

Abstract
A common feature in the structures of GT-A-fold-type glycosyltransferases is a mobile polypeptide loop that has been observed to participate in substrate recognition and enclose the active site upon substrate binding. This is the case for the human ABO(H) blood group B glycosyltransferase GTB, where amino acid residues 177–195 display significantly higher levels of disorder in the unliganded state than in the fully liganded state. Structural studies of mutant enzymes GTB/C80S/C196S and GTB/C80S/C196S/C209S at resolutions ranging from 1.93 to 1.40 Å display the opposite trend, where the unliganded structures show nearly complete ordering of the mobile loop residues that is lost upon substrate binding. In the liganded states of the mutant structures, while the UDP moiety of the donor molecule is observed to bind in the expected location, the galactose moiety is observed to bind in a conformation significantly different from that observed for the wild-type chimeric structures. Although this would be expected to impede catalytic turnover, the kinetics of the transfer reaction are largely unaffected. These structures demonstrate that the enzymes bind the donor in a conformation more similar to the dominant solution rotamer and facilitate its gyration into the catalytically competent form. Further, by preventing active-site closure, these structures provide a basis for recently observed cooperativity in substrate binding. Finally, the mutation of C80S introduces a fully occupied UDP binding site at the enzyme dimer interface that is observed to be dependent on the binding of H antigen acceptor analog.
Keywords: human ABO(H) blood groups, glycosyltransferase, x-ray crystallography, kinetics
Introduction
The human ABO(H) blood group antigens are oligosaccharides synthesized by specific glycosyltransferases (GTs) that transfer monosaccharides from a donor to an acceptor molecule in a regio- and stereospecific manner. The H antigen disaccharide α-Fuc-(1 → 2)-β-D-Gal-OR is the common acceptor for the A and the B glycosyltransferases (GTA and GTB, respectively) and corresponds to the O blood type when the genes for the A and/or B enzymes code for truncated1 or inactive2,3 enzymes. The A antigen is formed by GTA, which transfers GalNAc from UDP-GalNAc to the H antigen, and similarly, the B antigen trisaccharide is formed by GTB, which transfers Gal from UDP-Gal to the same H antigen.4 The A and B trisaccharide antigens are nearly identical, differing only in the replacement of an acetamido group with a hydroxyl, yet this small difference can be sufficient to cause a lethal immune response in a mismatched blood transfusion, which led to the ABO blood group discovery by Landsteiner in 1901.5
GTA and GTB are the two most homologous, naturally occurring GTs that utilize two distinct naturally occurring donors, differing in only 4 of 354 amino acids: Arg/Gly176, Gly/Ser235, Leu/Met266, and Gly/Ala268.1 As these two enzymes share the H antigen as a common acceptor, it was first thought that these four ‘critical’ amino acids were involved strictly in donor recognition;1 however, subsequent kinetic and structural studies from our laboratories revealed that only Leu/Met266 and Gly/Ala268 affect donor recognition.6,7 Arg/Gly176 was found to be too distant from the active site to affect donor or acceptor recognition, although it has been implicated in enzyme turnover.3,6 Gly/Ser235 was found to have no impact on donor recognition6,7 but could significantly affect acceptor binding.8
GTA and GTB are both members of the GT-A-fold-type family, with two closely associated domains one of which contains a Rossmann fold responsible for nucleotide-donor binding. GT-A-fold-type enzymes have loops of polypeptide near the active site that have been observed to be disordered in the unliganded state,7,9–11 which become ordered or undergo a conformational shift in GT-A-fold-type enzymes to cover the active site upon the binding of substrate.12–19 We demonstrated previously that loops in GTA/GTB chimera (internal loop residues 177–195 and C-terminal tail residues 345–355) follow this paradigm, as they are normally disordered in the absence of substrate and gradually become more ordered as substrates are added to culminate in an enzyme that shows nearly complete order in the presence of donor and acceptor analogs.9 The mobile loop participates in substrate recognition in GTA/GTB but has been suggested to function in GT-A-fold-type enzymes in part by limiting the access of water molecules that may hydrolyze the nucleotide sugar donor.13 Recent kinetic studies of GTA and GTB reveal cooperative substrate binding, where the binding of donor is enhanced by the presence of acceptor20 and vice versa.21
In order to explore the effects of residues potentially involved in loop closure and concomitant substrate recognition, we have constructed the soluble, catalytically active domains of GTB cysteine-to-serine mutants GTB/C80S/C196S and GTB/C80S/C196S/C209S, and we now report their kinetic characterization and the high-resolution structures of these mutants in the absence of substrate and in combination with the ligands UDP, H antigen disaccharide, UDP-Gal, and a 3-deoxy-Gal acceptor analog α-L-Fucp-(1 → 2)-β-D-(3-deoxy)-Galp-OR.
Results
Crystallographic models
High-resolution structures of GTB/C80S/C196S and GTB/C80S/C196S/C209S were determined in their unliganded state as well as in the presence of UDP alone, of the H antigen acceptor alone, of both UDP and the H antigen acceptor, and of both UDP-Gal and the 3-deoxy-Gal inactive acceptor analog (DA) (Table 1). All crystals belong to space group C2221, with the biological homodimer’s individual units related by 2-fold crystallographic axes of symmetry. The mutations did not significantly impact their immediate environment but did affect the observed conformations of the internal mobile polypeptide loop.
Table 1.
