

Abstract
Helicobacter pylori (H. pylori) has co-evolved with humans to be transmitted from person to person and to colonize the stomach persistently. A well-choreographed equilibrium between the bacterial effectors and host responses permits microbial persistence and health of the host, but confers a risk for serious diseases including gastric cancer. During its long coexistence with humans, H. pylori has developed complex strategies to limit the degree and extent of gastric mucosal damage and inflammation, as well as immune effector activity. The present editorial thus aims to introduce and comment on major advances in the rapidly developing area of H. pylori/human gastric mucosa interaction (and its pathological sequelae), which is the result of millennia of co-evolution of, and thus of reciprocal knowledge between, the pathogen and its human host.
Keywords: Helicobacter pylori, Gastric mucosa, Pathogen/host interaction, Gastric diseases, Bacterial virulence factors, CagA, VacA
INTRODUCTION
Helicobacter pylori (H. pylori) is a Gram-negative microaerophilic, spiral bacterium that specifically colonizes the gastric mucosa, and it is the most common bacterial infection worldwide. Typically acquired during childhood, the infection can persist in the gastric ecosystem throughout the life span of the host, if untreated[1]. Colonization of the stomach by H. pylori causes chronic gastritis that, during the decades that follow initial infection, can remain silent, due to the dynamic equilibrium between the bacterium and its human host, or evolve into more severe diseases, such as atrophic gastritis, peptic ulcer, lymphoma of the mucosa-associated lymphoid tissue or gastric adenocarcinoma[2]. Gastric cancer, despite its declining incidence rate, remains the fourth most common cancer, the second leading cause of cancer-related death, and the 14th most common cause of death overall worldwide, which kills > 700 000 people each year[3,4]. Early stages of the disease are often clinically silent, with patients having advanced stage disease at the time of diagnosis, and reported 5-year survival rates are approximately 20%[5]. H. pylori infection is the strongest known risk factor for gastrointestinal malignancies that arise within the stomach, and epidemiological studies have determined that the attributable risk for gastric cancer conferred by H. pylori is approximately 75%[6]. While H. pylori infection increases the risk of developing both types of gastric cancer (i.e. diffuse and intestinal), chronic inflammation is not a prerequisite for development of diffuse-type cancer, thus suggesting that different mechanisms underlie the ability of H. pylori to induce gastric malignancies. Also, it is likely that H. pylori influences early stages in gastric carcinogenesis, as suggested by the demonstration that eradication of the infection significantly decreases the incidence of gastric cancer only in patients without premalignant lesions at the time of diagnosis[7].
Development of gastric adenocarcinoma occurs in < 1% of H. pylori-infected subjects[8]. Also, incidences of gastric carcinoma in H. pylori-infected individuals may vary dramatically among different geographical areas[9]. This might be accounted for by H. pylori strain diversity within different geographical areas and/or within different individuals[10], and further suggests that factors other than the bacterium may be involved in the carcinogenic process.
Evidence increasingly indicates that H. pylori-related gastric carcinogenesis is likely to be the result of a well-choreographed interaction between the pathogen and host, which is in turn, dependent on strain-specific bacterial factors, host genotypic traits and permissive environmental factors.
The present editorial thus aims to introduce and comment on major advances in the rapidly developing area of H. pylori/human gastric mucosa interaction (and its pathological sequelae), which is the result of millennia of co-evolution of, and thus of reciprocal knowledge between, the pathogen and its human host.
HOST FACTORS
The basic process that mediates H. pylori-induced damage is gastritis with its associated humoral and cell-mediated immune mechanisms. The outcome of H. pylori infection depends on the severity and the anatomical distribution of the gastritis induced by the bacterium. Individuals with corpus-predominant gastritis (so-called “gastric cancer phenotype”), which accounts for almost 1% of infected subjects, are more likely to develop hypochlorhydria, gastric atrophy, and eventually, gastric cancer; those with antrum-predominant gastritis (so-called “duodenal ulcer phenotype”), which accounts for up to 15% of infected subjects, have excessive acid secretion and are more likely to develop duodenal ulcer. Finally, subjects with mild, mixed antrum and corpus gastritis (so-called “benign gastritis phenotype”), which accounts for up to 85% of infected subjects, have almost normal acid secretion and, generally, no serious disease. These clinical outcomes seem to be mutually exclusive, and are largely influenced by a genetically regulated inflammatory response of the host gastric mucosa to the infection[5,6].
A combination of polymorphisms in the host interleukin-1 (IL1) gene cluster (i.e. in the IL1B gene, which encodes IL-1β, a pro-inflammatory cytokine and a powerful inhibitor of gastric secretion, and in IL1RN, which encodes IL-1ra, the naturally occurring receptor antagonist of IL-1), and in the genes that encode tumor necrosis factor (TNF)-α, and IL-10, which result in elevated levels of IL-1β and TNF-α and in low levels of IL-10 (which inhibits production of pro-inflammatory cytokines), confer a 27-fold increased risk of developing gastric cancer[11]. Also, it has recently been demonstrated that polymorphisms in the promoter of IL-8 gene, which enhance the transcriptional activity in response to IL-1β or TNF-α, are associated with increased risk of gastric cancer in patients with H. pylori infection[12,13]. This chemokine belongs to the CXC family and is a potent chemoattractant for neutrophils and lymphocytes, and has effects on cell proliferation, migration and tumor angiogenesis.
H. pylori infection is first handled by receptors of the innate immune response, and it is therefore conceivable that functionally relevant polymorphisms in genes of this arm of the immune system could affect the magnitude and subsequent direction of the host’s response against the infection. In particular, toll-like receptor (TLR)4 is a member of a family of pattern recognition receptors that activate pro-inflammatory signaling pathways in response to microbes or pathogen-associated molecular patterns[14]. TLR4 transduces signals that promote transcription of genes that are involved in immune activation, including nuclear factor (NF)-κB and mitogen-activated protein (MAP) kinase pathways[15]. A functional polymorphism at position +896 in exon 4 of the TLR4 gene has been demonstrated[16], which renders carriers hyporesponsive to lipopolysaccharide challenge by disrupting transport of TLR4 to the cell membrane, or impairing ligand binding or protein interactions[16]. The defective signaling through TLR4 leads to an exaggerated inflammatory response with severe tissue destruction that causes gastric atrophy and severe hypochlorhydria. Two independent case-control studies have demonstrated that TLR4+896G carriers have an almost eightfold increase in OR for hypochlorhydria and gastric atrophy, and a 2.5-fold increase for gastric cancer[17].
It is likely that subjects with an overall pro-inflammatory genetic makeup based on a combination of markers from the adaptive and innate immune systems, respond to H. pylori infection by creating an environment within the stomach that is chronically inflamed and with markedly reduced acidity. These environmental conditions favor the growth of other bacteria within the gastric milieu, which leads to sustained inflammation and decreased levels of vitamin C in gastric juice. This facilitates the formation of mutagenic N-nitroso compounds and reactive oxygen species (ROS)[6,11], which ultimately leads to increased oxidative/genotoxic stress. Moreover, the profound inhibition of acid secretion that is associated with these pro-inflammatory genotypes favors a shift from an antrum-predominant to corpus-predominant gastritis with the onset of gastric atrophy and intestinal metaplasia (i.e. precancerous lesions).
H. PYLORI-INDUCED CELL SIGNALING IN GASTRIC CARCINOGENESIS
The host response to H. pylori infection may contribute to gastric carcinogenesis by promotion of a chronic inflammatory response that contributes to mucosal cell damage, or by interference with the mechanisms of proliferation and/or survival that regulate epithelial cell homeostasis[2,6,10].
H. pylori proteins and induced responses play a major role in the increased disease risk associated with the infection, but they are not absolute determinants of gastric carcinogenesis. The chronic inflammation that develops in response to this organism greatly contributes to transformation. In this respect, bone-marrow-derived cells (BMDCs) have been demonstrated to home to and engraft the inflamed gastric mucosa of mice infected with Helicobacter felis. This phenomenon takes place within foci in which tissue injury induces excessive apoptosis and overwhelms the population of endogenous tissue stem cells[18]. Subsequently, BMDCs in the inflamed gastric environment degenerate into adenocarcinoma, thus suggesting that gastric adenocarcinoma originates from BMDCs.
It is generally accepted that H. pylori infection results in a Th1-dominant response and that gastric inflammation largely depends on Th1 cell response with increased production of IL-1β, TNF-α and IL-8, but not IL-4 and IL-10[2,6,10,19,20]. A novel subset of effector T cells, identified by secretion of IL-17, has been defined as Th17 cells, which are distinct from Th1 and Th2 cells in their differentiation and function[21]. TNF-α and IL-6 from activated macrophages/dendritic cells (DCs) are required for Th17 cell differentiation, whereas IL-12 and interferon-γ promote Th1 cell development, and Il-4 primes Th2 cell differentiation. Recently, it has been suggested that H. pylori infection mainly leads to a specific Th17/Th1 immune response that plays a major role in H. pylori infection, which promotes mucosal inflammation and contributes to bacterial colonization[22]. In fact, H. pylori burden and inflammation are both reduced when IL-17 activity is blocked in vivo or IL-17-/- mice are used[22]. The dynamics of Th cell immune responses to H. pylori suggest that Th17 cell responses are induced earlier than Th1 cell responses, thus implying that Th17 and Th1 cells promote inflammation at different stages. It is likely that the Th17/Il-17 pathway modulates Th1 cell responses, and Th17 and Th1 cells may act synergistically to induce gastritis during H. pylori infection, by triggering the recruitment of inflammatory cells, including Th1 cells, into the gastric mucosa through the induction of chemokines. Also, the activated Th17/Th1 pathway may destroy gastric tissue by inducing matrix metalloproteinase (MMP) production, which favors subsequent pathogen dissemination and persistent infection. This might have implications also for the carcinogenic process associated with H. pylori infection. In fact, Th17 cells have been reported to contribute to gastric cancer pathogenesis[23].
A concomitant helminthic infection that triggers a Th2 cell response may blunt the Th17/Th1 cell responses associated with H. pylori infection, thus limiting the pathological consequences of H. pylori gastric colonization; in particular, gastric atrophy. This might partially explain why in African countries, where the prevalence of H. pylori infection acquired during childhood is close to 80%, the prevalence of gastric cancer is very low and accounts for < 2% of all malignant tumors[24].
Variation in the ability of H. pylori strains to trigger the production of chemokines from gastric epithelium depends on the presence of a functional type IV secretion system (TFSS), which is encoded by the cag pathogenicity island (PAI). Although the cag PAI facilitates the translocation of CagA, the effect of this bacterial protein on cytokine synthesis is controversial; the majority of reports show no effect of CagA on cytokine synthesis, thus suggesting that other effectors are involved in the epithelial cytokine and chemokine response to H. pylori infection[10,25,26]. Indeed, it has been shown that induction of pro-inflammatory responses in epithelial cells infected by H. pylori is mediated by the host protein CARD4 (also known as Nod1), an intracellular pathogen-recognition molecule, and that the effect is dependent on the delivery of peptidoglycan to host cells by the TFSS[27]. Consistent with involvement of CARD4 in host defense, Card4-deficient mice are more susceptible to infection by H. pylori strains that contain the cag PAI than are wild-type mice[27]. However, two studies have demonstrated that CagA directly induces IL-8 release from gastric mucosal cells[28,29].
Although H. pylori elicits innate and acquired immune responses, the host is unable to eliminate the organism from the gastric mucosa, and chronic infection is the usual outcome[2,6,30]. H. pylori antigenic variation (i.e. modification of bacterial antigenic determinants due to mutation or intragenomic homologous recombination) and mimicry of host antigens (i.e. bacterial expression of antigens similar to those expressed by the host)[31], as well as induction of apoptosis by H. pylori in DC precursor monocytes[20] and the intracellular persistence of the bacteria in gastric epithelial progenitor cells[32], might be crucial for evasion of the immune response. Also, it has recently been demonstrated that H. pylori evades TLR5 innate immunity[33]. In fact H. pylori, although being a flagellated organism, does not release flagellin, and recombinant H. pylori flagellin is much less active than Salmonella typhimurium flagellin in activating TLR5-mediated IL-8 secretion[33].
