

Abstract
Crohn’s disease (CD) is a chronic inflammatory bowel disease. Research has identified genetic predisposition and environmental factors as key elements in the development of the disease. However, the precise mechanism that initiates immune activation remains undefined. One pathway for luminal antigenic molecules to enter the sterile lamina propria and activate an immune response is via transcytosis. Transcytosis, although tightly regulated by the cell, has the potential for transepithelial transport of bacteria and highly antigenic luminal molecules whose uncontrolled translocation into the lamina propria can be the source of immune activation. Viewed as a whole, the evidence suggests that unregulated intestinal epithelial transcytosis is involved in the inappropriate presentation of immunogenic luminal macromolecules to the intestinal lamina propria. Thus fulfilling the role of an early pre-morbid mechanism that can result in antigenic overload of the lamina propria and initiate an immune response culminating in chronic inflammation characteristic of this disease. It is the aim of this paper to present evidence implicating enterocyte transcytosis in the early etio-pathogenesis of CD.
Keywords: Crohn’s, Transcytosis, Endocytosis
INTRODUCTION
Crohn’s disease (CD) is a chronic, lifelong, and unrelenting inflammatory bowel disease that mainly strikes young people in the prime of their productive life[1]. There are over 600 000 individuals with CD in North America, with up to 40 000 new cases being diagnosed each year, and double that amount at risk, based on a monozygotic concordance rate of 50%[1-3]. The effects on the patient and their family are devastating, with lifelong medication and repeated abdominal surgeries to relieve obstructed bowel, intestinal bleeding, or abdominal pain. Studies have shown that the age adjusted mortality risk from CD is over 50% greater than in the general population[2,4].
Current treatment for CD consists of inhibiting the immune response with powerful immunosuppressive agents. These medications have serious side effects and can eventually lose their therapeutic effect[5]. More importantly, immunosuppressive agents do not alter the natural history of this disease, suggesting that the immune response in these individuals is a secondary reaction to an, as yet undiscovered, primary cause, which results in chronic immune activation within the intestinal wall[5,6].
The etiology of CD is currently unknown. It is generally attributed to a faulty immune system; however, despite decades of research, no antecedent immune abnormality has been identified in these individuals. A closer review of pertinent studies reveals that the immediate causal mechanism of CD may have its origin in a process of unregulated transcytosis of intestinal luminal contents by intestinal epithelial cells.
The data presented in this paper suggests that a primary inherited cell membrane defect may be responsible for the initiation and perpetuation of a pathological transcytotic state, allowing bacteria and other intestinal contents to persistently penetrate into the bowel wall and initiate a chronic life-long inflammatory response characteristic of this disease.
The aim of this paper, therefore, is to present evidence that unregulated intestinal transcytosis may have a significant role in the initiation of CD, and to provide data implicating an antecedent cell membrane abnormality as a primary inheritable factor capable of initiating this pathologically altered transcytotic state.
WHAT IS TRANSCYTOSIS?
Transcytosis is the transport of macromolecules (cargo) from one side of a cell to the other within a vesicular cell membrane-bound carrier. Conceptually, transcytosis can be divided into three distinct processes; absorption of molecules (endocytosis), conveyance of cargo through the cell (transcellular transport), and extrusion of cargo at the other side of the cell (exocytosis). During endocytosis, extracellular macromolecules converge upon the cell as a portion of the plasma membrane is invaginated at the point of contact and pinched off forming a cytoplasmic membrane-bounded vesicle called an endosome, which contains the engulfed cargo. Material packaged within the endosome may undergo processing by the cell, after which the cytoplasmic vesicle can move to the opposite side and fuse with the plasma membrane, releasing its contents to the extracellular space during the process of exocytosis (Figure 1).
Figure 1.
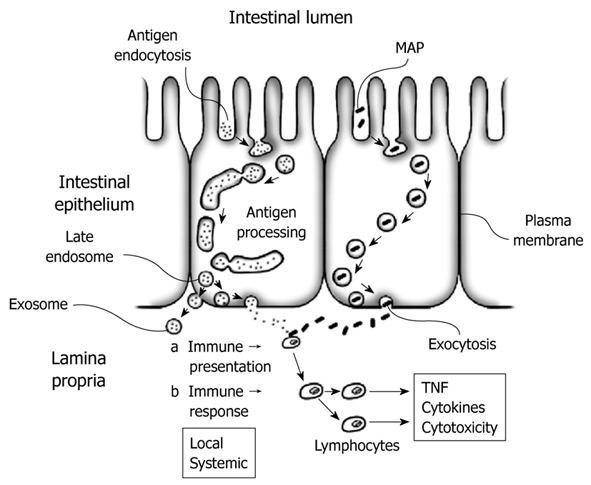
Antigenic exposure (a) of luminal antigens to the immune system can occur through either transcellular (through cells) or paracellular (between cells) pathways. Transcellular transport is mediated via transcytosis of processed antigen within discrete intracellular vesicles, termed endosomes, which are transported from apical to basal membrane prior to undergoing exocytosis, where immune recognition may occur within the lamina propria. Bacteria and macromolecules are transported in this fashion as they are too large to penetrate the tight intercellular space (paracellular pathway) maintained by tight junctional proteins. An immune response (b) can ensue subsequent to antigenic penetration into the lamina propria. Continuous unregulated transcytosis of luminal antigens and Mycobacterium avium paratuberculosis (MAP) into the lamina propria can initiate a chronic immune reaction. Inhibition of immune response (b) with immunosuppressive agents may temporarily restore epithelial integrity, but will not prevent transcytosis of luminal antigen, which continues upon discontinuation of immunosuppressive therapy. Mitigation of luminal antigenic exposure via restrictive diets or parenteral therapy cannot prevent subsequent transcytosis and relapse once normal dietary activity is resumed. A three pronged approach for acute therapy consisting of: (1) reduction of antigenic exposure via temporary dietary restriction; (2) temporary immunosuppression to inhibit immune mediated tissue damage and speed epithelial healing; and (3) Inhibition of transcytosis via endocytosis blocking agents to prevent future immune activation, may offer a therapeutic paradigm on which to base clinical decision making. Anti-MAP antibiotics may additionally be utilized to speed systemic removal of this bacterium and hasten downregulation of the immune response. Limited evidence suggests that inhibition of transcytosis may reduce intestinal antigenic exposure sufficiently to allow long term dietary activity with minimal restrictions. Maintenance therapy with endocytosis inhibiting agents may provide a safe and effective means of inducing long-term remission and interrupting the natural history of Crohn’s disease. TNF: Tumor necrosis factor.
Cytoplasmic material can also be extruded from the cell, forming plasma membrane bound extracellular vesicles called exosomes, in a similar, but reverse process, to endocytosis[7,8].
Endocytosis is required for a vast amount of cellular functions that are paramount to the survival and wellbeing of the cell. Internalizing thousands of molecules, and up to five times its surface area per minute, the cell membrane is a seething cauldron of continuous endocytotic invaginations involved in the non-stop absorption of nutrients and water, in addition to cell surface receptors, antigens, cell signaling molecules, protein homeostasis, and maintenance of plasma membrane lipids[9-14].
While inside the cell, the cargo is enclosed in a vesicle formed by the invagination of membrane lipids, called an endosome. The endosome can be destined for transport to areas within the cell or undergo transcellular transport to the opposite side, where fusion with the cell membrane results in exocytosis and deposition of the cargo (in the intestinal epithelium) into the lamina propria[15-17].
A multitasking membrane machine, the intestinal epithelium simultaneously integrates the transcytosis and processing of hundreds of distinct nutrient molecules into the lamina propria, while providing a single cell thickness physical barrier to luminal antigens that maintains sub-epithelial sterility, and does so while serving as a non-stop conveyer belt for the continual migration of new enterocytes to the surface epithelium.
Despite its highly dynamic physical state, the intestinal epithelium is very efficient at preventing translocation of luminal antigens into the lamina propria. This efficiency however notwithstanding, the translocation of intact proteins through the intestinal epithelium has been demonstrated and cell mechanisms exist for the endocytosis of entire bacteria. At least ten mechanistically distinct endocytotic mechanisms have been described, from phagocytosis and macropinocytosis, employed for the absorption of cargo over 500 nm, to clathrin coated pits and caveoli mediated endocytosis for smaller particulate macromolecules[9,15,18].
Endocytosis is highly dependent on the composition and organization of the cell membrane and experimentally induced changes in membrane properties can alter the cell’s ability to appropriately engage extracellular particulate matter leading to alterations in one or more mechanisms of endocytosis[9,15,19].
