

Abstract
The 5′-AMP-activated protein kinase (AMPK) functions as a metabolic fuel gauge that is activated in response to environmental stressors to restore cellular energy balance. In the heart, AMPK coordinates the activation of glucose and fatty acid metabolic pathways to ensure increased production of myocardial ATP when required, such as during cardiac ischemia/reperfusion and hypertrophy, causing an increase in AMPK activity that can be viewed as both protective and maladaptive. While we understand the basic regulation of AMPK activity by kinases, recent studies have introduced the concept that AMPK is regulated by other post-translational modifications, specifically ubiquitination. These studies reported that the ubiquitin ligase cell death–inducing DFFA-like effector a ubiquitinates the β subunit of AMPK to regulate its steady-state protein levels. Other investigators found that AMPK regulatory components, including the AMPK α subunit and AMPK kinases NUAK1 and MARK4, can be ubiquitinated with atypical ubiquitin chains. The USP9X-deubiquitinating enzyme was identified to remove ubiquitination from both NUAK1 and MARK4. Lastly, AMPK activation increases the expression of the ubiquitin ligases MAFBx/Atrogin-1 and MuRF1. These ubiquitin ligases regulate key cardiac transcription factors to control cardiomyocyte mass and remodeling, thus suggesting another mechanism by which AMPK may function in the heart. The relevance of AMPK ubiquitination in cardiac disease has yet to be tested directly, but it likely represents an important mechanism that occurs in common cardiac diseases that may be targeted for therapy.
The 5′-AMP-activated protein kinase (AMPK) functions as a metabolic fuel gauge that is activated in response to numerous environmental stressors to restore cellular and whole-body energy balance.1,2 AMPK is allosterically regulated by the competitive binding of AMP and ATP, thereby “sensing” cellular energy status and, on activation, triggers compensatory ATP-generating mechanisms while attenuating ATP-consuming processes.3 Perturbations in cardiac metabolism are closely linked to the onset and progression of cardiovascular diseases; given the central role of AMPK in regulating cellular energetics, there is considerable interest in the determination of the precise role(s) of AMPK in cardiac pathophysiological states and evaluation of the utility of modulating AMPK activity as a therapeutic intervention.4 This review discusses AMPK function in normal and diseased hearts, with emphases on AMPK and protein degradation via the ubiquitin proteasome pathway—a potential novel approach to treating cardiovascular disease. These are significant new findings as ubiquitination is emerging as a pivotal regulatory mechanism that rivals phosphorylation in its overall significance in biology.5
Structure and Regulation of AMPK
AMPK is a highly conserved heterotrimeric enzyme consisting of three subunits, α, β, and γ, with multiple genes encoding distinct subunit isoforms (ie, α1, α2, β1, β2, γ1, γ2, and γ3). The structure and regulation of AMPK is the subject of an extensive recent review6; therefore, this article presents a brief structural and regulatory overview and focuses on the heart-specific aspects of AMPK. The α subunit of AMPK contains the catalytic domain and the Thr172 residue targeted by upstream kinases required for subsequent activation.7 The β subunit contains a glycogen-binding domain that is important for kinase function and substrate definition8 and functions as scaffold for α and γ subunit binding.9 The γ subunit consists of four tandem or cystathione β-synthase (CBS) repeats, forming two basic functional units called Bateman domains that bind AMP or ATP in a mutually exclusive manner, depending on the particular intracellular energetic milieu.10,11
The α1 and β1 isoforms are ubiquitously expressed in mammals, whereas the α2 and β2 isoforms are enriched in the heart as well as skeletal muscle.12,13 Of the three γ subunits, γ1 is ubiquitously expressed,14 whereas γ2 is enriched in the heart (but not skeletal muscle), and γ3 is expressed exclusively in skeletal muscle.15 Together, these studies demonstrate distinct expression patterns for AMPK subunits; for example, the cardiac AMPK multi-protein complex predominantly consists of α2-β2-γ2 subunits, while α2-β2-γ3 is the major complex found in skeletal muscle.14 It is likely that different AMPK subunit isoform combinations play different roles in various tissues, depending on the prevailing intracellular energetic status; however, this interesting possibility requires further study.
