

Abstract
Lynch syndrome is an autosomal dominant cancer predisposition syndrome characterized by loss of function of DNA mismatch repair enzyme MLH1, MSH2, MSH6, or PMS2. Mutations in MLH1 and MSH2 account for ∼80% of the inherited cases. However, in up to 20% of cases suspected of having a germline mutation in MSH2 due to loss of MSH2 expression, a germline mutation is not identified. Recent studies have shown that some Lynch syndrome cases are due to 3′ EPCAM/TACSTD1 deletions that subsequently lead to MSH2 promoter hypermethylation. In this study, we examined the frequency of this novel mechanism for MSH2 inactivation in cases recruited through the Colon Cancer Family Registry and from the Mayo Clinic Molecular Diagnostics Laboratory. From the combined cohort, 58 cases were selected in which immunohistochemical staining suggested a mutation in MSH2 or MSH6, but no mutations were identified on follow-up testing. Of these 58 cases, 11 demonstrated a deletion of EPCAM/TACSTD1. Of cases with a deletion, the methylation status of the MSH2 promoter was confirmed in tumor tissue using methylation-sensitive PCR primers. One case showed MSH2 promoter hypermethylation in the absence of a detectable EPCAM/TACSTD1 deletion. These results indicate that approximately 20% to 25% of cases suspected of having a mutation in MSH2 but in which a germline mutation is not detected, can be accounted for by germline deletions in EPCAM/TACSTD1. These data also suggest the presence of other alterations leading to MSH2 promoter hypermethylation.
Lynch syndrome is an autosomal dominant predisposition syndrome in which patients have a propensity to develop colorectal adenocarcinoma, endometrial carcinoma, sebaceous neoplasms, upper urinary tract urothelial carcinomas, central nervous system neoplasms, and ovarian and hepatobiliary neoplasms.1–4 The underlying genetic basis for this syndrome is the presence of a mutation in one of the DNA mismatch repair genes MLH1, MSH2, MSH6 or PMS2.5–10 The defining phenotype of tumors from these patients is the presence of tumor microsatellite instability11–13 and loss of protein expression of the affected enzyme in the tumor nuclei as detected by immunohistochemical staining.14–16 The spectrum of mutations in these genes includes missense, nonsense, splice site alterations, insertion/deletions, and large gene rearrangements. Mutations in MLH1 and MSH2 are the most common and account for ∼80% of the inherited mismatch repair cases.17–19 Of the cases with loss of MSH2 immunohistochemical staining, approximately two-thirds of identifiable mutations in MSH2 are point mutations or small insertions and deletions, whereas the remaining one-third are large gene rearrangements and deletions.20,21 However, mutations in MSH2 are not identified in up to 20% of the cases expected to have such an alteration.
Recently, germline deletions involving the 3′ end of EPCAM (also known as TACSTD1), located approximately 16-kb upstream of MSH2, have been shown to result in hypermethylation of the MSH2 promoter region and subsequent loss of MSH2 expression from the affected allele.22 In this study, we determined the frequency of this novel mechanism for MSH2 inactivation and correlated results of multiplex ligation-dependent probe amplification (MLPA)23,24 testing with results of the MSH2 promoter hypermethylation test. Although specific endpoints of the deletions were not determined, the loss of material in the 3′ untranslated region is similar to the different size deletions previously reported.22
Materials and Methods
Case Selection
Colorectal cancer cases were selected from the Colon Cancer Family Registry (Colon CFR), a National Cancer Institute–supported consortium established in l997 to create a multinational comprehensive collaborative infrastructure for interdisciplinary studies in the genetic epidemiology of colorectal cancer. Detailed information about the Colon CFR, including method of ascertainment and molecular testing, can be found at http://epi.grants.cancer.gov/CFR/ (last accessed: March 19, 2010) as well as being described in detail by Newcomb et al25 and Poynter et al.26 Six Colon CFR sites used to identify eligible cases were: Seattle Familial Colorectal Cancer Registry, Hawaii Family Registry of Colon Cancer, Ontario Familial Colorectal Cancer Registry, Australasian Colorectal Cancer Family Study, University of Southern California Consortium, and Mayo Colorectal Cancer Family Registry. These sites use various strategies for recruitment such that the entire spectrum of colorectal cancer risk is represented, including participants recruited both from population-based sources and clinic-based sources. Inclusion criteria for the current study included: i) evidence of defective DNA mismatch repair by the loss of normal immunohistochemical staining of MSH2/MSH6 in tumor cell nuclei from diagnostic tumor samples; ii) the presence of the microsatellite instability-high tumor phenotype (if assessed); iii) no identifiable MSH2 or MSH6 mutation by sequencing and/or large gene rearrangement assays at the time of testing; and iv) sufficient clinical information and tumor material available for testing. Of 5927 cases in the Colon CFR database, 37 cases met these criteria. Approval from the institutional review boards of each participating CFR site was obtained. Colon CFR participants all provided informed consent.