This implies that pathological endocytosis of luminal antigenic material (endo cytopathy) may result in transcytotic antigenic overload of the lamina propria, with subsequent immune activation and the establishment of a chronic inflammatory state. An endo cytopathic state would, in theory, present as an increased permeability of the intestinal epithelium prior to the start of inflammation, such as that observed in CD. It is therefore reasonable to speculate that an inherited endo cytopathy may be involved as an early etiopathogenetic mechanism in CD, with the participation of one or more endocytotic mechanisms contributing to the disease process.
The following section examines studies of early lesions of Crohn’s epithelium for evidence of endocytopathy.
IS ABERRANT TRANSCYTOSIS INVOLVED IN CD?
Transcytosis is a normal endosomal generating process that is regulated by the cell. Pathologically triggered, unregulated, transcytosis can differ from its normal physiological counterpart by an increased endosomal generation rate; manifesting as an excess amount of cellular endosomes when compared to normal cells. Thus, a defining characteristic of unregulated transcytosis can be an increased cellular endosomal load.
This aspect of cellular function was evaluated by several studies, which documented multiple endosomes in 19 out of 19 cases of CD. No endosomes were observed in six control cases. Nor were they observed in radiation ileitis, celiac disease, experimental Yersinia enteritis, or in all of 155 cases of diagnosed ulcerative colitis[20]. Normal colonocytes had no visible endosomes.
The cellular location of endosomes in apical, basal, and lateral portions of the cell suggests a process of active transcytosis. Although diminished exocytosis could account for increased endosomes, the presence of concurrent inflammation suggests ongoing deposition of antigenic material into the lamina propria, along with active exocytosis. The absence of endosomes in both negative and active control groups suggests that the endosomal structures observed in all CD study patients are the result of a pathological transcytotic state, triggered by inappropriate endocytosis.
A more detailed analysis of the role of transcytosis in the pathogenesis of CD was undertaken in a study that examined the effects of autologous infusion of intestinal contents into the excluded ileum of three individuals who had undergone a resection of the terminal ileum with the creation of a temporary loop ileostomy. In this surgical procedure, the two openings from a bisected loop of ileum are brought through the abdominal wall to the surface[21]. After 3-6 mo of diversion, the patients were infused, via a catheter, with 60cc of ileal effluent (collected from the proximal limb) into the distal limb four times daily for 7 d.
Distal ileal biopsies obtained prior to infusion were histologically normal without evidence of inflammation. Biopsies obtained 1 d after the last infusion (day eight) revealed a moderate increase of mononuclear cells, eosinophils, and polymorphonuclear cells in the lamina propria. Neutrophils were also noted in the small vessels and epithelium without cryptitis or crypt abscesses.
Epithelial electron micrographs revealed dilation of rough endoplasmic reticulum (ER) and Golgi apparatus (GA), in addition to basally located transport vesicles (endosomes). Mitochondria appeared damaged and dilated.
These ultrastructural changes are characteristic of biosynthesis [i.e. major histocompatibility complexes (MHC) peptides] and antigen processing during transcytosis prior to presentation of antigen-MHC complexes on the cell surface[17,22]. Basal transport vesicles suggest late endosomes prior to exocytosis and exposure of antigen within the lamina propria (Figure 1).
Dilated mitochondria suggests sudden elevated metabolic activity, which is consistent with high ATP demand resulting from protein synthesis and the processing of a large influx of antigen by TAP (transporter associated peptide) during intracellular conjugation of processed antigen to MHC molecules. This process is estimated to consume 50 000 molecules of ATP per second/cell in prokaryotes during normal antigen processing, and is likely to consume more in eukaryotic (human) cells that may be undergoing excessive antigenic processing[17,23-25]. The appearance of damaged mitochondria suggests that excess reactive oxygen species (i.e. superoxide, hydrogen peroxide, and hydroxyl radicals), generated during an acute excessive demand for ATP, had overwhelmed mitochondrial reductive (antioxidant) capacity, with resultant activation of the mitochondrial permeability transition pore leading to the observed structural mitochondrial and epithelial cell damage. The presence of minute collections of lymphocytes within the lamina propria (aphthous lesions), which are not associated with superficial erosions or lymphoid follicles in CD, is compatible with local antigenic presentation to the lamina propria[26].
The above studies suggest that excessive transcytosis of luminal antigens is an early concomitant in the pathogenesis of CD intestinal inflammation. However, are luminal antigens actually transcytosed in CD and is this transcytosis pathological and able to account for the initiation of inflammation in CD?
An elegant study designed to evaluate enterocyte transcytosis of luminal antigens in CD was performed on mucosal biopsies taken from ileal mucosa after in-vivo incubation with luminally applied ovalbumin (OVA) in patients undergoing ileoscopy[27]. The authors found OVA associated with MHC within cytoplasmic late endosomes (the stage prior to exocytosis) and with exosomes in the intercellular space (Figure 1). OVA was also found in the lamina propria. Importantly, OVA cytoplasmic trafficking and antigen processing showed no qualitative differences between CD patients (active disease or histologic remission) and controls.
This study indicated that a luminal antigen can be transcytosed by intestinal epithelial cells and the intracellular antigenic processing in both CD and normal controls is qualitatively similar, regardless of disease state. The absence of a significant demonstrable difference between normal and CD enterocyte antigenic processing suggests that a disorder may exist during the initiation of transcytosis at the level of the cell membrane. This is suggested by studies documenting multiple endosomes in CD enterocytes which were not present in normal controls implying a quantitative difference in antigenic transcytosis rather than a qualitative one[20].
This interpretation is supported by studies in individuals undergoing small intestinal allograft transplantation as a result of short bowel syndrome subsequent to surgery required to treat CD[28]. In a series of four CD patients receiving small intestinal allografts, early characteristic CD lesions were documented in two patients, via allograft biopsy, at 3 and 5 wk after transplantation. None of the four transplant recipients developed clinical or endoscopic recurrence during the follow-up period of 20-40 mo. No similar histological findings were observed in any of 57 non-CD patients receiving small intestinal allografts at this institution.
The appearance of lamina propria lymphoplasmacytosis and inter-epithelial neutrophil infiltration suggests antigenic presentation by the allograft to the recipient’s immune system. This would not appear to be the result of aberrant immune processing by allograft tissue, since allograft donors are carefully screened prior to transplantation, and studies have documented similar enterocyte antigenic processing in both CD and normal controls (above). Likewise, there is no indication of abnormally increased intestinal permeability in allograft donors and no antecedent immune abnormality has been identified in CD to account for this immune reaction in the allografts.
An allograft immune reaction so soon after transplantation suggests a normal constitutive process of antigen presentation to the recipient’s immune system and studies have shown the presence of a constitutive apical internalization pathway in enterocytes[16,27]. This suggests that the allograft immune reaction in CD recipients is due to a heightened immune response to normal intestinal antigenic presentation as a result of normal constitutive endocytosis in CD individuals hypersensitized as a result of previous excessive exposure to intestinal luminal antigens.
Therefore, if normal antigenic transcytosis from a normal transplanted gut can cause characteristic histological CD lesions in allograft recipients, then it is reasonable to speculate that excessive luminal antigenic exposure via unregulated transcytosis may play a significant role in early pathogenic events leading to the development of CD.
Why individuals with CD may have unregulated transcytosis is examined in the next section.
WHAT CAUSES UNREGULATED TRANSCYTOSIS?
The cell membrane is the gateway that mediates all interaction with the external environment. This is largely accomplished via membrane coated vesicles that constantly bud off from the cell membrane and enter the cytoplasm, while others arrive from the cytoplasm and fuse with the cell membrane (transcytosis). The composition, integrity, and 3-dimensional arrangement of plasma membrane components are crucial to control of the vesicular fission/fusion process, which in turn contributes to maintaining the composition of the plasma membrane as vesicles are recycled back into the cell membrane[10,15,29].
Luminal antigenic sampling by enterocytes depends upon a controlled transcytotic antigenic presentation to the immune system; therefore, any alteration in the composition, properties, or structure of enterocyte cell membranes can have a significant impact on the immunological functionality of the entire gastrointestinal tract. Too little antigenic presentation and the gut cannot fulfill its role of immune surveillance; too much presentation can lead to a heightened immune response and chronic inflammation[9].
Studies have shown that alterations in cell membrane properties can have a profound effect on endocytosis and transcytosis. Treatment of cells with the cell membrane intercalating agent phorbol ester can initiate spontaneous endocytosis[15,30]. Enhanced endocytosis of non-targeted membrane enzymes has been observed during fat absorption, suggesting that this physiological process, which increases cell membrane fatty acid content, disturbs local membrane organization[16,31]. A change in cell membrane fatty acid composition can modify membrane properties and its interaction with cytosolic proteins involved in endocytosis[18].