Although it fails to eliminate the organism, the inflammatory response induced by H. pylori increases cellular damage. Activation of the TNF receptor by TNF-α results in the induction of apoptosis and mucosal cell damage. Increased apoptosis through direct or cytochrome-c-mediated activation of caspases is also contributed to by CD95/FAS and VacA. Recently, we have shown that H. pylori infection upregulated IL-21 levels in gastric epithelial cells in vitro, as well as in the gastric mucosa of H. pylori-infected humans[34]. IL-21 overexpression is associated with increased production of MMP-2 and MMP-9, through an NF-κB-dependent mechanism. Increased MMP-2 and MMP-9 levels might contribute to chronic gastric damage and inflammation by degrading extracellular matrix proteins and by favoring the recruitment of circulating cells into inflamed tissue[35,36].
Other ways in which pro-apoptotic pathways are induced during H. pylori infection include superoxide production by infiltrating neutrophils, elevated nitric oxide production by inducible nitric oxide synthase (iNOS), which is overexpressed in infected gastric mucosa[2,6,37], and generation of ROS by bacterial secretion of γ-glutamyltranspeptidase (γ-GT) in the presence of glutathione (GSH) and transferrin, as a source of iron[38]. In fact, it has been shown that γ-GT is important for H. pylori-mediated apoptosis of AGS (a cell line derived from a human gastric adenocarcinoma) gastric epithelial cells[39]. It is also interesting to note that inflammatory stimuli that activate cytokine receptors and p38 (a stress-activated MAPK) can induce apoptosis. Conversely, activation of cytokine receptors and p38 might also inhibit apoptosis through NF-κB and c-Jun activation[6].
Inhibition or induction of apoptosis could both be relevant to H. pylori-related carcinogenesis. Induction of apoptosis might favor the development of atrophic gastritis and gastric gland recruitment of bone-marrow-derived precursor cells that might ultimately develop into intraepithelial cancer[18]. Inhibition of apoptosis, on the other hand, represents the loss of a physiological safeguard against perpetuating the acquisition of DNA damage that can lead to the malignant transformation of cells[2,6,10].
Proliferative response
Several signal transduction pathways are activated during the proliferative response of gastric epithelial cells to H. pylori-induced cell damage[2,6]. The compensatory hyperproliferative response of gastric epithelial cells during H. pylori infection might be sustained by hypergastrinemia, increased expression of epidermal growth factor (EGF)-related peptides, and activation of the EGF receptor (EGFR) signal transduction pathway in gastric epithelial cells[2,6,40-42]. In addition, translocation of CagA into gastric epithelial cells induces a growth-factor-like-response through activation of the Ras-MAPK pathway[26]. Finally, it has been shown that H. pylori upregulates the expression of cyclooxygenase (COX)-2, the inducible isoform of the enzyme that is responsible for prostaglandin production, in human gastric epithelial cells in vitro and in human gastric mucosa in vivo[37,43].
EGFR-related pathway is upregulated in a number of malignancies and is an important target for treatment of several neoplasms of the gastrointestinal tract. H. pylori has been shown to upregulate amphiregulin and HB-EGF, members of the EGFR ligands family, and to activate EGFR in MKN 28 cells[40]. Subsequently, it has been demonstrated that EGFR transactivation by H. pylori is mediated through metalloproteinase-dependent cleavage of HB-EGF. The required metalloproteinases are likely to be members of a disintegrin and metalloproteinase (ADAM) family. In particular ADAM17 is the ideal candidate enzyme for the regulation pathway[42].
H. pylori-induced upregulation of COX-2 and EGF-related peptide expression in human gastric epithelial cells depends on the bacterial production of γ-GT[38]. Activation of phosphatidylinositol-3’-kinase (PI3K)-dependent and/or p38-dependent pathways is responsible for H. pylori γ-GT-induced upregulation of COX-2 and EGF-related peptide expression in gastric mucosal cells[38]. However, another study has demonstrated that upregulation of HB-EGF by H. pylori in human gastric epithelial cells is dependent on MAP kinase kinase activation, thus suggesting that EGF-related peptide expression might be regulated by different signal transduction pathways[42]. In keeping with this, it has been demonstrated that COX-2 expression in gastric epithelial cells is also regulated by extracellular signal-regulated kinase (ERK)/MAPK activation[43,44].
The upregulation of growth factor and COX-2 expression might increase the mitogenic activity of H. pylori-infected gastric mucosa, and protect cells from H. pylori-induced cell damage, which might therefore be regarded as early events in the development of H. pylori-associated gastric carcinogenesis[2,6]. Upregulation of COX-2 and iNOS expression might also contribute to the high levels of oxidative DNA damage seen during H. pylori infection, and this could increase the mutation rate in infected hyperproliferative gastric mucosa[2,37]. More recently, blockade of EGFR activation by HB-EGF neutralizing antibody or by abrogating ADAM17 expression, has been shown to protect gastric epithelial cells from H. pylori-induced apoptosis. The anti-apoptotic effect of EGFR activation seems to depend on PI3K-dependent activation of the anti-apoptotic factor Akt, increased expression of the anti-apoptotic factor Bcl-2, and decreased expression of the pro-apoptotic factor Bax[45]. Because EGFR activation is linked to increased proliferation, reduced apoptosis, disruption of cell polarity and enhancement of cell migration, transactivation of EGFR by H. pylori might represent an attractive target for studying early events that may precede transformation. Also, the EGFR-related pathway might be regarded as a molecular target for treatment of gastric cancer[46]. However, surveys of human gastric cancer specimens for evidence of overexpression or mutations of EGFR have found both events to be rare[47].
The activation of a pathway mediated by EGFR, MAPK and COX-2 is also responsible for the induction of vascular endothelial growth factor (VEGF) expression in H. pylori-infected gastric epithelial cells[44]. This effect is specifically related to the VacA toxin of H. pylori[44], and is associated with a significant increase in blood vessel formation, suggesting that neoangiogenesis might contribute to tumor growth in H. pylori-related gastric carcinogenesis[48]. H. pylori infection also promotes gastric epithelial cell invasion by inducing the production of MMP-7 through MAPK activation[49], and MMP-9 and VEGF expression through an NF-κB- and COX-2-mediated pathway[50].
Another host effector that is aberrantly activated during H. pylori-induced gastric carcinogenesis is β-catenin, a ubiquitously expressed molecule that regulates the expression of several genes, including c-myc, the cyclin D genes, MMP7 and PTGS2 (which encodes COX-2)[51]. Membrane-bound β-catenin is a component of adherens junctions that link cadherin receptors to the actin cytoskeleton. Cytoplasmic β-catenin is a downstream component of the Wnt signal transduction pathway. In the absence of Wnt ligand, the inhibitory complex composed of axin, adenomatous polyposis coli (APC) and glycogen synthase kinase 3β (GSK3β) induces the degradation of β-catenin, and maintains low steady-state levels of free β-catenin in the cytoplasm or nucleus. After binding of Wnt to its receptor Frizzled, Dishevelled is activated, thus preventing GSK3β from phosphorylating β-catenin. This allows β-catenin to translocate to the nucleus and activate the transcription of target genes that are involved in carcinogenesis. Increased β-catenin expression or APC mutations are present in up to 50% of gastric adenocarcinomas[52]. Moreover, an oncogenic H. pylori strain can induce nuclear translocation of β-catenin and activation of the LEF/TCF transcription factor that regulates the expression of β-catenin-responsive genes[51]. β-catenin activation is dependent on the translocation of CagA into host epithelial cells, which reinforces the evidence that cagA+ H. pylori strains induce stronger activation of the signal transduction pathways that regulate the proliferation, invasion and survival of gastric epithelial cells[51]. Recently, activation of PI3K and Akt has been shown to induce phosphorylation and inactivation of GSK3β, which permits β-catenin to accumulate in the cytosol and nucleus[53]. Sustained induction of PI3K/Akt signaling in response to H. pylori infection with subsequent β-catenin activation has been demonstrated to be due to the interaction of CagA with Met, the hepatocyte growth factor receptor, via CRPIA (for conserved repeat responsible for phosphorylation-independent activity), a conserved motif in the C-terminal region of CagA[53]. Also, EGFR transactivation by H. pylori leads to activation of PI3K/Akt signaling, which ultimately leads to β-catenin activation[45,54].
H. pylori has co-evolved with humans to be transmitted from person to person and to colonize the stomach persistently. A well-choreographed equilibrium between the bacterial effectors and host responses permits microbial persistence and health of the host, but confers a risk for serious diseases including gastric cancer. During its long coexistence with humans, H. pylori has evolved complex strategies to limit the degree and extent of gastric mucosal damage and inflammation, as well as of immune effector activity. Severe disease, associated with bacterial colonization, might reflect loss of this control[31]. In this respect, we have recently demonstrated that Hp(2-20), a cationic a-helical peptide that has been isolated from the N-terminal region of the H. pylori ribosomal protein, L1, by interacting with formyl peptide receptors (FPRs), stimulates migration and proliferation of gastric epithelial cells in vitro and accelerates gastric mucosal healing in vivo[55]. This raises the intriguing possibility that H. pylori, through the production of Hp(2-20) and its interaction with FPRs is also able to modulate the capacity of gastric mucosa to maintain or recover its integrity.
H. PYLORI INVASIVENESS AND INTERACTIONS WITH NON-EPITHELIAL CELLS: IN VIVO VERITAS
H. pylori is commonly considered an essentially extracellular, non-invasive bacterium. In an infection, 80%-90% of the bacteria freely swim into the mucus layer of the stomach, while the residual 10%-20% are in intimate contact with surface epithelial cells[54,56]. As a consequence, a prominent immune-inflammatory response is invariably mounted in the underlying lamina propria. An intact epithelium should form a structural barrier that prevents direct contact between the bacterium on the luminal side and reactive inflammatory cells on the stromal side. Therefore, to explain how a strong mucosal and systemic reaction may be elicited, H. pylori-induced functional changes in the epithelium have been considered, such as a bacterial activation of the accessory immune competence inherent in the gastric epithelium, which in turn may modulate underlying stroma cells. Among pertinent epithelial changes so far documented are de novo or increased expression of pro-inflammatory cytokines, proteases (like cathepsins E, B, L, S and D) known to be involved in antigen processing, or HLA-DR and co-stimulatory molecules like B7-1 and B7-2 that are involved in antigen presentation[57]. However, a few early light and electron microscopy studies have suggested the presence of H. pylori cells inside the gastric mucosa, either in epithelial cells and intraepithelial intercellular spaces, or in the underlying lamina propria[57-59]. Nevertheless, the scientific community has exhibited a widespread reluctance to accept this published evidence. By contrast, a lot of in vitro studies have been carried out to investigate epithelial cell invasion by H. pylori, as well as roles and molecular mechanisms of direct interactions between H. pylori or H. pylori products/virulence factors and immune cells such as T or B lymphocytes, DCs, macrophages, monocytes, and mast cells[57,59,60]. However, despite this interest, the importance of these in vitro studies has been questioned because of a lack of convincing evidence that H. pylori actually invades the gastric mucosa and interacts with non-epithelial cells, thus it is highly questionable whether any of these observations have clinical relevance[60]. In this respect, Lu et al[60] have strongly stressed the fact that in vitro studies are in theory designed to allow deeply detailed molecular study of in vivo events, whereas it is easy but meaningless to generate in vitro data irrespective of whether the experiment has an in vivo correlation. Similarly, other leading investigators[61,62] have underlined the problem that in-vitro-derived findings and considerations may make sense in terms of cell biology, but it remains to be established how and to what extent they apply to the in vivo situation, which is now needed to be investigated in detail and in quantitative terms. Thus, only in vivo veritas!
Indeed, recently there has been a re-emerging interest for in vivo investigations of H. pylori/human gastric mucosa interaction. By microscopy and molecular investigations, Semino-Mora et al[63] have demonstrated the presence of intracellular and interstitial H. pylori in preneoplastic as well as in neoplastic lesions of human gastric mucosa, with persistent intracellular expression of H. pylori virulence genes. These findings may substantially expand current concepts on the role of the bacterium in gastric carcinogenesis.