The clinical Microsatellite instability/Lynch syndrome database of the Molecular Genetics Laboratory at Mayo Clinic containing 5598 cases (all tumors that had been clinically tested for Lynch syndrome, predominantly but not exclusively colorectal cancer was also used to select cases for this study. This dataset (case series) was collected from 2001 to 2008 and contains Lynch syndrome screening results from microsatellite instability and immunohistochemical tests, demographic information provided to the laboratory, and follow-up testing results including germline analysis as provided by the ordering physician or as clinically performed at Mayo Clinic. The same inclusion and exclusion criteria as described for the Colon CFR were applied to the cases from the Mayo database. Of the 5598 cases, 344 had evidence of abnormal MSH2 protein expression, with 100 having evidence of at least partial follow-up germline testing. In 21 of these cases, no alterations were identified. Mayo Clinic Institutional Review Board approval was obtained.
Microsatellite Instability Immunohistochemistry and Germline Testing
Microsatellite instability data from the Colon CFR and from Mayo Clinic were determined using the same panel of 10 microsatellite markers using standard procedures, techniques, and classification as described previously.16,25 For some of the more recent Mayo Clinic cases, MSI testing was performed with the commercially available Promega MSI kit (Promega Corp., Madison, WI). Immunohistochemistry was performed according to previously described and standard protocols.16,27 Germline analysis was performed using a variety of methods including screening by denaturing high-performance liquid chromatography followed by sequencing of abnormal bands, direct Sanger sequencing, and Southern blot analysis or MLPA for large genomic deletions and insertions.24,25,28,29
MSH2 Promoter Hypermethylation Assay
Ten-micron-thick sections were cut from formalin-fixed, paraffin-embedded tissue from the selected cases. Normal and tumor tissue was macrodissected using a hematoxylin and eosin template. The slides were all tested at a single institution (Mayo Clinic). Bisulfite treatment was performed using the EZ DNA Methylation Kit (Zymo Research, Orange, CA) with modification of the method. Briefly, selected tissue was scraped into digestion buffer with Proteinase K and digested overnight at 50°C. This lysate was spun down, 5 μl of M-Dilution buffer and 5 μl of water were added and incubated at 37°C for 15 minutes. CT Conversion Reagent (100 μl) was added and incubated overnight at 50°C. The next day, 400 μl of M-Binding Buffer was added to Zymo-Spin I columns, followed by the sample. The columns were washed with 200 μl of M-Wash Buffer; 200 μl of M-Desulphonation Buffer was added and incubated at room temperature for 15 minutes, followed by two washes with 200 μl of M-Wash Buffer and elution of DNA into 10 μl of M-Elution Buffer.
The DNA methylation status of the MSH2 promoter was tested using methylation-specific PCR primers based on the ref Gene-NM-000251 by Ligtenberg et al22 These primers target two regions of the MSH2 promoter (regions 1 and 3) and result in expected fragment sizes of 145 and 137 bp for the unmethylated and methylated amplicons of region 1, and 216 and 209 bp of region 3, respectively.22 One microliter of bisulfite-treated DNA was used in each PCR reaction. Cycling conditions were modified as follows. For methylated and unmethylated primers for region 1 and the unmethylated primer for region 3, an initial denaturation step of 94°C for 10 minutes followed by 35 cycles of 92°C for 45 seconds, 65°C to 62°C for 45 seconds an extension of 72°C for 30 seconds, and a final extension of 72°C for 10 minutes. The region 3 methylated primers were run for 40 cycles with a similar thermocycler program except the annealing temperature was 62°C. Fragment analysis was performed on an ABI 3130X automated sequencer (Applied Biosystems, Foster City, CA) using standard methods and internal size marker ladder and instrument protocol 400-500bp-POP7-D.
Scoring of the MSH2 promoter hypermethylation assay was performed as follows. The interpretation cutoff for fluorescent intensity was set at 500 relative fluorescent units for the untreated DNA tube, otherwise the sample was considered failed for that region. A sample was equivocal if the untreated DNA signal was >500 but the bisulfite-treated signal was greater than 0 and less than 500. Cases in which both primers gave a signal >500 were called positive for MSH2 hypermethylation for that promoter region. Both regions 1 and 3 were taken into account for the final DNA methylation call. Only one positive region was needed for a final positive call. Equivocal and negative calls were conservatively called negative, and if one of the two regions failed, the score from the other region was used.
EPCAM/TACSTD1 Deletion
EPCAM/TACSTD1 deletion analysis was performed on DNA extracted from peripheral blood using a commercially available MLPA kit (P072 version 6; MRC Holland, Amsterdam, the Netherlands) at one institution (Mayo Clinic).23,24 This kit contains oligonucleotide probes targeting EPCAM/TACSTD1 exons 3, 8, 9, and two probes in the intervening region between EPCAM/TACSTD1 and MSH2: one 3 kb downstream and one 2.5 kb upstream from the MSH2 gene. Further delineation of the deletion breakpoints was not performed.
Results
Of the 11,525 cases from the Colon CFR and Mayo Clinic's clinical database, 37 and 21 cases, respectively, were identified in which loss of MSH2/MSH6 immunohistochemical stains could not be explained by follow-up germline testing. Of these 58 cases, 11 (19%) demonstrated a deletion within EPCAM/TACSTD1: 6 of 21 (29%) Mayo Clinic cases and 5 of 37 (14%) Colon CFR cases (Table 1). Examples of typical positive and negative cases are shown in Figure 1. All 11 deletions encompassed EPCAM/TACSTD1 exons 8 and 9 (del 8/9), whereas five cases showed larger deletions extending at least 3 kb downstream of the EPCAM/TACSTD1 coding region (del 8/9/3kb). None of the deletions involved exon 3 of EPCAM/TACSTD1 or the coding region of the MSH2 gene.