Analysis of synthetic liposome transcytosis across cell membranes revealed that cell membrane fluidity is the most important factor influencing transcytosis, followed by surface charge[19]. Other significant cell surface properties affecting transcytosis, such as lipid composition and surface density, have been described by other researchers in the field[19]. These studies indicate that biochemical and biophysical properties of cell membranes are major factors controlling cellular uptake and transcytosis. Combined with data suggesting unregulated intestinal transcytosis in CD, it is reasonable to consider the possibility of enterocyte cell membrane abnormalities as a contributing factor to unregulated transcytosis in the early pathogenesis of CD.
The next section will explore this aspect.
ARE CELL MEMBRANE ABNORMALITIES PRESENT IN CD?
Significant decreases in cell membrane fluidity, in addition to disturbances in cell membrane lipid composition, were observed in a study conducted on erythrocytes of individuals with active and inactive CD[32]. A separate study examining the mucosal fatty acid profile in uninvolved (never inflamed) colonic mucosa in individuals with CD demonstrated altered lipid composition compared to healthy controls[33].
Studies regarding cell membrane properties and lipid composition in CD are rare. Abnormal lipid profiles in uninvolved (never inflamed) colonic mucosa raises the possibility of an antecedent metabolic defect. Erythrocyte cell membrane fluidity abnormalities in CD are consistent with the possibility that membrane abnormalities may be present in other tissues, including the intestinal epithelium, resulting in deleterious effects on enterocyte transcytosis.
Further basic scientific data are necessary to identify potential cell membrane abnormalities and their role in enterocyte transcytosis. However, if unregulated transcytosis is involved in the pathogenesis of CD, then endocytosis blocking agents should have a beneficial therapeutic effect on the clinical parameters and progression of disease.
In the next section we evaluate the therapeutic effect of endocytosis blocking agents upon CD.
DO ENDOCYTOSIS BLOCKING AGENTS REDUCE INFLAMMATION IN CD?
Therapeutic measures available for the treatment of CD can be divided into three general categories; dietary measures, antibiotics, and immunosuppressive agents. No mechanism of action employed by these therapeutic interventions, either singly or in combination, has been proven to modify the natural history of CD. This suggests that an unrecognized pathogenetic mechanism is involved in the development of this disease. Clinical reports of complete remission in refractory CD after administration of non-conventional agents have been documented. One of these agents, thalidomide, a known endocytosis blocker, has been shown to be effective for induction and long term maintenance of remission in both intestinal and extraintestinal CD[34-44].
Studies have demonstrated a reduction in inflammation and inflammatory parameters in CD using the cholesterol lowering agent atorvastatin[45,46]. These agents inhibit the biosynthesis of cholesterol, a critical membrane lipid constituent required for the formation of endosomal vesicles[29].
The polyene antibiotic Nystatin, which inhibits endocytosis by cholesterol sequestration within cell membranes, has been used in combination therapy to reduce inflammatory activity in CD[29,47].
Azithromycin, a macrolide antibiotic, was observed to markedly inhibit endocytosis and has also been used in therapeutic regimens to reduce inflammation in CD[48-50].
Macrolide antibiotics, including azithromycin, are considered among the most effective therapeutic agents for the treatment of CD[51,52]. The rationale for the use of endocytosis blocking agents in CD is to prevent enterocyte transcytosis of luminal antigens into the lamina propria. A reduction in intestinal transcytotic antigenic load is also consistent with the mucosal healing and decrease in inflammatory cytokines, mucosal permeability, and clinical disease activity observed with the use of specific dietary exclusion measures in the treatment of active CD[53-56].
To date, no study has evaluated the effect of endocytosis blocking agents in the treatment of CD. The limited amount of data in which therapeutic agents with adjunct endocytotic blocking activity show a beneficial effect in the treatment of CD is consistent with the involvement of transcytosis in the pathogenesis of this disease. Further research is required to determine if specific endocytosis blocking agents are beneficial in the treatment of CD.
Certain questions, however, remain unanswered. For instance, what is the role of Mycobacterium avium paratuberculosis (MAP), a bacterium that has been uniquely associated with CD, and what is the genetic nature of the putative membrane abnormality proposed for CD?
A transcytosis mechanism for CD suggests answers to these questions, which are explored in following section.
THE ROLE OF MAP IN CD?
CD is the result of a complex interaction between the body’s immune system and environmental (luminal) factors, played out at the gastrointestinal epithelial interface. The data presented in this paper suggests that unregulated transcytosis of luminal antigens plays a significant role in the early pathogenesis of this disease.
The adult mammalian intestinal epithelium is normally very selective regarding the absorption of luminal molecules, with macromolecules being degraded prior to entering the bloodstream. The neonatal intestinal epithelium, however, has a greater capacity for non-selective absorption, and undergoes a gradual change in permeability that restricts the uptake of macromolecules. This process of decline in intestinal permeability to immunologically recognizable molecules is called intestinal closure and, for most species, is complete by the end of the perinatal period 1 to 4 wk after birth[16,57,58].
The process of intestinal closure is age dependent, and is accompanied by developmental changes and remodeling in membrane phospholipids, which become a potential regulator of intestinal transport[57]. Thus, alterations in the composition of cell membrane lipids can alter membrane properties, such as hydrophobicity, molecular structure, and fluidity. These alterations can lead to dysfunction of cell membrane dependent processes, such as intestinal epithelial transcytosis resulting in enhanced uptake of immunologically active molecules or luminal organisms.
One organism that stands out regarding its association with CD is Mycobacterium avium paratuberculosis (MAP). Since its initial isolation from CD patients in 1984, detection studies have shown that up to 95% of CD patients harbor this bacterium[59-61]. The association with CD is unique to MAP, and has not been described for other species of mycobacteria.
MAP is ubiquitously present in the environment, the water supply, and the human food chain[60,62,63]. It infects many wild and domesticated animals, including up to 68% of milk producing dairy herds in any geographical area. Infected animals can develop chronic diarrhea and wasting called Johne’s disease[52,64]. Although MAP can be acquired from drinking water, contaminated vegetables, and aerosol droplets, the principal reservoir of MAP for transmission to humans is the intestinal tract of infected cattle, which serves as a distribution point for dissemination of MAP into diary products such as milk, cheese, and other dairy by-products[60,63,65].
Studies suggest that, once in the GI tract, the interaction of MAP with CD patients is specific and not the result of a random generic process[66-69]. In other words, once contact is made, unique characteristics inherent in both Crohn’s intestinal epithelium and the MAP cell wall favor continued adherence and transcytosis into the lamina propria, leading to an immune reaction.
Exposure of normal human, 12 wk old, fetal intestinal epithelia to MAP revealed almost no internalization of MAP by enterocytes, with uptake limited to goblet cells[70]. This is consistent with resistance of normal human intestinal epithelium to MAP transcytosis, with a transitory gestational permeability effect on goblet cells, since oral Crohn’s lesions can appear on stratified squamous gingival mucosa, which is devoid of both dendritic and goblet cells. Goblet cells have not been observed to serve as unique foci of inflammation in early Crohn’s lesions[21,71-76]. This suggests that specific inherited cell membrane abnormalities in Crohn’s intestinal epithelium interact with unique MAP cell wall constituents that are not present in other mycobacterial species, conferring upon MAP the role of a transcytosis triggering agent when in contact with Crohn’s intestinal epithelium. Once processed within intestinal epithelial cells, luminal derived antigens are deposited within the lamina propria, which evokes an immune response characteristic of CD. This implies a predisposing disease genotype whose phenotypic expression is associated with cell membrane composition; a consideration that is further developed below.
WHAT ARE THESE DISTINCTIVE CELL WALL COMPONENTS AND WHAT CELL WALL PROPERTIES MIGHT ENHANCE MAP TRANSCYTOSIS BY CROHN’S INTESTINAL EPITHELIUM?
MAP is unique in having a cell wall that differs significantly from that of other bacteria[77,78]. The cell wall is composed of a thick layer of extremely hydrophobic lipid molecules. Long chain α branched lipids (mycolic acids) and species-specific glycopeptidolipids contribute to the extreme hydrophobicity of this organism[77,78].
MAP is also highly negatively charged, and studies have demonstrated a greater attachment of MAP to like-charged particles compared to similarly charged bacteria because of the extreme hydrophobic nature of the MAP cell wall[77]. Additional studies have demonstrated that MAP is highly predisposed to surface adherence, being the primary colonizers on a variety of external surfaces, the degree of which varies with the characteristics of the surface material being colonized[79].