AMPK activation is dependent on the cellular AMP/ATP ratio and the phosphorylation status of αThr172 that continually cycles between phosphorylated and dephosphorylated.16 Several cardiac stressors, such as ischemia, result in increased AMP and decreased ATP levels.17 With a rise in the AMP/ATP ratio, AMP displaces the ATP bound to the γ regulatory subunit, leading to three different functional outcomes.3,14 First, AMP allosterically activates AMPK activity by a factor of 10. Additionally, AMP binding causes a conformational change, thereby enhancing access by upstream AMPK kinases (AMPKK) and further increasing AMPK activation18 and relieving the inhibition of the autoinhibitory domain.19 Last, AMP binding inhibits αThr172 dephosphorylation by upstream phosphatases, thus promoting increased AMPK phosphorylation.20
However, there is also AMP/ATP-independent activation of cardiac AMPK by upstream AMPKKs, such as the serine-threonine liver kinase B1 (LKB1) and Ca2+/calmodulin-dependent protein kinase kinase (CAMKK)21,22 as well as the hormones insulin23–25 and adiponectin.26,27 LKB1 is highly expressed in the heart and is thought to be constitutively active under basal conditions.4 Accordingly, LKB1-deficient mouse hearts are unable to activate cardiac AMPKα2.22 CAMKK is expressed at relatively low levels in the heart and is triggered by an increase in cytosolic Ca2+ levels and subsequently activates AMPK without altered AMP/ATP ratios.21 Transforming growth factor-β (TGF-β)–activated kinase-1 is also expressed in the heart and activates AMPK, although its role in this process is less well understood.28
Interestingly, well-described cardiometabolic drugs, such as metformin and statins, can also activate cardiac AMPK, although likely through indirect means.29 For example, it has been suggested that metformin inhibits mitochondrial respiratory chain complex I, increasing the AMP/ATP ratio and subsequent AMPK activation.30,31 However, others have found that metformin can activate AMPK independent of changes in the AMP/ATP ratio.32 This discrepancy may be explained by differences in metformin dosing and length of exposure; further studies are required to elucidate the precise mechanism of metformin on cardiac AMPK activity. AICA riboside (AICAR) is an adenosine analog that allosterically activates AMPK and is often used in experimental studies; however, the short half-life of AICAR and effects on glycemic regulation limits AICAR as an effective agent for long-term activation of AMPK in vivo.29
AMPK Function in the Heart
The normal mammalian heart has tremendous energy requirements; as such, cardiac fuel substrate selection is a complex process that is orchestrated by a number of factors that include circulating metabolite and hormone levels, myocardial substrate uptake rates, and intracellular signaling cascades that regulate metabolic pathway flux at multiple levels.33 AMPK impacts cardiac metabolism at multiple levels: (1) increasing glucose metabolism through increased glucose transport and activation of phosphofructokinase (PFK); (2) enhancing mitochondrial fatty acid β-oxidation (FAO) by increasing fatty acid uptake and phosphorylating acetyl-CoA carboxylase (ACC), a key regulator of fatty acid oxidation; (3) increasing mitochondrial biogenesis by activation of peroxisome proliferator-activated receptor-γ co-activator-1α (PGC-1α), a key transcriptional modulator of mitochondrial biogenesis34; and (4) inhibiting energy-consuming pathways, such as the mammalian target of rapamycin (mTOR) and downstream targets p70S6K and 4EBP-1, decreasing cardiac protein synthesis.35 Together, AMPK activation of myocardial glucose and fatty acid metabolic pathways are coordinated at multiple levels: fuel substrate availability, uptake, and breakdown, ultimately ensuring increased production of myocardial ATP when required.
Metabolic derangements are closely linked to the onset of numerous cardiac pathophysiologies. Accordingly, a multitude of studies focus on understanding the role of AMPK under such conditions, such as myocardial ischemia-reperfusion (I/R), cardiomyopathies, and cardiac hypertrophy. The main observations of these studies are highlighted in Table 1.14,22,26,36–49
Table 1.
The Role of AMPK in Cardiac Disease Models
| Ischemia-reperfusion | Observational events in mouse models |
| Cardio-protective role for AMPK | |
| Some genetic mouse models support a beneficial role for AMPK activation during ischemia-reperfusion: | |
| Maladaptive role for AMPK | |
| AMPK activation also increases fatty acid oxidation, leading to detrimental consequences: | |
| PRKAG2 cardiac syndrome | Mutations within human AMPKγ2 gene (PRKAG2) |
| Human phenotype supported by multiple mouse models | |
| Pathological cardiac hypertrophy | There are conflicting reports as to the role of AMPK activation in response to pathological hypertrophy stimuli:
|
Gene encoding the 5′-AMP-activated protein kinase subunit gamma-2 (which makes up AMPK). Glut4, glucose transporter 4; PFK2, phosphofructokinase 2; PRKAG2, protein kinase, AMP-activated, gamma-2 non-catalytic subunit.
Regulation of AMPK by Post-Translational Modification of Ubiquitin
Post-translational modification of proteins is the process by which chemical modifications are made to a protein after it is translated. There is a wide range of post-translational modifications that can be made to proteins. One of the best-studied modifications is phosphorylation, whereby enzymes add a phosphate group to a protein, often at specific serine, threonine, or tyrosine residues, to change that protein's activity or fate. Phosphorylation of the α subunit of AMPK is the best-delineated system of AMPK regulation to date. However, several recent studies have implicated the modification of AMPK with ubiquitin, suggesting another layer of AMPK regulation in the heart. With many reports demonstrating a significant role of AMPK in heart disease (Table 114,22,26,36–49) and our increasing appreciation of the ubiquitin proteasome system in the heart (for a comprehensive review, see Rodriguez et al 200950), the significance of AMPK ubiquitination is just beginning to be appreciated.
Ubiquitin Proteasome System
Post-translational modification of proteins with ubiquitin is a multistep enzymatic process that reacts to the carboxylic acid of the ubiquitin to protein lysines to form a covalent amide bond (Figure 1A). The small protein ubiquitin is first activated in a two-step process. First, the E1 ubiquitin-activating enzyme interacts with ubiquitin in an ATP-dependent process to form a thioester linkage between the C-terminal carboxyl group of ubiquitin and the E1 sulfhydryl moiety. Ubiquitin is then transferred to the active cysteine of the E2 ubiquitin-conjugating enzyme. The specificity of the ubiquitination process is found in the E3 ubiquitin ligases. E3 ubiquitin ligases function to recognize specific substrates and to transfer ubiquitin to their final lysine on target proteins (Figure 1A). There are hundreds of ubiquitin ligases that have been identified, with at least nine identified in the heart to date.50 The single E1 is able to bind to dozens of E2s, which bind hundreds of E3s to regulate a host of cellular processes. After the addition of a single ubiquitin, additional ubiquitin molecules can be added to form a polyubiquitin chain. Ubiquitin has seven lysine residues by which this can occur [ie, Lys 6, Lys 11, Lys 27, Lys 29, Lys 33, Lys 48, and Lys 63 (Figure 1B)]. The more common chains that have been described to date are through the lysine 48, which are recognized by the 26S proteasome (Figure 1A). Polyubiquitinated chains can be removed by a family of enzymes called de-ubiquitinating enzymes (DUBs), consisting of a large group of proteases that counter the ubiquitin-dependent metabolic pathways by cleaving the bonds between ubiquitin and proteins. The human genome encodes approximately 100 DUBs specific for ubiquitin.51 DUBs function to recycle and proofread protein ubiquitination and act in the disassembly of chains that inhibit protein activity.
