

Abstract
AIM: To analyze the antiviral mechanism of Epigallocatechin gallate (EGCG) against hepatitis B virus (HBV) replication.
METHODS: In this research, the HBV-replicating cell line HepG2.117 was used to investigate the antiviral mechanism of EGCG. Cytotoxicity of EGCG was analyzed by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Hepatitis B virus e antigen (HBeAg) and hepatitis B virus surface antigen (HBsAg) in the supernatant were detected by enzyme-linked immunosorbent assay. Precore mRNA and pregenomic RNA (pgRNA) levels were determined by semi-quantitative reverse transcription polymerase chain reaction (PCR) assay. The effect of EGCG on HBV core promoter activity was measured by dual luciferase reporter assay. HBV covalently closed circular DNA and replicative intermediates of DNA were quantified by real-time PCR assay.
RESULTS: When HepG2.117 cells were grown in the presence of EGCG, the expression of HBeAg was suppressed, however, the expression of HBsAg was not affected. HBV precore mRNA level was also down-regulated by EGCG, while the transcription of precore mRNA was not impaired. The synthesis of both HBV covalently closed circular DNA and replicative intermediates of DNA were reduced by EGCG treatment to a similar extent, however, HBV pgRNA transcripted from chromosome-integrated HBV genome was not affected by EGCG treatment, indicating that EGCG targets only replicative intermediates of DNA synthesis.
CONCLUSION: In HepG2.117 cells, EGCG inhibits HBV replication by impairing HBV replicative intermediates of DNA synthesis and such inhibition results in reduced production of HBV covalently closed circular DNA.
Keywords: Covalently closed circular DNA, Epigallocatechin gallate, Hepatitis B virus e antigen, Hepatitis B virus, Precore mRNA, Replicative intermediates of DNA
INTRODUCTION
Hepatitis B virus (HBV) infection remains a major world health problem despite the availability of an effective vaccine. Approximately 350 million people are chronically infected with HBV worldwide[1,2]. HBV possesses a 3.2 kb relaxed circular partially double-stranded DNA (RC-DNA) genome[3]. After infection, this viral genome is released into cytoplasm and converted into covalently closed circular DNA (cccDNA) in the host cell nucleus. The cccDNA is the template for production of all viral RNAs. Among HBV RNAs, the pregenomic RNA (pgRNA) is packaged into new capsid and serves as the template for HBV DNA synthesis. The nucleocapsid acquires an envelope and then buds as a mature virion. Progeny nucleocapsids may redeliver their RC-DNA genomes to the nucleus for accumulation of a cccDNA pool[4].
Currently, there is no reliable treatment that completely eliminates HBV infection in all chronic carriers. Several nucleos(t)ide analogues (lamivudine, adefovir, entecavir, and telbivudine) and interferon-α (IFN-α) have been approved as antiviral drugs against HBV infection, however, they have their own limitations, e.g. drug resistance and side effects[5]. Several non-nucleoside natural products have also been reported to inhibit HBV replication using various mechanisms. For example, oxymatrine was shown to block HBV DNA synthesis and cccDNA formation[6]. A helioxanthin derivative, 8-1, was demonstrated to diminish HBV promoter activities and thus suppress HBV gene expression and DNA synthesis[7,8].
Epigallocatechin gallate (EGCG) is one of the most abundant polyphenols present in green tea[9]. EGCG is well known for its anti-tumor activity while other pharmaceutical effects were discovered subsequently[10-14]. Recently, it has also been reported that EGCG could inhibit HBV replication in a HBV stably replicating cell line - HepG2-N10[15]. However, the mechanism by which EGCG inhibits HBV replication is unclear. In fact, it is difficult to dissect the detailed anti-HBV mechanism of EGCG using HepG2-N10 cells, because the processes from cccDNA to antigen expression are severely affected by transcription from integrated HBV DNA[16].