Data collection and refinement statistics for GTB double and triple mutants –15/GTB/C80S/C196S and –15/GTB/C80S/C196S/C209S in their unliganded and acceptor-bound states
| Enzyme |
C80S/C196S |
C80S/C196S/C209S |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Substrates | Unliganded | H antigen | UDP | UDP + H | UDP-Gal + DA | Unliganded | H antigen | UDP | UDP + H | UDP-Gal + DA |
| Resolution (Å)a,b | 20–1.55 (1.61–1.55) | 20–1.81 (1.87–1.81) | 20–1.90 (1.97–1.90) | 20–1.56 (1.62–1.56) | 20–1.40 (1.45–1.40) | 20–1.79 (1.85–1.79) | 20–1.48 (1.53–1.48) | 20–1.49 (1.54–1.59) | 20–1.93 (2.00–1.93) | 20–1.60 (1.66–1.60) |
| No. of observations | 193,697 | 125,758 | 106,079 | 197,444 | 257,813 | 134,916 | 187,580 | 216,207 | 111,381 | 169,278 |
| Unique reflections | 43,878 | 27,621 | 24,723 | 43,699 | 58,949 | 28,441 | 51,095 | 49,738 | 22,845 | 39,424 |
| Completeness (%)a | 96.4 (98.1) | 95.6 (93.9) | 99.0 (99.7) | 97.3 (99.6) | 95.3 (76.9) | 96.2 (98.1) | 97.2 (99.4) | 96.7 (96.7) | 95.8 (98.5) | 94.6 (89.5) |
| Rmerge (%)a | 4.2 (28.8) | 7.4 (31.7) | 7.8 (31.4) | 4.1 (30.0) | 3.5 (32.5) | 5.1 (29.9) | 3.8 (24.9) | 3.5 (39.7) | 5.1 (29.8) | 4.6 (26.9) |
| I/σ(I) | 17.1 (3.8) | 12.0 (4.1) | 10.5 (4.2) | 17.3 (3.6) | 19.2 (3.2) | 14.0 (4.0) | 14.8 (4.1) | 17.2 (2.8) | 15.2 (4.1) | 15.3 (4.1) |
| Rwork (%) | 18.98 | 17.32 | 18.41 | 18.85 | 19.45 | 16.52 | 20.19 | 16.92 | 17.45 | 18.40 |
| Rfree (%) | 22.76 | 22.23 | 22.94 | 21.72 | 22.96 | 21.97 | 23.82 | 22.57 | 22.81 | 22.28 |
| RMS | ||||||||||
| Bond lengths (Å) | 0.028 | 0.025 | 0.025 | 0.030 | 0.030 | 0.023 | 0.027 | 0.028 | 0.023 | 0.030 |
| Bond angles (°) | 2.305 | 1.931 | 2.189 | 2.247 | 2.572 | 1.80 | 2.178 | 1.961 | 2.202 | 2.447 |
| Mean B (Å2) | 21.44 | 21.24 | 26.57 | 20.93 | 18.88 | 24.51 | 21.08 | 22.77 | 27.62 | 19.79 |
| Protein | 19.54 | 19.63 | 25.69 | 19.24 | 16.93 | 23.75 | 19.69 | 21.55 | 25.97 | 18.01 |
| Ligandsc | NA | 22.85 | 42.39 | 22.68 | 20.94 | NA | 22.49 | 39.98 | 33.92 | 20.89 |
| Waterc | 34.41 | 32.12 | 35.50 | 33.19 | 30.79 | 33.01 | 31.79 | 33.05 | 32.51 | 31.74 |
| No. of water | 320 | 301 | 210 | 306 | 344 | 202 | 269 | 227 | 143 | 317 |
| PDB ID | 3I0C | 3I0E | 3I0D | 3I0F | 3I0G | 3I0H | 3I0J | 3I0I | 3I0K | 3I0L |
Values in parentheses are for the highest-resolution shell.
All crystals belonged to space group C2221, with unit cell dimensions ranging from 52.1 to 52.9 Å, from 148.6 to 150.4 Å, and from 79.3 to 79.7 Å.
All ligand and water molecules are refined at 100% occupancy.
Internal loop structure
Both the double and triple mutants show differing levels of disorder depending on their liganded state, with the level of disorder increasing when substrates are bound (Table 2). The double mutant shows disorder over residues 177–180 in the unliganded and H-antigen-bound forms, with disorder increasing to residues 176–185 for the UDP−, UDP + H-, and UDP-Gal + DA-bound structures. The unliganded triple mutant shows a nearly complete internal loop (Fig. 1a–c), with residues 176–181 disordered for the H-antigen-bound structure and 177–179 disordered for the UDP-bound structure. The UDP + H- and UDP-Gal + DA-bound structures of the triple mutant show an internal loop completely disordered over residues 176–184.
Table 2.
Observed internal loop residue electron density for GTB double and triple mutants GTB/C80S/C196S and GTB/C80S/C196S/C209S compared to wild-type GTB (as reported in Alfaro et al.9)
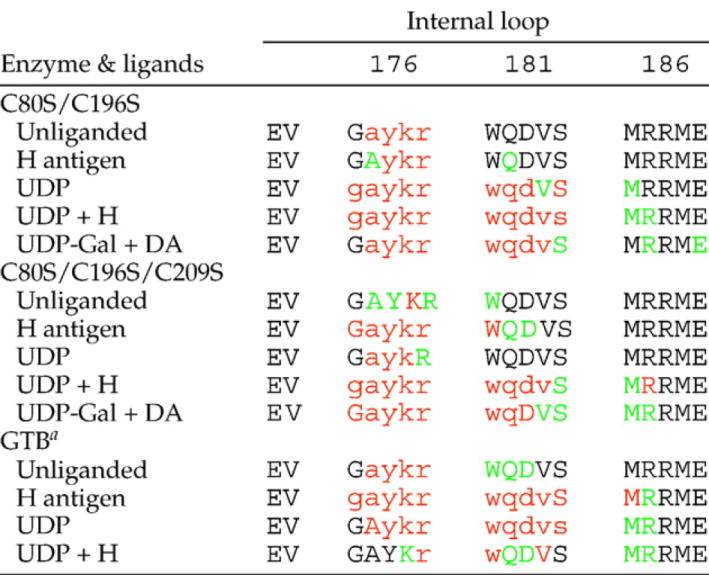 |
Black one-letter amino acid codes correspond to unambiguous electron density for main-chain and side-chain atoms; green correspond to unambiguous electron density for main-chain atoms only; red letters correspond to weak or ambiguous electron density for main-chain and side-chain atoms; residues with lowercase amino acid codes have not been included in the refined models.
as reported in Alfaro et al., 20089
Fig. 1.