In a transmission electron microscopy study, Necchi et al[57] have unequivocally detected H. pylori in intraepithelial, intercellular and stromal sites of the majority of human gastric biopsies, and have shown bacteria on their luminal side (Figures 1 and 2). In keeping with in vitro demonstration that H. pylori can functionally disrupt tight junctions, possibly through intraepithelial delivery of CagA[64], CagA has been found to accumulate over and around tight junctions of colonized gastric epithelium, as well as ultrastructural alterations of the tight junctions that cover intercellular spaces that have been penetrated by H. pylori cells[57]. These in vivo findings support the hypothesis that functional and structural alterations of the tight junctions may open the way to bacterial penetration into deep intercellular spaces, up to the underlying lamina propria. Consistent transepithelial penetration of H. pylori into infected gastric mucosa may help understand how the colonized mucosa invariably mounts a prominent local immune-inflammatory response, as well as a systemic immune reaction and, under special genetic conditions, even extra-gastric autoimmune pathology. In fact, direct contact (including intracellular penetration in some cases) between H. pylori or its remnants and immune-inflammatory stroma cells has been observed[57].
Figure 1.
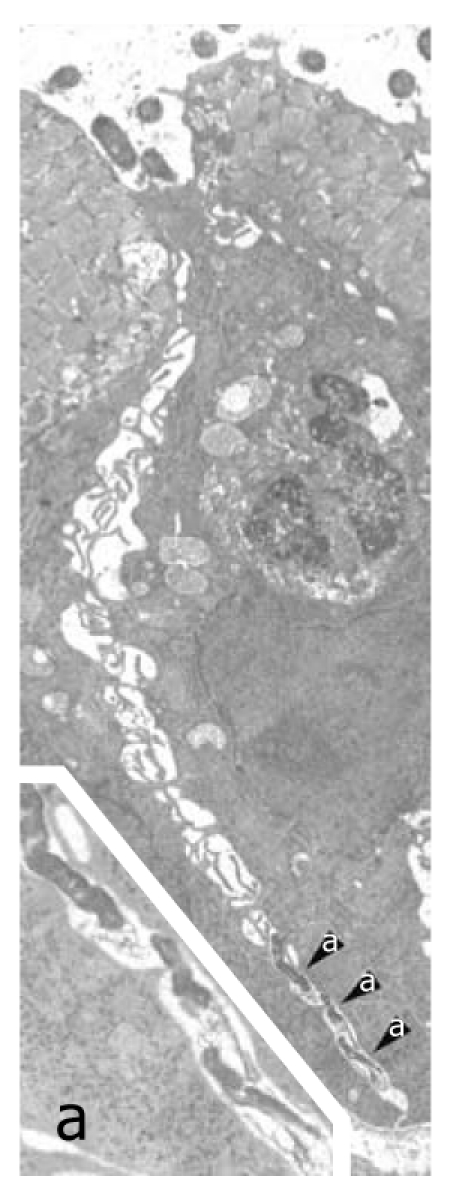
Helicobacter pylori penetration in human gastric epithelium in vivo. Three Helicobacter pylori (H. pylori) organisms (enlarged in a; 16 800 ×) lying in the deep intercellular intraepithelial space, just above the basal membrane. Note also luminal bacteria (top) overlying an apparently preserved tight junction, dilation of the underlying intercellular space, filled with lateral membrane plications, and an intraepithelial granulocyte (middle right, 6300 ×). Reprinted from Necchi et al[57], with permission from Elsevier.
Figure 2.
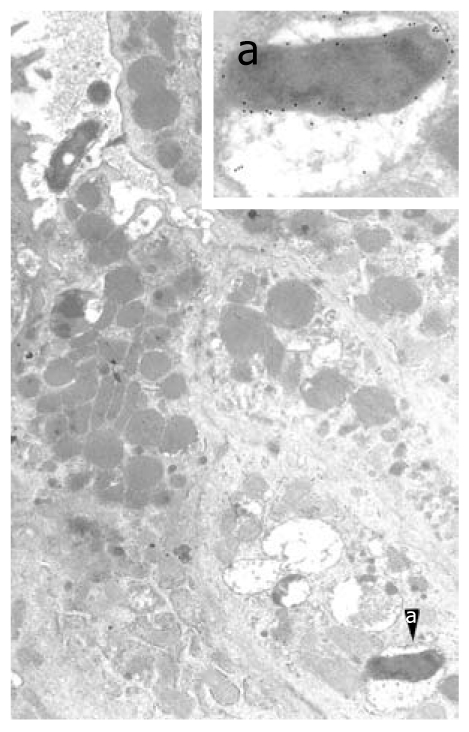
Intracellular Helicobacter pylori in human gastric epithelium in vivo. A well-preserved Helicobacter pylori (H. pylori) organism in a cytoplasmic vacuole, enlarged in a (55 200 ×; 15 nm gold particles) to show immunoreactivity with anti-VacA antibody. Note two H. pylori in a luminal cleft (top left, 13 500 ×). Reprinted from Necchi et al[57], with permission from Elsevier.
The morphological and cytochemical detection of H. pylori (often well preserved, VacA- and CagA-storing, and apparently vital) inside intact epithelial cells of human gastric mucosa (Figure 2) has confirmed the intracellular pathogenic potential of the bacterium. This observation confirms and extends previous in vitro observations that H. pylori exhibits an invasion frequency similar to that of Yersinia and greater than that of Shigella[65]; both bona fide invasive pathogens. This is also in keeping with the expression of Nod1, a known intraepithelial pathogen-recognition molecule, by H. pylori-colonized epithelial cells[27], as well as with the predominantly Th1 type of immune response that is elicited. In addition, confirmation of the intracellular occurrence of H. pylori is crucial when choosing appropriate antibiotics for bacterial eradication, as well as when monitoring their in vivo effectiveness after completion of treatment.
Of high interest is the presence of H. pylori in intestinal metaplasia (IM). Indeed, this finding substantially extends the possible interaction between H. pylori and IM in gastric carcinogenesis, from an early preneoplastic step that corresponds to epithelial progenitors[32] and IM genesis, to the whole process of its progression to neoplasia. Indeed, it supports a persistent in vivo activity of several H. pylori-activated molecular mechanisms of gastric carcinogenesis such as CagA-mediated intracellular disruption of growth regulation[26,66], inflammation-elicited NF-κB transcription factor activation[67], and silencing of DNA mismatch repair genes[68]. The demonstration by Necchi et al[57] of H. pylori occurrence in dysplastic as well as in neoplastic growths, in agreement with that of Semino-Mora et al[63], further substantiates and extends the H. pylori contribution to gastric carcinogenesis, and may explain some beneficial effects of bacterial eradication on IM-related cancer risk, as well as on cancer recurrence[57].
Another in vivo study[69] has recently shown that, in human superficial-foveolar gastric epithelium and its metaplastic or dysplastic foci, H. pylori products/virulence factors (sometimes coupled with bacterial bodies or remnants) accumulate in a discrete cytoplasmic structure that is characterized by 13-nm-thick cylindrical particles of regular punctate-linear substructure. This structure resembles the proteasome complex in size and structure, while being different from VacA-induced vacuoles, phagosomes, aggresomes or related bodies. Inside this novel cell compartment, named PaCS (for particle-rich cytoplasmic structure), co-localization of VacA, CagA, urease and outer membrane proteins (OMPs) with NOD1 receptor, ubiquitin-activating enzyme E1, polyubiquitinated proteins, proteasome components and potentially oncogenic proteins like SHP2 and ERKs has been demonstrated[69]. These findings suggest that PaCS is a novel, proteasome-enriched structure arising in ribosome-rich cytoplasm at sites of H. pylori product accumulation. As a site of selective concentration of bacterial virulence factors, the ubiquitin-proteasome system and interactive proteins, PaCS is likely to modulate immune-inflammatory and proliferative responses of the gastric epithelium of potential pathological relevance; also taking the mounting evidence for a role of the ubiquitin-proteasome system in cancer origin or progression[70].
A hotly debated question waiting for a definitive answer is whether human gastric DCs are able to send cell processes that cross the epithelium, reach the lumen and directly contact and engulf H. pylori, as shown to occur for DC-mediated pathogen-sensing at the intestinal level. The dendritic, epithelial, granulocytic or macrophagic nature of the cell that first interacts with the bacterium seems to be important, given the different type and cellular distribution of microbial product receptors shown by different immunocompetent cells, and the different chemokines and interleukins that they release when activated[27,62,71]. In fact, DCs have been found to respond differently when interacting directly with bacteria rather than secondarily to epithelium-bacteria contact, with IL-12 secretion and Th1 response preferentially activated only in the former case[72].
By ultrastructural immunocytochemistry on endoscopic biopsy samples, clear-cut in vivo evidence has been provided[73] of direct DC contact with H. pylori in the human gastric mucosa (Figure 3), which greatly extends the relevance of previous in vitro studies carried out on purified DCs. DCs have been shown to be present inside superficial-foveolar epithelium of H. pylori-infected (but not H. pylori-free) human gastric mucosa, and to send cytoplasmic extensions to the lumen, to which bacteria preferentially adhere (Figure 3). In addition, intraepithelial DCs are found to accumulate bacterial products like VacA, urease and OMPs in their cytoplasm. The importance of intraepithelial, lumen-contacting DCs lies in the well-known crucial role of these cells as sensors of pathogens, and as the first line of antibacterial immune defense of both innate and adaptive type[74]. In fact, DCs have been shown to act as main processing and presenting cells of internalized antigens, and major regulators of cells involved in the mucosal inflammatory response. Depending on their mode of activation, they may secrete pro-inflammatory or anti-inflammatory cytokines and chemokines that dictate the composition of the cellular infiltrate, in addition to activating NK and T cells with either Th1 or Th2 effector and T regulatory cell responses[73]. Thus, the direct interaction of DCs with H. pylori during active gastritis may be of key relevance. Also worth noting is the finding of a close adherence of DCs to surrounding epithelial cells along all or most of their confronting membranes, with involvement of lateral folds and formation of focal interdigitating complexes. It has been speculated that this pattern, possibly related to the capacity of immature DCs to produce tight and adherens junction proteins, may be finalized to preserve the barrier function of the epithelium[73].
Figure 3.

Intraepithelial dendritic cells in Helicobacter pylori-positive human gastric biopsies with active inflammation. A (5000 ×): Luminal ending of a dendritic cell (DC) process abutting on a collection of Helicobacter pylori (H. pylori); enlarged in a1 (16 000 ×) to show envelopment of a bacterium by caliceal veils (right); note in a2 (28 000 ×; detail of a1), close adherence of a clubbed process (bottom left) to another bacterium showing VacA immunoreactivity of outer membrane and flagella (upper right); B (3000 ×): Intraepithelial DC with a narrow luminal process directly contacting bacteria; note close membrane adhesion to surrounding epithelial cells, focal interruption of the basal lamina (arrow), and outer membrane protein (OMP) immunoreactivity of cytoplasmic vacuoles (b1, 32 000 ×) and a multivesicular late endosome/lysosome body (b2, 22 000 ×); C (8000 ×): DC with clear cytoplasm, close adherence to surrounding epithelial cells, numerous mitochondria, sparse ribosomes and small rough endoplasmic reticulum (RER) cisternae; cytoplasmic vesicles and membranous remnants of an intracellular bacterium, enlarged in c (42 000 ×), show VacA immunoreactivity; D (2000 ×): Nucleated DC with abundant supranuclear mitochondria, no secretory granules, small tubular RER cisternae (enlarged in d1, 18 000 ×), several cytoplasmic vesicles, scattered vacuoles (v), and close adhesion to epithelial cells. On the left, two DC processes (arrows) are contacting the basal membrane (arrowheads). Several cross or longitudinal sections of partly swollen cell processes are observed in the lamina propria; the largest of which (enlarged in d2, 12 000 ×) shows ultrastructural homology with DC cytoplasm, including scattered, small RER cisternae and vesicles; the precise cells of origin of such lamina propria processes could not be assessed. BV: Blood vessel; fg: Foveolar granules; E (2000 ×): Base of foveolar epithelium showing an immature monocytoid cell (DC precursor?) with kidney-shaped nucleus, scattered ribosomes, a few juxtanuclear lysosomes, and no vesicles or granules; note a caliceal process embracing a mast cell (enlarged in e1, 15 000 ×; scroll bodies, arrow), a clubbed process adhering to H. pylori (enlarged in e2, 55 000 ×), a lymphoid cell (Ly) crossing the epithelium basal membrane and another intercellular bacterium (arrow). Reprinted from Necchi et al[73], with permission from John Wiley.