Figure 1.
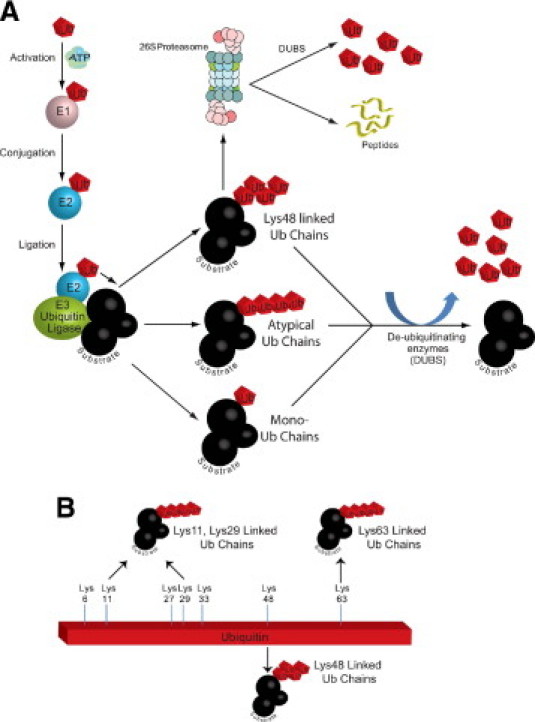
The ubiquitin proteasome system. A: Specific proteins are recognized by E3 enzymes (ubiquitin ligases) and ubiquitinated in a multistep process. The E1 enzyme activates ubiquitin, which is then transferred to one of the dozens of E2 enzymes. The E3 then catalyzes the transfer of the ubiquitin from the E2 to the recognized substrate in an ATP-dependent manner. Ubiquitinated substrates with canonical Lys48 chains are then recognized by the 26S proteasome and targeted for degradation or de-ubiquitinated as part of the ubiquitin proteasome system regulation and ubiquitin chain quality control. Substrate activity and/or location may be altered if mono-ubiquitin or atypical polyubiquitin chains are added. B: Polyubiquitin chains can be formed by any of the seven lysines within ubiquitin as well as by the N-terminal lysine to form atypical (Lys11, Lys29, and Lys63), canonical (Lys48), or linear (N-terminal lysine) chains. Our understanding of the significance of all of the non-canonical chains is incomplete.
While the canonical polyubiquitin (lys48) chains target their substrates for degradation, linkages through the other available lysines, including the N-terminus,52 and branched chains containing multiple linkage types53 also occur. These atypical chains generally do not target proteins for degradation, but they do play important roles in regulating cellular processes.54 Atypical ubiquitination is most studied in nuclear factor κB signaling, receptor endocytosis, and DNA repair processes55,56; therefore, it is not surprising that atypical ubiquitination regulates AMPK.
Emerging Concept: AMPK Is Regulated by Ubiquitin
Recent studies identified both direct and indirect mechanisms by which the ubiquitin proteasome system regulates AMPK activity. While LKB1 is the best-characterized AMPK kinase, there are others that regulate the activity of AMPK, including the protein NUAK1 (AMPK-related kinase 5) and MARK4 (microtubule-affinity-regulating kinase 4). Recent studies demonstrated that the de-ubiquitinating enzyme USP9X interacts with endogenous NUAK1 and MARK4 in HEK293 cells by immunoprecipitation.57 The AMPK kinases NUAK1 and MARK4 are also polyubiquitinated in vivo.57 Inhibiting the deubiquitinating enzyme enhances NUAK and MARK4 ubiquitination, implicating the role of USP9X in ubiquitinating these AMPK kinases in vitro.57 In vitro deubiquitination studies confirmed the ability of USP9X to directly deubiquitinate NUAK1 and MARK4; conversely, knock-down of USP9X in vitro enhanced their ubiquitination.57 Expressing mutant NUAK and MARK4 constructs unable to interact with the USP9X DUB causes hyperubiquitination of NUAK and MARK4, resulting in decreased AMPK catalytic activity.57 Ubiquitin-dependent inhibition of AMPK kinase activity is thought to be due to the interference of ubiquitin with phosphorylation of the activation (T-loop) residues.57 The polyubiquitination of NUAK and MARK4 is formed predominantly by linkages at lys29 and lys33, but not other lysines, indicative of atypical polyubiquitin chains.57 The mechanisms by which ubiquitination regulates the NUAK and MARK4 AMPKKs, which then affect AMPK activity, are summarized in Figure 2.58
Figure 2.
