

Abstract
The termination of epoxide-initiated cascade cyclizations with a range of “protected” phenols is described. When the protecting group can be lost as a stabilized electrophile, the cascade process continues beyond ring closure to afford products which have undergone a tandem electrophilic aromatic substitution. A number of groups have proven viable in this process and the regiochemistry of their substitution reactions has been studied. Application of this methodology in the first total synthesis of (+)-schweinfurthin A, a potent anti-proliferative agent, has been achieved.
Keywords: cascade cyclization, tandem reaction, polyene cyclization, cascade reaction, schweinfurthin
Introduction
As the field of organic synthesis has evolved, the laboratory synthesis of ever more complex natural products has become feasible.1–3 For such syntheses to be practical however, it is very helpful to assemble multiple bonds in a single transformation, and in this context catalytically driven tandem4 or cascade reactions can be particularly appealing.5–10 Epoxides have proven their utility as cascade components6–9 and arguably one of the most powerful cascade classes is based upon epoxide initiated, cationic polyene cyclizations.10–12 Although pioneering descriptions of such cascade cyclizations were set forth in the 1950’s,10–12 modern chemists continue to explore polyene cyclizations and recent studies have lead to many significant advances in novel modes of cascade initiation.13–25 In contrast, very little has been done to extend cationic cascade cyclizations beyond terminal ring closure.
Our group has been interested in epoxide initiated cascade cyclizations as they pertain to the formation of hexahydroxanthenes and particularly as they apply to synthesis of the schweinfurthins.26–31 Previous studies have shown that “MOM-protected” phenols are sufficiently nucleophilic to terminate a cascade sequence (e.g. conversion of epoxide 1 to hexahydroxanthene 2, Scheme 1), and also have discovered that the MOM group could be transferred to the A-ring hydroxyl group during the cyclization (e.g. yielding compound 3).29 If transfer of the MOM group is the result of a transient electrophilic species, then one might envision other applications of this electrophile.32 In an initial report on extension of this cascade process,33 our lab demonstrated that a MOM group can be transferred to an adjacent aromatic ring with high efficiency. This represents a cationic cascade ultimately terminated with a tandem electrophilic aromatic substitution reaction (e.g. cyclization of epoxide 4 to compound 5). In this initial example, the reaction sequence continues beyond ring closure to electrophilic aromatic substitution, and a new carbon-carbon bond is the result. That tandem process allowed a highly efficient synthesis of (+)-angelichalcone.33
Scheme 1.

Cascade Cyclization Routes to Hexahydroxanthenes.
The genesis of the present work resides in the concept that a new tandem cascade sequence might be used in the synthesis of several schweinfurthins via a single late stage intermediate. This report describes: 1) issues of regioselectivity that arise during application of the MOM-based cascade cyclization/aromatic substitution process; 2) exploration of tandem cascade cyclization/aromatic substitution with other “protecting” groups; and 3) application of a new tandem cascade cyclization/aromatic substitution sequence to the formal synthesis of schweinfurthins B, E, F, and G, and the first total synthesis of (+)-schweinfurthin A.29, 30, 34, 35
Results and Discussion
Several schweinfurthins and numerous analogues have been prepared through synthesis,27, 29, 30 but one of the most potent congeners34, 35 of the natural family (schweinfurthin A, 9) has remained elusive until now. As part of an ongoing program to determine their mode of action as antiproliferative agents,36–38 we have attempted to optimize access to a key intermediate en route to schweinfurthin A. Previous work has utilized vanillin as an inexpensive, commercial starting material.26, 27, 30 This reagent fits especially well into syntheses of schweinfurthin B, E, and F (10–12), where the methoxy group derived from vanillin remains on the C-5 position of the natural products, but for synthesis of schweinfurthin A or G (9 and 13) the use of this starting material would require demethylation early in the sequence. In an effort to develop a more versatile late-stage intermediate, a tantalizing synthetic proposal exploiting cascade cyclization terminated by electrophilic aromatic substitution33 was envisioned to allow a diversity oriented synthesis (Scheme 2). This approach would be based upon a tandem cascade cyclization/aromatic substitution of the “MOM-protected” epoxide 6 to obtain the hexahydroxanthene 7 followed by elaboration of the benzylic position at C-5 to phenolic functionality via standard oxidation protocols. If an efficient route could be developed, the intermediate 8 would represent a single keystone intermediate which could be converted into both C-5 hydroxy- and methoxy-substituted schweinfurthins (Scheme 2) as well as new analogues.
Scheme 2.

Synthetic Plan for a Divergent Route to the Schweinfurthins.
To explore this possibility, methyl 4-hydroxybenzoate (14) was elaborated rapidly to the requisite R epoxide 15 by standard methods (Scheme 3).39, 40 To our delight, when compound 15 was treated with BF3·OEt2 under standard cyclization conditions, the major product obtained had undergone both cyclization and electrophilic aromatic substitution. Unfortunately, even though the hexahydroxanthene 16 was obtained in reasonable yield as a single stereo- and regioisomer, efforts directed at further elaboration met with limited success. While oxidation, hydrolysis, and substitution reactions demonstrated high regioselectivity, they uniformly favored the benzylic position para to the hexahydroxanthene oxygen, rather than the position ortho. Thus, for example, treatment of compound 16 with excess DDQ afforded the single aldehyde 17 in nearly quantitative yield.
Scheme 3.

Synthesis and Regioselective Oxidation of Compound 16.
In an effort to sway this natural selectivity through modification of steric or electronic factors, a more extended sequence was pursued. Reduction of aldehyde 17 and protection of the resulting alcohol was readily accomplished and allowed preparation of a variety of protected alcohols. Of those prepared, including the Boc derivative, the p-nitrobenzoate ester, the MOM acetal, and the TBS ether, only derivatives with acyl groups were found to allow preparation of the desired C-5 aldehyde. For example, after preparation of the Boc derivative 18 or the p-nitrobenzoate 19, subsequent reaction with DDQ gave the desired aldehydes 20 and 21 in ~75% yield. While this longer sequence allowed preparation of the desired aldehyde regioisomer, the incompatibility of carbonyl-containing substituents with the organolithium intermediate necessary for the synthesis of epoxide 15 rules out early introduction of these protecting groups. Given the inherent impracticality and non-ideality41–43 of the oxidation/reduction and protection/deprotection sequences described above, it became prudent to explore alternative approaches. Based on the hypothesis that an ortho-selective differentiation of the benzylic positions could be accomplished if a group other than the MOM acetal were induced to undergo a cascade cyclization/electrophilic aromatic substitution, this process was examined with other phenolic substituents.
Exploration of Cascade Cyclizations Terminated with Aromatic Substitution
Because it contains unsubstituted ortho and para positions and because it is readily available,33 4-geranylresorcinol was used as the starting point to explore the viability of different phenolic substituents in cascade cyclization/aromatic substitution reactions. Numerous racemic epoxides bearing different groups at the phenolic oxygens (22–27) were prepared by standard methods and then treated with BF3·OEt2 under typical cyclization conditions.33 Because the electrophilic species generated from the previously employed MOM acetal would be stabilized by the adjacent heteroatom, other acetals of similar structures were explored first (Table 1). As one might predict, other common alkoxymethyl protecting groups undergo parallel cascade cyclization/aromatic substitution reactions. The BOM (23), SEM (24), and MEM (25) derivatives all favored formation of the substituted products 29a–31a in yields comparable to that observed in the MOM case (28a).33 All of these products were obtained as single diastereo- and regioisomers. Furthermore, when a competition reaction was conducted with a 1:1 mixture of compounds 23 and 24, the major products were compounds 29a and 30a, both were found in yields comparable to those for the individual experiments, and no evidence for a crossed product was detected. The pivaloyloxymethyl (or POM) group, which features an acyloxy rather than an alkoxy substituent, was resistant to a complete cascade cyclization and the reaction of compound 26 instead gave the bridged ether 32c as the only major product.44–49 The surprising case in this series was the cyclization involving the 4-chlorophenoxymethyl group (27), which arose from commercially available α,4-dichloroanisole. This reaction provided compound 33b as the major product and none of compound 33a was isolated. Because all of the other substrates in Table 1 favored formation of either the a- or c-type products, this is unique among the groups surveyed. The use of secondary acetals was less productive under standard conditions,40 and the modest mass balance reflected in the isolated yields for some of these cyclizations may result from formation of trace amounts of the b-type structure as well as any of several epoxide rearrangement pathways.12, 31, 50
Table 1.
Cyclizations of Alkoxymethyl-substituted Phenols a
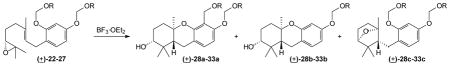 | |||||
|---|---|---|---|---|---|
| Entry | substrate (R =) | common abbreviation | A % yieldb | b % yieldb | c % yieldb |
| 133 | CH3 (22) | MOM | 52 (28a) | 30 (28b) | |
| 2 | Bn (23) | BOM | 62 (29a) | ||
| 3 | CH2CH2TMS (24) | SEM | 57 (30a) | ||
| 4 | CH2CH2OCH3 (25) | MEM | 53 (31a) | 28 (31b) | |
| 5 | C(O)C(CH3)3 (26) | POM | 47 (32c) | ||
| 6 | 4-ClC6H4 (27) | 56 (33b) | 37 (33c) | ||
See Supporting information for experimental details.
Isolated yields.
Other common protecting groups also were explored, including the acetate (34), benzoate, and Boc protected compounds. In all of these cases however, the major products were the bridged A-ring ethers as shown for the acetate 35 (Figure 1).40 This type of structure has been observed in several earlier studies and in some natural products.44–49, 51, 52 A crystalline sample of the product 35 was subjected to X-ray diffraction analysis to assign the relative configuration unambiguously, and acetate 35 was found to have an exo (trans) structure. On this basis, the structures of the parallel products from cyclization of the benzoate and Boc derivatives were assigned analogous stereochemistry.40,20, 53
Figure 1.

ORTEP of Compound 35.
The tandem cascade cyclization/aromatic substitution process then was explored with various alkyl groups as the phenol substituents (36–43, Table 2). Not surprisingly, the methyl compound 36 gave bridged ether 44c as the major product. The allyl derivative 37, which would add stabilization to the presumed intermediate through π delocalization, proved more interesting. Exposure of the diallyl epoxide 37 to BF3·OEt2 resulted in a mixture of products, with the bridged ether 45c predominating, and the cyclization/aromatic substitution product (45a) isolated as a minor component. This suggests that a group that could provide at least some stabilization to a formal cation is required for a tandem cascade cyclization/aromatic substitution process to occur.