Granulocytes have also been seen to contact and heavily phagocytose luminal H. pylori, while macrophages remain confined to basal epithelium, although taking up bacteria and bacterial products[73]. The inhibitory role of VacA on phagolysosome formation and acidification with resulting persistence of engulfed H. pylori has been outlined[75]; this may well allow macrophages and DCs to retain and translocate vital H. pylori to gastric lymph nodes, from which they have been cultivated[76]. The substantial restriction of intraepithelial DCs to active, granulocyte-rich gastritis seems of interest, especially considering the dominant role of granulocytes in early, first-contact H. pylori gastritis. Although no direct morphological interactions have been observed between DCs and granulocytes, both have shown coexistence in the same mucosa and direct interaction with H. pylori, resulting in phagocytosis and intracellular accumulation of bacterial virulence factors[73]. DC intracellular accumulation of H. pylori virulence factors like VacA, urease or OMPs, as documented in this paper, may also be of relevance for DC function and integrity; either as a source of antigenic material promoting the immune response or of cytotoxic damage. Indeed, signs of cellular damage have been observed in several intraepithelial DCs, including focal mitochondrial swelling, cytoplasmic edema and vacuolation and formation of autophagic vacuoles. These findings raise the issue of a possible impairment of DC function by accumulated bacterial toxins, which may reduce the effectiveness of the immune response and favor persistence of bacterial infection. Thus, this complex in vivo interaction of DCs, granulocytes and macrophages with H. pylori and H. pylori-infected epithelium is likely to have an important role in the origin, type specification, and outcome of the innate and adaptive immune responses.
VacA TOXIN AND ITS FUNCTIONAL INTERACTION WITH CagA: THE ART OF PRESSING THE BRAKE AND GAS PEDALS AT ONCE
VacA, the “vacuolating cytotoxin”, and CagA, the product of the cytotoxin-associated gene A, are virulence factors playing a pivotal role in H. pylori-induced pathogenesis. While VacA is defined as an A-B toxin (even though no enzymatic activity has been so far identified for any of its domain), CagA is usually defined as an effector protein: a bacterial protein delivered into host cells by a specialized bacterial machine so as to modulate a variety of cellular functions. Indeed, CagA is supposed to be directly injected into host cells by a TFSS. Many excellent reviews have been recently published on these two H. pylori virulence factors[77-80], thus, we do not intend to discuss the well-known characteristics of these proteins in detail. Here, we focus on the most recent experimental data that shed new light on their structure/activity relationships, on their internalization and trafficking in the host cells, and finally, on the apparent paradox of why H. pylori simultaneously produces two different virulence factors counteracting the effect of each other on host cells.
VacA toxin
VacA has three well-confirmed cell activities, namely: cell vacuolation, apoptosis, and CD4+ T-lymphocyte activation and proliferation[77,81]. This toxin was discovered as a secreted protein of H. pylori (at that time known as Campylobacter pylori) that induces a massive cytoplasmic vacuolation in vitro[82]. About 55% of isolates of H. pylori are able to induce that cytopathic effect[82]. VacA was shown later to form anionic channels into artificial lipid membranes, as well as cell membranes[77]. VacA channel drives first an hyperacidification of late endocytic compartments by inducing, via the transport of anions into the lumen of these organelles, overactivity of the V-ATPase. Accumulation, by protonation, of weak bases such as ammonia (produced by H. pylori urease activity) into these compartments creates an osmotic imbalance that results in their swelling[77,81].
Deep-etch electron microscopy of VacA preparations has revealed that the toxin forms regular oligomers with either six- or sevenfold radial symmetry[77]. Oligomerization of the toxin molecules creates a cavity in the center of the oligomer and mimics the structure of a pore-forming toxin (Figure 4) such as the α hemolysin of Staphylococcus aureus[83]. In its mature state, VacA is a 88-kDa protein that can be cleaved into two subunits (Figure 4). According to their molecular mass, the N-terminal subunit is denominated “p33”, whereas “p55” is the C-terminal one (Figure 4). The N terminus of the mature p33 toxin subunit consists of a stretch of 32 uncharged hydrophobic amino acids (found in the VacA s1 subtype[77,80]). The presence of additional residues to p33 (found in the s2 subtype), ablation deletions within the hydrophobic residues, or specific point mutations of amino acids in this stretch inhibit the vacuolating activity of VacA. From these results, it has been concluded that VacA channel formation involves the N-terminal 32 amino acids of p33[77,80,84,85]. Other observations have led to the hypothesis that, in addition to the p33 N-terminal part and p33 itself[85], a toxin domain localized in about the first 110 residues of the p55 is also required for the proper oligomerization of the toxin, for channel formation. This is supported by the fact that the minimal intracellular active domain of VacA contains the full p33 domain and about 110 residues of the N-terminal part of p55[86,87]. In these experiments, the p33 toxin subunit, produced alone within cells, does not induce vacuolation[86,87].
Figure 4.
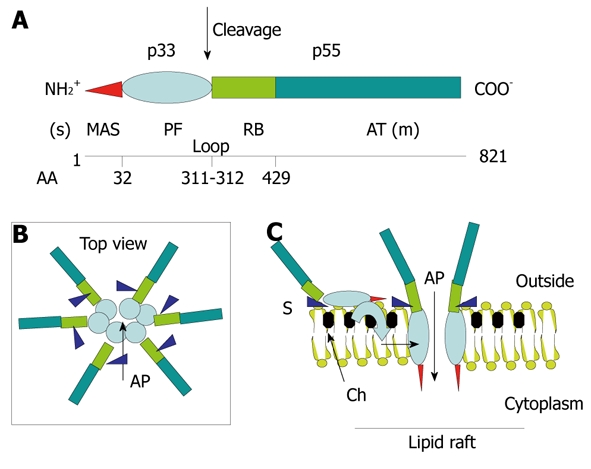
Structure-function relationships in the VacA toxin. A: VacA is an 88-kDa protein that can be cleaved into two subunits designated p33 (red and light blue) and p55 (light green and dark green). The cleavage between the subunit occurs in a flexible loop between residues 311 and 312. The two subunits are probably attached by non-covalent bonds between the p33 carboxy terminus and the N-p55 terminus. The amino-terminal part of p33 consists of 32 hydrophobic amino-acid stretch that is involved in the recognition of mitochondria (red) (MAS) (where the “s” toxin subtype site is located) followed by the p33 “core” subunit that forms, upon entry into the lipid membrane and oligomerization, an anionic channel (PF) (light blue). The toxin nicking site is located in the flexible loop domain. The amino-terminal part of p55 contains the cell receptor domain (about 110 amino acids) (RB; light green) followed by the type Va autotransporter domain (AT; dark green) that is involved in the secretion of the toxin by the bacterium (where the “m” toxin subtype site is located); B and C: VacA binds as a monomer to its cell surface receptor sphingomyelin (S) with low affinity, then the p33 core progressively is embedded (light blue curve arrow) in the lipid membrane bilayers, at the level of lipid rafts [containing saturated lipid such as sphingomyelin and cholesterol (Ch)] and forms an anionic channel (AP) by oligomerization of p33. This multiplies by 6 the number of toxin cell receptors associated with the oligomerized VacA, thus increasing greatly the toxin affinity for the target cells.
In addition to its vacuolating activity, VacA exhibits apoptotic activity[88-90]. Importantly, these studies have revealed that the p33 VacA subunit is targeted to mitochondria and only is required for apoptosis[88], and that a toxin mutant without vacuolating activity is non-apoptotic[89]. Collectively, these data indicate that VacA induces, through the p33 subunit alone, apoptosis by acting on mitochondria, and most likely involves the toxin channel activity. Recently, it has been demonstrated that p33 oligomerizes without the need of p55 and forms an anionic channel with characteristics identical to that of the whole VacA molecule[91]. Furthermore, ablation of the 32 N-terminal residues of p33 does not alter the pore-forming activity of the VacA subunit, but impairs the mitochondrial targeting activity of p33[91]. Altogether, these new data indicate that the portion of p33 without its first 32 N-terminal residues (that we call the “core p33”) oligomerizes and contains all the channel activity of VacA. Domańska et al[91] have clarified the following, formerly puzzling, observations: (1) that a mutant toxin with a major deletion in the p33 N-terminal hydrophobic amino acids (VacA delta 6-27), albeit inhibiting the vacuolation process, nonetheless induces anionic channels in artificial bilayers, even though exhibiting a longer delay of formation than the wild-type toxin[85]; and (2) that a single deletion (δ 49-57) in the p33 core domain inhibits the oligomerization of the toxin and the vacuolation[92]. Indeed, in the light of the study of Domańska et al[91], these observations are well explained by the role of the p33 core without the N-terminal hydrophobic amino-acid stretch in forming the toxin channel by oligomerization.
As depicted in Figure 4, the structure-activity of VacA would thus be the following: starting from the N terminus, the 32-amino-acid stretch consists of a membrane/mitochondrial targeting motif (which however does not participate in the toxin channel formation)[91]. Then, the p33 channel core forms the channel activity, either in endosomal membranes or in the inner mitochondrial membrane (IMM)[91]. At the end of the p33 subunit, there is the loop domain (58 residues) in which the toxin-nicking site is located (between residues 311 and 312). We speculate that a peptide motif just after the nicking site (i.e. residing in the 110 residues of the N-terminal part of p55 subunit) is implicated in the recognition of the toxin cell receptor. This is at odds with the current view that the toxin receptor domain is located in the so-called mid-region (m1/m2; i.e. residues 470-662) of p55[77,80]. How to reconcile the recent data of Domańska et al[91] with previous results indicating that the intracellular minimum portion of the VacA molecule active in the cytosol requires, in addition to p33, the 110 N-terminal residues of p55 for VacA oligomerization[84,86,87]? Taking in account that the p33 toxin subunit oligomerizes alone and supports all the pore-forming ability of VacA[91], why does expression of this toxin subunit alone into cells not lead to cell vacuolation[86,87]? We speculate that the different intracellularly expressed (by cDNA transfection) toxin domains might induce their vacuolating activities not after intracellular synthesis, but rather only after leaking out of the producing cells, followed by endocytosis via the VacA cognate receptor. This would point to the 110 residues present at the N-terminal part of p55 as containing the VacA cell receptor motif (Figure 4). This position would indeed be ideally suited to recognize a cell surface molecule close to the lipid bilayers (Figure 4). The 3D structure of the p55 VacA subunit has been recently resolved[93] and shows that a large part of this toxin subunit has the classical fold of autotransporter structures of the bacterial Va secretion system (by which the toxin is transported), which is mostly implicated in the bacterial secretion of VacA. This is supported by previous experiments showing that several deletions in the p55 subunit abolish the production of VacA by the bacterium[85]. Gangwer et al[93] have proposed that a conserved pocket, located in the m1/m2 regions of p55, is the receptor domain of VacA, which becomes fully accessible to the cell receptor when the toxin is assembled in either a single- or double-layered oligomeric structure. However, it is well known that it is necessary to disrupt the oligomeric structure of VacA (by acidic or alkaline treatments) to render it able to bind to its cell receptor. Furthermore, the mutant toxin δ 49-57, which does not form oligomeric structures, does not require to be acid-activated to bind the VacA cell receptor[92]. This demonstrates that the toxin receptor domain is probably not fully accessible in the VacA oligomeric form. More recently, González-Rivera et al[94] have shown that a mixture of purified p33 and p55 does not form oligomeric structures in aqueous solvent, but only monomeric structures, whereas, upon addition of a detergent that mimicks a lipid membrane environment, there is formation of oligomeric structures. This result suggests that the hydrophobic environment induces the oligomerization, most likely by the p33-dependent formation of the channel. Importantly, it has also been shown that the p33 subunit markedly increases the cell binding of p55[94]. Thus, VacA as a monomer first binds one cell receptor with low affinity, while upon oligomerization, bound to six receptors, it exhibit a very high affinity (Figure 4).