Effect of mutation on loop stability and on the second acceptor-dependent UDP binding site. (a) Secondary-structure overlap of wild-type GTB (white) and GTB/C80S/C196S/C209S (red) displaying the locations of the three Cys-to-Ser mutations and the internal disordered loop spanning residues 166–185 (circled). (b) Close-up of this loop displaying the open (white) and closed (cyan) conformations of wild-type GTB overlapped with GTB/C80S/C196S/C209S (red) and GTB/C80S/C196S (magenta), which display an ordered open conformation with UDP-Gal displayed in the classic ‘tucked under’ conformation described in Ref. 9. (Fo − Fc)-omit map generated after refinement of the initial molecular replacement solution and before modeling of ligand (left, contoured at 1.0 σ) and modeled 2Fo − Fc maps after the completion of refinement (right, contoured at 1.0 σ) of (c) the internal loop’s ordinarily disordered residues 180–185 from PDB 3I0H and from PDB 3I0L, (d) the deoxy-acceptor H antigen analog α-L-Fucp-(1 → 2)-β. -D-(3-deoxy)-Galp-OR (DA), (e) UDP-Gal observed in donor binding site, and (f) UDP observed at the dimer interface. (g) Stereoview of the UDP molecule observed at the dimer interface shown in its binding site with Pro89 of the symmetry-related molecule shown in yellow. (h) The dimer interface observed between [x,y,z] and [1 − x,y,1/2−z] z] 2-fold symmetry-related molecules showing the entire protein backbone of both dimer subunits and the observed bridging UDP molecules as well as UDP-Gal and DA in the active sites represented as van der Waals spheres. (i) Relative motion of the residues lying between the second UDP and acceptor binding sites of the unliganded and UDP + H mutant enzyme structures.
Substrate binding
Both mutants in complex with UDP alone show excellent electron density about the nucleotide ribose and phosphate moieties but poor density about uracil C5–6. Appropriate electron density for the entire acceptor is seen in each structure soaked with the H antigen or the DA analog (Fig. 1d). Structures in complex with UDP-Gal and DA show excellent electron density for the entirety of both substrates, with the Gal moiety in a previously unseen conformation (Fig. 1e).
Unambiguous electron density corresponding to a second UDP molecule can be seen in a pocket formed by the biological assembly dimer interface between two GTB molecules related by a crystallographic 2-fold axis in structures soaked with UDP or UDP-Gal (Fig. 1f–i).
Kinetic data
The kinetic parameters for these enzymes are given in Table 3. The – 15/GTB construct (residues 68–354) has slightly increased kcat and Km values for acceptor (KA) and slightly decreased Km for UDP-Gal donor (KB) compared to the previously characterized – 10/GTB construct (residues 63–354) reported in Ref. 22, and this small additional truncation further into the stem region of the enzyme has not adversely affected enzyme activity. The double Cys-to-Ser mutant has a reduction in Km for both donor and acceptor substrates. The triple Cys-to-Ser mutant has an acceptor Km elevated approximately five times over wild type, while the donor Km has doubled. The mutations appear to have very little effect on kcat. For all mutants, the transfer of UDP-GalNAc is much slower than that of UDP-Gal with elevations in Km for both donor and acceptor substrates.
Table 3.
Kinetic constants for wild-type and mutant – 15/GTB UDP-Gal and UDP-GalNAc glycosyltransfer and UDP-Gal hydrolysis
| Substrate | UDP-Gal |
UDP-Gal hydrolysis |
UDP-GalNAc |
|||||
|---|---|---|---|---|---|---|---|---|
| KAa (μM) | KBb (μM) | kcat(s−1) | Km (μM) | kcat (s−1) | KAa (μM) | KBb (μM) | kcat (s−1) | |
| – 15 GTB (amino acids 68–354) | 55±13 | 45±6 | 7.3±0.7 | 47±20 | 0.0022±0.0002 | 300±55 | 145±20 | 0.41±0.04 |
| GTB/C80S/C196S | 32±6 | 23±6 | 5.6±0.4 | 22±9 | 0.0027±0.0003 | 300±70 | 210±40 | 0.70±0.02 |
| GTB/C89S/C196S/C209S | 350±80 | 78±13 | 8±1.0 | 120±40 | 0.0012±0.0001 | 200±40 | 320±70 | 0.15±0.03 |
KA is the Michaelis-Menten constant Km for the acceptor [Fucα1–2Galβ-O-(CH2)7CH3].
KB is the Michaelis-Menten constant Km for the donor (UDP-Gal or UDP-GalNAc).
The hydrolysis of UDP-Gal is very slow compared to the GT reaction with a kcat 0.02–0.05% of the rate of transfer for the mutants and – 15/GTB enzyme (Table 3). The Km for donor hydrolysis is generally comparable to the respective Km for donor in the UDP-Gal GT reaction. Both mutants produce the correct B antigen trisaccharide as determined by 1H NMR (data not shown).
Discussion
C209S reorders the active site
The C209S mutation results in significant structural deviance from wild type by causing internal loop residues 184 to 195 in the unliganded form of the triple mutant to change from the disordered state observed in the wild-type enzyme to an ordered state (Fig. 2, right). While the Cys209 side chain is tucked into a hydrophobic pocket lined by Ile192, Leu207, and Val277 in both the unliganded and liganded structures of wild-type GTB (Fig. 2e and f), the side chain of Ser209 in the mutant has rotated into the active site where its hydrophilicity allows the residence of two water molecules not observed in the unliganded wild-type structures (Fig. 2g). Ser209 forms hydrogen bonds to both of these water molecules, one of which also forms a strong hydrogen bond to Arg188 in the center of the internal loop. The binding of Arg188 to these novel water molecules mimics the observed effect of the donor binding to Arg188 in the wild type and is accompanied by the formation of salt bridges to Asp302 and Asp211 that are only observed in wild-type structures when donor is bound (Fig. 2f). Cys209 forms no hydrogen bonds, and there is no clear-cut steric hindrance to the mobile loop; thus, this effect appears to be caused by the inclusion of new solvent molecules by Ser209.