Table 1.
Summary of MLPA and MSH2 Promoter Hypermethylation Results
| Case source (n) | # Tested by MLPA | Deletion identified | # Tested by methylation |
MSH2 promoter hypermethylation final call |
||
|---|---|---|---|---|---|---|
| Pos | Neg | No amp | ||||
| Colon CFR (37) | 36 | 5 | 35 | 5⁎ | 19 | 11† |
| Mayo Clinic (21) | 21 | 6 | 15 | 6 | 9 | 0 |
| Total (58) | 57 | 11 | 50 | 11 | 28 | 11 |
MLPA, multiplex ligation-dependent probe amplification; Pos, positive; Neg, negative; amp, amplification; Colon CFR, Colon Cancer Family Registry.
One case with MSH2 promoter hypermethylation did not show an EPCAM/TACSTD1 deletion.
One case with a deletion failed for the MSH2 promoter hypermethylation assay.
Figure 1.
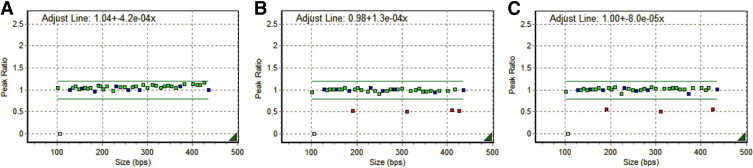
Analysis of EPCAM/TACSTD1 for large deletions and duplications by MLPA, two normal copies of each probe (green squares) and control probes (blue squares) are represented by a peak ratio of one. Loss of one allele at the probe site results in a decrease in the peak ratio to 0.5 and is represented by a red square. A: Shows a normal result, with all EPCAM/TACSTD1 probes showing normal dosage. B: Shows a deletion that includes exons 8 and 9 of EPCAM/TACSTD1 and two probes located downstream of the gene including a probe 3 kb away. C: Shows a deletion that includes exons 8 and 9 only.
Hypermethylation of the MSH2 promoter region was also tested in tissue samples from 50 of the 58 cases in which tissue was available (example shown in Figure 2). Eleven tissue samples (22%) demonstrated MSH2 promoter hypermethylation. Of the eleven cases with an EPCAM/TACSTD1 deletion, 10 demonstrated the presence of MSH2 promoter hypermethylation, including all six cases from the Mayo Clinic group and four of the Colon CFR cases. One case with a deletion failed the MSH2 hypermethylation assay due to poor amplification. Finally, there was one case within the Colon CFR cohort that showed MSH2 promoter hypermethylation without a detectable EPCAM/TACSTD1 deletion.
Figure 2.
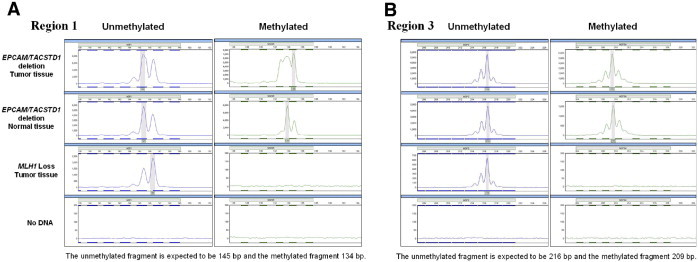
Results of MSH2 hypermethylation test by methylation-specific PCR and fragment length analysis. A: Shows the results of the ABI-based fragment analysis for methylation-specific primers in region 1 of the MSH2 promoter. MSH2 hypermethylation can be seen in tumor and normal tissue in a case with a EPCAM/TACSTD1 deletion. In contrast, the negative control case (loss of MLH1 expression) does not show MSH2 methylation. B: Shows similar data for region 3 of the MSH2 promoter.
We compared demographic information between individuals in the study in which an EPCAM/TACSTD1 deletion/MSH2 promoter hypermethylation (N = 12) was identified with those in which such a deletion was not identified (N = 46) (Table 2). The average age of diagnosis for those cases with a deletion was 53 years compared to 51 years (age at the time tumor was being tested) for those in the study without these abnormalities. The sex distribution (male/female) was approximately 6:5 and 1:1 for the cases with and without a deletion.
Table 2.
Summary of Demographic and Tumor Characteristics in Cohort
|
EPCAM/TACSTD1 deletion or MSH2 hypermethylation |
||||
|---|---|---|---|---|
| Present |
Absent |
|||
| Sample source | Mayo Clinic n = 6 | Colon CFR n = 6 | Mayo Clinic n = 15 | Colon CFR n = 23 |
| Average age (years) | 54 | 52 | 52 | 51 |
| Sex ratio | 3 M/3 F | 4 M/2 F | 6 M/9 F | 13 M/10 F |
| Tumor tested in proband | 6 Colorectal | 6 Colorectal | 8 Colorectal | 22 Colorectal |
| 4 Sebaceous | 1 Breast | |||
| 3 Uterine | ||||
| Meet revised Bethesda Guidelines | 5 of 5 | 5 of 6 | 10 of 13 | NA |
Colon CFR, Colon Cancer Family Registry; M, male; F, female.