An important breakthrough, seminal for the identification of the VacA receptor, has been the finding that the toxin requires the integrity of lipid rafts and glycosylphosphatidylinositol-anchored proteins (GPI-APs) for its full vacuolating activity, which suggests that VacA follows the GPI-AP pathway of endocytosis by a lipid raft-dependent, clathrin-independent pathway[95]; later described in detail by Sabharanjak et al[96]. The first vacuolar organelles collecting the incoming GPI-APs involve new intracellular compartments that have been named GPI-AP-enriched early endosomal compartments (GEECs), which differ from classical early endosomes[96]. This endocytic pathway, which also requires the small Rho GTPase Cdc42[96], is currently named the GEEC endocytic pathway[97,98], and has been confirmed to be exploited by VacA for its internalization[99-102] (Figure 5). It was later shown that the GEEC pathway requires the presence, in the cell plasma membrane, of the unsaturated lipid sphingomyelin, and that the length of the sphingomyelin acyl chain determines its endocytic intracellular trafficking[103]. This prompted an investigation that has demonstrated the role of sphingomyelin as the VacA cell receptor[103]. Accordingly, it has been demonstrated that the length of the sphingomyelin acyl chain also determines the VacA intracellular trafficking[102]. Normal cells contain in their lipid membrane a high proportion of long acyl chain sphingomyelin (C18), and VacA bound to this lipid follows the GEEC pathway[102]. In contrast, when cells are artificially enriched in short-acyl-chain sphingomyelin (C2), the toxin does not follow the GEEC pathway but is recycled back to the plasma membrane in a Cdc42-independent fashion[102].
Figure 5.
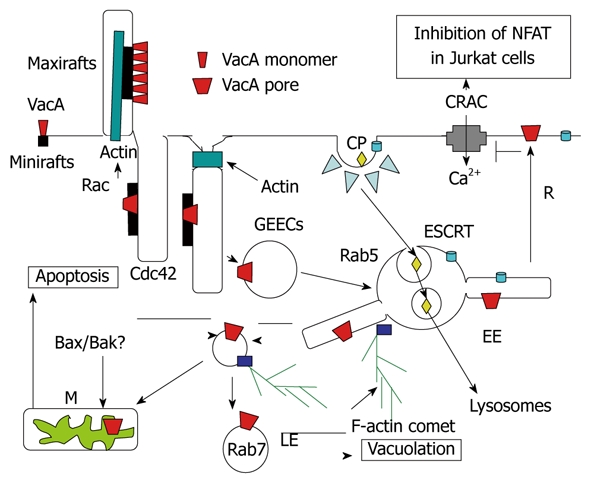
Endocytosis and intracellular trafficking of VacA. Monomeric VacA may bind sphingomyelin on small rafts. Then, by formation of membrane extensions by actin polymerization via Rac activation, VacA is clustered in macrorafts where the p33 subunit oligomerizes and forms a channel that enters the membrane lipid bilayers. By a Cdc42-dependent process, VacA bound to sphingomelin associated to lipid rafts is transferred into cell membrane invaginations (tubules?), which are then pinched out from the membrane, with the help of F-actin filament, which leading to the formation of the glycosylphosphatidylinositol-anchored protein-enriched early endosomal compartment (GEEC) compartment that contains VacA. The toxin is then transferred to Rab5-positive early endosomes (EEs). In EEs, VacA is selectively addressed to EE tubular extensions that are formed by an F-actin process. These tubular extensions are pinched out from the EEs and form highly motile vacuoles that are propelled by F-actin comets. These motile vesicles then fuse with mitochondria (M) or late endosomes (LEs), where VacA induces apoptosis or vacuolation. The pro-apoptotic channels Bax and Bak may be brought to mitochondria by binding on VacA-containing motile vacuoles. Some VacA molecules may be recycled (R) back to the plasma membrane where the channel activity of the toxin, by altering the electric transmembrane potential, inhibits the voltage-dependent Ca2+ release-activated Ca2+ (CRAC) channel. This blocks entry of calcium that activates the calcineurin protease, which is required for processing of the nuclear factor (NFAT), and therefore inhibits the transcription of the interleukin 2 (IL-2) gene in Jurkat T-lymphocytes. Ligands entering the coated-pit pathway (CP) are directed, by the endosomal sorting complex required for transport (ESCRT) complex, towards internal vesicles of EEs and form the multivesicular body multivesicular body (MVB). MVBs are directed to lysosomes, by being propelled along microtubules, where the contents of EE internal vesicles are transferred and degraded.
Upon VacA treatment, transformed human CD4+ T lymphocytes (Jurkat cells) block their constitutive production of IL-2[104]. It is well established that, in T lymphocytes, the nuclear factor NFAT, upon processing by the calcium-activated calcineurin protease, activates the transcription of the IL-2 gene. Massive entry of calcium, through the voltage-dependent Ca2+ release-activated Ca2+ (CRAC) channels, is required to activate calcineurin. However, a VacA mutant without vacuolating activity has no effect on NFAT inhibition in Jurkat cells[105]. Possibly, inhibition of NFAT by VacA is due to the electric depolarization of the lymphocytic plasma membrane, induced by the toxin anionic channel, which inhibits CRAC channels. Human primary T lymphocytes are, however, not sensitive to the deactivation of NFAT induced by VacA[106]. Treatment of primary human T lymphocytes by phorbol myristate acetate, which induces their migratory activity, together with the expression of the CD18 β-integrin at their surface, restores the VacA-induced inhibition of NFAT[107]. Furthermore, the direct expression of CD18 into primary human T lymphocytes renders them able to respond to VacA[107]. It has therefore been concluded that CD18 is the receptor for VacA in immune cells[107]. This result is puzzling because, for all the different toxins studied so far, the cell binding activity always depends on a unique molecule (or a family of closely related molecules). However, a few observations do not support the role of CD18 as the sole VacA receptor in immune cells. Indeed, it has been demonstrated that, although human primary lymphocytes do not deactivate the nuclear factor NFAT, they respond perfectly to VacA by blocking their induced proliferation (i.e. blockage of their cell cycle) due to a mitochondria-induced depletion of ATP[106,108,109]. This fact clearly indicates that VacA binds and penetrates into primary lymphocytes without needing CD18 (probably using the sphingomyelin receptor). The fact that VacA, entering via sphingomyelin in primary T cells, does not inactivate NFAT can be explained by the possibility that the toxin channel is probably not recycled back (or only weakly recycled) to the plasma membrane, and therefore, is unable to block calcium cell entry through CRAC channel inhibition. We speculate that CD18 may not be a real receptor for VacA, but rather activates a signaling pathway (perhaps via the direct activation of CD18 by VacA, because association of VacA with CD18 has been reported[107]) that modifies the intracellular trafficking of the toxin. For instance, stimulation of the migratory activity of T lymphocytes by CD18 signaling might activate Cdc42 boosting of the entry of VacA[99], and increasing its recycling to the cell surface. It has recently been reported that the GEEC pathway is pivotal during cell migration[98]. Another possibility would be that CD18-driven signaling drastically changes the lipid membrane content of long-acyl-chain sphingomyelin molecules toward short ones forcing the toxin to recycle back to the plasma membrane as recently described[102].
How is VacA transferred from early endosomes to mitochondria and/or late endosomes to induce apoptosis and vacuolation? It is well known that ligands or membrane receptors taken up and routed for degradation into lysosomes are endocytosed by the clathrin-dependent pathway and targeted to the luminal vesicles of endosome-forming multivesicular bodies (MVBs) by the endosomal sorting complex required for transport (ESCRT) complex[110] (Figure 5). Propelled along microtubules, MVBs then move to lysosomes where the content of MVB internal vesicles is selectively delivered for degradation. Several lines of evidences indicate that MVBs and lysosomes are not the cell compartments that undergo vacuolation by VacA activity: (1) no internal vesicles are observed by electron microscopy in VacA-induced vacuoles and the enlarged compartment does not have the structure of lysosomes[95,111]; (2) the vacuolated compartment contains markers of both late endosomes and lysosomes[112]; and (3) disruption of microtubules does not block VacA-induced vacuolation[113]. Thus, the organelles that are vacuolated by VacA are likely post-lysosomal compartments that are required to recycle the membrane of MVBs from lysosomes. VacA is probably transferred directly to that compartment by an F-actin-dependent mechanism of endosome motility[101]. Indeed, upon entry of VacA into early endosomes via the GEEC pathway, VacA-containing early endosomes exhibit F-actin comet tails associated with their cytosolic membrane surface, and are rapidly moving in the cytosol[101]. Inhibition of these F-actin structures blocks the VacA transfer into the late endosomal compartment and vacuole genesis[101], but also VacA-induced apoptosis and the localization of toxin molecules in mitochondria[114]. These findings suggest that VacA is addressed to late endosomes and mitochondria by the same mechanism that relies on F-actin-driven vesicular motility. F-actin structures have recently been shown to take place at the level of the early endosomal surface to induce budding of tubular structures followed by their cleavage to yield vesicles that retain F-actin tails[115]. It is thus tempting to speculate that VacA might be first transferred from GEECs to the early endosomal limiting membrane, then into these tubular extensions, and finally into the membrane of the ensuing F-actin motile vesicles (Figure 5). These vesicles are then brought to late endosomes and/or mitochondria via stochastic fusion events by F-actin-driven vesicular motility (Figure 5). In this model, the transfer of VacA (or only of its p33 subunit) from the donor membrane to the recipient membrane (late endosomes or mitochondria) would be achieved by the membrane/mitochondrial N-terminal 32 residues of p33[91] that may stick out of the membrane (Figure 4). This would explain why point mutations, or deletion or modification of the length of this amino-acid stretch might modulate the vacuolating process without altering the pore-forming capacity of the toxin[91]. Importantly, according to this model, the toxin would never be released free in the cytosol[90], which differs from the enzymatic subunits of canonical A-B toxin. By remaining associated with lipid membranes, out of reach of degradative processes, the VacA channel would keep its full activity for a long time, as observed several years ago[111].
Inside mitochondria, the p33 subunit (or the full VacA molecule) induces, probably by the rupture of IMM integrity[91], the release of cytochrome c (cyt c) and induction of apoptosis[88]. However, pro-apoptotic channels must be activated or transferred into the outer membrane of mitochondria to ensure the release into the cytosol of cyt c, which, by activation of the Apaf1 molecule, induces the stimulation of the caspase cascade that results in cell death. Accordingly, the pro-apoptotic channels Bax and Bak have been shown to be activated during VacA-induced apoptosis[116,117], and VacA is unable to induce apoptosis in Bax- and Bak-deficient mouse embryonic fibroblasts[117]. It has been recently proposed that Bax and Bak might be directly recruited from the cytosol by endosomes containing functional VacA channels in their membranes, which allows their juxtaposition with mitochondria[117]. This would be an ingenious way to combine the transfer of VacA channels to the IMM (and thereby the release of cyt c in the mitochondrial intermembrane space) and the leakage of cyt c in the cytosol.
Thus VacA is probably not really a multifunctional toxin, as previously defined[77]. The diversity of VacA activities observed may be simply explained by the channel formed by the p33 subunit that is addressed to different cell membranes by the intracellular trafficking of the toxin, which may be different in different cell types or during cell differentiation (Figure 5).
Functional relationship between VacA and CagA
The gene encoding VacA is present in all the H. pylori strains, although only 55% express an active vacuolating toxin. This is due to either additions or deletions of peptide sequences in the toxin that probably impair the VacA intracellular trafficking or its binding to the cell surface, as we have described above. It is now well-established that the most severe gastric pathology (i.e. peptic ulcer and gastric cancer) is restricted to H. pylori type 1 strains, which contain in their chromosome a PAI named cag PAI, and which secrete an active vacuolating VacA toxin[10]. The cag PAI encodes for the CagA protein and for the TFSS machinery that is implicated in the transport of CagA out of the bacterium, and its transfer to the host cell cytosol[78,79]. CagA is a 135-145-kDa protein that contains its full intracellular signaling activity in its 35-45-kDa C-terminal part[78,79,118] (Figure 6). Nevertheless, it is still unclear how and where the cag TFSS recognizes and target gastric epithelial cells so as to transfer CagA into their cytosol. Recently, two mechanisms have been proposed. One points to the recognition and binding of the TFSS to an integrin[119,120], then, through a mechanical sliding due to bound integrins, the TFSS might punch the cell surface and deliver CagA into the cytosol[120]. One caveat for this mechanism is that H. pylori might deliver CagA at the apical pole of colonized epithelial gastric cells, whereas integrins are localized on the basolateral pole. On the other hand, it has been proposed that the TFSS does not punch the cell surface but induces the flipping out of the lipid phosphatidylserine (PS) from the inner (where it normally resides) to the outer leaflet of the target cell membrane[121]. In this model, CagA, which is secreted constitutively by the TFSS, binds to PS through a lipid-binding peptide motif that is present in its 100-kDa N-terminal part[121]. Following this binding step, a novel type of endocytosis (not yet better characterized nor defined) is induced that carries CagA, associated with PS, into epithelial gastric cells[121]. If this is true, CagA should no longer be defined as an effector protein, but rather as a new type of A-B toxin that is released only in close proximity to the target cells.