Table 2.
Cascade Cyclization/Aromatic Substitution of Alkyl-substituted Phenols.a
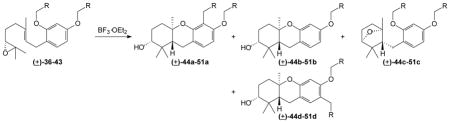 | |||||
|---|---|---|---|---|---|
| Entry | Substrate (R =) | a % yieldb | b % yieldb | c % yieldb | d % yieldb |
| 1 | H (36) | 72 (44c) | |||
| 2 | CH=CH2 (37) | 8 (45a) | 32 (45c) | ||
| 3 | 4-NO2C6H4 (38) | 20 (46c) | |||
| 4 | C6H5 (39) | 43 (47a) | 18 (47b) | 2 (47d) | |
| 5 | 2-BrC6H4 (40) | 49 (48c) | |||
| 6 | 4-CH3OC6H4 (41) | 33 (49a) | 12 (49b)c | 19 (49d) | |
| 7 | 3,4,5-trimethoxyphenyl (42) | 41 (50b)d | 28 (50d) | ||
| 8 | 3-furyl (43) | 49 (51a) | |||
See Supporting Information for experimental details.
Isolated yields.
Isolated as the A-ring PMB ether.
Isolated as the A-ring alcohol (32%) and A-ring ether (9%).
To strengthen a hypothesis regarding the impact of the displaced electrophile’s stability on the results of cascade cyclization/aromatic substitution reactions, several benzyl groups with varied substituents were examined. As one might expect, addition of withdrawing or donating groups to modify electron density at the benzylic position leads to stark differences in the product distribution (Table 2). With the parent system, benzyl ether 39, standard reaction conditions afforded the product of the tandem process as the major product (47a), along with a significant amount of the cascade-only product 47b. A trace of the para-substituted product 47d was found in this reaction, which marks the first observation of electrophilic aromatic substitution at the para position during the cascade process. Addition of an electron-withdrawing 4-nitro substituent to the phenyl ring (38) resulted in an unusually complicated reaction mixture, and the bridged ether 46c was the only significant product found. While reaction of the 2-bromophenyl derivative 40 gave a much less complicated reaction mixture, again only the bridged product (48c) was observed. In contrast, addition of an electron donating 4-methoxy substituent (41) resulted in cascade cyclization/aromatic substitution to afford the ortho product 49a, but a significant amount of the para regioisomer 49d also was observed. Further increasing the electron density of the aromatic system with the trimethoxybenzyl group (42) completely reversed the ortho regioselectivity, and only substitution at the para position was observed (50d) along with the cascade-only product (50b). Finally, the 3-furyl group was examined as a representative case of a heteroaryl system because it is found as a component of numerous natural terpenoids.54–57 The furan derivative 43 was found to undergo cascade cyclization/aromatic substitution and afford compound 51a as the major product. Thus it appears that a variety of neutral or electron rich arenes will afford a major product that has undergone cascade cyclization/aromatic substitution, but as the ability of the arene to stabilize a benzylic cation increases, less regioselectivity is observed. Ultimately, if there is minimal stabilization for a benzylic cation, the cascade process stops at the A-ring bridged ethers.
Based on the observations described above, at least a partial mechanistic picture of this process can be offered (Scheme 4). Initial complexation of BF3 to the epoxide oxygen, generalized as structure 52, could be followed by a cascade cyclization to the oxonium ion 53.58, 59 If loss of the primary alkyl group from the oxonium ion would afford an unstabilized carbocation (e.g. a methyl group), then the reaction may continue through the tertiary carbocation 54 instead. In this case, attack of the A-ring oxygen on the tertiary cation would explain formation of ether 55. The bridged ether 55 would have the stereochemistry shown, as found in the diffraction analysis of compound 35, because the C-6’ stereochemistry is set in the original epoxide and the C-2’ stereochemistry would be established through the chair-chair transition state11, 12, 27 that leads to oxonium ion 53. If oxonium ion 53 instead undergoes loss of a semi- or highly-stabilized carbocation, the adjacent arene may serve as a Lewis base and quickly trap this electrophile. This sequence would rapidly afford a π complex such as structure 56. This branch of the reaction manifold would be consistent with the lack of cross-over products observed in earlier studies with isotopically labeled MOM groups,33 as well as the fidelity observed in the competition experiment with compounds 23 and 24. Rapid formation of an ipso complex would be expected for any relatively short-lived cationic species (e.g. allyl or BOM), which would lead to the proximal substitution product at C-2. In the case of longer-lived cationic species, partial diffusion would lead to formation of the distal ipso complex and subsequently substitution at C-6. Finally, in those cases where dissociation of the oxonium ion 53 gives the longest lived cations (e.g. trimethoxybenzyl) a greater percentage of the unsubstituted hexahydroxanthene product would be more likely.
Scheme 4.

A Possible Mechanistic Picture.
Taken together, these results indicate that the cascade cyclization/aromatic substitution is a general process for those cases where the phenolic substituent can form a reasonably stabilized carbocation. With a touch of reflection, it should be clear that this process is a powerful strategy: this tandem reaction sequence closed two rings, generated two additional stereogenic centers with complete control of the relative stereochemistry, and forged a new functionalized carbon-carbon bond in a single operation. In addition, this cascade sequence has proven its viability in forming new benzylic positions with a range of substituent groups. With this improved understanding of the scope of this process, application of a tandem cascade cyclization/aromatic substitution sequence to a schweinfurthin synthesis was reexamined.
Application to the Synthesis of Schweinfurthins A, B, E, F, and G
Application of the cascade cyclization/aromatic substitution strategy to the synthesis of key intermediates en route to schweinfurthins now could proceed as shown in Scheme 5. The expanded scope of the tandem reaction sequence was exploited to differentiate the newly generated benzylic position at C-5 from the original benzyl methyl ether at an early stage in the synthesis (vide supra). To accomplish this objective, a BOM acetal was chosen as the phenolic substituent, both because of its commercial availability and the assumption that the benzyl ether could be selectively cleaved by hydrogenolysis late in the synthesis. The synthetic sequence was initiated with bromination of 4-hydroxybenzoate (58) followed by BOM protection. Reduction of the intermediate ester and methylation all proceeded smoothly to afford arene 59 in high overall yield on a 50 gram scale. Halogen metal exchange, transmetalation, and addition of (R)-6,7-epoxygeranyl bromide (up to 93% ee)28, 29 afforded epoxide 60 in excellent yield, again on a multi-gram scale. The crucial cascade cyclization/aromatic substitution proceeded smoothly to afford the desired tricycle 62 along with the unsubstituted analogue 61 (which had been obtained earlier from reaction of the corresponding MOM compound). Although compounds 61 and 62 were obtained as an inseparable mixture, each was a single regio- and diastereoisomer. Separation of these products was readily accomplished after selective hydrogenolysis, which afforded the benzylic alcohol 63 and recovered hexahydroxanthene 61, provided the reaction was monitored frequently. With excessive hydrogen pressure or extended reaction duration, over reduction was observed.
Scheme 5.

Formal Synthesis of Schweinfurthins B, E, F, and G.
Once alcohol 63 was in hand, installation of the requisite C-5 phenol proceeded smoothly (Scheme 5). Chemoselective oxidation of benzyl alcohol 63 to the corresponding aldehyde 64 was accomplished upon treatment with MnO2. Subsequent Baeyer-Villiger oxidation60 with m-CPBA and hydrolysis of the resultant formate provided phenol 65 in excellent yield. This reaction sequence extends cascade cyclization/aromatic substitution strategy to allow the introduction of the C-5 phenol central to the schweinfurthins. In fact, phenol 65 serves as the keystone divergent intermediate which intersects most of the previously synthesized schweinfurthins. Alkylation of phenol 65 with either methyl iodide or MOMCl proceeded with excellent selectivity to afford intermediates 66 and 67, respectively. The synthesis of compound 66 constitutes a formal synthesis of schweinfurthins B, E, and F as well as most of the presently known schweinfurthin analogues.27, 29, 30, 36, 38, 61–63 In a similar sense, preparation of compound 67 accomplishes a formal synthesis of schweinfurthin G.29 More importantly, the ready availability of compound 67 has provided the means to pursue the synthesis of the parent compound in this family, (+)-schweinfurthin A.
With gram amounts of intermediate 67 now in hand, the total synthesis of (+)-schweinfurthin A was pursued (Scheme 6).30 Intermediate 67 was oxidized with TPAP/NMO64 to provide the new ketone 68. Condensation of ketone 68 with benzaldehyde provided enone 69, which was to be utilized as a latent carbonyl group.65, 66 Reduction proceeded smoothly under Luche conditions67 to afford alcohol 70 in excellent yield. The relative stereochemistry was assigned based on consideration of models and precedence,30 and ultimately confirmed by comparison of the final product to natural material. Alcohol 70 was subjected to Upjohn dihydroxylation68 to provide triol 71. Subsequent glycolytic cleavage was accomplished in a separate step upon reaction with NaIO4. This reaction was highly selective for cleavage of the exocyclic diol, and provided ketone 72 in very good yield. In comparison to the earlier synthesis of schweinfurthin B where attempted oxidation in the presence of a C-2 MOM group was too sluggish to be useful,30 the C-2 hydroxyl group in this series greatly accelerated the desired oxidation. Diastereoselective reduction of ketone 72 provided diol 73, where the C-2 stereochemistry could be assigned based on precedents30, 69 and a straightforward analysis of the relevant coupling constants in the 1H NMR spectrum. Treatment of benzyl ether 73 with DDQ provided aldehyde 74, demonstrating once again the value of a benzyl methyl ether as a latent benzaldehyde.30, 70, 71 Unfortunately, ensuing attempts at a direct HWE condensation of aldehyde 74 with phosphonate 76 proved troublesome. To circumvent this problem, the A-ring diol of aldehyde 74 was protected by treatment with excess MOMCl and base to afford aldehyde 75. Subsequent attempts at the HWE condensation of aldehyde 75 with phosphonate 7672, 73 in the presence of KHMDS afforded stilbene 77 in moderate yield. Global hydrolysis of the MOM acetals provided schweinfurthin A in an acceptable yield. In direct NMR analyses, synthetic (+)-schweinfurthin A proved identical to an authentic sample of the natural material and displayed a specific rotation of the same sign and magnitude as that reported for the natural product ([α]25D = +47 (c 1.0, EtOH) from epoxide of 90% ee; lit ([α]25D = +51.8 (c 2.0, EtOH).35 Therefore, the natural antipode of schweinfurthin A now can be assigned as (+)-(2S, 3R, 4aR, 9aR)-schweinfurthin A.