Thus, the combination of uniquely strong hydrophobicity and high electronegative charge present on the MAP outer surface can aid in its adherence to a genetically altered Crohn’s intestinal epithelium, facilitating its transcytosis and ultimate dispersal from a negatively charged basal surface epithelium into the lamina propria, where an immune reaction can ensue[80]. Studies have shown that cell surface hydrophobicity may play an important in the rate of internalization of bacteria[81].
Consequently, MAP can be considered an environmental response modifier that interacts with the biological expression (phenotype) of certain susceptibility genes, whose genotype contributes to the composition of cell membranes[82-84]. To be consistent with this interpretation, alterations in cell membrane function should elicit a Crohn’s inflammatory phenotype. This has been observed with epidemiological studies linking the ingestion of aspirin, a nonsteroidal anti-inflammatory drug (NSAID) with increased risk of developing CD[85]. Aspirin, which is strongly lipophilic, binds to, and accumulates within, cell membranes altering their microviscosity, molecular structure, physical state, and biological function[86,87]. Aspirin, and other NSAIDs, have also been demonstrated to induce disorders of cell membrane lipid assembly, as well as rearrangements in membrane protein patterns[88,89].
NSAID-induced enteropathy is reported to have close similarities to CD and is not infrequently reported by the pathologist as consistent with CD[90,91]. NSAID-induced alterations in model cell membranes provoke membrane fusion, suggesting the possibility that pre-existing cell membrane anomalies may also contribute to the high occurrence of intestinal fistulas in CD[92]. NSAIDs increase the risk of developing de-novo CD, induce an enteropathy that can be histologically indistinguishable from CD, and alter cell membrane properties known to be involved in transcytosis (fluidity, viscosity, composition). It is reasonable, therefore, to speculate that the genetic predisposition in CD is an inherited membrane abnormality that gives rise to unregulated enterocyte transcytosis, which ultimately leads to chronic intestinal inflammation.
IF A CELL MEMBRANE ABNORMALITY DOES EXIST IN CD; IS THERE ANY EVIDENCE THAT MIGHT PROVIDE SOME IDENTIFYING CHARACTERISTICS FOR A CANDIDATE GENE?
It has been known for some time that smoking increases the risk of developing CD, and will worsen existing disease[93,94]. A clue to a potential underlying mechanism is the clinical observation of significant disease worsening above a threshold of 10-15 cigarettes per day[93,95]. A phenotypic threshold effect is characteristic of mitochondrial involvement in disease pathogenesis[96,97].
Smoking has been documented to cause considerable mitochondrial dysfunction, with up to 80% inhibition of electron transport chain activity and significant decreases in ATP production and availability[98-105]. Conversely, smoking cessation is associated with both normalization of mitochondrial function and clinical improvement in CD activity, suggesting a cause and effect relationship between mitochondrial bioenergetics and CD severity[93,95,106]. This unveils the possibility that an energy (ATP)-requiring enzyme participating in a cell membrane lipid biosynthetic pathway may be involved in CD pathogenesis.
A candidate enzyme fulfilling these criteria is long chain acyl Co-A fatty acid synthetase isoform 6 (ACSL6, E.C.6.2.1.3). The gene for this enzyme is located on 5q31, which has been designated as IBD susceptibility locus 5 (IBD5)[107-109].
These isoenzymes, located on the peroxisomal surface membrane, have a crucial role in plasma membrane phospholipid turnover, de novo lipid biosynthesis, fatty acid catabolism, and remodeling of biological membranes. They also use ATP to convert fatty acids into an activated form that can be incorporated into cell membranes[110-116].
Studies have shown a decrease in peroxisomal frequency in Crohn’s mucosal biopsies[117]. This suggests a mechanism in which a genetically dysfunctional or depleted ACSL6 enzyme may be further compromised by smoking-induced depletion of ATP, leading to cell membrane abnormalities that can enhance transcytosis of luminal antigens, resulting in development or worsening of the disease.
Smoking, however, does not affect all individuals with CD[93]. This suggests the involvement of other, non-ATP-requiring enzymes involved in cell membrane biogenesis, such as LPCAT2 (Lysophosphatidylcholine acyltransferase2, E.C. 2.3.1.67)[118]. The gene for this enzyme is located on 16q12, which has been designated IBD susceptibility locus 1 (IBD1); a locus that also contains the NOD2/CARD15 gene, which has been linked to CD. LPCAT2 is involved in the biosynthesis of membrane lipids, suggesting the possibility of membrane abnormalities as a result of a compromised LPCAT2 enzyme, that may disrupt cell membrane properties regulating transcytosis leading to enhanced antigen deposition in the intestinal lamina propria and a chronic immune reaction[119].
Finally, studies demonstrating that up to 50% of healthy individuals harbor MAP in their blood suggest a spectrum of intestinal mucosal affinity for MAP, in addition to variations in MAP exposure and interindividual immune response, as determining factors contributing to disease development and severity (Figure 1)[120,121].
CONCLUSION
Evidence presented in this paper provides a reasonable basis for the premise that unregulated transcytosis may be fundamentally involved in the development and early pathogenesis of CD. Unregulated transcytosis is compatible with the repeated clinical observation that mucosal healing does not alter the fundamental disorder present within the intestinal epithelium, leading to disease relapse upon discontinuation of treatment[5,122]. Present at birth, a genetic predisposition towards unregulated transcytosis (transcellular defect) can increase local concentrations of inflammatory cytokines that are known to trigger tight junctional barrier (paracellular) defects, which have been reported in CD patients and healthy first-degree relatives[123]. The presence of MAP in a significant percentage of mucosal biopsies and blood of children with CD is consistent with an inherited congenital mucosal anomaly favoring mucosal adherence and transcytosis of MAP[124].
Within this pathogenetic chronology, the immune-mediated paracellular permeability defect present in CD arises as a consequence of a primary inherited pathological transcellular transport of luminal antigens, resulting in a vicious cycle of paracellular antigenic overload, which is exacerbated by continuous unregulated transcytosis of immunogenic luminal macromolecules. Increased intestinal permeability to polyethylene glycol (a transcellular permeability probe) has been demonstrated in CD[125-128].
Supporting this interpretation is a report of prodromal symptoms, including fever, occurring from 7-10 years prior to a diagnosis of CD in almost 28% of 29 patients enrolled in a questionnaire based study. In contrast, none of the 15 ulcerative colitis patients enrolled in the study reported fever occurring prior to diagnosis[129].
This is compatible with a continuous stream of transcytosed immunogenic macromolecules resulting in systemic subclinical immune activation. Unregulated transcytosis is also consistent with other studies that implicated an inherent cell membrane permeability defect independent of inflammation[130,131].
Since the term “natural history” was introduced for CD in 1965, there has been no hard evidence for change in disease outcome[4,6]. The urgent need for a natural-history modifying therapy remains a priority. Although immune-mediated tissue damage is clearly evident, in a complex condition such as CD it may not be what you see when you look, but how you look at what you see that provides the answer.
Acknowledgments
I wish to thank Dr. William W Taylor for his continuous constructive feedback and support. I am deeply indebted to Kimberly D Shepherd whose courageous existence made this work possible.