In order to investigate the antiviral mechanism of EGCG against HBV in detail, we adopted a newly reported HepG2.117 cell line[17]. HBV replication is controllable using doxycycline in this cell line. More importantly, due to the delicate design, HBV precore mRNA can only be transcripted from replicated HBV DNA but not the integrated HBV DNA. As a result, hepatitis B virus e antigen (HBeAg) can only be translated from precore mRNA that is solely produced from replicated HBV DNA. Therefore, the precore mRNA and HBeAg level represents HBV DNA synthesis. On the other hand, PreS1/S2 mRNAs, which are templates for the translation of hepatitis B virus surface antigen (HBsAg), are mainly transcribed from integrated HBV DNA rather than replicated HBV DNA in this cell line, thus, HBsAg has little correlation to HBV DNA synthesis level.
In this study, we investigated the inhibition of HBV replication by EGCG in HepG2.117 cells. Our results showed that HBV replicative intermediates of DNA (RI-DNA) synthesis was significantly inhibited by EGCG, which resulted in less cccDNA production. In contrast, the production of HBV pgRNA, precore mRNA and the translation of HBeAg were not affected by EGCG.
MATERIALS AND METHODS
Cell line and cell culture
HepG2.117 (a gift from Dr. Dian-Xing Sun, University Hospital Freiburg, Germany) is an inducible HBV-replicating cell line[17]. It contains a slight over-length HBV genome under the control of a tetracycline responsive CMV promoter. Transcription of HBV pgRNA is induced upon doxycycline removal from the culture medium, leading to capsid assembly and DNA synthesis. A small portion of the de novo synthesized RC-DNA is transported into the nucleus and converted to cccDNA which serves as a transcriptional template for the production of viral RNAs.
The HBeAg start codon and precore mRNA transcription initiation point are inexistent in chromosome integrated HBV genome in HepG2.117 cells[17]. Therefore, HBeAg and precore mRNA can not be generated from the integrated genome. However, once HBV starts to replicate, a functional precore mRNA would be produced from cccDNA in this cell line, since the promoter and transcription initiation point would be restored in the process of cccDNA formation[17]. Subsequently, HBeAg would be produced from this precore mRNA where HBeAg start codon is also restored.
HepG2.117 cells were cultured in DMEM medium (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin and 100 μg/mL streptomycin (Sigma). When needed, doxycycline (Sigma) was routinely added at 1.5 μg/mL to suppress HBV pgRNA transcription.
Plasmid construction
To generate pHBVCP-Luc reporter plasmid, a 442 bp fragment corresponding to the HBV core promoter region containing enhancer II was amplified from pCH9-3091 (an ayw subtype HBV over-length genomic plasmid, kindly provided by Professor Michael Nassal, University Hospital, Freiburg)[18] by polymerase chain reaction (PCR) (forward primer: GGGGTACCCGCGGGACGTCCTTTG; reverse primer: CCAAGCTTACAAGAGATGATTAGGCAG). This fragment was then inserted into the pGL3-basic vector (Promega, Madison, WI, USA), producing pHBVCP-Luc reporter construct.
Drug treatment and cytotoxicity assay
Three days after the removal of doxycycline, HepG2.117 cells were seeded into 24-well plates. Twenty-four hours later, cells were treated with fresh DMEM medium containing various concentrations of EGCG (Hubei Institute of Chemistry, Wuhan), or lamivudine (NIH AIDS Research and Reference Reagent Program, Rockville, MD, USA), respectively. The drug-containing media were replaced each day for 3 d. The medium was then collected for antigen detection and attached cells were used for DNA and RNA analysis (see below) or EGCG toxicity analysis using the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells cultured with DMEM medium without EGCG were used as a negative control.
Enzyme-linked immunosorbent assay detection of HBsAg and HBeAg
Collected media were subjected to 10 000 × g centrifugation to remove cellular debris. Secreted HBsAg and HBeAg were quantified by enzyme-linked immunosorbent assay (ELISA) kits (Kehua, Shanghai) following the manufacturer’s instructions. All the data on HBsAg or HBeAg are presented as percentage of antigen level detected in the negative control samples.