Scheme 6.

Total Synthesis of (+)-Schweinfurthin A
Conclusions
In conclusion, the scope of cascade cyclizations terminated by electrophilic aromatic substitution has been expanded to include an array of substituents that ultimately become incorporated on the arene. This tandem process appears to be general as long as the original protecting group affords an electrophile that can be viewed as a stabilized cation. The diether 16 was found to undergo a highly regioselective para oxidation upon treatment with DDQ, but migration of other stabilized motifs has allowed rapid generation of complex polycycles that lead to the ortho oxidized products. To demonstrate the potential of this approach, a cascade cyclization/aromatic substitution strategy has been used to provide a late stage schweinfurthin intermediate that represents a formal synthesis of four natural products. Furthermore, this intermediate has been elaborated to complete the first synthesis of natural (+)-schweinfurthin A, which makes the rare natural product available by synthesis. Efforts to exploit the utility of this reaction manifold in other natural product syntheses are in progress and will be reported in due course.74–76
Experimental Section
Epoxide 15
To a solution of the corresponding olefin40 (6.2 g, 19 mmol) and Shi’s catalyst (1.4 g, 5.0 mmol) in aq buffer (80 mL, 2 M K2CO3 and 4 mM EDTA) and organic phase (100 mL, 1:1:1 CH2Cl2/MeCN/EtOH) at 0 °C was added hydrogen peroxide (7 mL, 30%) via syringe pump over 20 h. The reaction was then quenched by addition of aq Na2SO3. The resulting solution was extracted with ethyl acetate, and the organic phase was washed with brine. After the organic phase was dried (MgSO4), and concentrated in vacuo, final purification by column chromatography (20% ethyl acetate in hexanes) afforded recovered starting material (2.8 g, 44%) and epoxide 15 (2.9 g, 44%, ee ranged from 88% – 92%) as a colorless oil: 1H NMR (CDCl3) δ 7.11 – 7.08 (m, 2H), 7.01 (d, J = 8.0 Hz, 1H), 5.35 (m, 1H), 5.19 (s, 2H), 4.35 (s, 2H), 3.45 (s, 3H), 3.34 (s, 3H), 3.35 (m, 2H), 2.69 (t, J = 6.4 Hz, 1H), 2.19 – 2.11 (m, 2H), 1.73 (s, 3H), 1.68 – 1.60 (m, 2H), 1.25 (s, 3H), 1.23 (s, 3H); 13C NMR (CDCl3) δ 154.4, 134.8, 131.1, 130.4, 129.3, 126.6, 123.0, 113.6, 94.2, 74.3, 64.0, 58.2, 57.8, 55.8, 36.3, 28.5, 27.3, 24.7, 18.6, 16.0; HRMS (EI) m/z calcd for C20H30O4 (M+) 334.2144, found 334.2135.
Ether 16
To a solution of epoxide 15 (1.15 g, 3.4 mmol) in CH2Cl2 (350 mL) at −78 °C was added BF3·OEt2 (2.2 mL, 20 mmol). After 9 min, the reaction was quenched by addition of excess Et3N (5 mL). The resulting solution was concentrated in vacuo and the resulting oil was dissolved in ethyl acetate which was washed with 1N HCl followed by brine. The organic phase was dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (30% ethyl acetate in hexanes) afforded the cyclized product without a C-5 substituent40 (229 mg, 23%) along with ether 16 (620 mg, 54%) as a yellow oil: [α]25D = +39 (c 0.9, CHCl3, 82% ee by HPLC); 1H NMR (CDCl3) δ 7.12 (s, 1H), 6.97 (s, 1H), 4.40 (s, 2H), 4.30 (s, 2H), 3.38 (s, 3H), 3.34 (s, 3H), 3.09 (dd, J = 11.2, 4.0 Hz, 1H), 2.85 (br, 1H), 2.66 – 2.62 (m, 2H), 1.93 – 1.89 (m, 1H), 1.72 – 1.52 (m, 4H), 1.13 (s, 3H), 0.97 (s, 3H), 0.79 (s, 3H); 13C NMR (CDCl3) δ 150.0, 128.7, 128.5, 126.3, 125.7, 121.2, 77.1, 76.2, 74.5, 68.9, 58.1, 57.7, 46.5, 37.9, 37.5, 27.9, 27.0, 22.8, 19.8, 14.0; HRMS (EI) m/z calcd for C20H30O4 (M+) 334.2144, found 334.2134.
Aldehyde 17
To a solution of benzyl ether 16 (400 mg, 1.2 mmol) in CH2Cl2 (10 mL) and water (1.0 mL) at rt was added DDQ (308 mg, 1.4 mmol). After 2 hr, the reaction was quenched by addition of NaHCO3. The resulting mixture was extracted with CH2Cl2. The organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo afforded aldehyde 17 as orange oil which was advanced to the next step without further purification: 1H NMR (CDCl3) δ 9.79 (s, 1H), 7.70 (d, J = 1.2 Hz, 1H), 7.56 (d, J = 1.2 Hz, 1H), 4.46 (s, 2H), 3.41 (s, 3H), 3.40 (m, 1H), 2.76 – 2.71 (m, 2H), 2.25 (br, 1H), 2.01 (ddd, J = 12.4, 3.6, 3.6 Hz, 1H), 1.84 (dq, J = 12.8, 3.6 Hz, 1H), 1.73 (dd, J = 13.0, 3.8 Hz, 1H), 1.65 (dd, J = 11.8, 5.8 Hz, 1H), 1.59 (m, 1H), 1.19 (s, 3H), 1.07 (s, 3H), 0.85 (s, 3H); 13C NMR (CDCl3) δ 191.3, 156.1, 130.7, 128.5, 128.4, 127.0, 122.1, 78.0, 77.5, 68.7, 58.5, 46.3, 38.3, 37.4, 27.9, 27.1, 22.8, 20.2, 14.2; HRMS (EI) m/z calcd for C19H26O4 (M+) 318.1831, found 318.1813.
Boc Carbonate 18
To a solution of the parent alcohol40 (179 mg, 0.56 mmol) in THF in an ice bath, was added NaH (190 mg, 60% in oil, 4.8 mmol) followed by Boc2O (153 mg, 0.70 mmol). After 12 hr, the reaction was quenched by addition of water, the resulting solution was extracted with ethyl acetate, and the organic phases were washed with brine. The organic phase was dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (40% ethyl acetate in hexanes) afforded Boc carbonate 18 (102 mg, 43%) as a colorless oil: 1H NMR (CDCl3) δ 7.17 (d, J = 2.0 Hz, 1H), 7.03 (d, J = 1.6 Hz, 1H), 4.97 (s, 2H), 4.39 (s, 2H), 3.39 (s, 3H), 3.39 – 3.33 (m, 1H), 2.69 – 2.65 (m, 2H), 2.09 (br, 1H), 1.96 (dt, J = 12.8, 3.4 Hz, 1H), 1.81 (dq, J = 8.8, 3.6 Hz, 1H), 1.72 (dd, J = 14.0, 4.0 Hz, 1H), 1.63 (dd, J = 12.0, 6.4 Hz, 1H), 1.58 – 1.50 (m, 1H), 1.46 (s, 9H), 1.15 (s, 3H), 1.04 (s, 3H), 0.83 (s, 3H); 13C NMR (CDCl3) δ 156.4, 153.4, 150.7, 129.4, 127.0, 126.2, 121.6, 81.9, 77.7, 76.4, 69.0, 68.8, 58.3, 46.5, 38.2, 37.6, 28.0, 27.7 (3C), 27.2, 22.9, 19.9, 14.1; HRMS (EI) m/z calcd for C24H36O6 (M+) 420.2512, found 420.2507.
p-Nitrobenzoate 19
To a solution of the parent alcohol40 (222 mg, 0.69 mmol) in THF (10 mL) at rt was added 4-nitrobenzoyl chloride (160 mg, 0.86 mmol) followed by py (0.12 mL, 1.5 mmol). After 3.5 hr, the reaction was quenched by addition of water and the resulting mixture was extracted with ethyl acetate. The organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (30% to 50% ethyl acetate in hexanes) afforded ester 19 (209 mg, 64%) as colorless oil: [α]25D = +41 (c 0.6, CHCl3, 82% ee by HPLC); 1H NMR (CDCl3) δ 8.20 – 8.05 (m, 4 H), 7.19 (d, J = 1.6 Hz, 1H), 7.06 (d, J = 2.0 Hz, 1H), 5.20 (s, 2H), 4.35 (d, J = 12.4 Hz, 1H), 4.30 (d, J = 12.4 Hz, 1H), 3.34 (s, 3H), 3.30 – 3.27 (m, 1H), 2.65 – 2.62 (m, 2H), 1.94 – 1.91 (m, 1H), 1.78 – 1.50 (m, 4H), 1.11 (s, 3H), 1.02 (s, 3H), 0.80 (s, 3H); 13C NMR (CDCl3) δ 165.6, 152.0, 151.5, 136.5, 131.6 (2C), 130.8, 128.2, 127.4, 127.1, 124.3 (2C), 123.0, 78.3, 77.7, 70.0, 68.6, 58.7, 47.7, 39.2, 38.7, 28.7, 27.8, 23.9, 20.4, 14.8; HRMS (EI) m/z calcd for C26H31NO7 (M+) 469.2101, found 469.2096.