Since the putative mechanism of MSH2 gene inactivation involves the expression of another gene, EPCAM/TACSTD1, the question of whether or not individuals and families with this alteration would be affected by the same spectrum of tumors as typically seen in Lynch syndrome was raised. First, we looked at tumors that were initially tested for Lynch syndrome, the primary site was available in 50 cases. Thirty-two tumors sent for testing were colorectal primaries, 12 (38%) of which had evidence of EPCAM/TACSTD1 deletion or MSH2 promoter hypermethylation. None of the 18 non-colon tumors sent for testing were found to have an EPCAM/TACSTD1 abnormality (Table 2). Second, to further investigate the spectrum of cancers in those families demonstrating a deletion in EPCAM/TACSTD1, additional personal and family history was examined from the two respective databases (available in 10 of 12 cases). For these 10 available families, 95 neoplasms were recorded among 83 individuals. Of the 95 neoplasms, there were 62 Lynch-related colorectal lesions (adenocarcinomas and adenomatous polyps), as well as 10 non-colon malignancies (stomach, uterine, ovarian, sebaceous, and pancreatic cancer). Finally, 23 tumors not typically associated with Lynch syndrome were observed (Table 3). Testing for the presence of defective mismatch repair (microsatellite instability and/or immunohistochemistry) was not performed on any of these additional tumors either from the proband or from family members. Third, we also looked at whether or not the probands would meet revised Bethesda Guidelines.30 Based on available clinical and family history, 11 of the 12 cases met revised Bethesda Guidelines.
Table 3.
Demographics and Characteristics of Cases with EPCAM/TACSTD1 Deletion /MSH2 Promoter Hypermethylation
| Sex | Age⁎ | Number of probes deleted | MSH2 hypermethylation | Meets revised Bethesda Guidelines | Tumor tested in proband | Other tumors in proband | Tumors in family members (<50 years) | Tumors in family members (>50 years or age not available) |
|---|---|---|---|---|---|---|---|---|
| M | 52 | 4 | Identified | Yes | CRC | — | — | CRC and stomach cancer |
| M | 49 | 3 | Identified | Yes | CRC | CRC | Melanoma, CRC ×2 | CRC ×3, breast, bone, stomach, prostate and upper limb cancer† |
| M | 58 | 0 | Identified | Yes | CRC | — | — | Pancreatic and uterine cancer |
| F | 68 | 3 | Identified | Yes | CRC | — | CRC | CRC ×6, melanoma |
| F | 35 | 3 | Identified | Yes | CRC | — | NA | NA |
| M | NA | 4 | Identified | Yes | CRC | CRC ×2 | CRC | ovarian cancer |
| M | 59 | 3 | Identified | Cannot determine | CRC | NA | NA | NA |
| F | 47 | 3 | Identified | Yes | CRC | — | CRC ×4 | — |
| F | 40 | 4 | Identified | Yes | CRC | — | CRC ×3, extra-colonic tumor,† stomach, testicular, lung and gynecologic cancers | CRC, BCC, breast, prostate ×2, and liver cancer |
| M | 69 | 4 | Identified | Yes | CRC | CRC | CRC ×3 and pancreatic cancer | CRC ×3, and stomach cancer |
| F | 60 | 4 | Identified | Yes | CRC | Sebaceous carcinoma, bladder papillary urothelial carcinoma, SCC | CRC ×4, and colonic polyps | CRC ×2, and breast cancer ×2 |
| M | 51 | 3 | Identified | Yes | CRC | CRC | Sarcoma† | CRC ×7, colonic polyps ×3, prostate cancer ×2, breast and renal cancer |
M, male; CRC, colorectal adenocarcinoma; F, female; NA, not available; BCC, basal cell carcinoma; SCC, squamous cell carcinoma; —, none.
Age at diagnosis of the sample being tested as reported at the time of testing.
Histologic type and tumor location not specified.
Discussion
In this study, we have demonstrated that deletions in the EPCAM/TACSTD1 gene account for a significant fraction (approximately 20%) of cases suspected of having an MSH2 germline mutation but in which a mutation was not identified using standard laboratory-based approaches. This result is consistent with previously published reports showing that this alteration has been observed in approximately 19% to 27% of cases with unidentified germline abnormalities.31,32
The presence of the EPCAM/TACSTD1 gene deletion correlates with DNA hypermethylation of the MSH2 promoter. Heritable DNA methylation abnormalities have been identified in two Lynch syndrome–associated genes, MLH133–40 and MSH2.22,31,32,41,42 Heritable MLH1 promoter hypermethylation appears to be a relatively rare event, and the mechanism for this alteration has not yet been determined. Evidence presented in the literature22 proposes that the hypermethylation of the MSH2 promoter region results from the production of an abnormal EPCAM/TACSTD1 RNA; specifically, a deletion of the EPCAM/TACSTD1 transcriptional termination signal results in a fusion transcript that includes at least part of MSH2. This abnormal transcript has been postulated to mediate the DNA hypermethylation of the MSH2 promoter in cis.22,31 This transcription-mediated inactivation of gene expression via epigenetic regulation has been previously reported in the α-globin gene locus.43,44
In this study, the MSH2 methylation-specific PCR confirmed the association between the upstream EPCAM/TACSTD1 deletion and MSH2 promoter hypermethylation in cases with available DNA. In addition, the MSH2 promoter hypermethylation assay can also identify rare cases in which MLPA for the EPCAM/TACSTD1 3′ region is not deleted. Indeed, one example of such a case was identified in the Colon CFR cohort in which MSH2 methylation was detected but a deletion was not found by MLPA. The mechanism for the MSH2 promoter hypermethylation in this case has not yet been elucidated. To rule out the possibility of specimen misidentification, DNA from both peripheral blood and tissue were genotyped, which showed that the two samples did indeed belong to the same individual. One explanation for the discordant results could be that this individual is tissue mosaic for the deletion—MLPA was not performed on DNA from the formalin-fixed, paraffin-embedded tissue. However, a more likely explanation for this discordant result is that this proband has either a small deletion, not detectable by the current MLPA probes, or a point mutation in the transcription termination signal of EPCAM/TACSTD1 resulting in the same RNA read through that is currently thought to mediate the abnormal promoter hypermethylation of MSH2. This case demonstrates that the currently available MLPA test may not be 100% sensitive for EPCAM/TACSTD1-mediated events.