Footnotes
Peer reviewers: Enzo Ierardi, Professor, Section of Gastroenterology, Department of Medical Sciences, University of Foggia, AOU Ospedali Riuniti, Viale Pinto, 71100 Foggia, Italy; Yanfang Guan, PhD, Research Scientist, Department of Molecular and Cellular Physiology, University of Cincinnati College of Medicine, Cincinnati, OH 45267, United States; Reiko Miyazawa, Department of Pediatrics, Gunma University Graduate School of Medicine, 3-39-22, Showa-machi, Maebashi, Gunma 371-8511, Japan
S- Editor Tian L L- Editor Stewart GJ E- Editor Zheng XM
References
- 1.Loftus EV Jr, Schoenfeld P, Sandborn WJ. The epidemiology and natural history of Crohn's disease in population-based patient cohorts from North America: a systematic review. Aliment Pharmacol Ther. 2002;16:51–60. doi: 10.1046/j.1365-2036.2002.01140.x. [DOI] [PubMed] [Google Scholar]
- 2.Canavan C, Abrams KR, Mayberry JF. Meta-analysis: mortality in Crohn's disease. Aliment Pharmacol Ther. 2007;25:861–870. doi: 10.1111/j.1365-2036.2007.03276.x. [DOI] [PubMed] [Google Scholar]
- 3.Tysk C, Lindberg E, Järnerot G, Flodérus-Myrhed B. Ulcerative colitis and Crohn's disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut. 1988;29:990–996. doi: 10.1136/gut.29.7.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peyrin-Biroulet L, Loftus EV Jr, Colombel JF, Sandborn WJ. The natural history of adult Crohn's disease in population-based cohorts. Am J Gastroenterol. 2010;105:289–297. doi: 10.1038/ajg.2009.579. [DOI] [PubMed] [Google Scholar]
- 5.Vermeire S, van Assche G, Rutgeerts P. Review article: Altering the natural history of Crohn's disease--evidence for and against current therapies. Aliment Pharmacol Ther. 2007;25:3–12. doi: 10.1111/j.1365-2036.2006.03134.x. [DOI] [PubMed] [Google Scholar]
- 6.Wolters FL, Russel MG, Stockbrügger RW. Systematic review: has disease outcome in Crohn's disease changed during the last four decades? Aliment Pharmacol Ther. 2004;20:483–496. doi: 10.1111/j.1365-2036.2004.02123.x. [DOI] [PubMed] [Google Scholar]
- 7.Tuma PL, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83:871–932. doi: 10.1152/physrev.00001.2003. [DOI] [PubMed] [Google Scholar]
- 8.Sircar S. Principles of medical physiology. New York, NK: Thieme Medical Publishers; 2008. pp. 36–42. [Google Scholar]
- 9.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 10.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowler BC. Endocytosis: structural components, functions and pathways. Hauppauge, NY: Nova Science Publishers; 2010. [Google Scholar]
- 12.Dunn WA, Hubbard AL. Receptor-mediated endocytosis of epidermal growth factor by hepatocytes in the perfused rat liver: ligand and receptor dynamics. J Cell Biol. 1984;98:2148–2159. doi: 10.1083/jcb.98.6.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawyer ST, Krantz SB. Transferrin receptor number, synthesis, and endocytosis during erythropoietin-induced maturation of Friend virus-infected erythroid cells. J Biol Chem. 1986;261:9187–9195. [PubMed] [Google Scholar]
- 14.Miaczynska M, Stenmark H. Mechanisms and functions of endocytosis. J Cell Biol. 2008;180:7–11. doi: 10.1083/jcb.200711073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20:256–275. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen GH, Rasmussen K, Niels-Christiansen LL, Danielsen EM. Endocytic trafficking from the small intestinal brush border probed with FM dye. Am J Physiol Gastrointest Liver Physiol. 2009;297:G708–G715. doi: 10.1152/ajpgi.00192.2009. [DOI] [PubMed] [Google Scholar]
- 17.Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. doi: 10.1016/j.coi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Sandvig K, Torgersen ML, Raa HA, van Deurs B. Clathrin-independent endocytosis: from nonexisting to an extreme degree of complexity. Histochem Cell Biol. 2008;129:267–276. doi: 10.1007/s00418-007-0376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orthmann A, Zeisig R, Koklic T, Sentjurc M, Wiesner B, Lemm M, Fichtner I. Impact of membrane properties on uptake and transcytosis of colloidal nanocarriers across an epithelial cell barrier model. J Pharm Sci. 2010;99:2423–2433. doi: 10.1002/jps.22001. [DOI] [PubMed] [Google Scholar]
- 20.Roediger WE. A new hypothesis for the aetiology of Crohn's disease--evidence from lipid metabolism and intestinal tuberculosis. Postgrad Med J. 1991;67:666–671. doi: 10.1136/pgmj.67.789.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D'Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn's disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–267. doi: 10.1016/s0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 22.Pichler H, Riezman H. Where sterols are required for endocytosis. Biochim Biophys Acta. 2004;1666:51–61. doi: 10.1016/j.bbamem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Abele R, Tampé R. The ABCs of immunology: structure and function of TAP, the transporter associated with antigen processing. Physiology (Bethesda) 2004;19:216–224. doi: 10.1152/physiol.00002.2004. [DOI] [PubMed] [Google Scholar]
- 24.Procko E, Gaudet R. Antigen processing and presentation: TAPping into ABC transporters. Curr Opin Immunol. 2009;21:84–91. doi: 10.1016/j.coi.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, Abele R, Tampé R. Peptides induce ATP hydrolysis at both subunits of the transporter associated with antigen processing. J Biol Chem. 2003;278:29686–29692. doi: 10.1074/jbc.M302757200. [DOI] [PubMed] [Google Scholar]
- 26.Okada M, Maeda K, Yao T, Iwashita A, Nomiyama Y, Kitahara K. Minute lesions of the rectum and sigmoid colon in patients with Crohn's disease. Gastrointest Endosc. 1991;37:319–324. doi: 10.1016/s0016-5107(91)70723-2. [DOI] [PubMed] [Google Scholar]
- 27.Hundorfean G, Zimmer KP, Strobel S, Gebert A, Ludwig D, Büning J. Luminal antigens access late endosomes of intestinal epithelial cells enriched in MHC I and MHC II molecules: in vivo study in Crohn's ileitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G798–G808. doi: 10.1152/ajpgi.00135.2007. [DOI] [PubMed] [Google Scholar]
- 28.Harpaz N, Schiano T, Ruf AE, Shukla D, Tao Y, Fishbein TM, Sauter BV, Gondolesi GE. Early and frequent histological recurrence of Crohn's disease in small intestinal allografts. Transplantation. 2005;80:1667–1670. doi: 10.1097/01.tp.0000184621.63238.ec. [DOI] [PubMed] [Google Scholar]
- 29.Ivanov AI. Pharmacological inhibition of endocytic pathways: is it specific enough to be useful? Methods Mol Biol. 2008;440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- 30.Goel G, Makkar HP, Francis G, Becker K. Phorbol esters: structure, biological activity, and toxicity in animals. Int J Toxicol. 2007;26:279–288. doi: 10.1080/10915810701464641. [DOI] [PubMed] [Google Scholar]
- 31.Hansen GH, Niels-Christiansen LL, Immerdal L, Nystrøm BT, Danielsen EM. Intestinal alkaline phosphatase: selective endocytosis from the enterocyte brush border during fat absorption. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1325–G1332. doi: 10.1152/ajpgi.00379.2007. [DOI] [PubMed] [Google Scholar]
- 32.Aozaki S. Decreased membrane fluidity in erythrocytes from patients with Crohn's disease. Gastroenterol Jpn. 1989;24:246–254. doi: 10.1007/BF02774321. [DOI] [PubMed] [Google Scholar]
- 33.Bühner S, Nagel E, Körber J, Vogelsang H, Linn T, Pichlmayr R. Ileal and colonic fatty acid profiles in patients with active Crohn's disease. Gut. 1994;35:1424–1428. doi: 10.1136/gut.35.10.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avalos-Díaz E, Guzmán-Enríquez L, López-Robles E, Herrera-Esparza R. Inhibitory effect on endocytosis in polymorphonuclear cells caused by thalidomide. Arch Invest Med (Mex) 1985;16:139–143. [PubMed] [Google Scholar]
- 35.Plamondon S, Ng SC, Kamm MA. Thalidomide in luminal and fistulizing Crohn's disease resistant to standard therapies. Aliment Pharmacol Ther. 2007;25:557–567. doi: 10.1111/j.1365-2036.2006.03239.x. [DOI] [PubMed] [Google Scholar]
- 36.Kabuki T, Ogimi C, Tanaka R, Ikematsu K, Joh K, Kagimoto S, Oh-Ishi T. [Thalidomide therapy for infantile-onset Crohn's disease] Nihon Rinsho Meneki Gakkai Kaishi. 2005;28:92–98. doi: 10.2177/jsci.28.92. [DOI] [PubMed] [Google Scholar]
- 37.Kolivras A, De Maubeuge J, André J, Song M. Thalidomide in refractory vulvar ulcerations associated with Crohn's disease. Dermatology. 2003;206:381–383. doi: 10.1159/000069963. [DOI] [PubMed] [Google Scholar]
- 38.Hegarty A, Hodgson T, Porter S. Thalidomide for the treatment of recalcitrant oral Crohn's disease and orofacial granulomatosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;95:576–585. doi: 10.1067/moe.2002.113. [DOI] [PubMed] [Google Scholar]