Figure 6.
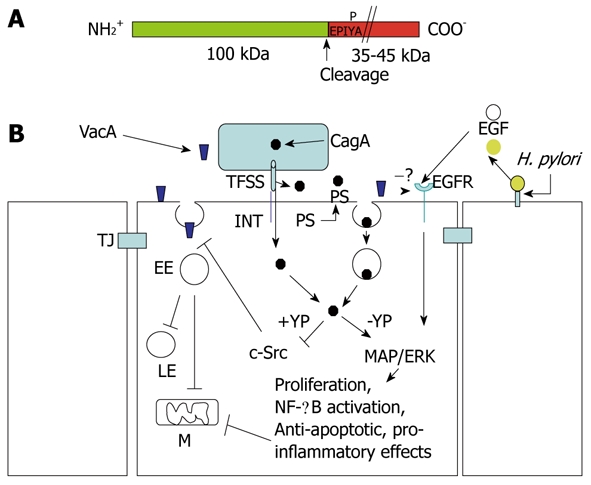
Structure of CagA and signaling cross-talk between VacA and CagA. A: CagA encompasses two fragments: the N-terminal 100-kDa fragment may contain the cell binding domain [to phosphatidylserine (PS)?]. Cleavage of CagA may take place just at the beginning of the first EPIYA motif, which can be tyrosine-phosphorylated by the c-Src tyrosine kinase. The C-terminal portion of CagA, which contains all the signaling activity of the molecule, may have a different molecular mass (up to 45 kDa) due to the repetition of the EPIYA-containing domain; B: CagA produced within the bacterium is transferred in the external medium by the type IV secretion system (TFSS) machinery. Two possibilities for CagA transfer into the target epithelial cell: (a) by binding to an integrin (INT), the TFSS punches the cell membrane and injects CagA; or (b) the TFSS induces the flipping of PS on the outer cell surface. By its 100-kDa N-terminal fragment, CagA binds PS and, upon endocytosis, which is transferred into the gastric cells. In the cytosol, the CagA signaling domain can be tyrosine-phosphorylated (+YP) and inhibits the c-Src kinase activity that is required to allow the transfer of VacA from glycosylphosphatidylinositol-anchored protein-enriched early endosomal compartments (GEECs) to early endosomes (EEs). This blocks vacuolation in late endosomes (LEs) and mitochondria (M)-dependent apoptosis induced by VacA. In an unphosphorylated (-YP) state, CagA activates mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) and nuclear factor (NF)-κB anti-apototic and pro-inflammatory pathways, which also counteract VacA-induced apoptosis. VacA interferes with epidermal growth factor (EGF) receptor (EGFR) activation and endocytosis, thus impairing the signaling pathway that is triggered by this receptor. Free EGFR ligands (EGF) are liberated from the cell-surface-bound molecules via cleavage triggered by Helicobacter pylori (H. pylori). TJ: Tight junction.
Inside the cell, CagA may be tyrosine phosphorylated by the p60 c-Src kinase[118]. Phosphorylation occurs on the 35-45-kDa C-terminal part of CagA on specific five-amino-acid motifs called EPIYA, which can be repeated several-fold and increase the size of this fragment[26,78]. CagA can be split into two fragments by proteases[122,123]. This generates a 100-kDa N-terminal fragment and a C-terminal 35-45-kDa one that contain all the cell signaling activity of CagA[114]. Proteolytic cleavage of CagA does not require its tyrosine phosphorylation, and the nicking site is localized just at the onset of the first EPIYA motif (Boquet and Ricci, unpublished data). The 100-kDa N-terminal part of CagA might be involved in the cell recognition and transfer of the 35-45-kDa C-terminal part to the cytosol, as suggested by recent data[121]. Therefore, CagA would indeed behave as a typical A-B toxin.
Two main intracellular activities of CagA are now well established. In its tyrosine phosphorylated state, CagA may act, either directly or upon association and activation of the tyrosine phosphatase SHP2[78], to inhibit the c-Src kinase and therefore to affect many regulating proteins that depend on this enzyme, among which are those involved in the actin cytoskeleton assembly/regulation[124]. In its non-tyrosine-phosphorylated form (or independent of the phosphorylation state), CagA activates the MAP/ERK and NF-κB pathways[53,125], which induce cell proliferation, inhibition of apoptosis, a typical cellular elongation called “hummingbird phenotype”[66], and inflammatory response[28] (Figure 6).
An increasing body of evidence suggests that CagA and VacA have several opposing cellular effects. It has been shown that, while CagA activates the NFAT nuclear factor, VacA inhibits it[126]. Then, it has been demonstrated that CagA decreases VacA-induced vacuolation, while in turn, VacA reduces CagA-induced hummingbird phenotype formation[127]. It has also been shown that VacA can downregulate CagA effects on epithelial cells by interfering with activation and endocytosis of EGFR, thus impairing the signaling pathway triggered by this receptor, and which plays a pivotal role in cell proliferation and hummingbird phenotype formation[128]. Taken together, these findings raise the hypothesis that VacA and CagA may downregulate the cell effects of the other, allowing H. pylori interaction with epithelial cells, while avoiding excessive cell damage.
We have recently shown that CagA interferes with VacA action by two complementary mechanisms[114]. In its tyrosine-phosphorylated state, CagA inhibits the c-Src kinase and, in doing so, blocks VacA in the GEECs and thus its delivery to early and late endosomes (inhibition of the vacuolation) or mitochondria (inhibition of apoptosis). In its unphosphorylated state, CagA stimulates the NF-κB pathway and, in doing so, induces anti-apoptotic activity (mediated possibly by the anti-apoptotic factor Bcl2) that also blocks VacA-induced apoptosis. We hypothesize that, once bacteria have colonized the gastric niche, the apoptotic action of VacA might be detrimental for the survival of H. pylori that are adherent to the mucosa, and thus the CagA counteracting action gives a rationale for the association of these two virulence factors in the most pathogenic H. pylori strains[114]. This would be a new, highly ingenious mechanism by which a bacterium locally protects its ecological niche against the action of one of its own virulence factors. However, while exerting a beneficial role for survival and growth of the bacterium by counteracting VacA toxin, CagA injection in the gastric epithelial cells triggers pro-inflammatory and anti-apoptotic responses that are detrimental for the human host in the long-term, because they favor the development of ulcer and cancer.
Because it simultaneously delivers to its host two independent virulence factors that antagonize each other, H. pylori appears to act like a driver that presses the brake and gas pedals at once. This apparently paradoxical behavior is now emerging as an intriguing strategy to achieve the best fit between the bacterium and the hostile gastric environment that represents its ecological niche.
CONCLUSION
With regard to the pathogen, illness is often inadvertent; the result of exquisite tricks learned long ago and played on human cells to achieve the paramount goal of the microorganism: conservation of the species[56,129]. Taken together, all the new findings described above strongly reinforce the notion that H. pylori is a skilled bacterium that has been smart enough to: (1) learn about the physiology of its host; (2) keep this information in its genetic library; (3) share the gene book with other organisms; (4) be “open minded” about acquiring new knowledge and discarding obsolete information; and, of key importance; and (5) know how to communicate with its human host, realizing that killing the host would oblige it to find a new one very quickly to ensure its reproductive success[56,129,130]. H. pylori makes a long and hard, but also successful, journey in the human stomach, given that H. pylori is widespread and has probably been part of the human biota since time immemorial[31,56,130]. Although H. pylori seeking survival may be sometimes achieved at the expense of the well-being of the stomach and the host, human physiology and immunology have co-evolved in the presence of persistent gastric H. pylori colonization, and so are disrupted by its absence[130]. As emphasized by Atherton et al[130], this imbalance of a long-lasting pathogen/host equilibrium, caused by progressive disappearance of H. pylori from some populations, may however result in an increased incidence of other diseases such as reflux esophagitis and esophageal carcinoma, as well as obesity, type 2 diabetes, and allergic disorders.
Footnotes
Peer reviewer: Dr. Nawfal Hussein, PhD, Centre for Biomolecular Sciences, University of Nottingham, University Park, Nottingham, NG7 2RD, United Kingdom
S- Editor Tian L L- Editor Kerr C E- Editor Zheng XM
References
- 1.Everhart JE. Recent developments in the epidemiology of Helicobacter pylori. Gastroenterol Clin North Am. 2000;29:559–578. doi: 10.1016/s0889-8553(05)70130-8. [DOI] [PubMed] [Google Scholar]
- 2.Zarrilli R, Ricci V, Romano M. Molecular response of gastric epithelial cells to Helicobacter pylori-induced cell damage. Cell Microbiol. 1999;1:93–99. doi: 10.1046/j.1462-5822.1999.00018.x. [DOI] [PubMed] [Google Scholar]
- 3.Herszényi L, Tulassay Z. Epidemiology of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci. 2010;14:249–258. [PubMed] [Google Scholar]
- 4.Herrera V, Parsonnet J. Helicobacter pylori and gastric adenocarcinoma. Clin Microbiol Infect. 2009;15:971–976. doi: 10.1111/j.1469-0691.2009.03031.x. [DOI] [PubMed] [Google Scholar]
- 5.Correa P, Piazuelo MB, Camargo MC. The future of gastric cancer prevention. Gastric Cancer. 2004;7:9–16. doi: 10.1007/s10120-003-0265-0. [DOI] [PubMed] [Google Scholar]
- 6.Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 7.Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, Lai KC, Hu WH, Yuen ST, Leung SY, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–194. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
- 8.Ernst PB, Peura DA, Crowe SE. The translation of Helicobacter pylori basic research to patient care. Gastroenterology. 2006;130:188–206; quiz 212-213. doi: 10.1053/j.gastro.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 9.Van Doorn LJ, Figueiredo C, Mégraud F, Pena S, Midolo P, Queiroz DM, Carneiro F, Vanderborght B, Pegado MD, Sanna R, et al. Geographic distribution of vacA allelic types of Helicobacter pylori. Gastroenterology. 1999;116:823–830. doi: 10.1016/s0016-5085(99)70065-x. [DOI] [PubMed] [Google Scholar]
- 10.Peek RM Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–248. doi: 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 11.El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, Stanford JL, Mayne ST, Goedert J, Blot WJ, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193–1201. doi: 10.1016/s0016-5085(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 12.Lu W, Pan K, Zhang L, Lin D, Miao X, You W. Genetic polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor necrosis factor {alpha} and risk of gastric cancer in a Chinese population. Carcinogenesis. 2005;26:631–636. doi: 10.1093/carcin/bgh349. [DOI] [PubMed] [Google Scholar]
- 13.Ohyauchi M, Imatani A, Yonechi M, Asano N, Miura A, Iijima K, Koike T, Sekine H, Ohara S, Shimosegawa T. The polymorphism interleukin 8 -251 A/T influences the susceptibility of Helicobacter pylori related gastric diseases in the Japanese population. Gut. 2005;54:330–335. doi: 10.1136/gut.2003.033050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol. 2005;560:11–18. doi: 10.1007/0-387-24180-9_2. [DOI] [PubMed] [Google Scholar]
- 15.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 16.Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 17.Hold GL, Rabkin CS, Chow WH, Smith MG, Gammon MD, Risch HA, Vaughan TL, McColl KE, Lissowska J, Zatonski W, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132:905–912. doi: 10.1053/j.gastro.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 18.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–1571. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 19.Lindholm C, Quiding-Järbrink M, Lönroth H, Hamlet A, Svennerholm AM. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–5971. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galgani M, Busiello I, Censini S, Zappacosta S, Racioppi L, Zarrilli R. Helicobacter pylori induces apoptosis of human monocytes but not monocyte-derived dendritic cells: role of the cag pathogenicity island. Infect Immun. 2004;72:4480–4485. doi: 10.1128/IAI.72.8.4480-4485.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, Wu C, Mao XH, Jia KR, Wang FJ, et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol. 2010;184:5121–5129. doi: 10.4049/jimmunol.0901115. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B, Rong G, Wei H, Zhang M, Bi J, Ma L, Xue X, Wei G, Liu X, Fang G. The prevalence of Th17 cells in patients with gastric cancer. Biochem Biophys Res Commun. 2008;374:533–537. doi: 10.1016/j.bbrc.2008.07.060. [DOI] [PubMed] [Google Scholar]