The resulting clinical phenotype of an EPCAM/TACSTD1 deletion is dependent on the co-expression of EPCAM/TACSTD1 and MSH2. Thus, we questioned whether the tumor spectrum in cases with a deletion would be different from those without EPCAM/TACSTD1 involvement. EPCAM/TACSTD1 and MSH2 have somewhat different expression patterns in normal and tumor tissues. The expression pattern of EPCAM/TACSTD1 in normal tissues is primarily associated with epithelial tissues. MSH2 is more ubiquitous in normal tissue, although the levels of protein expression vary in different tissue types (GeneCards ID GC02P047572 at http://www.genecards.org, last accessed: April 6, 2010). High levels of expression have been associated with organs typically at risk in Lynch syndrome (colon, small bowel, stomach, endometrium, ovary, and transitional epithelium of the upper urinary tract). However, MSH2 expression can be seen in a range of other normal tissues including squamous epithelium, lung bronchi, pancreas, brain, testis, thyroid, prostate, and breast.45
Does the overlap of expression patterns for these two genes affect the tumor spectrum for this subset of Lynch patients? Based on the initial tumor sites tested (Table 2), and on previous work,31 the data suggested that patients with a EPCAM/TACSTD1 deletion may be over-represented with colorectal cancer only. Given the bias in the selection of tumors being tested in this albeit large cohort, additional personal and family history data were evaluated to determine what other tumors had occurred in the probands and their family members. On the basis of a review of available family history from the Mayo Clinic and Colon CFR databases, 95 tumors were reported among 83 individuals. The majority of tumors reported are colorectal. One family appears to be affected with colon cancer only, and several other families are predominantly affected with CRC, with only one or two non-colon tumors. However, there were four families that showed a range of tumors, having at least three non-colon tumors. This observation suggests that patients with an EPCAM/TACSTD1 deletion may be at an increased risk of malignancies other than colon cancer.
There are a number of limitations regarding the tumor data. First, slides and/or pathology reports were not available for review for other tumors in the probands or for tumors reported in their families. In some cases, it is not clear whether some of the tumors reported represent second primaries, treatment-related tumors, or metastatic tumors. Some cancers (both Lynch and non-Lynch associated) occurred at older ages and may represent sporadic cancers in members of the family. Second, because tumor tissue from other Lynch and non-typical Lynch tumors was not available to us for additional studies, we were not able to test for microsatellite instability or expression of MSH2 protein by immunohistochemistry. Thus, we cannot determine whether these non-typical Lynch tumors are indeed associated with the germline defect or simply sporadic tumors within these families. Finally, the small number of cases identified with EPCAM/TACSTD1 deletions (and MSH2 promoter hypermethylation) precludes any definitive conclusion from being drawn regarding the spectrum of tumors in these patients. Nevertheless, the fact that nearly a quarter of tumors in these families were not typical Lynch syndrome tumors raises the possibility that affected individuals may be predisposed to a broader distribution of tumors and warrants additional study.
In summary, this study provides additional information regarding the frequency and spectrum of EPCAM/TACSTD1 deletions and MSH2 promoter hypermethylation in Lynch syndrome. This mechanism accounted for ∼20% of cases in which colorectal cancer had loss of expression of MSH2/MSH6 but no previously identified germline mutation. Overall, this mechanism would account for approximately 5% of Lynch syndrome cases with abnormal MSH2/MSH6 expression. The presence of one discordant case in which MSH2 promoter hypermethylation was detected in the tumor but a deletion was not identified by the current MLPA panel suggests that other mutations and/or somatic events may occur in rare cases. Although not definitive, our data suggest that patients with an EPCAM/TACSTD1 deletion are at risk for malignancies in a variety of tissue types.
Footnotes
Supported by the National Cancer Institute, National Institutes of Health under RFA #CA-95-011 and through cooperative agreements with the members of the Colon Cancer Family Registry (CFR) and PIs. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CFRs, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the CFR. Collaborating centers include the Australian Colorectal Cancer Family Registry (UO1 CA097735), the USC Familial Colorectal Neoplasia Collaborative Group (UO1 CA074799), Mayo Clinic Cooperative Family Registry for Colon Cancer Studies (UO1 CA074800), Ontario Registry for Studies of Familial Colorectal Cancer (UO1 CA074783), Seattle Colorectal Cancer Family Registry (UO1 CA074794), University of Hawaii Colorectal Cancer Family Registry (UO1 CA074806), and University of California, Irvine Informatics Center (UO1 CA078296).