Fig. 2.

Effects of Cys-to-Ser mutations on substrate binding and loop order. Residue 196 (left column) cannot participate in a proximal hydrogen-bond cascade as cysteine in wild-type GTB (a and b), however, does when mutated to serine (c and d). This reordering of the proximal hydrogen-bond network in turn moves Glu197 of the mobile loop into a previously unseen conformation forming a salt bridge to Arg282, which helps stabilize the normally disordered internal loop. Residue 209 (right column) as wild-type cysteine (e and f) normally has no interaction with Arg188 of the mobile loop. C209S (g and h) introduces two water molecules not normally present, which shift Arg188 slightly to reinforce its salt bridge to Asp302, further stabilizing the loop.
The combinative effect of the C196S and C209S mutations is to cause the internal mobile loop observed to be disordered in unliganded wild-type GTB to become ordered in the absence of ligand (Fig. 1a–c). Significantly, the observed conformation of a random coil in the internal loop of the mutant enzyme differs considerably (Fig. 3a) from the helical structures observed in the donor-bound state of the wild-type enzyme (Fig. 3b). The observation that this region remains helical in the C80S/C196S mutant of this study, as well as a previously described C209A mutant,8 also shows that this new conformation is due to the combined effects of the C196S and C209S mutations. The main-chain rotation of Ser196 noted above that brings Glu197 within hydrogen-bonding distance of Arg282 (Fig. 2a and b), together with the stabilization of the remainder of the internal loop by the introduction of water into the active site by Ser209, pulls the internal loop away from the position observed for the liganded wild-type enzyme, resulting in a cascade of misplaced hydrophobic moieties (Fig. 3). The side chain of Phe195 is displaced from its place in a grouping of hydrophobic residues (including Ile192 and Leu201) and replaced by the side chain of Phe200, while Leu201 on the mutant replaces Phe200. The side chain of Phe195 in turn replaces the side chain of Arg198, which is bumped to occupy the position held by Arg199 in the wild-type enzyme.
Fig. 3.

SETOR23 diagrams depicting the rearrangement of hydrophobic clusters and secondary structure. The triple mutant GTB/C80S/C196S/C209S (a, red) has reordered a short amphipathic helix spanning residues 200–203 in the wild-type structure (b, white) by rearrangement of hydrophobic (orange circles) and hydrophilic (cyan circles) patches releasing the Glu203 helix dipole cap.
C196S stabilizes a helix adjacent to the internal mobile loop
The mutation of Cys196 to serine also results in some rearrangement of side-chain and main-chain hydrogen bonds from the wild-type enzyme in both the double and triple mutants in their unliganded and liganded states (Fig. 2, left). Similar stabilization of mobile sections of a GT-A GT by mutagenesis has been achieved before for β4Gal-T1, in which a C342T mutation stabilizes a region homologous to GTB’s labile C-terminal tail.19
Ser196 forms hydrogen bonds to the main-chain carbonyl atoms of Ile192 and Ser193 in both the liganded and unliganded triple mutant structures, which precludes the formation of hydrogen bonds from Ser193 to the main-chain carbonyl groups of Met189 and Glu190 seen in the wild type (Fig. 2a and b). Further, while Glu197 is usually disordered in the unliganded wild-type enzyme,9 it forms a salt bridge to residue Arg282 in the unliganded forms of both the double and triple mutants, which brings complete order to this helical segment of polypep-tide (residues 188–196) (Fig. 2c and d).
Novel donor substrate binding conformation
Binding of either UDP or both donor and acceptor in both mutants results in a significant increase in disorder in the internal loop (Table 2). Only residue 179 is disordered in the unliganded C80S/C196S/C209S structure, which expands to include residues 176–183 when in complex with UDP-Gal and DA. As stated above, this is in sharp contrast to what is observed for other GT-A-fold-type enzymes,12–19 including wild-type GTB9 (Table 2), which show a marked decrease in disorder and/or closing of the active site going from the unliganded to the liganded forms.
Surprisingly, the UDP-Gal donor molecule is bound in the active site in an orientation significantly different from that reported for the wild-type enzyme9 (Fig. 4a and b). While the uracil moiety remains in the position observed previously, the galactosyl residue has shifted by as much as 3.3 A resulting in different hydrogen-bonding partners for each and every sugar oxygen atom (as seen in Fig. 4). Asp211 and Arg188 are no longer involved in binding the galatosyl moiety, Asp302 has traded binding partners from Gal O4 to Gal O2, and His301, which previously bound only Gal O6, now makes two contacts to Gal O5 as well as one to Gal O3. An intramolecular contact between βPO4 and Gal O5 has also been established. This large shift appears to be at least partially due the introduction of a water molecule (Wat3) stabilized by Arg188. The shifted Gal residue also differs from the conformation previously observed in that the β-phosphate moiety has rotated about the Mn2+ coordination site such that it forms a direct hydrogen bond to Asp211 and to Wat3.
Fig. 4.

Stereoviews of enzyme bound to UDP-galactose. (a) Wild-type GTB9 and (b) Cys-to-Ser mutant enzymes. Cyan spheres represent Mn2+. (c) Compared to wild type (yellow), this novel UDP-Gal conformation (green) displaces the closed conformation of the internal loop by steric hindrance of Trp181 and inhibition of C-termini closure by rotation of β-phosphate out of Arg352 access.