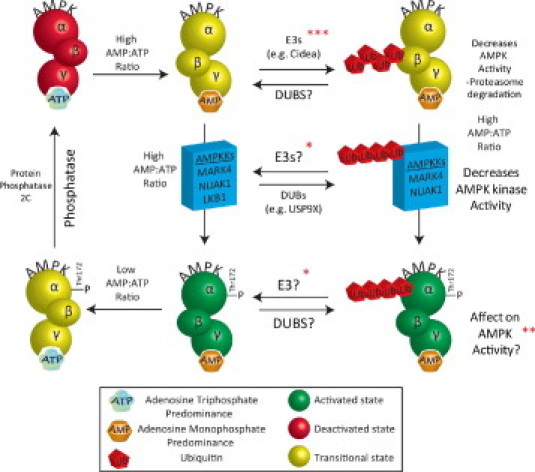
Post-translational regulation of AMPK activity. The AMP-activated protein kinase (AMPK) is a metabolite-sensing serine-threonine kinase that acts as a master regulator of cellular energy metabolism, with the ability to regulate lipid, glucose, and protein metabolism in response to decreasing ATP levels. The AMPK holoenzyme–consisting of α, β, and γ subunits–is regulated by AMP:ATP ratios, whereby AMP binds the Bateman (CBS) domains in the γ subunit, resulting in a conformational change which allows the α catalytic domain to be phosphorylated (Thr172) by one of many AMPK kinases. Activation (green) and deactivation (red) has canonically been described by AMPK kinases and phosphatases (left column). Protein phosphatases, such as protein phosphatase 2C, also counteract AMPK.58 The role of the ubiquitin proteasome system in regulating AMPK activity is just beginning to be understood (right column). Polyubiquitination and proteasome-dependent degradation has been reported to be mediated by the E3 ubiquitin ligase Cidea (top, right), which inhibits downstream AMPK activity. The AMPK kinases MARK4 and NUAK1 are de-ubiquitinated by the USP9X DUB, affecting the ability of these enzymes to activate AMPK (middle, right). Last, atypical ubiquitin chains can be placed on the α subunit of AMPK have been described, and that may affect its activity. *A DUB has been identified, although competing E3 not identified (See text for details). **Ubiquitination of the alpha1 subunit of AMPK is enhanced in the presence of a DUB inhibitor (See text for details). ***Cidea interacts with the beta subunit of AMPK to target (polyubiquitinate) it for proteasome-mediated degradation (See text for details).
These fascinating studies raise many additional questions about how the ubiquitin proteasome system regulates AMPK activity. Most of the work to date on the ubiquitin proteasome system has focused on the E1, E2, and particularly the E3 ubiquitin ligases that regulate substrate activities. Much less focus has been put on identifying de-ubiquitinases and their role in regulating cellular processes (recently reviewed59–61). However, the therapeutic potential of deubiquitinating enzyme inhibitors has been a new line of investigation in cancer chemotherapies.62 So it is interesting that a specific DUB has been identified in regulating AMPK kinases without the identification of the E3 ubiquitin ligases they are competing with (Figure 258). Moreover, the alpha1 subunit of AMPK is found to be ubiquitinated in the presence of DUB inhibitors (Figure 258). In the presence of NEM, an inhibitor of de-ubiquitination, enhanced polyubiquitination of immunoprecipitated AMPK alpha1 subunit has been observed.57 Interestingly, the polyubiquitin chains on this subunit were predominantly atypical (lys29 and lys33) linkages. This observation indicates an important role for DUBs in the direct regulation of AMPK and raises the question of what E3 ubiquitin ligase(s) are placing the atypical polyubiquitin chains on the AMPK alpha1 subunit. It also remains to be determined if this modification affects AMPK activity.
To date, only one study has identified an E3 ubiquitin ligase that mediates the ubiquitination and degradation of AMPK. In brown adipose tissue, the Cidea (cell death-inducing DFFA-like effector a) protein ubiquitinates and degrades AMPK.63 Cidea is found in many tissues, including heart, and is found to regulate metabolism through its interaction with AMPK. Cidea co-localizes in the endoplasmic reticulum and forms a complex with AMPK in vivo.63 Cidea specifically interacts with the β subunit of AMPK, but not the α or γ (Figure 258).63 When Cidea is co-expressed with AMPK β subunit, the stability of AMPK β is reduced due to ubiquitin-dependent degradation.63 Cidea-/- mice have an enhanced AMPK stability and enzymatic activity, which is consistent with both the role of Cidea as an E3 ubiquitin ligase that regulates AMPK activity as well as with the increased energy expenditure and lean phenotype that could be explained by this enhanced AMPK activity.63 While the type of polyubiquitination that Cidea placed on the AMPK β subunit was not identified in these studies, it is likely that the polyubiquitination is of the canonical (lys48)-type that targets proteins for proteasome-dependent degradation. This contrasts to the atypical lysine chains found on the α subunit in previous studies.63 However, atypical lys29 ubiquitin chains can also promote protein degradation via the lysosome, so parallels may exist between these two systems.64 Ubiquitination of AMPK by the ubiquitin ligase Cidea plays a role in inhibiting AMPK activity, likely by targeting the AMPK holo-enzyme or one of its subunits (β) for degradation by the proteasome.
AMPK Interacts with the Proteasome?
Last, a recent study identified that AMPK interacts with the proteasome itself. In yeast two-hybrid studies, it has been reported that the PSMD11 proteasome subunit interacts with AMPK.65 This finding suggests that AMPK may physically interact with this proteasome subunit to affect its phosphorylation status and potential function. However, the effects of AMPK on the proteasome (and vice versa) have not been directly tested. With the realization of the importance of proteasome function in cardiac health and disease, these findings may prove to be significant as we better understand how AMPK regulates the proteasome, which may be important for its own (ie, AMPK's) activity described above.
AMPK Activation Increases the Expression of Ubiquitin Ligases Relevant to Cardiac Disease
While the regulation of AMPK activity by ubiquitination is just beginning to be elucidated, much more is known about how AMPK activation regulates the ubiquitination machinery, in particular the E3 ubiquitin ligases, the proteins that give specificity to the system and mediate the ubiquitination of specific substrates. This work has exclusively been performed in skeletal muscle. However, like many lines of study of the ubiquitin ligases MuRF1 and MAFBx/Atrogin-1, applicability to the heart is likely relevant.
AMPK Activation Increases Expression of MuRF1 and MAFBx/Atrogin-1
AMPK activation in striated muscle has dual effects. It enhances cellular processes involved in ATP production, including glucose uptake and fatty acid oxidation.66,67 It also inhibits processes that consume energy, such as protein synthesis.68–70 AMPK activation inhibits protein synthesis by decreasing the mTOR activation.68 Three studies have recently investigated how AMPK activation affects MuRF1 and MAFBx/Atrogin-1 expression, both in vitro and in vivo. They have generally found that AMPK activation increases MuRF1 and MAFBx/Atrogin-1 expression.