Semi-quantitative reverse transcription polymerase chain reaction detection of HBV mRNA
Total cellular RNA was extracted with Trizol reagents (Invitrogen) and treated with RQ1 RNase-free DNase I (Promega) to digest residual DNA molecules. For the semi-quantification of precore mRNA and pgRNA, 1 μg total cellular RNA was subjected to cDNA synthesis using MMLV reverse transcriptase (Promega) and random hexamer as a primer based on the manufacturer’s instructions. The cDNAs were then measured by semi-quantitative PCR using Ex Taq (Takara, Dalian) and subsequently analyzed by agarose gel electrophoresis. The specific primers for semi-quantitative detection of precore mRNA were as follows: forward, 5'-TCTGCGCACCAGCACCATG; reverse, 5'-TGCCTCGTCGTCTAACAA. The forward primer for pgRNA amplification was 5'-TCGGGAAGCCTTAGAGTC, while the reverse primer was 5'-TGCCTCGTCGTCTAACAA. Primers used for Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA amplification were as previously reported[19].
Dual luciferase reporter assay detection of HBV core promoter activity
Three days after the removal of doxycycline, HepG2.117 cells were seeded into 24-well plates. Twenty-four hours later, the cells were transiently co-transfected with pHBVCP-Luc reporter plasmid and normalizing plasmid pRL-TK with FuGENE-HD reagent according to the manufacturer’s instructions (Roche Applied Science, Manheim, Germany). Twelve hours after transfection, cells were subjected to a 3-d period of treatment with EGCG. HBV core promoter activity was determined by measuring luciferase activity using the Dual Luciferase Reporter Assay System (Promega).
Real-time PCR detection of HBV DNA
Extraction of cccDNA was carried out using a modified Hirt extraction procedure[16,20]. Briefly, cells from one well in the 24-well plate were lysed in 300 μL of 10 mmol/L Tris-HCl, 10 mmol/L EDTA and 0.7% SDS. After 30 min incubation at room temperature, the lysate was mixed with 80 μL 5 mol/L NaCl and incubated at 4°C overnight. The lysate was clarified by centrifugation at 12 000 × g for 30 min at 4°C, and then extracted twice with phenol and once with phenol:chloroform. DNAs were precipitated with ethanol overnight at -20°C and dissolved in TE buffer, followed by further treatment of plasmid-safe DNase (Epicenter) to digest residual linear and open circular DNA molecules. When the reaction was finished, the plasmid-safe DNase was removed by phenol:chloroform extraction and the DNA was precipitated with ethanol at -20°C overnight and dissolved in TE buffer. Total cellular HBV DNA was extracted from the cell monolayer with the total DNA extraction kit (Takara, Dalian) according to the manufacturer’s instructions.
A pair of primers (corresponding to HBV S ORF) introduced by Liu et al[21] was applied to quantify HBV DNA copies. In order to quantify cccDNA, extracted cccDNA samples were subjected to real-time PCR and the quantification was normalized by cell numbers. The total HBV DNA samples, which contained several types of HBV DNA forms (99% of them were HBV DNA replicative intermediates), were analyzed by real-time PCR and the quantification was normalized to the GAPDH DNA copies.
RESULTS
HBeAg expression level in HepG2.117 cells was down-regulated by EGCG
In order to choose an appropriate initial concentration of EGCG in the HBV inhibition assay, the MTT assay was used to determine the cytotoxicity of EGCG to HepG2.117 cells. As shown in Figure 1, the cytotoxic effects of EGCG were undetectable when the EGCG concentration was below 100 μmol/L, while slight cytotoxicity (18% inhibition of cell viability) and significant cytotoxicity (60% inhibition of cell viability) were observed when 200 and 400 μmol/L of EGCG were used, respectively. Therefore, the maximum non-toxic concentration was 100 μmol/L, and this was used as the highest concentration of EGCG in the HBV inhibition assay.