- 39.Sabate JM, Villarejo J, Lemann M, Bonnet J, Allez M, Modigliani R. An open-label study of thalidomide for maintenance therapy in responders to infliximab in chronically active and fistulizing refractory Crohn's disease. Aliment Pharmacol Ther. 2002;16:1117–1124. doi: 10.1046/j.1365-2036.2002.01273.x. [DOI] [PubMed] [Google Scholar]
- 40.Fishman SJ, Feins NR, D' Amato RJ, Folkman J. Long-term remission of Crohn's disease treated with thalidomide: a seminal case report. Angiogenesis. 1999;3:201–204. doi: 10.1023/a:1009027315912. [DOI] [PubMed] [Google Scholar]
- 41.Ehrenpreis ED, Kane SV, Cohen LB, Cohen RD, Hanauer SB. Thalidomide therapy for patients with refractory Crohn's disease: an open-label trial. Gastroenterology. 1999;117:1271–1277. doi: 10.1016/s0016-5085(99)70276-3. [DOI] [PubMed] [Google Scholar]
- 42.Vasiliauskas EA, Kam LY, Abreu-Martin MT, Hassard PV, Papadakis KA, Yang H, Zeldis JB, Targan SR. An open-label pilot study of low-dose thalidomide in chronically active, steroid-dependent Crohn's disease. Gastroenterology. 1999;117:1278–1287. doi: 10.1016/s0016-5085(99)70277-5. [DOI] [PubMed] [Google Scholar]
- 43.Weinstein TA, Sciubba JJ, Levine J. Thalidomide for the treatment of oral aphthous ulcers in Crohn's disease. J Pediatr Gastroenterol Nutr. 1999;28:214–216. doi: 10.1097/00005176-199902000-00025. [DOI] [PubMed] [Google Scholar]
- 44.Odeka EB, Miller V. Thalidomide in oral Crohn's disease refractory to conventional medical treatment. J Pediatr Gastroenterol Nutr. 1997;25:250–251. [PubMed] [Google Scholar]
- 45.Grip O, Janciauskiene S, Bredberg A. Use of atorvastatin as an anti-inflammatory treatment in Crohn's disease. Br J Pharmacol. 2008;155:1085–1092. doi: 10.1038/bjp.2008.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grip O, Janciauskiene S. Atorvastatin reduces plasma levels of chemokine (CXCL10) in patients with Crohn's disease. PLoS One. 2009;4:e5263. doi: 10.1371/journal.pone.0005263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saverymuttu S, Hodgson HJ, Chadwick VS. Controlled trial comparing prednisolone with an elemental diet plus non-absorbable antibiotics in active Crohn's disease. Gut. 1985;26:994–998. doi: 10.1136/gut.26.10.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fa N, Ronkart S, Schanck A, Deleu M, Gaigneaux A, Goormaghtigh E, Mingeot-Leclercq MP. Effect of the antibiotic azithromycin on thermotropic behavior of DOPC or DPPC bilayers. Chem Phys Lipids. 2006;144:108–116. doi: 10.1016/j.chemphyslip.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 49.Tyteca D, Schanck A, Dufrêne YF, Deleu M, Courtoy PJ, Tulkens PM, Mingeot-Leclercq MP. The macrolide antibiotic azithromycin interacts with lipids and affects membrane organization and fluidity: studies on Langmuir-Blodgett monolayers, liposomes and J774 macrophages. J Membr Biol. 2003;192:203–215. doi: 10.1007/s00232-002-1076-7. [DOI] [PubMed] [Google Scholar]
- 50.Gui GP, Thomas PR, Tizard ML, Lake J, Sanderson JD, Hermon-Taylor J. Two-year-outcomes analysis of Crohn's disease treated with rifabutin and macrolide antibiotics. J Antimicrob Chemother. 1997;39:393–400. doi: 10.1093/jac/39.3.393. [DOI] [PubMed] [Google Scholar]
- 51.Mendoza JL, Lana R, Díaz-Rubio M. Mycobacterium avium subspecies paratuberculosis and its relationship with Crohn's disease. World J Gastroenterol. 2009;15:417–422. doi: 10.3748/wjg.15.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenstein RJ. Is Crohn's disease caused by a mycobacterium? Comparisons with leprosy, tuberculosis, and Johne's disease. Lancet Infect Dis. 2003;3:507–514. doi: 10.1016/s1473-3099(03)00724-2. [DOI] [PubMed] [Google Scholar]
- 53.Fell JM, Paintin M, Arnaud-Battandier F, Beattie RM, Hollis A, Kitching P, Donnet-Hughes A, MacDonald TT, Walker-Smith JA. Mucosal healing and a fall in mucosal pro-inflammatory cytokine mRNA induced by a specific oral polymeric diet in paediatric Crohn's disease. Aliment Pharmacol Ther. 2000;14:281–289. doi: 10.1046/j.1365-2036.2000.00707.x. [DOI] [PubMed] [Google Scholar]
- 54.Sanderson IR, Boulton P, Menzies I, Walker-Smith JA. Improvement of abnormal lactulose/rhamnose permeability in active Crohn's disease of the small bowel by an elemental diet. Gut. 1987;28:1073–1076. doi: 10.1136/gut.28.9.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamamoto T, Nakahigashi M, Umegae S, Kitagawa T, Matsumoto K. Acute duodenal Crohn's disease successfully managed with low-speed elemental diet infusion via nasogastric tube: a case report. World J Gastroenterol. 2006;12:649–651. doi: 10.3748/wjg.v12.i4.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto T, Nakahigashi M, Umegae S, Kitagawa T, Matsumoto K. Impact of elemental diet on mucosal inflammation in patients with active Crohn's disease: cytokine production and endoscopic and histological findings. Inflamm Bowel Dis. 2005;11:580–588. doi: 10.1097/01.mib.0000161307.58327.96. [DOI] [PubMed] [Google Scholar]
- 57.Pácha J. Development of intestinal transport function in mammals. Physiol Rev. 2000;80:1633–1667. doi: 10.1152/physrev.2000.80.4.1633. [DOI] [PubMed] [Google Scholar]
- 58.Sanderson I, Walker W. Development of the gastrointestinal tract. Hamilton Ontario: B.C. Decker Inc; 1999. pp. 123–146, 245-260. [Google Scholar]
- 59.Chiodini RJ, Van Kruiningen HJ, Merkal RS, Thayer WR Jr, Coutu JA. Characteristics of an unclassified Mycobacterium species isolated from patients with Crohn's disease. J Clin Microbiol. 1984;20:966–971. doi: 10.1128/jcm.20.5.966-971.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hermon-Taylor J, El-Zaatari FAK. The mycobacterium avium subspecies paratuberculosis problem and its relation to the causation of Crohn disease. In: Bartram J, Cotruvo J, Dufour A, Rees G, Pedley S, et al., editors. Pathogenic mycobacterium in water: a guide to public health consequences, monitoring and management. London: IWA Publishing; 2004. pp. 77–94. [Google Scholar]
- 61.Karp SM, Koch TR, Pang G. Is there a MAP ( Mycobacterium Avium Subspecies Paratuberculosis) for treating Crohn's disease? Practical Gastroenterol. 2007;31:40–50. [Google Scholar]
- 62.Eltholth MM, Marsh VR, Van Winden S, Guitian FJ. Contamination of food products with Mycobacterium avium paratuberculosis: a systematic review. J Appl Microbiol. 2009;107:1061–1071. doi: 10.1111/j.1365-2672.2009.04286.x. [DOI] [PubMed] [Google Scholar]
- 63.Pierce ES. Possible transmission of Mycobacterium avium subspecies paratuberculosis through potable water: lessons from an urban cluster of Crohn's disease. Gut Pathog. 2009;1:17. doi: 10.1186/1757-4749-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hermon-Taylor J. Mycobacterium avium subspecies paratuberculosis, Crohn's disease and the Doomsday scenario. Gut Pathog. 2009;1:15. doi: 10.1186/1757-4749-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abubakar I, Myhill DJ, Hart AR, Lake IR, Harvey I, Rhodes JM, Robinson R, Lobo AJ, Probert CS, Hunter PR. A case-control study of drinking water and dairy products in Crohn's Disease--further investigation of the possible role of Mycobacterium avium paratuberculosis. Am J Epidemiol. 2007;165:776–783. doi: 10.1093/aje/kwk067. [DOI] [PubMed] [Google Scholar]
- 66.Pott J, Basler T, Duerr CU, Rohde M, Goethe R, Hornef MW. Internalization-dependent recognition of Mycobacterium avium ssp. paratuberculosis by intestinal epithelial cells. Cell Microbiol. 2009;11:1802–1815. doi: 10.1111/j.1462-5822.2009.01372.x. [DOI] [PubMed] [Google Scholar]
- 67.Abubakar I, Myhill D, Aliyu SH, Hunter PR. Detection of Mycobacterium avium subspecies paratuberculosis from patients with Crohn's disease using nucleic acid-based techniques: a systematic review and meta-analysis. Inflamm Bowel Dis. 2008;14:401–410. doi: 10.1002/ibd.20276. [DOI] [PubMed] [Google Scholar]
- 68.Feller M, Huwiler K, Stephan R, Altpeter E, Shang A, Furrer H, Pfyffer GE, Jemmi T, Baumgartner A, Egger M. Mycobacterium avium subspecies paratuberculosis and Crohn's disease: a systematic review and meta-analysis. Lancet Infect Dis. 2007;7:607–613. doi: 10.1016/S1473-3099(07)70211-6. [DOI] [PubMed] [Google Scholar]
- 69.Bull TJ, McMinn EJ, Sidi-Boumedine K, Skull A, Durkin D, Neild P, Rhodes G, Pickup R, Hermon-Taylor J. Detection and verification of Mycobacterium avium subsp. paratuberculosis in fresh ileocolonic mucosal biopsy specimens from individuals with and without Crohn's disease. J Clin Microbiol. 2003;41:2915–2923. doi: 10.1128/JCM.41.7.2915-2923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Golan L, Livneh-Kol A, Gonen E, Yagel S, Rosenshine I, Shpigel NY. Mycobacterium avium paratuberculosis invades human small-intestinal goblet cells and elicits inflammation. J Infect Dis. 2009;199:350–354. doi: 10.1086/596033. [DOI] [PubMed] [Google Scholar]
