zhuang S.H., Block N.L., Binderup L. Inhibition of prostate cancer metastasis in vivo: a comparison of 1,23-dihydroxyvitamin D (calcitriol) and EB1089. Cancer Epidemiol Biomarkers Prev. 1999;8:241–248. [PubMed] [Google Scholar]
- 18.Halbleib J.M., Nelson W.J. Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006;20:3199–3214. doi: 10.1101/gad.1486806. [DOI] [PubMed] [Google Scholar]
- 19.Gumbiner B., Stevenson B., Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol. 1988;107:1575–1587. doi: 10.1083/jcb.107.4.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semb H., Christofori G. The tumor-suppressor function of E-cadherin. Am J Hum Genet. 1998;63:1588–1593. doi: 10.1086/302173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thiery J.P. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 22.Wojciechowski J.C., Narasipura S.D., Charles N., Mickelsen D., Rana K., Blair M.L., King M.R. Capture and enrichment of CD34-positive haematopoietic stem and progenitor cells from blood circulation using P-selectin in an implantable device. Br J Haematol. 2008;140:673–681. doi: 10.1111/j.1365-2141.2007.06967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ting H.J., Bao B.Y., Reeder J.E., Messing E.M., Lee Y.F. Increased expression of corepressors in aggressive androgen-independent prostate cancer cells results in loss of 1alpha,25-dihydroxyvitamin D3 responsiveness. Mol Cancer Res. 2007;5:967–980. doi: 10.1158/1541-7786.MCR-06-0318. [DOI] [PubMed] [Google Scholar]
- 24.Thoreson M.A., Anastasiadis P.Z., Daniel J.M., Ireton R.C., Wheelock M.J., Johnson K.R., Hummingbird D.K., Reynolds A.B. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol. 2000;148:189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gebbink M.F., Zondag G.C., Wubbolts R.W., Beijersbergen R.L., van Etten I., Moolenaar W.H. Cell-cell adhesion mediated by a receptor-like protein tyrosine phosphatase. The Journal of biological chemistry. 1993;268:16101–16104. [PubMed] [Google Scholar]
- 26.Taichman R.S., Cooper C., Keller E.T., Pienta K.J., Taichman N.S., McCauley L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–1837. [PubMed] [Google Scholar]
- 27.Malek A.M., Alper S.L., Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282:2035–2042. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 28.Skowronski R.J., Peehl D.M., Feldman D. Vitamin D and prostate cancer: 1,25 dihydroxyvitamin D3 receptors and actions in human prostate cancer cell lines. Endocrinology. 1993;132:1952–1960. doi: 10.1210/endo.132.5.7682937. [DOI] [PubMed] [Google Scholar]
- 29.Bao B.Y., Hu Y.C., Ting H.J., Lee Y.F. Androgen signaling is required for the vitamin D-mediated growth inhibition in human prostate cancer cells. Oncogene. 2004;23:3350–3360. doi: 10.1038/sj.onc.1207461. [DOI] [PubMed] [Google Scholar]
- 30.Hidalgo A., Peired A.J., Wild M.K., Vestweber D., Frenette P.S. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity. 2007;26:477–489. doi: 10.1016/j.immuni.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmer H.G., Gonzalez-Sancho J.M., Espada J., Berciano M.T., Puig I., Baulida J., Quintanilla M., Cano A., de Herreros A.G., Lafarga M., Munoz A. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schweitzer K.M., Drager A.M., van der Valk P., Thijsen S.F., Zevenbergen A., Theijsmeijer A.P., van der Schoot C.E., Langenhuijsen M.M. Constitutive expression of E-selectin and vascular cell adhesion molecule-1 on endothelial cells of hematopoietic tissues. Am J Pathol. 1996;148:165–175. [PMC free article] [PubMed] [Google Scholar]
- 33.Giavazzi R., Foppolo M., Dossi R., Remuzzi A. Rolling and adhesion of human tumor cells on vascular endothelium under physiological flow conditions. J Clin Invest. 1993;92:3038–3044. doi: 10.1172/JCI116928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peled A., Grabovsky V., Habler L., Sandbank J., Arenzana-Seisdedos F., Petit I., Ben-Hur H., Lapidot T., Alon R. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J Clin Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naiyer A.J., Jo D.Y., Ahn J., Mohle R., Peichev M., Lam G., Silverstein R.L., Moore M.A., Rafii S. Stromal derived factor-1-induced chemokinesis of cord blood CD34(+) cells (long-term culture-initiating cells) through endothelial cells is mediated by E-selectin. Blood. 1999;94:4011–4019. [PubMed] [Google Scholar]