Figure 1.

The cytotoxic effect of Epigallocatechin gallate on HepG2.117 cells. Twenty four hours after seeding, cells were incubated with medium containing different concentrations of Epigallocatechin gallate (EGCG) for 3 d, and cell viability was determined by the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Cell viability of the control well not treated with EGCG was determined and arbitrarily designated as 1. Error bars indicate standard deviation of 3 independent experiments.
EGCG was reported to inhibit the expression of both HBeAg and HBsAg in HepG2-N10 cells[15]. To investigate the antiviral mechanisms of EGCG against HBV in HepG2.117 cells, a cell-based assay was used. The levels of HBeAg and HBsAg in the supernatant of EGCG-treated HepG2.117 cells were measured by ELISA. As shown in Figure 2A, HBeAg expression was dose-dependently inhibited by EGCG with an IC50 of 39.4 μmol/L. However, the expression level of HBsAg was largely unaffected (Figure 2B). These results indicated that EGCG could down-regulate HBeAg level but not HBsAg level in HepG2.117 cells.
Figure 2.
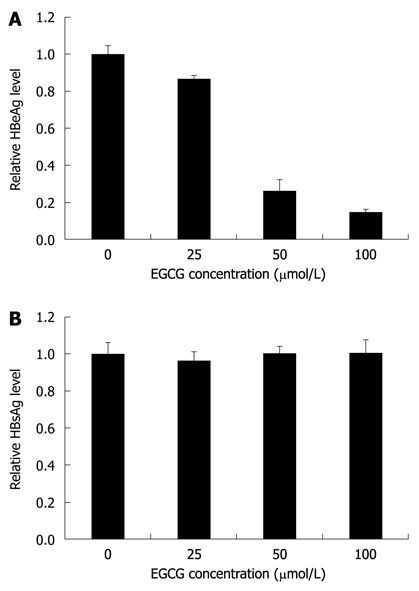
Epigallocatechin gallate inhibited hepatitis B virus e antigen expression level but not hepatitis B virus surface antigen expression level in the HepG2.117 cell line. Twenty four hours after seeding, cells were incubated with medium containing different concentrations of Epigallocatechin gallate (EGCG) for 3 d. The hepatitis B virus e antigen (HBeAg) and hepatitis B virus surface antigen (HBsAg) expression levels in the supernatants were measured using enzyme-linked immunosorbent assay kits for measuring HBeAg and HBsAg, respectively. The level of HBeAg (A) and HBsAg (B) from cells not treated with EGCG (the control) were arbitrarily designated as 1. The levels of HBeAg (A) and HBsAg (B) in EGCG-treated cells were normalized with the control and expressed as relative levels. Error bars indicate standard deviation of 3 independent experiments.
HBV precore mRNA level was down-regulated in the presence of EGCG
Unlike several HBV DNA stably transfected cell lines (e.g. HepG2.2.15) whose HBeAg is mainly translated from the long early RNA that is transcribed from the inserted exogenous promoter (e.g. CMV promoter), in the HepG2.117 cell line, HBeAg is exclusively translated from precore mRNA that is transcribed from cccDNA, akin to the actual HBV life cycle in HBV-infected hepatocytes. Since EGCG down-regulates HBeAg level but not HBsAg level in HepG2.117 cells, we determined whether the down-regulation of HBeAg by EGCG was due to inhibition of the transcription of precore mRNA.
Precore mRNA and pgRNA share the same sequence, except that precore mRNA is 35 nt longer at the 5’ terminal[16]. In order to distinguish precore mRNA from pgRNA, a specific forward primer complementary to the 5’ terminal of precore mRNA was designed for the precore mRNA specific reverse transcription PCR (RT-PCR) assay. Briefly, the 19 nt forward primer was designed to anneal to the 5’ terminal of precore RNA (nt 1800-1818) which is absent in pgRNA. The reverse primer was complementary to a sequence (nt 2345-2362) present in both precore mRNA and in pgRNA. Thus, a 563 bp fragment could be amplified from precore mRNA but not pgRNA.