CME Disclosure: The authors did not disclose any relevant financial relationships.
References
- 1.Lynch H.T., de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 2.Aarnio M., Sankila R., Pukkala E., Salovaara R., Aaltonen L.A., de la Chapelle A., Peltomaki P., Mecklin J.P., Jarvinen H.J. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–218. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 3.Lynch H.T., de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–818. [PMC free article] [PubMed] [Google Scholar]
- 4.Watson P., Lynch H.T. The tumor spectrum in HNPCC. Anticancer Res. 1994;14:1635–1639. [PubMed] [Google Scholar]
- 5.Fishel R., Lescoe M.K., Rao M.R., Copeland N.G., Jenkins N.A., Garber J., Kane M., Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 6.Leach F.S., Nicolaides N.C., Papadopoulos N., Liu B., Jen J., Parsons R., Peltomaki P., Sistonen P., Aaltonen L.A., Nystrom-Lahti M., Guan X.Y., Zhang J., Meltzer P.S., Yu J.W., Kao F.T., Chen D.J., Cerosaletti K.M., Fournier R.E.K., Todd S., Lewis T., Leach R.J., Naylor S.L., Weissenbach J., Mecklin J.P., Järvinen H., Petersen G.M., Hamilton S.R., Green J., Jass J., Watson P., Lynch H.T., Trent J.M., de la Chapelle A., Kinzler K.W., Vogelstein B. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 7.Bronner C.E., Baker S.M., Morrison P.T., Warren G., Smith L.G., Lescoe M.K., Kane M., Earabino C., Lipford J., Lindblom A., Tannergard P., Bollag R.J., Godwin A.R., Ward D.C., Nordenskjold M., Fishel R., Kolodner R., Liskay R.M. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 8.Miyaki M., Konishi M., Tanaka K., Kikuchi-Yanoshita R., Muraoka M., Yasuno M., Igari T., Koike M., Chiba M., Mori T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271–272. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 9.Nicolaides N.C., Papadopoulos N., Liu B., Wei Y.F., Carter K.C., Ruben S.M., Rosen C.A., Haseltine W.A., Fleischmann R.D., Fraser C.M., Adams M.D., Venter J.C., Dunlop M.G., Hamilton S.R., Petersen G.M., de la Chapelle A., Vogelstein B., Kinzler K.W. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- 10.Wijnen J., de Leeuw W., Vasen H., van der Klift H., Moller P., Stormorken A., Meijers-Heijboer H., Lindhout D., Menko F., Vossen S., Moslein G., Tops C., Brocker-Vriends A., Wu Y., Hofstra R., Sijmons R., Cornelisse C., Morreau H., Fodde R. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet. 1999;23:142–144. doi: 10.1038/13773. [DOI] [PubMed] [Google Scholar]
- 11.Aaltonen L.A., Peltomaki P., Leach F.S., Sistonen P., Pylkkanen L., Mecklin J.P., Jarvinen H., Powell S.M., Jen J., Hamilton S.R., Petersen G.M., Kinzler K.W., Vogelstein B., de la Chapelle A. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 12.Lothe R.A., Peltomaki P., Meling G.I., Aaltonen L.A., Nystrom-Lahti M., Pylkkanen L., Heimdal K., Andersen T.I., Moller P., Rognum T.O., Fossa S.D., Haldorsen T., Langmark F., Brogger A., de la Chapelle A., Borresen A.L. Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res. 1993;53:5849–5852. [PubMed] [Google Scholar]
- 13.Thibodeau S.N., Bren G., Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 14.Thibodeau S.N., French A.J., Roche P.C., Cunningham J.M., Tester D.J., Lindor N.M., Moslein G., Baker S.M., Liskay R.M., Burgart L.J., Honchel R., Halling K.C. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res. 1996;56:4836–4840. [PubMed] [Google Scholar]
- 15.Muller W., Burgart L.J., Krause-Paulus R., Thibodeau S.N., Almeida M., Edmonston T.B., Boland C.R., Sutter C., Jass J.R., Lindblom A., Lubinski J., MacDermot K., Sanders D.S., Morreau H., Muller A., Oliani C., Orntoft T., Ponz De Leon M., Rosty C., Rodriguez-Bigas M., Ruschoff J., Ruszkiewicz A., Sabourin J., Salovaara R., Moslein G., Icg H. The reliability of immunohistochemistry as a prescreening method for the diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC): results of an international collaborative study. Fam Cancer. 2001;1:87–92. doi: 10.1023/a:1013840907881. [DOI] [PubMed] [Google Scholar]
- 16.Lindor N.M., Burgart L.J., Leontovich O., Goldberg R.M., Cunningham J.M., Sargent D.J., Walsh-Vockley C., Petersen G.M., Walsh M.D., Leggett B.A., Young J.P., Barker M.A., Jass J.R., Hopper J., Gallinger S., Bapat B., Redston M., Thibodeau S.N. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol. 2002;20:1043–1048. doi: 10.1200/JCO.2002.20.4.1043. [DOI] [PubMed] [Google Scholar]