Stimulating C2C12 myotubes with AICAR (5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside) to enhance AMPK activity, Nakashima and Yakabe71 identified an enhanced protein degradation and increased MAFBx/Atrogin-1 and MuRF1. In subsequent studies, Krawiec et al72 found that AICAR, metformin, or 2-deoxyglucose could all enhance MAFBx/atrogin-1and MuRF1 expression over time. Krawiec et al also found that when these AMPK activity enhancers were given in parallel with dexamethasone, a synergistic increase in these ubiquitin ligases were seen.72 Dexamethasone is a commonly used, potent inducer of experimental atrophy that highly up-regulates the ubiquitin ligases MuRF1 and MAFBx/atrogin-1 that directly mediate muscle atrophy. AICAR, metformin, and 2-deoxyglucose treatment (with and without dexamethasone treatment) did not enhance the expression of other UBR box E3 ubiquitin ligases, including UBR1/E3 alphaI and UBR2/E3 alphaII, suggesting that AMPK was specifically activating ubiquitin ligases that are known to mediate muscle atrophy.72 Inhibiting AICAR with compound C prevented MAFBx/atrogin-1 and MuRF1 expression increases in response to serum deprivation, AICAR treatment, or AICAR and dexamethasone treatment together.72 These findings were also seen in in vivo models. When mice were challenged with AICAR, an increase in skeletal muscle MAFBx/atrogin-1 and MuRF1 mRNA was identified, suggesting a role of AMPK in the regulation of these ubiquitin ligases in vivo. Since these ubiquitin ligases are critical components of the complex network of signaling pathways activated in skeletal muscle atrophy, AMPK may play key roles in regulating atrophy in response to catabolic challenges.
Insulin-like growth factor 1 (IGF-1) stimulation enhances downstream signaling through activating (phosphorylating) Akt, which in turn phosphorylates the transcription factor FoxO3a, resulting in a reduction in the ubiquitin ligases MAFBx/atrogin-1 and MuRF1.73–75 Recent studies have identified that AICAR synergizes with IGF-1 induced Akt activation without affecting (inhibiting) the expression of the MAFBx/atrogin-1 and MuRF1 ubiquitin ligases.76 It was identified that AICAR inhibited Fox03a phosphorylation in the cytoplasm and induced Fox03a nuclear relocation.76 Inhibiting mTOR increased basal MAFBx/atrogin-1 expression and reversed the inhibitor effect of IGF-1 on ubiquitin ligase expression. These studies demonstrate that AICAR activation of AMPK stimulates MAFBx/atrogin-1and MuRF1 expression despite the activation of Akt signaling, which may be due to the inhibition of Fox0 phosphorylation by AMPK via Akt.76 AMPK inhibition of mTOR may additionally provide a mechanism by which AMPK enhances the expression of ubiquitin ligases.76
These studies uniformly demonstrate that enhancing AMPK activity increases the expression of ubiquitin ligases fundamentally involved in skeletal muscle and cardiac atrophy (Figure 3). And specific ubiquitin ligases ubiquitinate AMPK directly, such as Cidea (Figure 3). Whether there is a feedback loop by which the MuRF1 and MAFBx/Atrogin-1 ubiquitin ligases limit AMPK has not been determined. This is a logical possibility, given the several ways in which ubiquitination regulates AMPK, either directly or indirectly, as outlined in the previous section (Figure 258). MuRF1 has been reported to have the ability to place atypical ubiquitin chains, like those found on AMPK experimentally.53 While these studies have been uniformly performed in skeletal muscle, the ubiquitin ligases MAFBx/atrogin-1 and MuRF1 have proven to have a significant role in cardiac disease (see recent review50). For example, MuRF1 inhibits cardiac hypertrophy, is necessary for regression of cardiac hypertrophy, and is cardioprotective in I/R injury by inhibiting transcription factors such as SRF and cJun.50 Similarly, MAFBx/atrogin-1 inhibits pressure-overload induced cardiac hypertrophy and increases susceptibility to I/R induced apoptosis by proteasome-dependent degradation of MKP-1, a negative regulator of JNK.50 With the diverse regulation of AMPK during various cardiac stresses (Table 114,22,26,36–49), it is certainly possible that AMPK upstream regulation of MAFBx/atrogin-1 and MuRF1 is involved in the heart’s adaptive responses to injury. However, this remains to be directly tested.
Figure 3.
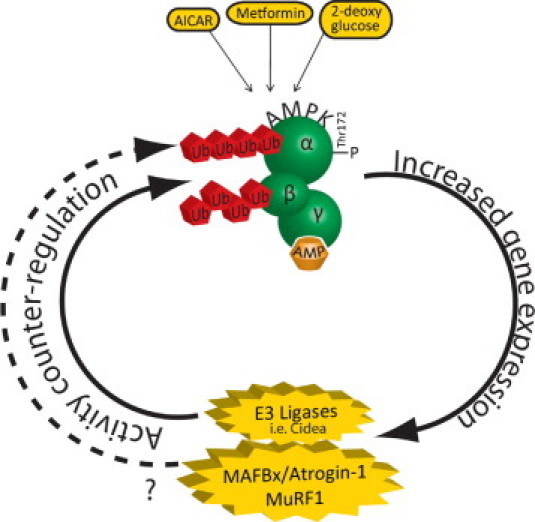
AMPK agonists enhance the expression of the ubiquitin ligases MAFBx/Atrogin-1 and MuRF1. Enhancing AMPK activity using AMPK agonists in C2C12 myotubes increases the expression of MAFBx/Atrogin-1 and MuRF1 (discussed in the text). With evidence that AMPK is ubiquitinated (outlined in Figure 258), it raises the possible counterregulation of AMPK yet-to-be-determined E3 ligases, or possibly MAFBx/Atrogin-1 and MuRF1, although this remains to be investigated directly.