- 36.Forlow S.B., White E.J., Barlow S.C., Feldman S.H., Lu H., Bagby G.J., Beaudet A.L., Bullard D.C., Ley K. Severe inflammatory defect and reduced viability in CD18 and E-selectin double-mutant mice. J Clin Invest. 2000;106:1457–1466. doi: 10.1172/JCI10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunne J.L., Ballantyne C.M., Beaudet A.L., Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99:336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 38.Hafezi-Moghadam A., Thomas K.L., Prorock A.J., Huo Y., Ley K. L-selectin shedding regulates leukocyte recruitment. J Exp Med. 2001;193:863–872. doi: 10.1084/jem.193.7.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ley K. Integration of inflammatory signals by rolling neutrophils. Immunol Rev. 2002;186:8–18. doi: 10.1034/j.1600-065x.2002.18602.x. [DOI] [PubMed] [Google Scholar]
- 40.Updyke T.V., Nicolson G.L. Malignant melanoma cell lines selected in vitro for increased homotypic adhesion properties have increased experimental metastatic potential. Clin Exp Metastasis. 1986;4:273–284. doi: 10.1007/BF00133592. [DOI] [PubMed] [Google Scholar]
- 41.Umbas R., Schalken J.A., Aalders T.W., Carter B.S., Karthaus H.F., Schaafsma H.E., Debruyne F.M., Isaacs W.B. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992;52:5104–5109. [PubMed] [Google Scholar]
- 42.Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Cheng J.C., Klausen C., Leung P.C. Hydrogen peroxide mediates EGF-induced down-regulation of E-cadherin expression via p38 MAPK and snail in human ovarian cancer cells. Mol Endocrinol. 2010;24:1569–1580. doi: 10.1210/me.2010-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong W.M., Hofer H., Ellinger A., Peterlik M., Cross H.S. Mechanism of antimitogenic action of vitamin D in human colon carcinoma cells: relevance for suppression of epidermal growth factor-stimulated cell growth. Oncol Res. 1999;11:77–84. [PubMed] [Google Scholar]
- 45.Gulliford T., English J., Colston K.W., Menday P., Moller S., Coombes R.C. A phase I study of the vitamin D analogue EB 1089 in patients with advanced breast and colorectal cancer. Br J Cancer. 1998;78:6–13. doi: 10.1038/bjc.1998.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans T.R., Colston K.W., Lofts F.J., Cunningham D., Anthoney D.A., Gogas H., de Bono J.S., Hamberg K.J., Skov T., Mansi J.L. A phase II trial of the vitamin D analogue seocalcitol (EB1089) in patients with inoperable pancreatic cancer. Br J Cancer. 2002;86:680–685. doi: 10.1038/sj.bjc.6600162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalhoff K., Dancey J., Astrup L., Skovsgaard T., Hamberg K.J., Lofts F.J., Rosmorduc O., Erlinger S., Bach Hansen J., Steward W.P., Skov T., Burcharth F., Evans T.R. A phase II study of the vitamin D analogue seocalcitol in patients with inoperable hepatocellular carcinoma. Br J Cancer. 2003;89:252–257. doi: 10.1038/sj.bjc.6601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beer T.M. Development of weekly high-dose calcitriol based therapy for prostate cancer. Urol Oncol. 2003;21:399–405. doi: 10.1016/s1078-1439(03)00170-4. [DOI] [PubMed] [Google Scholar]
- 49.Beer T.M., Lemmon D., Lowe B.A., Henner W.D. High-dose weekly oral calcitriol in patients with a rising PSA after prostatectomy or radiation for prostate carcinoma. Cancer. 2003;97:1217–1224. doi: 10.1002/cncr.11179. [DOI] [PubMed] [Google Scholar]
- 50.Moreno J., Krishnan A.V., Swami S., Nonn L., Peehl D.M., Feldman D. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res. 2005;65:7917–7925. doi: 10.1158/0008-5472.CAN-05-1435. [DOI] [PubMed] [Google Scholar]