Aldehyde 20
To a solution of carbonate 18 (95 mg, 0.23 mmol) in CH2Cl2 (5 mL) and water (0.5 mL) at rt was added DDQ (110 mg, 0.48 mmol). After 2 hr, the reaction was quenched by addition of NaHCO3, and the resulting mixture was extracted with CH2Cl2. The organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo to afford aldehyde 20 (71 mg, 78%) as an orange oil: [α]25D = +42 (c 0.7, CHCl3, 82% ee by HPLC); 1H NMR (CDCl3) δ 10.37 (s, 1H), 7.64 (d, J = 1.6 Hz, 1H), 7.34 (d, J = 2.0 Hz, 1H), 4.98 (s, 2H,), 3.44 (dd, J = 11.2, 4.4 Hz, 1H), 2.75 – 2.71 (m, 2H), 2.07 – 2.04 (m, 1H), 1.48 (s, 9H), 1.25 (s, 3H), 1.07 (s, 3H), 0.88 (s, 3H); 13C NMR (CDCl3) δ 189.8, 156.2, 153.3, 136.3, 126.8, 126.3, 124.2, 123.8, 82.4, 80.0, 77.7, 68.0, 46.3, 38.3, 37.4, 28.1, 27.7 (3C), 27.2, 22.8, 20.1, 14.2; HRMS (EI) m/z calcd for C23H32O6 (M+) 404.2199, found 404.2192.
Aldehyde 21
According to the procedure described above, compound 19 was treated with DDQ followed by standard work up. Concentration of the resulting solution afforded aldehyde 21 (140 mg, 71%) as an orange oil: 1H NMR (CDCl3) δ 10.38 (s, 1H), 8.24 (d, J = 8.6 Hz, 2H), 8.18 (d, J = 8.6 Hz, 2H), 7.71 (d, J = 2.4 Hz, 1H), 7.41 (d, J = 2.0 Hz, 1H), 5.28 (s, 2H), 3.42 (dd, J = 11.6, 4.4 Hz, 1H), 2.77 – 2.74 (m, 2H), 2.05 (dt, J = 12.8, 3.2 Hz, 1H), 2.04 (br, 1H), 1.89 (dq, J = 12.8, 3.2 Hz, 1H), 1.78 (dd, J = 13.2, 3.6 Hz, 1H), 1.70 (dd, J = 11.6 Hz, 6.4 Hz, 1H), 1.64 – 1.60 (m, 1H), 1.23 (s, 3H), 1.10 (s, 3H), 0.87 (s, 3H); 13C NMR (CDCl3) δ 189.7, 164.4, 156.3, 150.4, 136.4, 135.3, 130.7 (2C), 126.4, 126.4, 124.2, 124.0, 123.4 (2C), 78.1, 77.5, 66.9, 46.2, 38.3, 37.4, 28.0, 27.1, 22.8, 20.1, 14.2; HRMS (EI) m/z calcd for C25H27NO7 (M+) 453.1788, found 453.1782.
BOM Epoxide 23
To the parent geranyl arene40 (411 mg, 0.88 mmol) in CH2Cl2 (15 mL) at −20 °C was added m-CPBA (200 mg, 0.89 mmol) slowly. The reaction was allowed to stir for 1 hour and then quenched by addition of Na2SO3 (sat.) and extracted with CH2Cl2. The organic extracts were washed with 0.5 M NaOH, brine, dried (MgSO4), filtered, and concentrated in vacuo. Final purification by flash column chromatography (4% to 5% ethyl acetate in hexanes) afforded the external epoxide 23 (131 mg, 31%) as a colorless oil: 1H NMR (CDCl3) δ 7.33 – 7.27 (m, 10H), 7.04 (d, J = 8.3 Hz, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.71 (dd, J = 8.3, 2.3 Hz, 1H), 5.36 (t, J = 7.3 Hz, 1H), 5.29 (s, 2H), 5.24 (s, 2H), 4.72 (s, 2H), 4.71 (s, 2H), 3.32 (d, J = 7.3 Hz, 2H), 2.71 (t, J = 6.3 Hz, 1H), 2.22 – 2.12 (m, 2H), 1.74 (s, 3H), 1.74 – 1.61 (m, 2H), 1.27 (s, 3H), 1.24 (s, 3H); 13C NMR (CDCl3) δ 156.5, 155.5, 137.2, 137.2, 134.7, 129.7, 128.4 (2C), 128.4 (2C), 127.9 (2C), 127.9 (2C), 127.8, 127.7, 123.9, 123.3, 108.8, 103.5, 92.4, 92.2, 69.9, 69.7, 64.2, 58.4, 36.3, 27.9, 27.3, 24.8, 18.7, 16.1; HRMS (EI) m/z calcd for C32H38O5 (M+) 502.2719; found 502.2716.
Benzyl Ether 29a
To epoxide 23 (121 mg, 0.24 mmol) in CH2Cl2 (50 mL) at −78 °C was added BF3·OEt2 (0.15 mL, 1.19 mmol). After 8 minutes the reaction was quenched by addition of Et3N, diluted with water, and extracted with CH2Cl2. The organic layers were washed with brine, dried (MgSO4), filtered, and concentrated in vacuo. Final purification by flash column chromatography (30% ethyl acetate in hexanes) afforded ether 29a (75 mg, 62%) as a colorless oil: 1H NMR (CDCl3) δ 7.39 – 7.21 (m, 10H), 6.98 (d, J = 8.4 Hz, 1H), 6.71 (d, J = 8.5 Hz, 1H), 5.28 (s, 2H), 4.70 (s, 2H), 4.65 (d, J = 10.0 Hz, 1H), 4.61 (d, J = 9.9 Hz, 1H), 4.58 (s, 2H), 3.41 (dd, J = 11.5, 4.1 Hz, 1H), 2.67 – 2.64 (m, 2H), 2.04 – 1.97 (m, 1H), 1.85 – 1.75 (m, 2H), 1.70 – 1.56 (m, 3H), 1.20 (s, 3H), 1.08 (s, 3H), 0.86 (s, 3H); 13C NMR (CDCl3) δ 155.4, 152.6, 139.3, 137.4, 129.8, 128.4 (2C), 128.1 (2C), 127.9 (2C), 127.7, 127.2 (2C), 127.2, 115.6, 115.0, 106.6, 92.7, 78.1, 76.4, 72.0, 69.9, 60.8, 46.7, 38.3, 37.7, 28.2, 27.3, 22.7, 20.0, 14.2; HRMS (EI) m/z calcd for C32H38O5 (M+) 502.2719; found 502.2721.
Competition experiment
To a solution of epoxide 23 (77 mg, 0.15 mmol) and epoxide 24 (85 mg, 0.16 mmol) in CH2Cl2 (40 mL) at −78 °C was added BF3·OEt2 (0.20 mL, 1.6 mmol). After 10 min, the reaction was quenched by addition of Et3N (0.4 mL), allowed to warm to rt, and then extracted with CH2Cl2. The organic extracts were washed with water, dried (MgSO4), and filtered, and then the filtrate was concentrated in vacuo. Final purification by flash column chromatography (7.5% to 20% ethyl acetate in hexanes) afforded compound 29a as a colorless oil (52 mg, 68%) and compound 30a (58 mg, 68%) as a colorless oil.
Epoxide 34
To a solution of the geranyl arene40 (304 mg, 0.92 mmol) in CH2Cl2 (40 mL) at −30 °C was added m-CPBA (228 mg, 77% maximum by mass, 1.0 mmol). After 1 h the reaction was quenched by addition of NaHCO3 and the resulting solution was extracted with CH2Cl2. The organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (8% to 25% ethyl acetate in hexanes) afforded epoxide 34 (226 mg, 71%) as colorless oil: 1H NMR (CDCl3) 7.17 (d, J = 8.4 Hz, 1H), 6.89 (dd, J = 8.4, 2.4 Hz, 1H), 6.83 (d, J = 2.4 Hz, 1H), 5.24 (td, J = 7.2, 1.2 Hz, 1H), 3.20 (d, J = 7.2 Hz, 2H), 2.67 (t, J = 6.2 Hz, 1H), 2.24 (s, 3H), 2.21 (s, 3H), 2.20 – 2.09 (m, 2H), 1.68 (s, 3H), 1.65 – 1.59 (m, 2H), 1.24 (s, 3H), 1.22 (s, 3H); 13C NMR (CDCl3) δ 170.1, 169.9, 150.2, 150.0, 137.2, 131.8, 131.2, 122.9, 120.2, 117.0, 65.0, 59.3, 37.4, 29.3, 28.4, 25.9, 22.1, 21.8, 19.8, 17.2: HRMS (EI) m/z calcd for C20H26O5 (M+) 346.1780, found 346.1772.
Bridged Ether 35
To a solution of epoxide 34 (95 mg, 0.27 mmol) in CH2Cl2 (50 mL) at −78 °C was added BF3·OEt2 (0.18 mL, 1.4 mmol). After 7 min the reaction was quenched by addition of Et3N (0.35 mL, 2.5 mmol) and concentrated in vacuo. The residue was dissolved in ethyl acetate and washed with water, 1N HCl, and brine. The organic phase was then dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (20% to 50% ethyl acetate in hexanes) afforded tricyclic ether 35 (44 mg, 46%) as a colorless oil: 1H NMR (CDCl3) δ 7.20 (d, J = 8.4 Hz, 1H), 6.86 (dd, J = 8.4, 2.4 Hz, 1H), 6.79 (d, J = 2.4 Hz, 1H), 3.68 (d, J = 5.2 Hz, 1H), 2.45 (d, J = 7.6 Hz, 2H), 2.23 (s, 3H), 2.18 (s, 3H), 1.88 – 1.84 (m, 1H), 1.81 (t, J = 7.6 Hz, 1H), 1.66 – 1.59 (m, 1H), 1.51 – 1.45 (m, 1H), 1.39 (dd, J = 12.2, 4.8 Hz, 1H), 1.20 (s, 3H), 0.90 (s, 3H), 0.90 (s, 3H); 13C NMR (CDCl3) δ 168.8, 168.6, 148.8, 148.7, 131.0, 129.7, 118.6, 115.9, 86.6, 86.0, 54.2, 45.4, 38.8, 26.8, 25.8, 25.8, 23.6, 21.0, 20.8, 19.0; HRMS (EI) m/z calcd for C20H26O5 (M+) 346.1780, found 346.1790.