- 71.Sanders DS. Mucosal integrity and barrier function in the pathogenesis of early lesions in Crohn's disease. J Clin Pathol. 2005;58:568–572. doi: 10.1136/jcp.2004.021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rehberger A, Püspök A, Stallmeister T, Jurecka W, Wolf K. Crohn's disease masquerading as aphthous ulcers. Eur J Dermatol. 1998;8:274–276. [PubMed] [Google Scholar]
- 73.Nagel E, Bartels M, Pichlmayr R. Scanning electron-microscopic lesions in Crohn's disease: relevance for the interpretation of postoperative recurrence. Gastroenterology. 1995;108:376–382. doi: 10.1016/0016-5085(95)90063-2. [DOI] [PubMed] [Google Scholar]
- 74.Schattenfroh S, Bartels M, Nagel E. Early morphological changes in Crohn's disease. Transmission electron-microscopic findings and their interpretation: an overview. Acta Anat (Basel) 1994;149:237–246. [PubMed] [Google Scholar]
- 75.Sankey EA, Dhillon AP, Anthony A, Wakefield AJ, Sim R, More L, Hudson M, Sawyerr AM, Pounder RE. Early mucosal changes in Crohn's disease. Gut. 1993;34:375–381. doi: 10.1136/gut.34.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dvorak AM, Dickersin GR. Crohn's disease: transmission electron microscopic studies. I. Barrier function. Possible changes related to alterations of cell coat, mucous coat, epithelial cells, and Paneth cells. Hum Pathol. 1980;11:561–571. [PubMed] [Google Scholar]
- 77.Bolster CH, Cook KL, Haznedaroglu BZ, Walker SL. The transport of Mycobacterium avium subsp. paratuberculosis through saturated aquifer materials. Lett Appl Microbiol. 2009;48:307–312. doi: 10.1111/j.1472-765X.2008.02519.x. [DOI] [PubMed] [Google Scholar]
- 78.Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 79.Cook KL, Britt JS, Bolster CH. Survival of Mycobacterium avium subsp. paratuberculosis in biofilms on livestock watering trough materials. Vet Microbiol. 2010;141:103–109. doi: 10.1016/j.vetmic.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 80.Biet F, Bay S, Thibault VC, Euphrasie D, Grayon M, Ganneau C, Lanotte P, Daffé M, Gokhale R, Etienne G, et al. Lipopentapeptide induces a strong host humoral response and distinguishes Mycobacterium avium subsp. paratuberculosis from M. avium subsp. avium. Vaccine. 2008;26:257–268. doi: 10.1016/j.vaccine.2007.10.059. [DOI] [PubMed] [Google Scholar]
- 81.Etienne G, Villeneuve C, Billman-Jacobe H, Astarie-Dequeker C, Dupont MA, Daffé M. The impact of the absence of glycopeptidolipids on the ultrastructure, cell surface and cell wall properties, and phagocytosis of Mycobacterium smegmatis. Microbiology. 2002;148:3089–3100. doi: 10.1099/00221287-148-10-3089. [DOI] [PubMed] [Google Scholar]
- 82.Booth FW, Shanely RA. The biochemical basis of the health effects of exercise: an integrative view. Proc Nutr Soc. 2004;63:199–203. doi: 10.1079/pns2004337. [DOI] [PubMed] [Google Scholar]
- 83.Tiret L. Gene-environment interaction: a central concept in multifactorial diseases. Proc Nutr Soc. 2002;61:457–463. doi: 10.1079/pns2002178. [DOI] [PubMed] [Google Scholar]
- 84.Beaudet AL, Scriver CR, Sly WS, Valle D. Genetics, biochemistry, and molecular basis of variant human phenotypes. In: Scriver CR, Beaudet AL, Sly WS, Valle D, et al., editors. The metabolic and molecular bases of inherited disease. 7th ed. New York, NY: McGraw-Hill; 1995. pp. 53–118. [Google Scholar]
- 85.Chan SS. Aspirin in the aetiology of Crohn's disease and ulcerative colitis - results from a european prospective cohort study. Gastroenterology. 2010;138:S17. doi: 10.1111/j.1365-2036.2011.04784.x. [DOI] [PubMed] [Google Scholar]
- 86.Kaur G, Kaur J, Mittal N, Nath Sanyal S. The effect of prostaglandin synthase inhibitor, aspirin on the rat intestinal membrane structure and function. Nutr Hosp. 2010;25:290–298. [PubMed] [Google Scholar]
- 87.Modi DN, Merchant MA. In vitro effects of aspirin and salicylate on erythrocytes: size and Na+/ K+ ATPase activity. Indian J Pharmacol. 2003;35:27–31. [Google Scholar]
- 88.Gamerdinger M, Clement AB, Behl C. Effects of sulindac sulfide on the membrane architecture and the activity of gamma-secretase. Neuropharmacology. 2008;54:998–1005. doi: 10.1016/j.neuropharm.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 89.Watała C, Gwoździński K. Effect of aspirin on conformation and dynamics of membrane proteins in platelets and erythrocytes. Biochem Pharmacol. 1993;45:1343–1349. doi: 10.1016/0006-2952(93)90288-8. [DOI] [PubMed] [Google Scholar]
- 90.Stolte M, Hartmann FO. Misinterpretation of NSAID-induced Colopathy as Crohn's disease. Z Gastroenterol. 2010;48:472–475. doi: 10.1055/s-0028-1109760. [DOI] [PubMed] [Google Scholar]
- 91.Bjarnason I, Peters TJ. Intestinal permeability, non-steroidal anti-inflammatory drug enteropathy and inflammatory bowel disease: an overview. Gut. 1989;30 Spec No:22–28. doi: 10.1136/gut.30.spec_no.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chakraborty H, Mondal S, Sarkar M. Membrane fusion: a new function of non steroidal anti-inflammatory drugs. Biophys Chem. 2008;137:28–34. doi: 10.1016/j.bpc.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 93.Cosnes J, Carbonnel F, Carrat F, Beaugerie L, Cattan S, Gendre J. Effects of current and former cigarette smoking on the clinical course of Crohn's disease. Aliment Pharmacol Ther. 1999;13:1403–1411. doi: 10.1046/j.1365-2036.1999.00630.x. [DOI] [PubMed] [Google Scholar]
- 94.Seksik P, Nion-Larmurier I, Sokol H, Beaugerie L, Cosnes J. Effects of light smoking consumption on the clinical course of Crohn's disease. Inflamm Bowel Dis. 2009;15:734–741. doi: 10.1002/ibd.20828. [DOI] [PubMed] [Google Scholar]
- 95.Reddy RP, Tremaine WJ. Is there a threshold for the deleterious effect of smoking in Crohn's disease? Inflamm Bowel Dis. 2008;14 Suppl 2:S16–S17. doi: 10.1002/ibd.20692. [DOI] [PubMed] [Google Scholar]
- 96.Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- 97.Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van der Toorn M, Rezayat D, Kauffman HF, Bakker SJL, Gans ROB, Koëter GH, Choi AMK, van Oosterhout AJM, Slebos DJ. Mitochondria are essential in cigarette smoke induced ROS generation. In: van der Toorn M, et al., editors. Cigarette smoke-induced mitochondrial dysfunction and oxidative stress in epithelial cells. Groningen, Netherlands: The University of Groningen; 2009. pp. 39–52. [Google Scholar]
- 99.van der Toorn M, Slebos DJ, de Bruin HG, Leuvenink HG, Bakker SJ, Gans RO, Koëter GH, van Oosterhout AJ, Kauffman HF. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1211–L1218. doi: 10.1152/ajplung.00291.2006. [DOI] [PubMed] [Google Scholar]
- 100.Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, Karlsson JM, Watkins SC, Kim HP, Wang X, et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol. 2007;36:409–417. doi: 10.1165/rcmb.2006-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Pravda J. Radical induction theory of ulcerative colitis. World J Gastroenterol. 2005;11:2371–2384. doi: 10.3748/wjg.v11.i16.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bouhours-Nouet N, May-Panloup P, Coutant R, de Casson FB, Descamps P, Douay O, Reynier P, Ritz P, Malthièry Y, Simard G. Maternal smoking is associated with mitochondrial DNA depletion and respiratory chain complex III deficiency in placenta. Am J Physiol Endocrinol Metab. 2005;288:E171–E177. doi: 10.1152/ajpendo.00260.2003. [DOI] [PubMed] [Google Scholar]
- 103.Miró O, Alonso JR, Jarreta D, Casademont J, Urbano-Márquez A, Cardellach F. Smoking disturbs mitochondrial respiratory chain function and enhances lipid peroxidation on human circulating lymphocytes. Carcinogenesis. 1999;20:1331–1336. doi: 10.1093/carcin/20.7.1331. [DOI] [PubMed] [Google Scholar]
- 104.Noronha-Dutra AA, Epperlein MM, Woolf N. Effect of cigarette smoking on cultured human endothelial cells. Cardiovasc Res. 1993;27:774–778. doi: 10.1093/cvr/27.5.774. [DOI] [PubMed] [Google Scholar]
- 105.Pryor WA, Arbour NC, Upham B, Church DF. The inhibitory effect of extracts of cigarette tar on electron transport of mitochondria and submitochondrial particles. Free Radic Biol Med. 1992;12:365–372. doi: 10.1016/0891-5849(92)90085-u. [DOI] [PubMed] [Google Scholar]
- 106.Cardellach F, Alonso JR, López S, Casademont J, Miró O. Effect of smoking cessation on mitochondrial respiratory chain function. J Toxicol Clin Toxicol. 2003;41:223–228. doi: 10.1081/clt-120021102. [DOI] [PubMed] [Google Scholar]
- 107.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Available from: http://www.ncbi.nlm.nih.gov/omim/266600.