- 17.Peltomaki P., Vasen H. Mutations associated with HNPCC predisposition: update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20:269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hampel H., Frankel W.L., Martin E., Arnold M., Khanduja K., Kuebler P., Nakagawa H., Sotamaa K., Prior T.W., Westman J., Panescu J., Fix D., Lockman J., Comeras I., de la Chapelle A. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 19.Barnetson R.A., Tenesa A., Farrington S.M., Nicholl I.D., Cetnarskyj R., Porteous M.E., Campbell H., Dunlop M.G. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354:2751–2763. doi: 10.1056/NEJMoa053493. [DOI] [PubMed] [Google Scholar]
- 20.Woods M.O., Williams P., Careen A., Edwards L., Bartlett S., McLaughlin J.R., Younghusband H.B. A new variant database for mismatch repair genes associated with Lynch syndrome. Hum Mutat. 2007;28:669–673. doi: 10.1002/humu.20502. [DOI] [PubMed] [Google Scholar]
- 21.van der Klift H., Wijnen J., Wagner A., Verkuilen P., Tops C., Otway R., Kohonen-Corish M., Vasen H., Oliani C., Barana D., Moller P., Delozier-Blanchet C., Hutter P., Foulkes W., Lynch H., Burn J., Moslein G., Fodde R. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2. MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC) Genes Chromosomes Cancer. 2005;44:123–138. doi: 10.1002/gcc.20219. [DOI] [PubMed] [Google Scholar]
- 22.Ligtenberg M.J.L., Kuiper R.P., Chan T.L., Goossens M., Hebeda K.M., Voorendt M., Lee T.Y.H., Bodmer D., Hoenselaar E., Hendriks-Cornelissen S.J.B., Tsui W.Y., Kong C.K., Brunner H.G., van Kessel A.G., Yuen S.T., van Krieken J.H.J.M., Leung S.Y., Hoogerbrugge N. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41:112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 23.Schouten J.P., McElgunn C.J., Waaijer R., Zwijnenburg D., Diepvens F., Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baudhuin L.M., Mai M., French A.J., Kruckeberg K.E., Swanson R.L., Winters J.L., Courteau L.K., Thibodeau S.N. Analysis of hMLH1 and hMSH2 gene dosage alterations in hereditary nonpolyposis colorectal cancer patients by novel methods. J Mol Diagn. 2005;7:226–235. doi: 10.1016/S1525-1578(10)60549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newcomb P.A., Baron J., Cotterchio M., Gallinger S., Grove J., Haile R., Hall D., Hopper J.L., Jass J., Le Marchand L., Limburg P., Lindor N., Potter J.D., Templeton A.S., Thibodeau S., Seminara D., Colon Cancer Family R. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:2331–2343. doi: 10.1158/1055-9965.EPI-07-0648. [DOI] [PubMed] [Google Scholar]
- 26.Poynter J.N., Siegmund K.D., Weisenberger D.J., Long T.I., Thibodeau S.N., Lindor N., Young J., Jenkins M.A., Hopper J.L., Baron J.A., Buchanan D., Casey G., Levine A.J., Le Marchand L., Gallinger S., Bapat B., Potter J.D., Newcomb P.A., Haile R.W., Laird P.W., Colon Cancer Family Registry I. Molecular characterization of MSI-H colorectal cancer by MLHI promoter methylation, immunohistochemistry, and mismatch repair germline mutation screening. Cancer Epidemiol Biomarkers Prev. 2008;17:3208–3215. doi: 10.1158/1055-9965.EPI-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gill S., Lindor N.M., Burgart L.J., Smalley R., Leontovich O., French A.J., Goldberg R.M., Sargent D.J., Jass J.R., Hopper J.L., Jenkins M.A., Young J., Barker M.A., Walsh M.D., Ruszkiewicz A.R., Thibodeau S.N. Isolated loss of PMS2 expression in colorectal cancers: frequency, patient age, and familial aggregation. Clin Cancer Res. 2005;11:6466–6471. doi: 10.1158/1078-0432.CCR-05-0661. [DOI] [PubMed] [Google Scholar]
- 28.Baudhuin L.M., Ferber M.J., Winters J.L., Steenblock K.J., Swanson R.L., French A.J., Butz M.L., Thibodeau S.N. Characterization of hMLH1 and hMSH2 gene dosage alterations in Lynch syndrome patients. Gastroenterology. 2005;129:846–854. doi: 10.1053/j.gastro.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 29.Baglietto L., Lindor N.M., Dowty J.G., White D.M., Wagner A., Gomez Garcia E.B., Vriends A.H., Dutch Lynch Syndrome Study Group. Cartwright N.R., Barnetson R.A., Farrington S.M., Tenesa A., Hampel H., Buchanan D., Arnold S., Young J., Walsh M.D., Jass J., Macrae F., Antill Y., Winship I.M., Giles G.G., Goldblatt J., Parry S., Suthers G., Leggett B., Butz M., Aronson M., Poynter J.N., Baron J.A., Le Marchand L., Haile R., Gallinger S., Hopper J.L., Potter J., de la Chapelle A., Vasen H.F., Dunlop M.G., Thibodeau S.N., Jenkins M.A. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2010;102:193–201. doi: 10.1093/jnci/djp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Umar A., Boland C.R., Terdiman J.P., Syngal S., de la Chapelle A., Ruschoff J., Fishel R., Lindor N.M., Burgart L.J., Hamelin R., Hamilton S.R., Hiatt R.A., Jass J., Lindblom A., Lynch H.T., Peltomaki P., Ramsey S.D., Rodriguez-Bigas M.A., Vasen H.F., Hawk E.T., Barrett J.C., Freedman A.N., Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kovacs M.E., Papp J., Szentirmay Z., Otto S., Olah E. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat. 2009;30:197–203. doi: 10.1002/humu.20942. [DOI] [PubMed] [Google Scholar]