Methyl Ether 59
To a solution of the parent alcohol40 (ca. 210 mmol from above) in THF (250 mL) cooled to 0 °C was added NaH (18 g, 60% in oil, 600 mmol) in small portions. After evolution of hydrogen ceased (ca. 10 min), CH3I (17 mL, 272 mmol) was added slowly. After an additional 5 hr, the reaction was slowly quenched by addition of water. The resulting solution was extracted with ethyl acetate. The organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (25% ethyl acetate in hexanes) afforded compound 59 (51 g, 73% over 4 steps) as colorless oil: 1H NMR (CDCl3) δ 7.61 (s, 1H), 7.38 – 7.37 (m, 5H), 7.24 – 7.23 (m, 2H), 5.37 (s, 2H), 4.79 (s, 2H), 4.39 (s, 2H), 3.39 (s, 3H); 13C NMR (CDCl3) δ 152.95, 136.7, 133.1, 132.5, 128.2, (2C), 127.8 (2C), 127.7, 127.7, 115.7, 112.5, 92.6, 73.1, 70.0, 57.7; HRMS (EI) m/z calcd for C16H17O3Br (M+) 336.0361, found 336.0362.
Epoxide 60
To a solution of TMEDA (3 mL, 20 mmol) in ether (40 mL) cooled in a brine bath was added BuLi (7.5 mL, 2.3 M in hexanes, 17.3 mmol). After 5 min, bromide 59 (4.86 g, 14.4 mmol) was added in ether (20 mL) via canula. After an additional 15 min, CuI (3.0 g, 16 mmol) was added as a solid in one portion, resulting in a slow blackening of the solution. After an additional 15 min at the same temperature, freshly prepared (R)-6,7-epoxy geranyl bromide, prepared from the corresponding alcohol (3.74g, 22 mmol, 93% ee), was added via canula as a solution in ether (5 mL). The reaction was allowed to warm slowly to rt and then was quenched by addition of NH4Cl after an additional 4 h. The resulting mixture was extracted with ethyl acetate, and the organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (18% ethyl acetate in hexanes) afforded epoxide 60 (4.2 g, 71%) as colorless oil: 1H NMR (CDCl3) δ 7.31 – 7.22 (m, 5H), 7.13 (s, 1H), 7.09 (m, 2H), 5.38 (t, J = 7.2 Hz, 1H), 5.26 (s, 2H), 4.67 (s, 2H), 4.33 (s, 2H), 3.37 (d, J = 7.2 Hz, 2H), 3.32 (s, 3H), 2.66 (t, J = 6.2 Hz, 1H), 2.20 – 2.12 (m, 2H), 1.74 (s, 3H), 1.67 – 1.60 (m, 2H), 1.23 (s, 3H), 1.21 (s, 3H); 13C NMR (CDCl3) δ 154.3, 137.0, 134.6, 131.0, 130.1, 129.0, 128.1 (2C), 127.6 (2C), 127.4, 126.4, 122.9, 113.5, 92.0, 74.1, 69.6, 63.6, 57.8, 57.5, 36.1, 28.4, 27.1, 24.5, 18.4, 15.8; HRMS (EI) m/z calcd for C26H34O4 (M+) 410.2457, found 410.2462.
Hexahydroxanthene 62
To a solution of epoxide 60 (774 mg, 1.89 mmol) in CH2Cl2 (200 mL) at −78 °C was added BF3·OEt2 (1.0 mL, 8.0 mmol). After 8 min, the reaction was quenched by addition of Et3N (2 mL), allowed to warm to rt, and concentrated in vacuo. Purification of the initial oil by column chromatography (30% ethyl acetate in hexanes) afforded compounds 61 and 62 as an inseparable mixture (520 mg, 1:2 61: 62 corresponding to compound 62 (384 mg, 50%) and compound 61 (136 mg, 25%)) as a colorless oil: For compound 62, HRMS (EI) m/z calcd for C26H34O4 (M+) 410.2457, found 410.2450.
Benzyl Alcohol 63
A mixture of compound 61 and compound 62 (185 mg, 2:1 62 to 61) was dissolved in CH3OH (0.5 mL) and 10% Pd/C (30 mg) was added. The reaction vessel was sealed and purged with H2 at rt. A stream of H2 then was provided to maintain 1 atm of H2, which resulted in the slow concentration of the solution to near dryness. After 23 hr, the resulting mixture was diluted with CH3OH, filtered through celite, and concentrated in vacuo. Final purification by column chromatography (50% ethyl acetate in hexanes) afforded alcohol 63 (81 mg, 76%) as colorless oil: 1H NMR (CDCl3) δ 7.03 (m, 2H), 4.63 (d, J = 12.8 Hz, 1H), 4.57 (d, J = 12.8, 1H), 4.34 (s, 2H), 3.41 (dd, J = 11.6, 4.0 Hz, 1H), 3.37 (s, 3H), 2.73 – 2.68 (m, 2H), 2.02 (dt, J = 12.8, 3.4 Hz, 1H), 1.88 – 1.60 (m, 4 H), 1.22 (s, 3H), 1.09 (s, 3H), 0.90 (s, 3H); 13C NMR (CDCl3) δ150.7, 129.4, 129.0, 128.5, 126.4, 121.8, 77.9, 74.5, 62.4, 58.0, 46.8, 38.4, 37.8, 28.2, 27.3, 22.9, 20.1, 14.2; 13C NMR (MeOD) δ 151.3, 130.1, 130.0, 129.8, 126.8, 122.8, 78.8, 77.7, 75.8, 60.3, 57.9, 39.4, 39.0, 29.0, 27.9, 24.0, 20.3, 14.8; HRMS (EI) m/z calcd for C19H28O4 (M+) 320.1988, found 320.1990.
Aldehyde 64
To a solution of alcohol 63 (13 mg, 0.04 mmol) in CH2Cl2 at rt was added activated MnO2 (95 mg, 0.87 mmol). After 20 h at rt, the solution was diluted with ethyl acetate, filtered through celite, and concentrated in vacuo which afforded aldehyde 64 (13 mg, 96%) as a yellow oil: 1H NMR (CDCl3) δ 10.40 (s, 1H), 7.58 (d, J = 2.0 Hz, 1H), 7.33 (d, J = 2.0 Hz, 1H), 4.35 (s, 2H), 3.42 (dd, J = 11.6, 3.6 Hz, 1H), 3.37 (s, 3H), 2.76 – 2.73 (m, 2H), 2.05 (dt, J = 12.6, 3.2 Hz, 1H), 1.90 – 1.60 (m, 5H), 1.26 (s, 3H), 1.10 (s, 3H), 0.88 (s, 3H); 13C NMR (CDCl3) δ 190.0, 155.8, 135.7, 129.5, 125.6, 125.1, 123.7, 77.8, 77.8, 74.0, 58.1, 46.4, 38.4, 37.5, 28.2, 27.2, 22.9, 20.1, 14.2; HRMS (EI) m/z calcd for C19H26O4 (M+) 318.1831, found 318.1839.
Phenol 65
To a solution of aldehyde 64 (18 mg, 0.056 mmol) in CH2Cl2 (2 mL) was added m-CPBA (35 mg, 77% maximum, 0.14 mmol) at rt. After 2 hr, the reaction was diluted with CH3OH (3mL) and solid K2CO3 (65 mg, 0.47 mmol) was added. After an additional 20 hr, the reaction was quenched by addition of 1N HCl and Na2SO3. The resulting mixture was neutralized by addition of NaHCO3 and extracted with CH2Cl2. After the organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo, final purification by column chromatography (30% to 50% ethyl acetate in hexanes) afforded phenol 65 (17 mg, 98%) as a colorless oil: 1H NMR (CDCl3) δ 6.73 (d, J = 1.6 Hz, 1H), 6.63 (d, J = 1.6, 1H), 5.46 (s, 1H), 4.31 (s, 2H), 3.40 (dd, J = 11.2, 3.6 Hz, 1H), 3.36 (s, 3H), 2.71 – 2.66 (m, 2H), 2.01 (dt, J = 12.8, 3.6 Hz, 1H), 1.89 – 1.59 (m, 5H), 1.22 (s, 3H), 1.09 (s, 3H), 0.87 (s, 3H); 13C NMR (CDCl3) δ 145.0, 139.6, 130.0, 121.9, 120.1, 112.0, 77.9, 77.7, 74.7, 57.9, 47.2, 38.4, 37.7, 28.2, 27.3, 22.7, 20.1, 14.3; HRMS (EI) m/z calcd for C18H26O4 (M+) 306.1831, found 306.1837.
Schweinfurthin B intermediate 66
To a solution of phenol 65 (9 mg, 0.03 mmol) in acetone (2 mL) was added K2CO3 (60 mg, 0.43 mmol) followed by CH3I (0.06 mL, 0.96 mmol) and this solution was heated to reflux. After 2 hrs, the solution was allowed to cool to rt, quenched by addition of water and extracted with CH2Cl2. The organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo to afford compound 66 (9 mg, 94%) as a colorless oil in sufficient purity for further use. The 1H MNR spectrum of this material was identical to that prepared via different methods.30
Schweinfurthin G intermediate 67
To a solution of phenol 65 (13 mg, 0.04 mmol) in CH2Cl2 (2mL), was added DIPEA (0.1 mL, 0.57 mmol) followed by MOMCl (0.02 mL, 0.26 mmol). After 2 h at rt, the reaction was quenched by addition of water and extracted with CH2Cl2. The resulting solution was washed with 1N HCl. The organic phase was dried (MgSO4), and concentrated in vacuo to afford compound 67 (13 mg, 89%) as a yellow oil. The 1H NMR spectrum of this material was identical to that prepared via different methods;29 [α]25D = +34 (c 1.6, CHCl3, 90% ee by HPLC).
Ketone 68
To a solution of hexahydroxanthene 67 (680 mg, 1.9 mmol) in CH2Cl2 at rt was added catalytic TPAP (73 mg, 0.21 mmol) and NMO (284 mg, 2.4 mmol). After 26 hr, the reaction mixture was diluted with ethyl acetate, filtered through celite and silica, and concentrated in vacuo. Final purification by column chromatography (50% ethyl acetate in hexanes) afforded ketone 68 (675 mg, 100%) as a colorless oil: [α]25D = +88 (c 1.1, CHCl3, 90% ee by HPLC); 1H NMR (CDCl3) δ 6.81 (s, 1H), 6.66 (s, 1H), 5.04 (d, J = 6.4 Hz, 1H), 5.02 (d, J = 6.8 Hz, 1H), 4.18 (s, 2H), 3.36 (s, 3H), 3.22 (s, 3H), 2.73 – 2.65 (m, 1H), 2.60 – 2.52 (m, 2H), 2.33 – 2.28 (m, 1H), 2.22 – 2.17 (m, 1H), 2.01 – 1.90 (m, 2H), 1.29 (s, 3H), 1.02 (s, 3H), 0.96 (s, 3H); 13C NMR (CDCl3) δ 212.5, 145.3, 142.7, 129.2, 122.4, 121.5, 115.2, 95.2, 74.9, 73.8, 57.3, 55.5, 46.9, 45.8, 37.5, 34.5, 23.9, 23.1, 20.2, 18.5; HRMS (EI) m/z calcd for C20H28O5 (M+) 348.1937, found 348.1940.