- 24.Holcombe C. Helicobacter pylori: the African enigma. Gut. 1992;33:429–431. doi: 10.1136/gut.33.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 26.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 27.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 28.Brandt S, Kwok T, Hartig R, König W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SY, Lee YC, Kim HK, Blaser MJ. Helicobacter pylori CagA transfection of gastric epithelial cells induces interleukin-8. Cell Microbiol. 2006;8:97–106. doi: 10.1111/j.1462-5822.2005.00603.x. [DOI] [PubMed] [Google Scholar]
- 30.Cooke CL, Huff JL, Solnick JV. The role of genome diversity and immune evasion in persistent infection with Helicobacter pylori. FEMS Immunol Med Microbiol. 2005;45:11–23. doi: 10.1016/j.femsim.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci USA. 2005;102:5186–5191. doi: 10.1073/pnas.0407657102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gewirtz AT, Yu Y, Krishna US, Israel DA, Lyons SL, Peek RM Jr. Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis. 2004;189:1914–1920. doi: 10.1086/386289. [DOI] [PubMed] [Google Scholar]
- 34.Caruso R, Fina D, Peluso I, Fantini MC, Tosti C, Del Vecchio Blanco G, Paoluzi OA, Caprioli F, Andrei F, Stolfi C, et al. IL-21 is highly produced in Helicobacter pylori-infected gastric mucosa and promotes gelatinases synthesis. J Immunol. 2007;178:5957–5965. doi: 10.4049/jimmunol.178.9.5957. [DOI] [PubMed] [Google Scholar]
- 35.Nagase H, Woessner JF Jr. Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 36.Kruidenier L, MacDonald TT, Collins JE, Pender SL, Sanderson IR. Myofibroblast matrix metalloproteinases activate the neutrophil chemoattractant CXCL7 from intestinal epithelial cells. Gastroenterology. 2006;130:127–136. doi: 10.1053/j.gastro.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 37.Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, Meltzer SJ, Wilson KT. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology. 1999;116:1319–1329. doi: 10.1016/s0016-5085(99)70496-8. [DOI] [PubMed] [Google Scholar]
- 38.Busiello I, Acquaviva R, Di Popolo A, Blanchard TG, Ricci V, Romano M, Zarrilli R. Helicobacter pylori gamma-glutamyltranspeptidase upregulates COX-2 and EGF-related peptide expression in human gastric cells. Cell Microbiol. 2004;6:255–267. doi: 10.1046/j.1462-5822.2004.00366.x. [DOI] [PubMed] [Google Scholar]
- 39.Shibayama K, Kamachi K, Nagata N, Yagi T, Nada T, Doi Y, Shibata N, Yokoyama K, Yamane K, Kato H, et al. A novel apoptosis-inducing protein from Helicobacter pylori. Mol Microbiol. 2003;47:443–451. doi: 10.1046/j.1365-2958.2003.03305.x. [DOI] [PubMed] [Google Scholar]
- 40.Romano M, Ricci V, Di Popolo A, Sommi P, Del Vecchio Blanco C, Bruni CB, Ventura U, Cover TL, Blaser MJ, Coffey RJ, et al. Helicobacter pylori upregulates expression of epidermal growth factor-related peptides, but inhibits their proliferative effect in MKN 28 gastric mucosal cells. J Clin Invest. 1998;101:1604–1613. doi: 10.1172/JCI1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keates S, Sougioultzis S, Keates AC, Zhao D, Peek RM Jr, Shaw LM, Kelly CP. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Biol Chem. 2001;276:48127–48134. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- 42.Wallasch C, Crabtree JE, Bevec D, Robinson PA, Wagner H, Ullrich A. Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem Biophys Res Commun. 2002;295:695–701. doi: 10.1016/s0006-291x(02)00740-4. [DOI] [PubMed] [Google Scholar]
- 43.Romano M, Ricci V, Memoli A, Tuccillo C, Di Popolo A, Sommi P, Acquaviva AM, Del Vecchio Blanco C, Bruni CB, Zarrilli R. Helicobacter pylori up-regulates cyclooxygenase-2 mRNA expression and prostaglandin E2 synthesis in MKN 28 gastric mucosal cells in vitro. J Biol Chem. 1998;273:28560–28563. doi: 10.1074/jbc.273.44.28560. [DOI] [PubMed] [Google Scholar]
- 44.Caputo R, Tuccillo C, Manzo BA, Zarrilli R, Tortora G, Blanco Cdel V, Ricci V, Ciardiello F, Romano M. Helicobacter pylori VacA toxin up-regulates vascular endothelial growth factor expression in MKN 28 gastric cells through an epidermal growth factor receptor-, cyclooxygenase-2-dependent mechanism. Clin Cancer Res. 2003;9:2015–2021. [PubMed] [Google Scholar]
- 45.Yan F, Cao H, Chaturvedi R, Krishna U, Hobbs SS, Dempsey PJ, Peek RM Jr, Cover TL, Washington MK, Wilson KT, et al. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology. 2009;136:1297–1307, e1-3. doi: 10.1053/j.gastro.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romano M, Ricci V, Zarrilli R. Mechanisms of disease: Helicobacter pylori-related gastric carcinogenesis--implications for chemoprevention. Nat Clin Pract Gastroenterol Hepatol. 2006;3:622–632. doi: 10.1038/ncpgasthep0634. [DOI] [PubMed] [Google Scholar]
- 47.Mammano E, Belluco C, Sciro M, Mencarelli R, Agostini M, Michelotto M, Marchet A, Nitti D. Epidermal growth factor receptor (EGFR): mutational and protein expression analysis in gastric cancer. Anticancer Res. 2006;26:3547–3550. [PubMed] [Google Scholar]
- 48.Tuccillo C, Cuomo A, Rocco A, Martinelli E, Staibano S, Mascolo M, Gravina AG, Nardone G, Ricci V, Ciardiello F, et al. Vascular endothelial growth factor and neo-angiogenesis in H. pylori gastritis in humans. J Pathol. 2005;207:277–284. doi: 10.1002/path.1844. [DOI] [PubMed] [Google Scholar]
- 49.Crawford HC, Krishna US, Israel DA, Matrisian LM, Washington MK, Peek RM Jr. Helicobacter pylori strain-selective induction of matrix metalloproteinase-7 in vitro and within gastric mucosa. Gastroenterology. 2003;125:1125–1136. doi: 10.1016/s0016-5085(03)01206-x. [DOI] [PubMed] [Google Scholar]
- 50.Wu CY, Wang CJ, Tseng CC, Chen HP, Wu MS, Lin JT, Inoue H, Chen GH. Helicobacter pylori promote gastric cancer cells invasion through a NF-kappaB and COX-2-mediated pathway. World J Gastroenterol. 2005;11:3197–3203. doi: 10.3748/wjg.v11.i21.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L, Perez-Perez GI, Hatakeyama M, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsukashita S, Kushima R, Bamba M, Nakamura E, Mukaisho K, Sugihara H, Hattori T. Beta-catenin expression in intramucosal neoplastic lesions of the stomach. Comparative analysis of adenoma/dysplasia, adenocarcinoma and signet-ring cell carcinoma. Oncology. 2003;64:251–258. doi: 10.1159/000069310. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, Nagai S, Koyasu S, Gilman RH, Kersulyte D, et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5:23–34. doi: 10.1016/j.chom.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 54.Polk DB, Peek RM Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10:403–414. doi: 10.1038/nrc2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Paulis A, Prevete N, Rossi FW, Rivellese F, Salerno F, Delfino G, Liccardo B, Avilla E, Montuori N, Mascolo M, et al. Helicobacter pylori Hp(2-20) promotes migration and proliferation of gastric epithelial cells by interacting with formyl peptide receptors in vitro and accelerates gastric mucosal healing in vivo. J Immunol. 2009;183:3761–3769. doi: 10.4049/jimmunol.0900863. [DOI] [PubMed] [Google Scholar]
- 56.Ricci V, Zarrilli R, Romano M. Voyage of Helicobacter pylori in human stomach: odyssey of a bacterium. Dig Liver Dis. 2002;34:2–8. doi: 10.1016/s1590-8658(02)80051-2. [DOI] [PubMed] [Google Scholar]
- 57.Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, Ricci V, Solcia E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132:1009–1023. doi: 10.1053/j.gastro.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 58.Petersen AM, Krogfelt KA. Helicobacter pylori: an invading microorganism? A review. FEMS Immunol Med Microbiol. 2003;36:117–126. doi: 10.1016/S0928-8244(03)00020-8. [DOI] [PubMed] [Google Scholar]
- 59.Dubois A, Borén T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell Microbiol. 2007;9:1108–1116. doi: 10.1111/j.1462-5822.2007.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu H, Yamaoka Y, Graham DY. Helicobacter pylori virulence factors: facts and fantasies. Curr Opin Gastroenterol. 2005;21:653–659. doi: 10.1097/01.mog.0000181711.04529.d5. [DOI] [PubMed] [Google Scholar]
- 61.Montecucco C, de Bernard M. Immunosuppressive and proinflammatory activities of the VacA toxin of Helicobacter pylori. J Exp Med. 2003;198:1767–1771. doi: 10.1084/jem.20031839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 63.Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Dis. 2003;187:1165–1177. doi: 10.1086/368133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilkinson SM, Uhl JR, Kline BC, Cockerill FR 3rd. Assessment of invasion frequencies of cultured HEp-2 cells by clinical isolates of Helicobacter pylori using an acridine orange assay. J Clin Pathol. 1998;51:127–133. doi: 10.1136/jcp.51.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. Altered states: involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci USA. 1999;96:14559–14564. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maeda S, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y, Akanuma M, Shiratori Y, Omata M. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. 2000;119:97–108. doi: 10.1053/gast.2000.8540. [DOI] [PubMed] [Google Scholar]
- 68.Kim JJ, Tao H, Carloni E, Leung WK, Graham DY, Sepulveda AR. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123:542–553. doi: 10.1053/gast.2002.34751. [DOI] [PubMed] [Google Scholar]
- 69.Necchi V, Sommi P, Ricci V, Solcia E. In vivo accumulation of Helicobacter pylori products, NOD1, ubiquitinated proteins and proteasome in a novel cytoplasmic structure. PLoS One. 2010;5:e9716. doi: 10.1371/journal.pone.0009716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol. 2005;23:4776–4789. doi: 10.1200/JCO.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 71.Uronen-Hansson H, Allen J, Osman M, Squires G, Klein N, Callard RE. Toll-like receptor 2 (TLR2) and TLR4 are present inside human dendritic cells, associated with microtubules and the Golgi apparatus but are not detectable on the cell surface: integrity of microtubules is required for interleukin-12 production in response to internalized bacteria. Immunology. 2004;111:173–178. doi: 10.1111/j.0019-2805.2003.01803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rimoldi M, Chieppa M, Larghi P, Vulcano M, Allavena P, Rescigno M. Monocyte-derived dendritic cells activated by bacteria or by bacteria-stimulated epithelial cells are functionally different. Blood. 2005;106:2818–2826. doi: 10.1182/blood-2004-11-4321. [DOI] [PubMed] [Google Scholar]
- 73.Necchi V, Manca R, Ricci V, Solcia E. Evidence for transepithelial dendritic cells in human H. pylori active gastritis. Helicobacter. 2009;14:208–222. doi: 10.1111/j.1523-5378.2009.00679.x. [DOI] [PubMed] [Google Scholar]
- 74.Pulendran B, Palucka K, Banchereau J. Sensing pathogens and tuning immune responses. Science. 2001;293:253–256. doi: 10.1126/science.1062060. [DOI] [PubMed] [Google Scholar]
- 75.Allen LA. Phagocytosis and persistence of Helicobacter pylori. Cell Microbiol. 2007;9:817–828. doi: 10.1111/j.1462-5822.2007.00906.x. [DOI] [PubMed] [Google Scholar]
- 76.Ito T, Kobayashi D, Uchida K, Takemura T, Nagaoka S, Kobayashi I, Yokoyama T, Ishige I, Ishige Y, Ishida N, et al. Helicobacter pylori invades the gastric mucosa and translocates to the gastric lymph nodes. Lab Invest. 2008;88:664–681. doi: 10.1038/labinvest.2008.33. [DOI] [PubMed] [Google Scholar]
- 77.Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005;3:320–332. doi: 10.1038/nrmicro1095. [DOI] [PubMed] [Google Scholar]