After HepG2.117 cells were treated with EGCG for 3 d, total RNAs were extracted and subjected to semi-quantitative RT-PCR analysis using the precore mRNA specific primers. As shown in Figure 3, the amount of 563 bp specific RT-PCR products amplified from precore mRNA decreased in a dose-dependent manner with increasing concentrations of EGCG. The total RNA sample for RT-PCR was free of DNA contamination as this specific product did not appear in reactions without reverse transcription (Figure 3, NC lane). These results indicated that after EGCG treatment, precore mRNA level was down-regulated in HepG2.117 cells, and the lowered precore mRNA level accounted for the decreased production of HBeAg.
Figure 3.
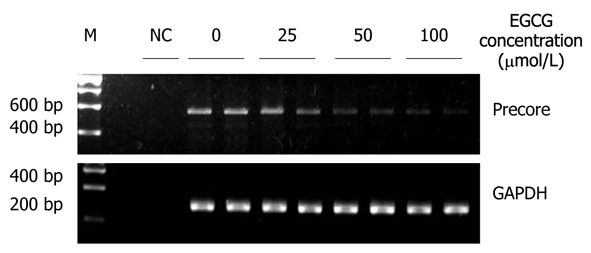
Epigallocatechin gallate inhibited the production of hepatitis B virus precore mRNA. Twenty four hours after seeding, HepG2.117 cells were treated with medium containing different concentration of Epigallocatechin gallate (EGCG) for 3 d. Total RNAs were prepared and were quantitated by semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) with a pair of precore mRNA selective primers. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA was semi-quantitatively RT-PCR amplified and used for normalization. NC: Total RNA sample was subjected to PCR without reverse transcriptase and used as the negative control.
Transcriptional efficiency of HBV precore mRNA was not affected by EGCG
Precore mRNA is transcribed from cccDNA and this process is controlled by HBV core promoter[22]. To investigate whether EGCG can inhibit HBV core promoter activity, a reporter plasmid, pHBVCP-Luc, was constructed by inserting HBV core promoter before the firefly luciferase gene in the pGL3-basic vector and thus the measured firefly luciferase activity represented core promoter activity. Twelve hours after HepG2.117 cells were transfected with pHBVCP-Luc, different concentrations of EGCG were added and maintained for 3 d, and the cells were then subjected to luciferase reporter assay. As shown in Figure 4, HBV core promoter activity was not inhibited even in the presence of 100 μmol/L of EGCG, in which the expression of HBeAg was down-regulated by 85.5%. Thus, the down-regulation of precore mRNA level was not attributed to the inhibition of core promoter activity by EGCG.
Figure 4.

Epigallocatechin gallate does not affect hepatitis B virus core promoter activity. Twelve hours after cotransfection with pHBVCP-Luc and pRL-TK, HepG2.117 cells were treated with media containing different concentrations of Epigallocatechin gallate (EGCG) for 3 d, and were lysed for the dual luciferase reporter analysis. Core promoter activity was normalized with control TK promoter activity and presented as relative luciferase units. Error bars indicate standard deviation of 3 independent experiments.
The production of HBV cccDNA and RI-DNA was dose-dependently inhibited by EGCG
Since the precore mRNA in the HepG2.117 cell line was exclusively transcribed from HBV cccDNA, and EGCG did not suppress HBV precore mRNA transcription efficiency, we speculated that the down-regulation of precore mRNA by EGCG may be due to decreased HBV cccDNA formation.
To quantitatively analyze the amount of HBV cccDNA, EGCG-treated HepG2.117 cells were collected to obtain cccDNA, and real-time PCR analysis was used to detect cccDNA production. The results showed that EGCG could decrease cccDNA formation in a dose-dependent manner (Figure 5A). The inhibitory rates of cccDNA formation were 36.3%, 63% and 72.4%, following treatment with 25, 50 and 100 μmol/L of EGCG, respectively. As a control, the HBV polymerase inhibitor, lamivudine, also showed dose-dependent inhibition of HBV cccDNA production (Figure 5B).