Enone 69
To a solution of ketone 68 (140 mg, 0.40 mmol) in ethanol at rt was added benzaldehyde (0.2 mL, 1.7 mmol) followed by KOH (177 mg, 3.2 mmol). After 25 min, the reaction was quenched by addition of NH4Cl, the resulting solution was extracted with ethyl acetate, and the organic extracts were washed with brine. After the organic phase was dried (MgSO4) and concentrated in vacuo, final purification of the residue by column chromatography (25% ethyl acetate in hexanes) afforded enone 69 (148 mg, 86%) as a colorless oil: [α]25D = +157 (c 1.0, CH3OH, 90% ee by HPLC); 1H NMR (CDCl3) δ 7.63 (d, J = 2.4 Hz, 1H), 7.45 – 7.32 (m, 5H), 6.96 (d, J = 2.0 Hz, 1H), 6.79 (d, J = 1.6 Hz, 1H), 5.20 (d, J = 6.8 Hz, 1H), 5.15 (d, J = 6.8 Hz, 1H), 4.32 (s, 2H), 3.49 (s, 3H), 3.50 – 3.40 (m, 1H), 3.36 (s, 3H), 2.98 (dd, J = 15.2, 2.8 Hz, 1H), 2.84 – 2.70 (m, 2H), 2.34 (dd, J = 12.6, 5.6, 1H), 1.31 (s, 3H), 1.20 (s, 3H), 1.16 (s, 3H); 13C NMR (CDCl3) δ 205.0, 145.5, 142.8, 138.6, 135.0, 132.2, 130.0 (2C), 129.5, 128.7, 128.3 (2C), 122.6, 121.8, 115.4, 95.6, 75.4, 74.3, 57.8, 56.0, 45.7, 45.3, 41.7, 28.6, 24.2, 22.1, 19.1; HRMS (EI) m/z calcd for C27H32O5 (M+) 436.2250, found 436.2257.
Alcohol 70
To a solution of ketone 69 (115 mg, 0.26 mmol) in CH3OH at rt was added CeCl3·7H2O (108 mg, 0.27 mmol) followed by NaBH4 (12 mg, 0.32 mmol). After 2 hr, the reaction was quenched by addition of water and concentrated in vacuo. The resulting solution was extracted with ethyl acetate, and the extracts were washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (20% ethyl acetate in hexanes) afforded alcohol 70 (110 mg, 96%) as white crystals: [α]25D = +62 (c 0.9, CHCl3, 90% ee by HPLC); 1H NMR (CDCl3) δ 7.37 – 7.22 (m, 5H), 6.94 (d, J = 2.0 Hz, 1H), 6.80 (m, 2H), 5.19 (d, J = 6.6 Hz, 1H), 5.14 (d, J = 6.6 Hz, 1H), 4.33 (s, 2H), 3.91 (s, 1H), 3.41 (s, 3H), 3.39 (s, 3H), 2.75 – 2.70 (m, 2H), 2.29 (m, 2H), 1.92 (dd, J = 12.2, 5.8 Hz, 1H), 1.20 (s, 3H), 1.06 (s, 3H), 0.85 (s, 3H); 13C NMR (CDCl3) δ 145.8, 143.5, 138.0, 137.4, 129.2, 128.8 (2C), 128.2 (2C), 126.3, 123.9, 123.2, 122.8, 115.3, 95.6, 79.8, 78.0, 74.6, 58.0, 56.0, 47.1, 41.2, 39.7, 27.2, 23.2, 19.8, 14.2; HRMS (EI) m/z calcd for C27H34O5 (M+) 438.2406, found 438.2408.
Triol 71
To a solution of alcohol 70 (115 mg, 0.26 mmol) in dioxane (2 mL) and water (0.1 mL) at rt was added OsO4 (0.2 mL, 0.002 M in tBuOH, 0.004 mmol) followed by NMO (68 mg, 0.58 mmol). After 17 hr, the reaction was diluted with water and extracted with ethyl acetate. The extracts were washed with brine, dried (MgSO4), and concentrated in vacuo, which afforded triol 71 as a crystalline solid: 1H NMR (CDCl3) δ 7.50 – 7.48 (m, 2H), 7.34 – 7.26 (m, 3H), 6.86 (s, 1H), 6.75 (s, 1H), 5.07 (s, 1H), 5.03 (s, 2H), 4.64 (br s, 1H), 4.36 (br s, 1H), 4.28 (s, 2H), 3.61 (br s, 1H), 3.40 (s, 3H), 3.36 – 3.30 (m, 1H), 3.33 (s, 3H), 2.69 – 2.66 (m, 2H), 2.14 (d, J = 14.4 Hz, 1H), 1.94 (dd, J = 10.8, 7.2 Hz, 1H), 1.72 (d, J = 14.4 Hz, 1H), 1.26 (s, 3H), 1.12 (s, 3H), 1.06 (s, 3H); 13C NMR (CDCl3) δ 145.8, 143.2, 139.4, 129.1 (2C), 129.1, 128.2 (2C), 128.1, 123.2, 123.0, 115.8, 95.8, 84.8, 77.8, 76.7, 75.1, 74.5, 57.9, 56.0, 46.0, 45.5, 37.8, 30.1, 23.2, 21.7, 16.5; HRMS (EI) m/z calcd for C27H36O7 (M+) 472.2461, found 472.2467.
Ketone 72
To a solution of partially purified triol 71 (ca. 0.26 mmol) in CH2Cl2 (5mL) and water (0.1 mL) at rt was added NaIO4 (720 mg, 3.3 mmol) and the resulting mixture was vigorously stirred. After 24 hr, the reaction was diluted with water and extracted with CH2Cl2 and the extracts were washed with brine, dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (30% ethyl acetate in hexanes) afforded ketone 72 (81 mg, 84% over 2 steps) as a colorless oil: 1H NMR (CDCl3) δ 6.95 (d, J = 1.6 Hz, 1H), 6.80 (s, 1H), 5.18 (d, J = 6.4 Hz, 1H), 5.15 (d, J = 6.4 Hz, 1H), 4.33 (s, 2H), 4.05 (d, J = 3.6 Hz, 1H), 3.50 (s, 3H), 3.46 (d, J = 4.4 Hz, 1H), 3.38 (s, 3H), 3.03 – 2.99 (m, 2H), 2.85 (dd, J = 16.4, 3.6 Hz, 1H), 2.77 (m, 1H), 2.34 (dd, J = 12.8, 5.2 Hz, 1H), 1.25 (s, 3H), 1.20 (s, 3H), 0.74 (s, 3H);13C NMR (CDCl3) δ 207.5, 146.0, 142.8, 130.2, 122.9, 122.5, 115.4, 95.8, 82.6, 78.8, 74.5, 58.1, 56.2, 52.5, 46.3, 41.5, 27.3, 23.2, 21.0, 15.1; HRMS (EI) m/z calcd for C20H28O6 (M+) 364.1886, found 364.1883.
Diol 73
To a solution of ketone 72 (81 mg, 0.22 mmol) in CH3OH (0.3 mL) and THF (3 mL) at rt was added NaBH4 (18 mg, 0.47 mmol). After 15 min, the reaction was quenched by addition of 1N HCl. The resulting solution was extracted with ethyl acetate, and the organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Immediate purification by column chromatography (80% ethyl acetate in hexanes) afforded diol 73 (78 mg, 96%) as a colorless oil: 1H NMR (CDCl3) δ 6.92 (d, J = 1.6 Hz, 1H), 6.77 (s, 1H), 5.18 (d, J = 6.0 Hz, 1H), 5.14 (d, J = 6.4 Hz, 1H), 4.31 (s, 2H), 4.15 (q, J = 3.2 Hz, 1H), 3.51 (s, 3H), 3.38 (s, 3H), 3.39 – 3.36 (m, 1H), 3.16 (brd, 1H), 2.77 – 2.69 (m, 3H), 2.42 (dd, J = 14.2, 3.0 Hz, 1H), 1.93 (dd, J = 14.4, 3.6 Hz, 1H), 1.70 (dd, J = 12.2, 5.4 Hz, 1H), 1.41 (s, 3H), 1.05 (s, 3H), 1.04 (s, 3H); 13C NMR (CDCl3) δ 145.9, 143.3, 129.1, 123.4, 123.2, 115.9, 95.9, 77.3, 76.3, 74.7, 70.8, 58.0, 56.2, 46.9, 43.3, 37.9, 28.8, 23.0, 21.5, 15.9; HRMS (EI) m/z calcd for C20H30O6 (M+) 366.2042, found 366.2041.
Aldehyde 74
To a solution of methyl ether 73 (116 mg, 0.32 mmol), in CH2Cl2/water (15:1) at rt was added DDQ (90 mg, 0.40 mmol). After 15 min, the reaction was quenched by addition of brine and NaHCO3. The resulting solution was extracted with CH2Cl2, and the organic extracts were washed with a small amount of water followed by brine. After the organic phase was dried (MgSO4) and concentrated in vacuo, aldehyde 74 (109 mg, 98%) was obtained as a faintly yellow wax that was used without further purification: 1H NMR (CDCl3) δ 9.79 (s, 1H), 7.46 (d, J = 1.6 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 5.23 (d, J = 6.4 Hz, 1H), 5.21 (d, J = 6.4 Hz,1 H), 4.26 (q, J = 3.2 Hz, 1H), 3.56 (s, 3H), 3.40 (m, 1H), 2.86 – 2.82 (m, 2H), 2.51 (dd, J = 14.2, 3.0 Hz, 1H), 2.25 (m, 2H), 2.03 (dd, J = 14.8, 3.4, Hz, 1H), 1.78 (dd, J = 11.8, 5.8 Hz, 1H), 1.47 (s, 3H), 1.13 (s, 3H), 1.09 (s, 3H); 13C NMR (CDCl3) δ 190.9, 149.6, 146.6, 128.8, 127.1, 123.5, 115.3, 95.7, 78.0, 77.4, 70.7, 56.3, 46.6, 43.2, 38.1, 28.9, 23.0, 21.8, 16.0; HRMS (EI) m/z calcd for C19H26O6 (M+) 350.1729, found 350.1736.