This small change in orientation for the Gal moiety has significant consequences to formation of the catalytically competent ‘closed’ conformation in the crystal state, which is generated by the ordering of both the internal loop and C-terminal residues to form specific contacts to the substrate.9 Firstly, the new position of Gal O6 (Fig. 4c, green) prevents the approach of Trp181 of the mobile loop necessary to form the ordered closed conformation, and secondly, the rotated position of the UDP-Gal β-phosphate sequesters it from Arg352 of the C-terminus as is observed in the chimeric structures9 (Fig. 4c, yellow). Residues in the internal loop and the C-terminus of GTB have been shown to be essential for catalysis,11,13,24 and an inability of the Cys-to-Ser mutants to bind the donor in this conformation is incompatible with catalytic turnover.
Multiple substrate conformations during enzyme docking are routinely observed through spectroscopic techniques such as NMR and Raman spectroscopy;25,26 however, this is the first report of a crystal structure of a GT where unambiguous electron density is seen for a substrate in a stable conformation intermediate to that required for catalysis.
While the conformation of UDP-Gal observed previously9 to bind in the catalytically competent classic ‘tucked-under’ form (where the oxygen atoms of the pyrophosphate moiety are eclipsed) is consistent with that determined by NMR,27 this second conformation is more similar to the observed dominant solution rotamer of UDP-Gal as determined by transferred nuclear Overhauser enhancement.27 It has been suggested that GTB selectively binds the sparse population of UDP-Gal in solution that is folded in the bioactive conformation;27 however, these findings suggest that the enzyme is able to bind more abundant rotamers and facilitate gyration to the active conformation.
C80S imparts a second acceptor-dependent UDP binding site
The C80S mutation has little structural impact on most aspects of the unliganded enzymes, with only the side chain of Ser80 rotated ~ 120° about χ1 with respect to Cys80 without forming any new interactions. However, Ser80 is observed to bind an additional UDP molecule in the N-terminal dimer interface in those structures where UDP or UDP-Gal has been soaked in the presence of acceptor (Fig. 1f–i). There is no persuasive density for the corresponding Gal moiety in the UDP-Gal-soaked structures, indicating that either UDP-Gal has hydrolyzed or the sugar moiety is disordered. In addition to forming a hydrogen bond to Ser80, the uracil moiety forms hydrogen bonds directly to the main-chain Phe100 and through bridging water molecules to Thr78 O, Thr99 N, and a weak van der Waal’s contact to Pro89 on the symmetry-related molecule of the dimer.
Interestingly, UDP is not present in this second binding site in either mutant when the crystals are soaked in the presence of UDP alone, indicating some dependence on acceptor binding. The binding of acceptor alone does not appear to induce a change in the relative orientations of the dimers, but the communication between the acceptor and second UDP binding sites can be seen in Fig. 1i, where main-chain and side-chain atoms that connect the acceptor and second UDP molecule binding site move by up to 1 Å between the liganded and unliganded structures.
The observation of 2-fold dimer interface in the crystal was an indication of potential physiological relevance.3 First, the dimer interface is substantial as it constitutes nearly 20% of the monomer surface area. Second, the dimer could exist in the membrane-bound form as the observed N-termini (which would contain a leucine-rich N-terminal transmembrane region in the full-length wild-type enzyme) of the symmetry-related molecules point in the same direction (Fig. 1h) (which is the face of the enzyme opposite the second observed UDP binding site). Partial density of a second UDP molecule has previously been observed for the structure of retaining enzyme α-1,3 galactosyltransferase, with physiological roles proposed.28 However, UDP has not been observed at this site in the wild-type GTA/GTB enzymes, and no such species has been detected by mass spectrometry analysis,20 suggesting that a physiological role for this UDP binding site in GTA and GTB is unlikely.
Implications for catalysis
The introduction of water into the active site, a donor conformation that precludes active-site closure, and changes in polypeptide loop and bound donor orientation would indicate that the mutants would have a kinetic behavior significantly different from that of the wild-type GTB enzyme; however, while changes are observed in Km for both donor (KB) and acceptor (KA), the rate of turnover kcat has not altered appreciably, and the observed loop stringency is no hindrance to enzyme turnover.
The lack of effect on catalytic turnover by the binding of the donor in a non-catalytically competent conformation suggests that it is common to the wild-type enzyme as well and that the enzyme binds the donor in a low-energy rotamer and then facilitates its gyration to the catalytic conformation. Further, the binding of donor in a conformation more similar to the most prevalent solution rotamer would favor the active site remaining in the ‘open’ form and facilitate acceptor binding and, thus, form the structural basis for the recently observed cooperativity in substrate binding.20,29
Finally, the existence of an additional binding site for UDP at the dimer interface might be expected to increase Km for the donor (KB). While this is true for GTB/C80S/C196S/C209S, it is not true for GTB/C80S/C196S where KB is only half that of the wild-type enzyme.
Conclusions
The GTB/C80S/C196S double and GTB/C80S/C196S/C209S triple mutations have affected the conformation of the enzyme about the binding site and have altered the apparent donor and acceptor Km with little consequence to kcat. Examination of the crystal structures shows significant differences between wild-type GTB and these mutants in the behavior of the internal polypeptide loop and C-terminal residues, the conformation of the bound donor molecules, and the appearance of a second UDP binding site.
These structures do not adhere to the existing GT-A-fold-type enzyme paradigm wherein the binding and recognition of substrate occur by concomitant conformational changes preceding catalysis. Further, the structures of wild-type enzyme chimera9 show that the donor sugar must lie in a precise conformation in the active site in order to form the catalytically competent closed form of the enzyme. The novel low-energy conformations of mobile loop and donor binding observed in these Cys-to-Ser mutants are inconsistent with catalysis and so must be transient; however, they can play a role in cooperative binding by holding the active site open to recruit the acceptor prior to formation of the new glycosidic linkage.
Though there is no evidence of a physiological role for the second UDP molecule, detailed analysis of residues involved in binding the second molecule demonstrate the existence of subtle changes in the dimer quaternary structure and conformation upon substrate binding.
Materials and Methods
Construction of GTB/C80S/C196S and GTB/C80S/C196S/C209S
The original report of GTA/GTB kinetics30 utilized a construct in which the N-terminal transmembrane domain was deleted. Further deletions of 10–15 amino acid residues resulted in more crystallizable protein. All cysteine-to-serine constructs in this article have the additional 15-residue deletion – 15/GTB residues 68–354.