Figure 5.
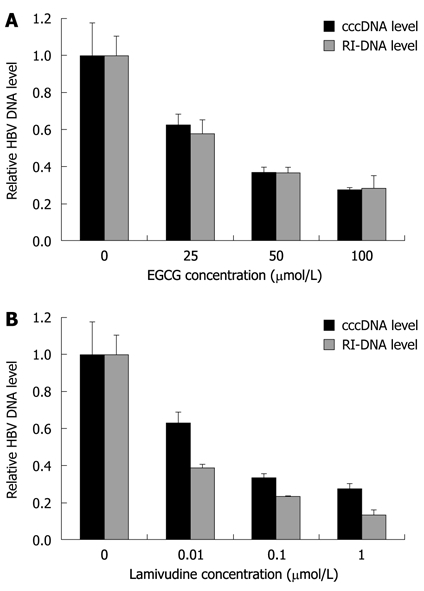
Epigallocatechin gallate down-regulates hepatitis B virus covalently closed circular DNA and replicative intermediates of DNA level. A: Epigallocatechin gallate (EGCG) treatment was the same as described in Figure 1. Hepatitis B virus (HBV) covalently closed circular DNA (cccDNA) and total cellular HBV DNA were extracted and subjected to real-time polymerase chain reaction analysis; B: Lamivudine, the potent HBV DNA synthesis inhibitor, was used as a control inhibitor in this analysis. DNA level in the no treatment control was arbitrarily designated as 1. Error bars indicate standard deviation of 3 independent experiments. RI-DNA: Replicative intermediates of DNA.
HBV cccDNA is repaired from RC-DNA. RC-DNA and other forms of HBV DNA (double-stranded linear DNA and single-stranded DNA), which are collectively known as HBV replicative intermediates of DNA (RI-DNA), represent HBV DNA synthesis efficiency. In HepG2.117 cells, RI-DNA molecules constitute 99% of HBV DNA molecules[17]. Therefore, total HBV DNA level can reflect HBV RI-DNA level as well as HBV DNA synthesis level.
We suspected that impairment of cccDNA production was a result of less RI-DNA molecules produced when the cells were treated with EGCG. To verify this, total HBV DNA was extracted from EGCG-treated HepG2.117 cells and then subjected to real-time quantitative PCR analysis. As shown in Figure 5A, EGCG inhibited the level of RI-DNA dose-dependently, and the inhibitory rates were 42.2%, 63.4% and 71.8% following treatment with 25, 50 and 100 μmol/L of EGCG, respectively. The inhibitory rates were almost the same as that for cccDNA production, suggesting that the inhibition of cccDNA formation was mainly due to the decrease in HBV RI-DNA template.
Interestingly, it was observed that when treated with the same concentration of lamivudine, the decline in RI-DNA level was slightly stronger than the decline in cccDNA level (Figure 5B), e.g. 1 μmol/L lamivudine decreased RI-DNA production by 86.8% and only decreased cccDNA production by 72.4%. The reason for this could be that part of the cccDNA can be repaired from a certain amount of pre-existing RC-DNA[23,24]. The observed minimal difference in the inhibition of cccDNA and RI-DNA production implies that EGCG may partly inhibit cccDNA formation.
Transcription of pgRNA from integrated HBV genome was not affected by EGCG
HBV RI-DNA is synthesized from pgRNA and catalyzed by HBV polymerase. In the HepG2.117 cell line, pgRNA is transcribed from chromosome-integrated HBV DNA genome. Since RI-DNA level was down-regulated by EGCG in this cell line, we determined whether this down-regulation was actually caused by inhibition of RI-DNA synthesis rather than suppression of pgRNA transcription.