Tris-MOM Aldehyde 75
To a solution of diol 74 (40 mg, 0.11 mmol) in CH2Cl2 (0.5 mL) at rt was added DIPEA (0.8 mL, 7.3 mmol) followed by slow addition of MOMCl (0.2 mL, 2.6 mmol) over 20 min. After 5 hr, the reaction was quenched by addition of water, the resulting solution was extracted with CH2Cl2, and the organic phases were washed with 1N HCl followed by brine. The organic phase was dried (MgSO4), and concentrated in vacuo. Final purification by column chromatography (45% ethyl acetate in hexanes) afforded aldehyde 75 (37 mg, 74%) as a colorless oil: 1H NMR (CDCl3) δ 9.78 (s, 1H), 7.46 (d, J = 1.6 Hz, 1H), 7.35 (d, J = 1.2 Hz, 1H), 5.24 (d, J = 6.4 Hz, 1H), 5.20 (d, J = 6.8 Hz, 1H), 4.85 (d, J = 6.8 Hz, 1H), 4.70 (s, 2H), 4.64 (d, J = 6.8 Hz, 1H), 4.21 (q, J = 3.4 Hz, 1H), 3.50 (s, 3H), 3.45 (s, 3H), 3.40 (s, 3H), 3.32 (d, J = 3.2 Hz, 1H), 2.84 – 2.81 (m, 2H), 2.50 (dd, J = 14.4, 3.6 Hz, 1H), 1.91 (dd, J = 14.0, 3.2 Hz, 1H), 1.78 (dd, J = 11.2, 6.8 Hz, 1H), 1.44 (s, 3H), 1.12 (s, 3H), 1.11 (s, 3H); 13C NMR (CDCl3) δ 190.9, 149.5, 146.6, 128.7, 127.1, 123.6, 115.0, 96.2, 95.9, 95.5, 82.7, 78.1, 72.9, 56.3, 56.2, 55.6, 47.1, 41.5, 38.3, 28.7, 22.9, 21.3, 16.2; HRMS (EI) m/z calcd for C23H34O8 (M+) 438.2254, found 438.2250.
Stilbene 77
To a solution of diisopropyl amine (0.05 mL, 0.36 mmol) in THF (0.3 mL) in an ice bath was added BuLi (2.4M in hexanes, 0.05 mL, 0.12 mmol). To this solution, phosphonate 76 (68 mg, 0.14 mmol) in THF (0.5 mL) was added via canula. After 5 min, aldehyde 75 (21 mg, 0.05 mmol) in THF (0.5 mL) was added via canula. After 2 hr, TLC analysis showed little reaction progress and additional base was added (KHMDS, 0.5 M in toluene, 0.3 mL, 0.15 mmol). After an additional 2 hr, the reaction was complete by TLC and the reaction was quenched by addition of NH4Cl. The resulting solution was extracted with ethyl acetate, and the organic phases were washed with 1N HCl followed by brine. After the organic phase was dried (MgSO4), and concentrated in vacuo, final purification by column chromatography (50% ethyl acetate in hexanes) afforded stilbene 77 (25 mg, 68%) as colorless oil: 1H NMR (CDCl3) δ 7.16 (d, J = 1.6 Hz, 1H), 6.96 (s, 1H), 6.94 – 6.89 (m, 4H), 5.27 (d, J = 6.8 Hz, 1H), 5.25 (s, 4H), 5.23 (d, J = 7.2 Hz, 1H), 5.20 (t, J = 5.2 Hz, 1H), 5.05 (t, J = 5.2 Hz, 1H), 4.87 (d, J = 6.8 Hz, 1H), 4.73 (d, J = 6.8 Hz, 1H), 4.68 (d, J = 7.2 Hz, 1H), 4.66 (d, J = 7.2 Hz, 1H), 4.20 (q, J = 3.2 Hz, 1H), 3.54 (s, 3H), 3.49 (s, 6H), 3.46 (s, 3H), 3.41 (s, 3H), 3.37 (d, J = 6.4 Hz, 2H), 3.35 (d, J = 3.6 Hz, 1H), 2.78 – 2.75 (m, 2H), 2.51 (dd, J = 14.0, 2.8 Hz, 1H), 2.06 – 1.88 (m, 6H), 1.78 (s, 3H), 1.64 (s, 3H), 1.56 (s, 3H), 1.43 (s, 3H), 1.11 (s, 3H), 1.09 (s, 3H); 13C NMR (CDCl3) δ 155.4 (2C), 146.1, 142.9, 136.3, 134.7, 131.4, 128.7, 127.6, 126.2, 124.1, 123.2, 122.1, 121.7, 118.9, 112.3, 105.3 (2C), 95.9, 95.4, 95.3, 93.9 (2C), 82.3, 77.2, 72.9, 56.2, 56.2, 55.9 (2C), 55.6, 46.9, 41.2, 39.7, 38.1, 28.6, 26.5, 25.7, 22.9, 22.4, 21.0, 17.6, 16.2, 16.0; HRMS (EI) m/z calcd for C44H64O11 (M+) 768.4449, found 768.4452.
Schweinfurthin A (9)
To a solution of stilbene 77 (12 mg, 0.016 mmol) in methanol (1 mL) at rt was added TsOH (16 mg, 0.08 mmol). After 48 hr, the reaction was quenched by addition of NaHCO3 and the resulting solution was extracted with ethyl acetate. The organic phase was dried (MgSO4) and concentrated in vacuo. Final purification by preparative thin layer chromatography (70% ethyl acetate in hexanes) afforded schweinfurthin A (9, 5 mg, 58%) as a yellow wax: [α]25D = +47 (c 1.0, EtOH, 90% ee by HPLC); 1H NMR (MeOD) δ 6.79 (s, 1H), 6.77 (d, J = 16.4 Hz, 1H), 6.72 (s, 1H), 6.72 (d, J = 16.4 Hz, 1H), 6.44 (s, 2H), 5.24 (t, J = 7.2 Hz, 1H), 5.07 (t, J = 7.2 Hz, 1H), 4.14 (q, J = 3.6 Hz, 1H), 3.30 (m, 3H), 2.75 (m, 2H), 2.36 (dd, J = 13.8, 3.0 Hz, 1H), 2.06 – 2.02 (m, 2H), 1.96 – 1.93 (m, 3H), 1.76 (s, 3H), 1.77 – 1.73 (m, 1H), 1.62 (s, 3H), 1.56 (s, 3H), 1.41 (s, 3H), 1.10 (s, 3H), 1.08 (s, 3H); 13C NMR (MeOD) δ 157.3 (2C), 147.1, 141.9, 137.5, 134.8, 132.0, 130.9, 128.6, 127.4, 125.5, 124.6, 124.2, 120.4, 115.8, 111.0, 105.6 (2C), 78.8, 78.1, 71.8, 48.9, 44.7, 41.0, 39.2, 29.4, 27.8, 25.9, 23.9, 23.2, 22.0, 17.7, 16.6, 16.3; HRMS (EI) m/z calcd for C34H44O6 (M+) 548.3138, found 548.3145.
Supplementary Material
Acknowledgments
We thank Dr. Nolan R. Mente for providing phosphonate 76, Dr. Dale C. Swenson for his assistance with the diffraction analysis of compound 12, and Dr. John A. Beutler (National Cancer Institute – Frederick) for an authentic sample of schweinfurthin A. Financial support from the ACS Division of Medicinal Chemistry (in the form of a predoctoral fellowship to J.J.T.), the University of Iowa Graduate College (in the form of a Presidential Fellowship to J. G. K.), and the National Cancer Institute (R41CA126020 via Terpenoid Therapeutics, Inc.) is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental details for all other compounds, and the 1H and 13C NMR spectra for compounds 15–21, 23–27, 29–51, 59–75, 77, and 9. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nicolaou KC, Sorensen EJ. Classics in Total Synthesis: Targets, Strategies, Methods. Vol. 1. Wiley-VCH; 1996. p. 821. [Google Scholar]
- 2.Nicolaou KC, Snyder SA. Classics in Total Synthesis II: More Targets, Strategies, Methods. Vol. 1. Wiley-VCH; 2003. p. 658. [Google Scholar]
- 3.Corey EJ, Cheng X. The Logic of Chemical Synthesis. Vol. 1. Wiley-Interscience; 1995. p. 436. [Google Scholar]
- 4.Many reaction sequences have been combined in tandem or domino processes, c.f.: Tietze LF. Chem Rev. 1996;96:115–136. doi: 10.1021/cr950027e.