The – 15/GTB construct was cloned by PCR amplification of GTB gene reported in Seto et al. as a template with the primers 5′-ATA TGA ATT CAT GAT GGT TTA CCC GCA GCC GAA AGT TCT GAC CCC ATG-3′ (MIN3) to introduce an EcoRI site in the 5′ end and 5′-ATA ATT AAG CTT CTA TCA CGG GTT ACG AAC AGC CTG GTG GTT TTT-3′ (PCR-3B), which introduced a HindIII site in the 3′ end using a 30 × 94 °C/30 s, 55 °C/30 s, and 72 °C/1 min cycle PCR profile.30
The GTB/C80S/C196S clone was made by recombinant PCR using GTB as a template. The first gene fragment was amplified using the primers 5′-ATA GAA TTC ATG GTT TAC CCG CAG CCG AAA GTT CTG ACC CCA TCC CGT AAA GAC GTT CTG GTT-3′ (Cys1) and 5′-GTA GTC AAC TTC GGA CAG GAA ACG ACG TTC GGA GAA GTC GCT GAT CAT TTC-3′ (Cys2). The second fragment was made using 5′-TTC CTG TCC GAA GTT GAC TAC CTG GTT TGC GTT GAC GTT GAC ATG GAG TTC-3′ (Cys3) and PCR-3B. The two amplified products were joined together by PCR 3′ extension without primers (94 °C/30 s, 50 °C/60 s, 72 °C/60 s) × 7 cycles followed by 30 cycles after addition of primers. Similarly, for GTB/C80S/C196S/C209S, the primers Cys1 and Cys2 were used to amplify the first fragment; 5′-TTC CTG TCC GAA GTT GAC TAC CTG GTT TCC GTT GAC GTT GAC ATG GAG TTC-3′ (Cys4) and PCR-3B were used for the second fragment.
After gel purification, the PCR products were digested with EcoRI and HindIII (New England Biolabs), ligated into the plasmid pCWAlac, and transformed into BL21 (Novagen) competent cells.
Protein expression and purification
Mutant enzymes were expressed and purified from Escherichia coli cultures as described previously for the wild-type constructs.22 Protein concentrations were estimated using the method of Bradford using bovine gamma globulin as a protein standard.31
Crystallization
GTB/C80S/C196S and GTB/C80S/C196S/C209S were crystallized in 10- to 15-μl drops with 5.5–6 mg/ml protein, 35 mM sodium acetate, pH 4.6, 45 mM N-(2-acetamido)-2-iminodiacetic acid, pH 7.5, 30 mM sodium chloride, 3–4 mM magnesium chloride, and 3–3.5% polyethylene glycol 4000 over a reservoir of 10% polyethylene glycol 4000, 50 mM N-(2-acetamido)-2-iminodiacetic acid, 50 mM sodium acetate, 100 mM ammonium sulfate, and 5 mM magnesium chloride grown at 4–6 °C over 3–5 days. Crystals were washed in reservoir solution containing 30% 2-methyl-2,4-pentanediol as cryoprotectant. Ligand complexes were soaked in drops consisting of reservoir solution containing 7 mM H antigen (H) or the deoxy-acceptor α-L-Fucp-(1 → 2)-β.-D-(3-deoxy)-Galp-OR (DA) overnight and/or 10–15 mM UDP or UDP-Gal for 30–60 min. Unlike the crystals of the donor-bound chimeric enzymes reported earlier from our laboratory,9 there was little significant cracking of the crystals when placed into reservoirs containing substrates.
Data collection
Data for all crystals were collected on a Rigaku RAXIS IV++ area detector at distances of 72 mm with exposure times from 4.0 to 5.0 min at 0.5° oscillations and processed with d*trek.32 X-rays were produced by an MM-002 generator (Rigaku/MSC) coupled to Osmic ‘Blue’ confocal x-ray mirrors. The crystals were frozen and maintained under cryogenic conditions at a temperature of −160 °C using a CryoStream 700 crystal cooler (Oxford Cryosystems).
Structure determination and analysis
All structures were solved by molecular replacement using MOLREP,33 initially using wild-type GTB [Protein Data Bank (PDB) ID 1LZ7] and its progeny as starting models. Model refinement used REFMAC534 of the CCP4 program suite35 and models were fitted with SetoRibbon (S.V.E., unpublished).
Kinetic characterization
Kinetics were measured at 37 °C with a radiochemical assay using a hydrophobic acceptor, α-Fuc-(1 → 2)-β.-D-Gal-O(CH2)7CH3, and UDP-[6-3H]Gal as donor.36 The total assay volume was 12 μl containing substrates and enzyme in 50 mM 4-morpholinepropanesulfonic acid buffer, pH 7.0, 20 mM MnCl2, and 1 mg/ml bovine serum albumin assay buffer. At least seven concentrations of acceptor or donor were used with 1 mM of the alternate substrate. At the end of assay, the mixture was applied to a Sep-Pak reverse-phase C18 cartridge that was washed with 50–100 ml of water, to remove radiolabeled donor. Unreacted acceptor and radiolabeled trisaccharide product were eluted from the cartridge with 3.5 ml of methanol and counted in a liquid scintillation counter after the addition of 10 ml of Ecolite+ cocktail. Less than 10% of the substrate was consumed in the assay to ensure initial rate conditions. The kinetic parameters kcat and Km were derived from the best fit of the data to the Michaelis-Menten equation using nonlinear regression analysis with the GraphPad Prism 3.0 program (GraphPad Software, San Diego, CA). For the kinetics of UDP-Gal hydrolysis, enzyme was added to assays (50–70 μg/ml final enzyme concentration) that contained assay buffer and radiolabeled donor without any acceptor substrate. At the end of reaction, the mixture was diluted with 600 μl of water and immediately applied to a 0.5-ml column of AG1-X8 resin (Cl− form) to bind the donor. The column eluent and three successive aqueous washes of the column (0.6, 0.8, and 1.0 ml) were added to 10 ml of Ecolite+ cocktail and counted. Control reactions lacking enzyme were carried out in parallel.