To analyze whether EGCG inhibited pgRNA transcription, doxycycline was removed from the culture medium, and EGCG was added at the same time. Three days later, total RNAs were extracted from HepG2.117 cells and pgRNAs were analyzed semi-quantitatively. As shown in Figure 6, EGCG treatment did not interfere with pgRNA production even when 100 μmol/L of EGCG was applied. Therefore, the transcription of pgRNA from integrated HBV DNA genome was not affected by EGCG. Taken together, we conclude that the decrease in RI-DNA resulted from the inhibition of RI-DNA synthesis, rather than the down-regulation of pgRNA production.
Figure 6.

Epigallocatechin gallate did not inhibit hepatitis B virus pregenomic RNA transcription. When doxycycline was removed from the cell culture medium, different concentrations of Epigallocatechin gallate (EGCG) were added to the HepG2.117 cells. After 3 d of culture, total RNA were extracted from the treated cells and the pregenomic RNAs (pgRNAs) were analyzed by semi-quantitative reverse transcription polymerase chain reaction. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNAs were used for normalization. NC: Total RNA sample was subjected to polymerase chain reaction without reverse transcription.
DISCUSSION
In this study, we investigated the anti-HBV mechanism of EGCG in HepG2.117 cells, an inducible HBV-replicating cell line. Our results showed that EGCG down-regulated HBeAg level, precore mRNA level, cccDNA level and RI-DNA level to a similar extent, however, the production of HBV pgRNA was not affected. Therefore, the inhibition of HBV replication mainly occurred in HBV RI-DNA synthesis. When comparing the down-regulation of HBV cccDNA and RI-DNA production mediated by EGCG and lamivudine, we speculated that EGCG may inhibit HBV cccDNA formation to a minor extent. Taken together, suppression of HBV DNA synthesis is the major anti-HBV mechanism of EGCG, while cccDNA formation might be slightly impaired in the presence of EGCG. Lowered RI-DNA level leads to decreased cccDNA formation, which in turn leads to a decline in precore mRNA level and HBeAg level. HBV precore mRNA transcription and HBeAg translation were unaffected by EGCG.
In a previous study, the anti-HBV activity of EGCG was reported to down-regulate extracellular HBsAg and HBeAg level, intracellular cccDNA level and RI-DNA level. That research was conducted in the HepG2-N10 cell line which was constructed by stable transfection of a 1.3-fold adw subtype HBV genome into HepG2 cells. We reasoned that EGCG could inhibit HBsAg production in HepG2-N10 but not in HepG2.117 cells, which may be due to the different HBV subtype (adw vs ayw) and DNA length (1.3-fold vs slightly more than 1.0-fold). The results obtained in HepG2-N10 cells were not adequate to elucidate the antiviral mechanism of EGCG against HBV replication, because all HBV mRNAs are mainly transcribed from integrated HBV genomic DNA in HepG2-N10, and thus the antigens (representing mRNA levels) can not truly reflect the levels of HBV RI-DNA synthesis, cccDNA formation and mRNA transcription in the cells.
On the other hand, in the HepG2.117 cell line, HBsAg could be produced from both integral HBV genome and cccDNA, however, cccDNA copy numbers constitute only 1% of the integrated HBV genome copy number. Thus, nearly all of the HBsAg produced from mRNA is transcripted from integrated HBV genome rather than from HBV cccDNA. Therefore, we speculate that the inability of EGCG to inhibit HBsAg production in the HepG2.117 cell line was attributed to the unchanged transcription level of HBsAg mRNA from integrated HBV DNA, even in the presence of EGCG. However, in actual infected hepatocytes, HBsAg can only be produced from cccDNA[25,26], like HBeAg in the HepG2.117 cell line. Thus, we speculate that EGCG is able to inhibit HBsAg in HBV infection because cccDNA can be suppressed by EGCG treatment.