- 5.a) Nicolaou KC, Edmonds DJ, Bulger PG. Angew Chem Int Ed. 2006;45:7134–7186. doi: 10.1002/anie.200601872. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Montagnon T, Snyder SA. Chem Commun. 2003:551–564. doi: 10.1039/b209440c. [DOI] [PubMed] [Google Scholar]
- 6.Rolfe A, Samarakoon TB, Hanson PR. Org Lett. 2010;12:1216–1219. doi: 10.1021/ol100035e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith AB, Kim W, Tong R. Org Lett. 2010;12:588–591. doi: 10.1021/ol902784q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith AB, Tong R. Org Lett. 2010;12:1260–1263. doi: 10.1021/ol100130x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith AB, Xian M. J Am Chem Soc. 2006;128:66–67. doi: 10.1021/ja057059w. [DOI] [PubMed] [Google Scholar]
- 10.Johnson WS. Acc Chem Res. 1968;1:1–8. [Google Scholar]
- 11.Johnson WS. Tetrahedron. 1991;47:xi–l. [Google Scholar]
- 12.Yoder RA, Johnston JN. Chem Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelsen DL, Gagne MR. Organometallics. 2009;28:950–952. doi: 10.1021/om801069s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullen CA, Campbell AN, Gagne MR. Angew Chem Int Ed. 2008;47:6011–6014. doi: 10.1002/anie.200801423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullen CA, Gagne MR. J Am Chem Soc. 2007;129:11880–11881. doi: 10.1021/ja073573l. [DOI] [PubMed] [Google Scholar]
- 16.Rendler S, MacMillan DWC. J Am Chem Soc. 2010;132:5027–5029. doi: 10.1021/ja100185p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snyder SA, Treitler DS. Angew Chem Int Ed. 2009;48:7899–7903. doi: 10.1002/anie.200903834. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Serena L, Li B, Li S, Loh T. Chem Commun. 2009:3738–3740. doi: 10.1039/b903696b. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y, Loh T. Chem Commun. 2008:1434–1436. doi: 10.1039/b714474a. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y, Loh T. J Am Chem Soc. 2008;130:10024–10029. doi: 10.1021/ja802896n. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Chng S, Loh T. J Am Chem Soc. 2007;129:492–493. doi: 10.1021/ja067660+. [DOI] [PubMed] [Google Scholar]
- 22.Gansäuer A, Justicia J, Rosales A, Worgull D, Rinker B, Cuerva JM, Oltra JE. Euro J Org Chem. 2006;2006:4115–4127. [Google Scholar]
- 23.Kumazawa K, Ishihara K, Yamamoto H. Org Lett. 2004;6:2551–2554. doi: 10.1021/ol049126h. [DOI] [PubMed] [Google Scholar]
- 24.Franck-Neumann M, Geoffroy P, Hanss D. Tetrahedron Lett. 2002;43:2277–2280. [Google Scholar]
- 25.Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]
- 26.Treadwell EM, Neighbors JD, Wiemer DF. Org Lett. 2002;4:3639–3642. doi: 10.1021/ol0266368. [DOI] [PubMed] [Google Scholar]
- 27.Neighbors JD, Beutler JA, Wiemer DF. J Org Chem. 2005;70:925–931. doi: 10.1021/jo048444r. [DOI] [PubMed] [Google Scholar]
- 28.Neighbors JD, Mente NR, Boss KD, Zehnder DW, II, Wiemer DF. Tetrahedron Lett. 2008;49:516–519. [Google Scholar]
- 29.Mente NR, Neighbors JD, Wiemer DF. J Org Chem. 2008;73:7963–7970. doi: 10.1021/jo800951q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topczewski JJ, Neighbors JD, Wiemer DF. J Org Chem. 2009;74:6965–6972. doi: 10.1021/jo901161m. [DOI] [PubMed] [Google Scholar]
- 31.Neighbors JD, Topczewski JJ, Swenson DC, Wiemer DF. Tetrahedron Lett. 2009;50:3881–3884. [Google Scholar]
- 32.Kartika R, Frein JD, Taylor RE. J Org Chem. 2008;73:5592–5594. doi: 10.1021/jo800704d. [DOI] [PubMed] [Google Scholar]
- 33.Topczewski JJ, Callahan MP, Neighbors JD, Wiemer DF. J Am Chem Soc. 2009;131:14630–14631. doi: 10.1021/ja906468v. [DOI] [PubMed] [Google Scholar]
- 34.Beutler JA, Jato JlCG, Wiemer DF, Neighbors JD, Salnikova MS, Hollingshead M, Scudiero DA, McCloud TG. The Schweinfurthins: Issues in Development of a Plant-Derived Anticancer Lead. In: Bogers RJ, editor. Medicinal and Aromatic Plants. Springer; 2006. pp. 301–309. [Google Scholar]
- 35.Beutler JA, Shoemaker RH, Johnson T, Boyd MR. J Nat Prod. 1998;61:1509–1512. doi: 10.1021/np980208m. [DOI] [PubMed] [Google Scholar]
- 36.Topczewski JJ, Kuder CH, Neighbors JD, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2010;18:6734–6741. doi: 10.1016/j.bmc.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulrich NC, Kuder CH, Hohl RJ, Wiemer DF. Bioorg Med Chem Lett. 2010;20:6716–6720. doi: 10.1016/j.bmcl.2010.08.143. [DOI] [PubMed] [Google Scholar]
- 38.Kuder CH, Neighbors JD, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2009;17:4718–4723. doi: 10.1016/j.bmc.2009.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basabe P, Diego A, Delgado S, Díez D, Marcos IS, Urones JG. Tetrahedron. 2003;59:9173–9177. [Google Scholar]
- 40.See Supporting Information.
- 41.Gaich T, Baran PS. J Org Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- 42.Shenvi RA, O’Malley DP, Baran PS. Acc Chem Res. 2009;42:530–541. doi: 10.1021/ar800182r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young IS, Baran PS. Nature Chemistry. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
- 44.Tsangarakis C, Arkoudis E, Raptis C, Stratakis M. Org Lett. 2007;9:583–586. doi: 10.1021/ol062798i. [DOI] [PubMed] [Google Scholar]
- 45.Janusz JM, Gardlik JM, Young PA, Burkes RV, Stoll SJ, Estelle AF, Riley CM. J Med Chem. 1990;33:1052–1061. doi: 10.1021/jm00165a027. [DOI] [PubMed] [Google Scholar]
- 46.Aziz M, Rouessac F. Tetrahedron. 1988;44:101–110. [Google Scholar]
- 47.Yamada Y, Sanjoh H, Iguchi K. J Chem Soc, Chem Commun. 1976:997–998. [Google Scholar]
- 48.Barrero AF, Alvarez-Manzaneda EJ, Mar Herrador M, Alvarez-Manzaneda R, Quilez J, Chahboun R, Linares P, Rivas A. Tetrahedron Lett. 1999;40:8273–8276. [Google Scholar]
- 49.Barrero AF, Alvarez-Manzaneda EJ, Palomino PL. Tetrahedron. 1994;50:13239–13250. [Google Scholar]
- 50.Fish PV, Sudhakar AR, Johnson WS. Tetrahedron Lett. 1993;34:7849–7852. [Google Scholar]
- 51.Marco JA, Sanz JF, Yuste A, Carda M, Jakupovic J. Phytochemistry. 1991;30:3661–3668. [Google Scholar]
- 52.Xu Y, Lai Y, Imiyabir Z, Goh S. J Nat Prod. 2001;64:1191–1195. doi: 10.1021/np0101393. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y, Loh T. Org Lett. 2008;10:2143–2145. doi: 10.1021/ol800499p. [DOI] [PubMed] [Google Scholar]
- 54.Savona G, Bruno M, Piozzi F, Barbagallo C. Phytochemistry. 1982;21:2132–2133. [Google Scholar]
- 55.Ono M, Sawamura H, Ito Y, Mizuki K, Nohara T. Phytochemistry. 2000;55:873–877. doi: 10.1016/s0031-9422(00)00214-4. [DOI] [PubMed] [Google Scholar]
- 56.Prisinzano TE. J Nat Prod. 2009;72:581–587. doi: 10.1021/np8005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shults EE, Velder J, Schmalz H-G, Chernov SV, Rubalova TV, Gatilov YV, Henze G, Tolstikov GA, Prokop A. Bioorg Med Chem Lett. 2006;16:4228–4232. doi: 10.1016/j.bmcl.2006.05.077. [DOI] [PubMed] [Google Scholar]
- 58.Lee HM, Nieto-Oberhuber C, Shair MD. J Am Chem Soc. 2008;130:16864–16866. doi: 10.1021/ja8071918. [DOI] [PubMed] [Google Scholar]
- 59.Mascal M, Hafezi N, Toney MD. J Am Chem Soc. 2010;132:10662–10664. doi: 10.1021/ja103880c. [DOI] [PubMed] [Google Scholar]
- 60.Baeyer A, Villiger V. Ber Dtsch Chem Ges. 1899;32:3625–3633. [Google Scholar]
- 61.Mente NR, Wiemer AJ, Neighbors JD, Beutler JA, Hohl RJ, Wiemer DF. Biorg Med Chem Lett. 2007;17:911–915. doi: 10.1016/j.bmcl.2006.11.096. [DOI] [PubMed] [Google Scholar]
- 62.Ulrich NC, Kodet JG, Mente NR, Kuder CH, Beutler JA, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2010;18:1676–1683. doi: 10.1016/j.bmc.2009.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neighbors JD, Salnikova MS, Beutler JA, Wiemer DF. Biorg Med Chem. 2006;14:1771–1784. doi: 10.1016/j.bmc.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 64.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639–666. [Google Scholar]
- 65.Wasserman HH, Parr J. Acc Chem Res. 2004;37:687–701. doi: 10.1021/ar0300221. [DOI] [PubMed] [Google Scholar]
- 66.Wasserman HH, Kuo G. Tetrahedron. 1992;48:7071–7082. [Google Scholar]
- 67.Luche JL. J Am Chem Soc. 1978;100:2226–2227. [Google Scholar]
- 68.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973–1976. [Google Scholar]
- 69.Wen X, Xia J, Cheng K, Zhang L, Zhang P, Liu J, Zhang L, Ni P, Sun H. Biorg Med Chem Lett. 2007;17:5777–5782. doi: 10.1016/j.bmcl.2007.08.057. [DOI] [PubMed] [Google Scholar]
- 70.Cook SP, Danishefsky SJ. Org Lett. 2006;8:5693–5695. doi: 10.1021/ol062067i. [DOI] [PubMed] [Google Scholar]
- 71.Liu H, Siegel DR, Danishefsky SJ. Org Lett. 2006;8:423–425. doi: 10.1021/ol052618p. [DOI] [PubMed] [Google Scholar]
- 72.Treadwell EM, Cermak SC, Wiemer DF. J Org Chem. 1999;64:8718–8723. [Google Scholar]
- 73.Neighbors JD, Salnikova MS, Wiemer DF. Tetrahedron Lett. 2005;46:1321–1324. [Google Scholar]
- 74.Singh SB, Zink DL, Williams M, Polishook JD, Sanchez M, Silverman KC, Lingham RB. Bioorg Med Chem Lett. 1998;8:2071–2076. doi: 10.1016/s0960-894x(98)00371-0. [DOI] [PubMed] [Google Scholar]
- 75.Hinkley SF, Fettinger JC, Dudley K, Jarvis BB. J Antibiot. 1999;52:988–997. doi: 10.7164/antibiotics.52.988. [DOI] [PubMed] [Google Scholar]
- 76.Pecchio M, Solis PN, Lopez-Perez J, Vasquez Y, Rodriguez N, Olmedo D, Correa M, San Feliciano A, Gupta MP. J Nat Prod. 2006;69:410–413. doi: 10.1021/np050338c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.