Small-scale enzymatic synthesis
Small-scale reactions to verify that mutant enzymes produced blood group B trisaccharide were carried out in reaction mixtures that contained 1.5 mg of α-Fuc-(1 → 2)-β-D-Gal-O(CH2)7CH3 acceptor, 4.4 mg of UDP-Gal (di-K+ salt), and 0.3 U of the respective enzymes in a total volume of 400 μl of 100 mM 4-morpholinepropanesulfonic acid buffer, pH 7.0, containing 2 mg/ml bovine serum albumin, 40 mM MnCl2, and 25 U of DNA-grade alkaline phosphatase. The reaction mixture was left at ambient temperature (22 °C) for 48 h, with the products being isolated as described previously37 and analyzed by 1H NMR.
Accession numbers
The atomic coordinates and structure factors (codes 3I0C, 3I0D, 3I0E, 3I0F, 3I0G, 3I0H, 3I0I, 3I0J, 3I0K, and 3I0L, as described in Table 1) have been deposited in the PDB, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ†.
Acknowledgments
We thank Nikki Robert, Kristina Marvin, Mette Bien, and Line Lund for enzyme isolation and Bent O. Petersen for NMR spectra of reaction products (not shown). Funding from the Canadian Institutes of Health Research and salary support from the Michael Smith Foundation for Health Research (to S.V.E.) and from the Natural Science and Engineering Research Council of Canada and the Danish Natural Science Research Council (FNU) (to M.M.P.) are also acknowledged.
Abbreviations used
- GTA
glycosyltransferase A
- GTB
glycosyltransferase B
- DA
deoxy-acceptor, 3-deoxy-Gal-H antigen disaccharide
- PDB
Protein Data Bank
Footnotes
References
- 1.Yamamoto F, Clausen H, White T, Marken J, Hakomori S. Molecular genetic basis of the histo-blood group ABO system. Nature. 1990;345:229–233. doi: 10.1038/345229a0. [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto F, McNeill PD, Yamamoto M, Hakomori S, Bromilow IM, Duguid JK. Molecular genetic analysis of the ABO blood group system: 4. Another type of O allele. Vox Sang. 1993;64:175–178. doi: 10.1111/j.1423-0410.1993.tb05158.x. [DOI] [PubMed] [Google Scholar]
- 3.Lee HJ, Barry CH, Borisova SN, Seto NO, Zheng RB, Blancher A, et al. Structural basis for the inactivity of human blood group O2 glycosyltransferase. J Biol Chem. 2005;280:525–529. doi: 10.1074/jbc.M410245200. [DOI] [PubMed] [Google Scholar]
- 4.Kobata A, Grollman EF, Ginsburg V. An enzymatic basis for blood type B in humans. Biochem Biophys Res Commun. 1968;32:272–277. doi: 10.1016/0006-291x(68)90380-x. [DOI] [PubMed] [Google Scholar]
- 5.Landsteiner K. Über Agglutinationserschei-nungen normalen menschlichen Blutes. Wien Klin Wochenschr. 1901;14:1132–1134. [PubMed] [Google Scholar]
- 6.Seto NO, Compston CA, Evans SV, Bundle DR, Narang SA, Palcic MM. Donor substrate specificity of recombinant human blood group A, B and hybrid A/B glycosyltransferases expressed in Escherichia coli. Eur J Biochem. 1999;259:770–775. doi: 10.1046/j.1432-1327.1999.00086.x. [DOI] [PubMed] [Google Scholar]
- 7.Patenaude SI, Seto NO, Borisova SN, Szpacenko A, Marcus SL, Palcic MM, Evans SV. The structural basis for specificity in human ABO(H) blood group biosynthesis. Nat Struct Biol. 2002;9:685–690. doi: 10.1038/nsb832. [DOI] [PubMed] [Google Scholar]
- 8.Letts JA, Persson M, Schuman B, Borisova SN, Palcic MM, Evans SV. The effect of heavy atoms on the conformation of the active-site polypeptide loop in human ABO(H) blood-group glycosyltransferase B. Acta Crystallogr Sect D: Biol. Crystallogr. 2007;63:860–865. doi: 10.1107/S0907444907026479. [DOI] [PubMed] [Google Scholar]
- 9.Alfaro JA, Zheng RB, Persson M, Letts JA, Polakowski R, Bai Y, et al. ABO(H) blood group A and B glycosyltransferases recognize substrate via specific conformational changes. J Biol Chem. 2008;283:10097–10108. doi: 10.1074/jbc.M708669200. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen LC, Darden TA, Negishi M. Crystal structure of beta 1,3-glucuronyltransferase I in complex with active donor substrate UDP-GlcUA. J Biol Chem. 2002;277:21869–21873. doi: 10.1074/jbc.M112343200. [DOI] [PubMed] [Google Scholar]
- 11.Yazer MH, Palcic MM. The importance of disordered loops in ABO glycosyltransferases. Transfus Med Rev. 2005;19:210–216. doi: 10.1016/j.tmrv.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Unligil UM, Zhou S, Yuwaraj S, Sarkar M, Schachter H, Rini JM. X-ray crystal structure of rabbit N-acetylglucosaminyltransferase I: catalytic mechanism and a new protein superfamily. EMBO J. 2000;19:5269–5280. doi: 10.1093/emboj/19.20.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, Strynadka NC. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat Struct Biol. 2001;8:166–175. doi: 10.1038/84168. [DOI] [PubMed] [Google Scholar]
- 14.Boix E, Zhang Y, Swaminathan GJ, Brew K, Acharya KR. Structural basis of ordered binding of donor and acceptor substrates to the retaining glycosyltransferase, alpha-1,3-galactosyltransferase. J Biol Chem. 2002;277:28310–28318. doi: 10.1074/jbc.M202631200. [DOI] [PubMed] [Google Scholar]