- 109. Available from: http://www.genenames.org/data/hgnc_data.php?hgnc_id=5334.
- 110.Soupene E, Dinh NP, Siliakus M, Kuypers FA. Activity of the acyl-CoA synthetase ACSL6 isoforms: role of the fatty acid Gate-domains. BMC Biochem. 2010;11:18. doi: 10.1186/1471-2091-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Soupene E, Kuypers FA. Mammalian long-chain acyl-CoA synthetases. Exp Biol Med (Maywood) 2008;233:507–521. doi: 10.3181/0710-MR-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soupene E, Kuypers FA. Multiple erythroid isoforms of human long-chain acyl-CoA synthetases are produced by switch of the fatty acid gate domains. BMC Mol Biol. 2006;7:21. doi: 10.1186/1471-2199-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marszalek JR, Kitidis C, Dirusso CC, Lodish HF. Long-chain acyl-CoA synthetase 6 preferentially promotes DHA metabolism. J Biol Chem. 2005;280:10817–10826. doi: 10.1074/jbc.M411750200. [DOI] [PubMed] [Google Scholar]
- 114.Mannaerts GP, van Veldhoven PP. Functions and organization of peroxisomal beta-oxidation. Ann N Y Acad Sci. 1996;804:99–115. doi: 10.1111/j.1749-6632.1996.tb18611.x. [DOI] [PubMed] [Google Scholar]
- 115.Hashimoto T. Peroxisomal beta-oxidation: enzymology and molecular biology. Ann N Y Acad Sci. 1996;804:86–98. doi: 10.1111/j.1749-6632.1996.tb18610.x. [DOI] [PubMed] [Google Scholar]
- 116. Available from: http://www.ncbi.nlm.nih.gov/gene/23305.
- 117.Aimone-Gastin I, Cable S, Keller JM, Bigard MA, Champigneulle B, Gaucher P, Gueant JL, Dauça M. Studies on peroxisomes of colonic mucosa in Crohn's disease. Dig Dis Sci. 1994;39:2177–2185. doi: 10.1007/BF02090368. [DOI] [PubMed] [Google Scholar]
- 118.Uniprot Protein Knowledge database. Protein ID Q7L5N7 (Lpcat2-Human) Available from: http://www.uniprot.org/uniprot/Q7L5N7.
- 119.Shindou H, Hishikawa D, Nakanishi H, Harayama T, Ishii S, Taguchi R, Shimizu T. A single enzyme catalyzes both platelet-activating factor production and membrane biogenesis of inflammatory cells. Cloning and characterization of acetyl-CoA:LYSO-PAF acetyltransferase. J Biol Chem. 2007;282:6532–6539. doi: 10.1074/jbc.M609641200. [DOI] [PubMed] [Google Scholar]
- 120.Juste RA, Elguezabal N, Garrido JM, Pavon A, Geijo MV, Sevilla I, Cabriada JL, Tejada A, García-Campos F, Casado R, et al. On the prevalence of M. avium subspecies paratuberculosis DNA in the blood of healthy individuals and patients with inflammatory bowel disease. PLoS One. 2008;3:e2537. doi: 10.1371/journal.pone.0002537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Naser SA, Ghobrial G, Romero C, Valentine JF. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn's disease. Lancet. 2004;364:1039–1044. doi: 10.1016/S0140-6736(04)17058-X. [DOI] [PubMed] [Google Scholar]
- 122.Rutgeerts P, Vermeire S, Van Assche G. Mucosal healing in inflammatory bowel disease: impossible ideal or therapeutic target? Gut. 2007;56:453–455. doi: 10.1136/gut.2005.088732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shen L, Su L, Turner JR. Mechanisms and functional implications of intestinal barrier defects. Dig Dis. 2009;27:443–449. doi: 10.1159/000233282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kirkwood CD, Wagner J, Boniface K, Vaughan J, Michalski WP, Catto-Smith AG, Cameron DJ, Bishop RF. Mycobacterium avium subspecies paratuberculosis in children with early-onset Crohn's disease. Inflamm Bowel Dis. 2009;15:1643–1655. doi: 10.1002/ibd.20967. [DOI] [PubMed] [Google Scholar]
- 125.Irvine EJ, Marshall JK. Increased intestinal permeability precedes the onset of Crohn's disease in a subject with familial risk. Gastroenterology. 2000;119:1740–1744. doi: 10.1053/gast.2000.20231. [DOI] [PubMed] [Google Scholar]
- 126.Bjarnason I, MacPherson A, Hollander D. Intestinal permeability: an overview. Gastroenterology. 1995;108:1566–1581. doi: 10.1016/0016-5085(95)90708-4. [DOI] [PubMed] [Google Scholar]
- 127.Bjarnason I. Intestinal permeability. Gut. 1994;35:S18–S22. doi: 10.1136/gut.35.1_suppl.s18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med. 1986;105:883–885. doi: 10.7326/0003-4819-105-6-883. [DOI] [PubMed] [Google Scholar]
- 129.Pimentel M, Chang M, Chow EJ, Tabibzadeh S, Kirit-Kiriak V, Targan SR, Lin HC. Identification of a prodromal period in Crohn's disease but not ulcerative colitis. Am J Gastroenterol. 2000;95:3458–3462. doi: 10.1111/j.1572-0241.2000.03361.x. [DOI] [PubMed] [Google Scholar]
- 130.Suenaert P, Bulteel V, Vermeire S, Noman M, Van Assche G, Rutgeerts P. Hyperresponsiveness of the mucosal barrier in Crohn's disease is not tumor necrosis factor-dependent. Inflamm Bowel Dis. 2005;11:667–673. doi: 10.1097/01.mib.0000168371.87283.4b. [DOI] [PubMed] [Google Scholar]
- 131.Söderholm JD, Peterson KH, Olaison G, Franzén LE, Weström B, Magnusson KE, Sjödahl R. Epithelial permeability to proteins in the noninflamed ileum of Crohn's disease? Gastroenterology. 1999;117:65–72. doi: 10.1016/s0016-5085(99)70551-2. [DOI] [PubMed] [Google Scholar]