- 32.Niessen R.C., Hofstra R.M.W., Westers H., Ligtenberg M.J.L., Kooi K., Jager P.O.J., de Groote M.L., Dijkhuizen T., Olderode-Berends M.J.W., Hollema H., Kleibeuker J.H., Sijmons R.H. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48:737–744. doi: 10.1002/gcc.20678. [DOI] [PubMed] [Google Scholar]
- 33.Hitchins M.P., Wong J.J.L., Suthers G., Suter C.M., Martin D.I.K., Hawkins N.J., Ward R.L. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356:697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- 34.Gazzoli I., Loda M., Garber J., Syngal S., Kolodner R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002;62:3925–3928. [PubMed] [Google Scholar]
- 35.Suter C.M., Martin D.I.K., Ward R.L. Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet. 2004;36:497–501. doi: 10.1038/ng1342. [DOI] [PubMed] [Google Scholar]
- 36.Hitchins M., Williams R., Cheong K., Halani N., Lin V.A.P., Packham D., Ku S., Buckle A., Hawkins N., Burn J., Gallinger S., Goldblatt J., Kirk J., Tomlinson I., Scott R., Spigelman A., Suter C., Martin D., Suthers G., Ward R. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129:1392–1399. doi: 10.1053/j.gastro.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Miyakura Y., Sugano K., Akasu T., Yoshida T., Maekawa M., Saitoh S., Sasaki H., Nomizu T., Konishi F., Fujita S., Moriya Y., Nagai H. Extensive but hemiallelic methylation of the hMLH1 promoter region in early-onset sporadic colon cancers with microsatellite instability. Clin Gastroenterol Hepatol. 2004;2:147–156. doi: 10.1016/s1542-3565(03)00314-8. [DOI] [PubMed] [Google Scholar]
- 38.Valle L., Carbonell P., Fernandez V., Dotor A.M., Sanz M., Benitez J., Urioste M. MLH1 germline epimutations in selected patients with early-onset non-polyposis colorectal cancer. Clin Genet. 2007;71:232–237. doi: 10.1111/j.1399-0004.2007.00751.x. [DOI] [PubMed] [Google Scholar]
- 39.Sheng J.-Q., Zhang H., Ji M., Fu L., Mu H., Zhang M.-Z., Huang J.-S., Han M., Li A.-Q., Wei Z., Sun Z.-Q., Wu Z.-T., Xia C- H., Li S.-R. Genetic diagnosis strategy of hereditary non-polyposis colorectal cancer. World J Gastroenterol. 2009;15:983–989. doi: 10.3748/wjg.15.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H., Taylor N.P., Sotamaa K.M., Mutch D.G., Powell M.A., Schmidt A.P., Feng S., Hampel H.L., de la Chapelle A., Goodfellow P.J. Evidence for heritable predisposition to epigenetic silencing of MLH1. Int J Cancer. 2007;120:1684–1688. doi: 10.1002/ijc.22406. [DOI] [PubMed] [Google Scholar]
- 41.Chan T.L., Yuen S.T., Kong C.K., Chan Y.W., Chan A.S.Y., Ng W.F., Tsui W.Y., Lo M.W.S., Tam W.Y., Li V.S.W., Leung S.Y. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet. 2006;38:1178–1183. doi: 10.1038/ng1866. [DOI] [PubMed] [Google Scholar]
- 42.Yu G., Zhang X., Wang H., Rui D., Yin A., Qiu G., He Y. CpG island methylation status in the EpCAM promoter region and gene expression. Oncol Rep. 2008;20:1061–1067. [PubMed] [Google Scholar]
- 43.Whitelaw E., Proudfoot N. Alpha-thalassaemia caused by a poly(A) site mutation reveals that transcriptional termination is linked to 3′ end processing in the human alpha 2 globin gene. EMBO J. 1986;5:2915–2922. doi: 10.1002/j.1460-2075.1986.tb04587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tufarelli C., Stanley J.A.S., Garrick D., Sharpe J.A., Ayyub H., Wood W.G., Higgs D.R. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet. 2003;34:157–165. doi: 10.1038/ng1157. [DOI] [PubMed] [Google Scholar]
- 45.Plevova P., Sedlakova E., Zapletalova J., Krepelova A., Skypalova P., Kolar Z. Expression of the hMLH1 and hMSH2 proteins in normal tissues: relationship to cancer predisposition in hereditary non-polyposis colon cancer. Virchows Arch. 2005;446:112–119. doi: 10.1007/s00428-004-1139-5. [DOI] [PubMed] [Google Scholar]