Conclusions
The 5′-AMP–activated protein kinase (AMPK) functions as a metabolic fuel gauge activated in response to numerous environmental stressors to restore cellular energy balance. In the heart, AMPK coordinates the activation of glucose and fatty acid metabolic pathways at multiple levels by regulating substrate availability, uptake, and breakdown—ultimately ensuring increased production of myocardial ATP when required. Cardiac AMPK activity is regulated in cardiac diseases such as ischemia/reperfusion injury and is implicated in regulating cardiac metabolism to exert both positive and negative roles in cardiac ischemia/reperfusion injury and cardiac hypertrophy (Table 114,22,26,36–49). Although the basic regulation of AMPK activity by kinases has been delineated, we are beginning to understand that other post-translational modifications may regulate AMPK activity as well. Recent reports determined that the ubiquitin ligase Cidea ubiquitinates the β subunit of AMPK, leading to degradation. Atypical ubiquitination has been identified on the α subunit as well as two of the regulatory AMPK kinases NUAK1 and MARK4, which regulate AMPK activity. Activation of AMPK using agonists such as AICAR, metformin, and 2-deoxyglucose increases the expression of key cardiac ubiquitin ligases MAFBx/Atrogin-1 and MuRF1. Given the potential of these ubiquitin ligases to ubiquitinate key cardiac transcription factors as well as AMPK itself suggests that AMPK's role in cardiovascular biology extends beyond metabolism and is important in MAFBx/Atrogin-1– and MuRF1–dependent remodeling and cell signaling regulation. The relevance of the ubiquitination of AMPK in cardiac disease has not been tested directly, but likely represents an important mechanism that occurs in common cardiac diseases and that potentially could be targeted for therapy. How the ubiquitin proteasome system regulates AMPK activity is an emerging concept with far-reaching implications.
Footnotes
Supported by the National Heart, Lung, and Blood Institute grant R01HL065619 (C.P.) and the American Heart Associations (Scientist Development Grant) (M.S.W.).
M.Z. and J.C.S. contributed equally to this manuscript.
References
- 1.Hardie D.G., Hawley S.A., Scott J.W. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Young L.H., Li J., Baron S.J., Russell R.R. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med. 2005;15:110–118. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Hardie D.G. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 4.Dyck J.R., Lopaschuk G.D. AMPK alterations in cardiac physiology and pathology: enemy or ally. J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kerscher O., Felberbaum R., Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 6.Steinberg G.R., Kemp B.E. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 7.Woods A., Vertommen D., Neumann D., Turk R., Bayliss J., Schlattner U., Wallimann T., Carling D., Rider M.H. Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem. 2003;278:28434–28442. doi: 10.1074/jbc.M303946200. [DOI] [PubMed] [Google Scholar]
- 8.Polekhina G., Gupta A., Michell B.J., van Denderen B., Murthy S., Feil S.C., Jennings I.G., Campbell D.J., Witters L.A., Parker M.W., Kemp B.E., Stapleton D. AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol. 2003;13:867–871. doi: 10.1016/s0960-9822(03)00292-6. [DOI] [PubMed] [Google Scholar]
- 9.Warden S.M., Richardson C., O'Donnell J., Jr, Stapleton D., Kemp B.E., Witters L.A. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J. 2001;354:275–283. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 11.Scott J.W., Hawley S.A., Green K.A., Anis M., Stewart G., Scullion G.A., Norman D.G., Hardie D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stapleton D., Mitchelhill K.I., Gao G., Widmer J., Michell B.J., Teh T., House C.M., Fernandez C.S., Cox T., Witters L.A., Kemp B.E. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 13.Thornton C., Snowden M.A., Carling D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J Biol Chem. 1998;273:12443–12450. doi: 10.1074/jbc.273.20.12443. [DOI] [PubMed] [Google Scholar]
- 14.Arad M., Seidman C.E., Seidman J.G. AMP-activated protein kinase in the heart: role during health and disease. Circ Res. 2007;100:474–488. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 15.Cheung P.C., Salt I.P., Davies S.P., Hardie D.G., Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000;346(Pt 3):659–669. [PMC free article] [PubMed] [Google Scholar]
- 16.Hardie D.G. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008;582:81–89. doi: 10.1016/j.febslet.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Rosenfeldt F. Heart physiology from cell to circulation. In: Opie L.H., editor. Lippincott Williams & Wilkins; Philadelphia: 2004. p. 154. [Google Scholar]
- 18.Suter M., Riek U., Tuerk R., Schlattner U., Wallimann T., Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 19.Nagata D., Hirata Y. The role of AMP-activated protein kinase in the cardiovascular system. Hypertens Res. 2010;33:22–28. doi: 10.1038/hr.2009.187. [DOI] [PubMed] [Google Scholar]