- 15.Ramakrishnan B, Balaji PV, Qasba PK. Crystal structure of beta1,4-galactosyltransferase complex with UDP-Gal reveals an oligosaccharide acceptor binding site. J Mol Biol. 2002;318:491–502. doi: 10.1016/S0022-2836(02)00020-7. [DOI] [PubMed] [Google Scholar]
- 16.Qasba PK, Ramakrishnan B, Boeggeman E. Substrate-induced conformational changes in glycosyltransferases. Trends Biochem Sci. 2005;30:53–62. doi: 10.1016/j.tibs.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Kubota T, Shiba T, Sugioka S, Furukawa S, Sawaki H, Kato R, et al. Structural basis of carbohydrate transfer activity by human UDP-GalNAc: polypeptide alpha-N-acetylgalactosaminyltransferase (pp-GalNAc-T10) J Mol Biol. 2006;359:708–727. doi: 10.1016/j.jmb.2006.03.061. [DOI] [PubMed] [Google Scholar]
- 18.Jamaluddin H, Tumbale P, Withers SG, Acharya KR, Brew K. Conformational changes induced by binding UDP-2F-galactose to alpha-1,3 galactosyltransferase—implications for catalysis. J Mol Biol. 2007;369:1270–1281. doi: 10.1016/j.jmb.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 19.Ramakrishnan B, Shah PS, Qasba PK. alpha-Lactalbumin (LA) stimulates milk beta-1,4-galactosyltransferase I (beta 4Gal-T1) to transfer glucose from UDP-glucose to N-acetylglucosamine. Crystal structure of beta 4Gal-T1 × LA complex with UDP-Glc. J Biol Chem. 2001;276:37665–37671. doi: 10.1074/jbc.M102458200. [DOI] [PubMed] [Google Scholar]
- 20.Shoemaker GK, Soya N, Palcic MM, Klassen JS. Temperature-dependent cooperativity in donor-acceptor substrate binding to the human blood group glycosyltransferases. Glycobiology. 2008;18:587–592. doi: 10.1093/glycob/cwn043. [DOI] [PubMed] [Google Scholar]
- 21.Sindhuwinata N, Munoz E, Munoz FJ, Palcic MM, Peters H, Peters T. Binding of an acceptor substrate analog enhances the enzymatic activity of human blood Group B galactosyltransferase. Glycobiology. 2010;20:718–723. doi: 10.1093/glycob/cwq019. [DOI] [PubMed] [Google Scholar]
- 22.Marcus SL, Polakowski R, Seto NO, Leinala E, Borisova S, Blancher A, et al. A single point mutation reverses the donor specificity of human blood group B-synthesizing galactosyltransferase. J Biol Chem. 2003;278:12403–12405. doi: 10.1074/jbc.M212002200. [DOI] [PubMed] [Google Scholar]
- 23.Evans SV. SETOR: hardware-lighted three-dimensional solid model representations of macromolecules. J Mol Graphics. 1993;11:134–138. 127–128. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]
- 24.Boix E, Swaminathan GJ, Zhang Y, Natesh R, Brew K, Acharya KR. Structure of UDP complex of UDP-galactose:beta-galactoside-alpha-1,3-galactosyltransferase at 1.53-Å resolution reveals a conformational change in the catalytically important C terminus. J Biol Chem. 2001;276:48608–48614. doi: 10.1074/jbc.M108828200. [DOI] [PubMed] [Google Scholar]
- 25.Mildvan AS, Sloan DL, Fung CH, Gupta RK, Melamud E. Arrangement and conformations of substrates at the active site of pyruvate kinase from model building studies based on magnetic resonance data. J Biol Chem. 1976;251:2431–2434. [PubMed] [Google Scholar]
- 26.Lee H, Storer AC, Carey PR. Conformational states of N-acylglycine dithioesters in solution: resonance Raman studies of isotopically substituted models for enzyme-substrate complexes. Biochemistry. 1983;22:4781–4789. doi: 10.1021/bi00289a026. [DOI] [PubMed] [Google Scholar]
- 27.Angulo J, Langpap B, Blume A, Biet T, Meyer B, Krishna NR, et al. Blood group B galactosyltransferase: insights into substrate binding from NMR experiments. J Am Chem Soc. 2006;128:13529–13538. doi: 10.1021/ja063550r. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Swaminathan GJ, Deshpande A, Boix E, Natesh R, Xie Z, et al. Roles of individual enzyme-substrate interactions by alpha-1,3-galactosyltransferase in catalysis and specificity. Biochemistry. 2003;42:13512–13521. doi: 10.1021/bi035430r. [DOI] [PubMed] [Google Scholar]
- 29.Soya N, Shoemaker GK, Palcic MM, Klassen JS. Comparative study of substrate and product binding to the human ABO(H) blood group glycosyltransferases. Glycobiology. 2009;19:1224–1234. doi: 10.1093/glycob/cwp114. [DOI] [PubMed] [Google Scholar]
- 30.Seto NO, Palcic MM, Hindsgaul O, Bundle DR, Narang SA. Expression of a recombinant human glycosyltransferase from a synthetic gene and its utilization for synthesis of the human blood group B trisaccharide. Eur J Biochem. 1995;234:323–328. doi: 10.1111/j.1432-1033.1995.323_c.x. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr., Sect D: Biol. Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect D: Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Collaborative Computational Project No. 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Palcic MM, Heerze LD, Pierce M, Hindsgaul O. The use of hydrophobic synthetic glycosides as acceptors in glycosyltransferase assays. Glycoconjugate J. 1988;5:49–63. [Google Scholar]
- 37.Seto NO, Compston CA, Szpacenko A, Palcic MM. Enzymatic synthesis of blood group A and B trisaccharide analogues. Carbohydr Res. 2000;324:161–169. doi: 10.1016/s0008-6215(99)00297-9. [DOI] [PubMed] [Google Scholar]