- 78.Hatakeyama M. SagA of CagA in Helicobacter pylori pathogenesis. Curr Opin Microbiol. 2008;11:30–37. doi: 10.1016/j.mib.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 79.Backert S, Tegtmeyer N, Selbach M. The versatility of Helicobacter pylori CagA effector protein functions: The master key hypothesis. Helicobacter. 2010;15:163–176. doi: 10.1111/j.1523-5378.2010.00759.x. [DOI] [PubMed] [Google Scholar]
- 80.Isomoto H, Moss J, Hirayama T. Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J Exp Med. 2010;220:3–14. doi: 10.1620/tjem.220.3. [DOI] [PubMed] [Google Scholar]
- 81.Boquet P, Ricci V, Galmiche A, Gauthier NC. Gastric cell apoptosis and H. pylori: has the main function of VacA finally been identified? Trends Microbiol. 2003;11:410–413. doi: 10.1016/s0966-842x(03)00211-7. [DOI] [PubMed] [Google Scholar]
- 82.Leunk RD, Johnson PT, David BC, Kraft WG, Morgan DR. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J Med Microbiol. 1988;26:93–99. doi: 10.1099/00222615-26-2-93. [DOI] [PubMed] [Google Scholar]
- 83.Gouaux E. Channel-forming toxins: tales of transformation. Curr Opin Struct Biol. 1997;7:566–573. doi: 10.1016/s0959-440x(97)80123-6. [DOI] [PubMed] [Google Scholar]
- 84.Ivie SE, McClain MS, Torres VJ, Algood HM, Lacy DB, Yang R, Blanke SR, Cover TL. Helicobacter pylori VacA subdomain required for intracellular toxin activity and assembly of functional oligomeric complexes. Infect Immun. 2008;76:2843–2851. doi: 10.1128/IAI.01664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vinion-Dubiel AD, McClain MS, Czajkowsky DM, Iwamoto H, Ye D, Cao P, Schraw W, Szabo G, Blanke SR, Shao Z, et al. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J Biol Chem. 1999;274:37736–37742. doi: 10.1074/jbc.274.53.37736. [DOI] [PubMed] [Google Scholar]
- 86.de Bernard M, Burroni D, Papini E, Rappuoli R, Telford J, Montecucco C. Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect Immun. 1998;66:6014–6016. doi: 10.1128/iai.66.12.6014-6016.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ye D, Willhite DC, Blanke SR. Identification of the minimal intracellular vacuolating domain of the Helicobacter pylori vacuolating toxin. J Biol Chem. 1999;274:9277–9282. doi: 10.1074/jbc.274.14.9277. [DOI] [PubMed] [Google Scholar]
- 88.Galmiche A, Rassow J, Doye A, Cagnol S, Chambard JC, Contamin S, de Thillot V, Just I, Ricci V, Solcia E, et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000;19:6361–6370. doi: 10.1093/emboj/19.23.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cover TL, Krishna US, Israel DA, Peek RM Jr. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–957. [PubMed] [Google Scholar]
- 90.Chiozzi V, Mazzini G, Oldani A, Sciullo A, Ventura U, Romano M, Boquet P, Ricci V. Relationship between Vac A toxin and ammonia in Helicobacter pylori-induced apoptosis in human gastric epithelial cells. J Physiol Pharmacol. 2009;60:23–30. [PubMed] [Google Scholar]
- 91.Domańska G, Motz C, Meinecke M, Harsman A, Papatheodorou P, Reljic B, Dian-Lothrop EA, Galmiche A, Kepp O, Becker L, et al. Helicobacter pylori VacA toxin/subunit p34: targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathog. 2010;6:e1000878. doi: 10.1371/journal.ppat.1000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Genisset C, Galeotti CL, Lupetti P, Mercati D, Skibinski DA, Barone S, Battistutta R, de Bernard M, Telford JL. A Helicobacter pylori vacuolating toxin mutant that fails to oligomerize has a dominant negative phenotype. Infect Immun. 2006;74:1786–1794. doi: 10.1128/IAI.74.3.1786-1794.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gangwer KA, Mushrush DJ, Stauff DL, Spiller B, McClain MS, Cover TL, Lacy DB. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc Natl Acad Sci USA. 2007;104:16293–16298. doi: 10.1073/pnas.0707447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.González-Rivera C, Gangwer KA, McClain MS, Eli IM, Chambers MG, Ohi MD, Lacy DB, Cover TL. Reconstitution of Helicobacter pylori VacA toxin from purified components. Biochemistry. 2010;49:5743–5752. doi: 10.1021/bi100618g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ricci V, Galmiche A, Doye A, Necchi V, Solcia E, Boquet P. High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol Biol Cell. 2000;11:3897–3909. doi: 10.1091/mbc.11.11.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell. 2002;2:411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 97.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8:603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Howes MT, Mayor S, Parton RG. Molecules, mechanisms, and cellular roles of clathrin-independent endocytosis. Curr Opin Cell Biol. 2010;22:519–527. doi: 10.1016/j.ceb.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 99.Gauthier NC, Monzo P, Kaddai V, Doye A, Ricci V, Boquet P. Helicobacter pylori VacA cytotoxin: a probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol Biol Cell. 2005;16:4852–4866. doi: 10.1091/mbc.E05-05-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gauthier NC, Ricci V, Landraud L, Boquet P. Helicobacter pylori VacA toxin: a tool to study novel early endosomes. Trends Microbiol. 2006;14:292–294. doi: 10.1016/j.tim.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 101.Gauthier NC, Monzo P, Gonzalez T, Doye A, Oldani A, Gounon P, Ricci V, Cormont M, Boquet P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J Cell Biol. 2007;177:343–354. doi: 10.1083/jcb.200609061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gupta VR, Wilson BA, Blanke SR. Sphingomyelin is important for the cellular entry and intracellular localization of Helicobacter pylori VacA. Cell Microbiol. 2010;12:1517–1533. doi: 10.1111/j.1462-5822.2010.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gupta VR, Patel HK, Kostolansky SS, Ballivian RA, Eichberg J, Blanke SR. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog. 2008;4:e1000073. doi: 10.1371/journal.ppat.1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–1102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 105.Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D'Elios MM, Telford JL, Baldari CT. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887–1897. doi: 10.1084/jem.20030621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci USA. 2004;101:7727–7732. doi: 10.1073/pnas.0401528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sewald X, Gebert-Vogl B, Prassl S, Barwig I, Weiss E, Fabbri M, Osicka R, Schiemann M, Busch DH, Semmrich M, et al. Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe. 2008;3:20–29. doi: 10.1016/j.chom.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 108.Oswald-Richter K, Torres VJ, Sundrud MS, VanCompernolle SE, Cover TL, Unutmaz D. Helicobacter pylori VacA toxin inhibits human immunodeficiency virus infection of primary human T cells. J Virol. 2006;80:11767–11775. doi: 10.1128/JVI.00213-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol. 2007;179:5433–5440. doi: 10.4049/jimmunol.179.8.5433. [DOI] [PubMed] [Google Scholar]
- 110.Luzio JP, Piper SC, Bowers K, Parkinson MD, Lehner PJ, Bright NA. ESCRT proteins and the regulation of endocytic delivery to lysosomes. Biochem Soc Trans. 2009;37:178–180. doi: 10.1042/BST0370178. [DOI] [PubMed] [Google Scholar]
- 111.Sommi P, Ricci V, Fiocca R, Necchi V, Romano M, Telford JL, Solcia E, Ventura U. Persistence of Helicobacter pylori VacA toxin and vacuolating potential in cultured gastric epithelial cells. Am J Physiol. 1998;275:G681–G688. doi: 10.1152/ajpgi.1998.275.4.G681. [DOI] [PubMed] [Google Scholar]
- 112.Molinari M, Galli C, Norais N, Telford JL, Rappuoli R, Luzio JP, Montecucco C. Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J Biol Chem. 1997;272:25339–25344. doi: 10.1074/jbc.272.40.25339. [DOI] [PubMed] [Google Scholar]
- 113.Li Y, Wandinger-Ness A, Goldenring JR, Cover TL. Clustering and redistribution of late endocytic compartments in response to Helicobacter pylori vacuolating toxin. Mol Biol Cell. 2004;15:1946–1959. doi: 10.1091/mbc.E03-08-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oldani A, Cormont M, Hofman V, Chiozzi V, Oregioni O, Canonici A, Sciullo A, Sommi P, Fabbri A, Ricci V, et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009;5:e1000603. doi: 10.1371/journal.ppat.1000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Derivery E, Sousa C, Gautier JJ, Lombard B, Loew D, Gautreau A. The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev Cell. 2009;17:712–723. doi: 10.1016/j.devcel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 116.Yamasaki E, Wada A, Kumatori A, Nakagawa I, Funao J, Nakayama M, Hisatsune J, Kimura M, Moss J, Hirayama T. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J Biol Chem. 2006;281:11250–11259. doi: 10.1074/jbc.M509404200. [DOI] [PubMed] [Google Scholar]
- 117.Calore F, Genisset C, Casellato A, Rossato M, Codolo G, Esposti MD, Scorrano L, de Bernard M. Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death Differ. 2010;17:1707–1716. doi: 10.1038/cdd.2010.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Backert S, Meyer TF. Type IV secretion systems and their effectors in bacterial pathogenesis. Curr Opin Microbiol. 2006;9:207–217. doi: 10.1016/j.mib.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 119.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, König W, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 120.Jiménez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, Rohde M, Pirch T, Jung K, Retta SF, et al. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog. 2009;5:e1000684. doi: 10.1371/journal.ppat.1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Murata-Kamiya N, Kikuchi K, Hayashi T, Higashi H, Hatakeyama M. Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell Host Microbe. 2010;7:399–411. doi: 10.1016/j.chom.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 122.Moese S, Selbach M, Zimny-Arndt U, Jungblut PR, Meyer TF, Backert S. Identification of a tyrosine-phosphorylated 35 kDa carboxy-terminal fragment (p35CagA) of the Helicobacter pylori CagA protein in phagocytic cells: processing or breakage? Proteomics. 2001;1:618–629. doi: 10.1002/1615-9861(200104)1:4<618::AID-PROT618>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 123.Odenbreit S, Gebert B, Püls J, Fischer W, Haas R. Interaction of Helicobacter pylori with professional phagocytes: role of the cag pathogenicity island and translocation, phosphorylation and processing of CagA. Cell Microbiol. 2001;3:21–31. doi: 10.1046/j.1462-5822.2001.00088.x. [DOI] [PubMed] [Google Scholar]
- 124.Selbach M, Moese S, Hurwitz R, Hauck CR, Meyer TF, Backert S. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 2003;22:515–528. doi: 10.1093/emboj/cdg050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe. 2007;2:250–263. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 126.Yokoyama K, Higashi H, Ishikawa S, Fujii Y, Kondo S, Kato H, Azuma T, Wada A, Hirayama T, Aburatani H, et al. Functional antagonism between Helicobacter pylori CagA and vacuolating toxin VacA in control of the NFAT signaling pathway in gastric epithelial cells. Proc Natl Acad Sci USA. 2005;102:9661–9666. doi: 10.1073/pnas.0502529102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Argent RH, Thomas RJ, Letley DP, Rittig MG, Hardie KR, Atherton JC. Functional association between the Helicobacter pylori virulence factors VacA and CagA. J Med Microbiol. 2008;57:145–150. doi: 10.1099/jmm.0.47465-0. [DOI] [PubMed] [Google Scholar]
- 128.Tegtmeyer N, Zabler D, Schmidt D, Hartig R, Brandt S, Backert S. Importance of EGF receptor, HER2/Neu and Erk1/2 kinase signalling for host cell elongation and scattering induced by the Helicobacter pylori CagA protein: antagonistic effects of the vacuolating cytotoxin VacA. Cell Microbiol. 2009;11:488–505. doi: 10.1111/j.1462-5822.2008.01269.x. [DOI] [PubMed] [Google Scholar]
- 129.Fasano A. Cellular microbiology: can we learn cell physiology from microorganisms? Am J Physiol. 1999;276:C765–C776. doi: 10.1152/ajpcell.1999.276.4.C765. [DOI] [PubMed] [Google Scholar]
- 130.Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]