- 20.Davies S.P., Helps N.R., Cohen P.T., Hardie D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase: Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 21.Anderson K.A., Means R.L., Huang Q.H., Kemp B.E., Goldstein E.G., Selbert M.A., Edelman A.M., Fremeau R.T., Means A.R. Components of a calmodulin-dependent protein kinase cascade: Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J Biol Chem. 1998;273:31880–31889. doi: 10.1074/jbc.273.48.31880. [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto K., Zarrinpashneh E., Budas G.R., Pouleur A.C., Dutta A., Prescott A.R., Vanoverschelde J.L., Ashworth A., Jovanovic A., Alessi D.R., Bertrand L. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab. 2006;290:E780–E788. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamble J., Lopaschuk G.D. Insulin inhibition of 5′ adenosine monophosphate-activated protein kinase in the heart results in activation of acetyl coenzyme A carboxylase and inhibition of fatty acid oxidation. Metabolism. 1997;46:1270–1274. doi: 10.1016/s0026-0495(97)90229-8. [DOI] [PubMed] [Google Scholar]
- 24.Kovacic S., Soltys C.L., Barr A.J., Shiojima I., Walsh K., Dyck J.R. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 25.Longnus S.L., Segalen C., Giudicelli J., Sajan M.P., Farese R.V., Van Obberghen E. Insulin signalling downstream of protein kinase B is potentiated by 5′AMP-activated protein kinase in rat hearts in vivo. Diabetologia. 2005;48:2591–2601. doi: 10.1007/s00125-005-0016-3. [DOI] [PubMed] [Google Scholar]
- 26.Shibata R., Ouchi N., Ito M., Kihara S., Shiojima I., Pimentel D.R., Kumada M., Sato K., Schiekofer S., Ohashi K., Funahashi T., Colucci W.S., Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–1389. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimano M., Ouchi N., Shibata R., Ohashi K., Pimentel D.R., Murohara T., Walsh K. Adiponectin deficiency exacerbates cardiac dysfunction following pressure overload through disruption of an AMPK-dependent angiogenic response. J Mol Cell Cardiol. 2010;49:210–220. doi: 10.1016/j.yjmcc.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie M., Zhang D., Dyck J.R., Li Y., Zhang H., Morishima M., Mann D.L., Taffet G.E., Baldini A., Khoury D.S., Schneider M.D. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A. 2006;103:17378–17383. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong A.K., Howie J., Petrie J.R., Lang C.C. AMP-activated protein kinase pathway: a potential therapeutic target in cardiometabolic disease. Clin Sci (Lond) 2009;116:607–620. doi: 10.1042/CS20080066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Mir M.Y., Nogueira V., Fontaine E., Averet N., Rigoulet M., Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 31.Owen M.R., Doran E., Halestrap A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 32.Hawley S.A., Gadalla A.E., Olsen G.S., Hardie D.G. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 33.Stanley W.C., Recchia F.A., Lopaschuk G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 34.Jorgensen S.B., Richter E.A., Wojtaszewski J.F. Role of AMPK in skeletal muscle metabolic regulation and adaptation in relation to exercise. J Physiol. 2006;574:17–31. doi: 10.1113/jphysiol.2006.109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolinsky V.W., Dyck J.R. Role of AMP-activated protein kinase in healthy and diseased hearts. Am J Physiol Heart Circ Physiol. 2006;291:H2557–H2569. doi: 10.1152/ajpheart.00329.2006. [DOI] [PubMed] [Google Scholar]
- 36.Lopaschuk G.D. AMP-activated protein kinase control of energy metabolism in the ischemic heart. Int J Obes (Lond) 2008;32(Suppl 4):S29–S35. doi: 10.1038/ijo.2008.120. [DOI] [PubMed] [Google Scholar]
- 37.Russell R.R., 3rd, Li J., Coven D.L., Pypaert M., Zechner C., Palmeri M., Giordano F.J., Mu J., Birnbaum M.J., Young L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altarejos J.Y., Taniguchi M., Clanachan A.S., Lopaschuk G.D. Myocardial ischemia differentially regulates LKB1 and an alternate 5′-AMP-activated protein kinase. J Biol Chem. 2005;280:183–190. doi: 10.1074/jbc.M411810200. [DOI] [PubMed] [Google Scholar]
- 39.Paiva M.A., Goncalves L.M., Providencia L.A., Davidson S.M., Yellon D.M., Mocanu M.M. Transitory activation of AMPK at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc Drugs Ther. 2010;24:25–32. doi: 10.1007/s10557-010-6222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishino Y., Miura T., Miki T., Sakamoto J., Nakamura Y., Ikeda Y., Kobayashi H., Shimamoto K. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res. 2004;61:610–619. doi: 10.1016/j.cardiores.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 41.Dyck J.R., Kudo N., Barr A.J., Davies S.P., Hardie D.G., Lopaschuk G.D. Phosphorylation control of cardiac acetyl-CoA carboxylase by cAMP-dependent protein kinase and 5′-AMP activated protein kinase. Eur J Biochem. 1999;262:184–190. doi: 10.1046/j.1432-1327.1999.00371.x. [DOI] [PubMed] [Google Scholar]
- 42.Essop M.F., Opie L.H. Metabolic therapy for heart failure. Eur Heart J. 2004;25:1765–1768. doi: 10.1016/j.ehj.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 43.Gollob M.H. Glycogen storage disease as a unifying mechanism of disease in the PRKAG2 cardiac syndrome. Biochem Soc Trans. 2003;31:228–231. doi: 10.1042/bst0310228. [DOI] [PubMed] [Google Scholar]
- 44.Arad M., Moskowitz I.P., Patel V.V., Ahmad F., Perez-Atayde A.R., Sawyer D.B., Walter M., Li G.H., Burgon P.G., Maguire C.T., Stapleton D., Schmitt J.P., Guo X.X., Pizard A., Kupershmidt S., Roden D.M., Berul C.I., Seidman C.E., Seidman J.G. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation. 2003;107:2850–2856. doi: 10.1161/01.CIR.0000075270.13497.2B. [DOI] [PubMed] [Google Scholar]
- 45.Sidhu J.S., Rajawat Y.S., Rami T.G., Gollob M.H., Wang Z., Yuan R., Marian A.J., DeMayo F.J., Weilbacher D., Taffet G.E., Davies J.K., Carling D., Khoury D.S., Roberts R. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation. 2005;111:21–29. doi: 10.1161/01.CIR.0000151291.32974.D5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arad M., Benson D.W., Perez-Atayde A.R., McKenna W.J., Sparks E.A., Kanter R.J., McGarry K., Seidman J.G., Seidman C.E. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002;109:357–362. doi: 10.1172/JCI14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gollob M.H., Seger J.J., Gollob T.N., Tapscott T., Gonzales O., Bachinski L., Roberts R. Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation. 2001;104:3030–3033. doi: 10.1161/hc5001.102111. [DOI] [PubMed] [Google Scholar]
- 48.Tian R., Musi N., D'Agostino J., Hirshman M.F., Goodyear L.J. Increased adenosine monophosphate-activated protein kinase activity in rat hearts with pressure-overload hypertrophy. Circulation. 2001;104:1664–1669. doi: 10.1161/hc4001.097183. [DOI] [PubMed] [Google Scholar]
- 49.Chan A.Y., Soltys C.L., Young M.E., Proud C.G., Dyck J.R. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–32779. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez J.E., Schisler J.C., Patterson C., Willis M.S. Seek and destroy: the ubiquitin—-proteasome system in cardiac disease. Curr Hypertens Rep. 2009;11:396–405. doi: 10.1007/s11906-009-0069-7. [DOI] [PubMed] [Google Scholar]
- 51.Reyes-Turcu F.E., Ventii K.H., Wilkinson K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kirisako T., Kamei K., Murata S., Kato M., Fukumoto H., Kanie M., Sano S., Tokunaga F., Tanaka K., Iwai K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim H.T., Kim K.P., Lledias F., Kisselev A.F., Scaglione K.M., Skowyra D., Gygi S.P., Goldberg A.L. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375–17386. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 54.Ikeda F., Dikic I. Atypical ubiquitin chains: new molecular signals: “Protein modifications: beyond the usual suspects” review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haglund K., Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hayden M.S., Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 57.Al-Hakim A.K., Zagorska A., Chapman L., Deak M., Peggie M., Alessi D.R. Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem J. 2008;411:249–260. doi: 10.1042/BJ20080067. [DOI] [PubMed] [Google Scholar]
- 58.Witczak C.A., Sharoff C.G., Goodyear L.J. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. 2008;65:3737–3755. doi: 10.1007/s00018-008-8244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katz E.J., Isasa M., Crosas B. A new map to understand deubiquitination. Biochem Soc Trans. 2010;38:21–28. doi: 10.1042/BST0380021. [DOI] [PubMed] [Google Scholar]
- 60.Shabek N., Ciechanover A. Degradation of ubiquitin: the fate of the cellular reaper. Cell Cycle. 2010;9:523–530. doi: 10.4161/cc.9.3.11152. [DOI] [PubMed] [Google Scholar]
- 61.Kimura Y., Tanaka K. Regulatory mechanisms involved in the control of ubiquitin homeostasis. J Biochem. 2010;147:793–798. doi: 10.1093/jb/mvq044. [DOI] [PubMed] [Google Scholar]
- 62.Colland F: The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem Soc Trans, 38:137–143 [DOI] [PubMed]
- 63.Qi J., Gong J., Zhao T., Zhao J., Lam P., Ye J., Li J.Z., Wu J., Zhou H.M., Li P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chastagner P., Israel A., Brou C. Itch/AIP4 mediates Deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep. 2006;7:1147–1153. doi: 10.1038/sj.embor.7400822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moreno D., Viana R., Sanz P. Two-hybrid analysis identifies PSMD11, a non-ATPase subunit of the proteasome, as a novel interaction partner of AMP-activated protein kinase. Int J Biochem Cell Biol. 2009;41:2431–2439. doi: 10.1016/j.biocel.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Henin N., Vincent M.F., Gruber H.E., Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9:541–546. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- 67.Park H., Kaushik V.K., Constant S., Prentki M., Przybytkowski E., Ruderman N.B., Saha A.K. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277:32571–32577. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- 68.Bolster D.R., Crozier S.J., Kimball S.R., Jefferson L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 69.Dubbelhuis P.F., Meijer A.J. Hepatic amino acid-dependent signaling is under the control of AMP-dependent protein kinase. FEBS Lett. 2002;521:39–42. doi: 10.1016/s0014-5793(02)02815-6. [DOI] [PubMed] [Google Scholar]
- 70.Horman S., Browne G., Krause U., Patel J., Vertommen D., Bertrand L., Lavoinne A., Hue L., Proud C., Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–1423. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- 71.Nakashima K., Yakabe Y. AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem. 2007;71:1650–1656. doi: 10.1271/bbb.70057. [DOI] [PubMed] [Google Scholar]
- 72.Krawiec B.J., Nystrom G.J., Frost R.A., Jefferson L.S., Lang C.H. AMP-activated protein kinase agonists increase mRNA content of the muscle-specific ubiquitin ligases MAFbx and MuRF1 in C2C12 cells. Am J Physiol Endocrinol Metab. 2007;292:E1555–E1567. doi: 10.1152/ajpendo.00622.2006. [DOI] [PubMed] [Google Scholar]
- 73.Sandri M., Sandri C., Gilbert A., Skurk C., Goldberg A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stitt T.N., Drujan D., Clarke B.A., Panaro F., Timofeyva Y., Kline W.O., Gonzalez M., Yancopoulos G.D., Glass D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 75.Lee S.W., Dai G., Hu Z., Wang X., Du J., Mitch W.E. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol. 2004;15:1537–1545. doi: 10.1097/01.asn.0000127211.86206.e1. [DOI] [PubMed] [Google Scholar]
- 76.Tong J.F., Yan X., Zhu M.J., Du M. AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. J Cell Biochem. 2009;108:458–468. doi: 10.1002/jcb.22272. [DOI] [PubMed] [Google Scholar]