HBV DNA stably transfected cell lines are frequently used in cell-based assays for the discovery of antiviral drugs against HBV[27,28]. In such cell lines, all HBV replicative intermediate DNA forms, cccDNA, nucleocapsid and infectious virion (Dane particle) can be detected, hence, it is generally accepted that the HBV life cycle is complete. However, all HBV RNAs and antigens can be produced from both chromosome-integrated HBV genomic DNA and replicated HBV genomic DNA. More importantly, the strong HBV DNA synthesis inhibitor, lamivudine, does not affect HBV RNA levels and antigen levels in such cell models[29], indicating that HBV RNA transcription from integrated HBV DNA is much stronger than that from cccDNA. The amount of HBV RNAs and antigens in these cell lines do not reflect the actual HBV DNA replication level. In contrast, in the HepG2.117 cell line, HBeAg and precore mRNA exclusively come from HBV cccDNA, consequently, the pathway from HBV cccDNA to precore mRNA and then HBeAg can be quantitatively analyzed. Although the authenticity of the HBeAg signal in antigen detection is controversial[30], the precore mRNA is a solid and positive parameter in this cell system. In summary, the HepG2.117 cell line is a better cell model to analyze the anti-HBV mechanism of potential drugs against HBV replication.
Currently, researchers have focused mainly on the potential application of EGCG in treating cancers. EGCG is known to block each stage of carcinogenesis by modulating signal transduction pathways involved in cell proliferation, transformation, inflammation, apoptosis, metastasis and invasion[31,32]. In this study, we investigated the anti-HBV mechanism of EGCG in detail and found that the suppression of HBV RI-DNA synthesis is the major anti-HBV mechanism of EGCG, while cccDNA formation might be slightly impaired in the presence of EGCG. Although the detailed replication mechanism of HBV remains unclear, it is estimated that multiple cellular factors are involved in HBV genome replication. We assume that EGCG may interact with certain cellular proteins and interfere with the function of the DNA replication machinery. As a result, HBV RI-DNA synthesis is impaired and HBV replication is inhibited. To fully understand the antiviral mechanism of EGCG against HBV and explore the potential use of EGCG as anti-HBV drug, further study needs to be done to determine how EGCG impairs HBV RI-DNA synthesis and which cellular factors are affected.
COMMENTS
Background
Hepatitis B virus (HBV) infection remains a major world health problem despite the availability of an effective vaccine. Currently, there is no reliable treatment that completely eliminates HBV infection in all chronic carriers. Epigallocatechin gallate (EGCG) is one of the most abundant polyphenols present in green tea. Recently, it has been reported that EGCG could inhibit HBV replication in a HBV stably replicating cell line. However, the mechanism by which EGCG inhibits HBV replication is unclear.
Research frontiers
Currently, there are very few cell lines which can be used to investigate HBV replication. HepG2.117 is a newly reported cell line in which HBV replication is controllable using doxycycline. Hepatitis B virus e antigen (HBeAg) can only be translated from precore mRNA solely produced from replicated HBV DNA. HBeAg level correlates well with HBV DNA synthesis, whereas hepatitis B virus surface antigen level does not. Hence, this cell line is more convenient when investigating the antiviral mechanisms of leading compounds against HBV.
Innovations and breakthroughs
Results from this report showed that HBV replicative intermediates of DNA synthesis was significantly inhibited by EGCG, which resulted in reduced covalently closed circular DNA (cccDNA) production. In contrast, the production of HBV pregenomic RNA and the translation of HBeAg were not affected by EGCG.
Applications
The finding that EGCG can inhibit HBV relaxed circular partially double-stranded DNA synthesis is important both when exploring the potential of new drugs against HBV and in understanding which targets might be valid in eliminating viral replication.
Peer review
The authors investigated the anti-viral mechanism of epigallocatechin gallate against HBV replication. It was observed that epigallocatechin gallate impairs replicative intermediate DNA synthesis, resulting in reduced production of cccDNA of the virus. The study is well conducted and the methodology was sound.
Footnotes
Supported by National Technology and Science Key Project (2008ZX10002-010) and the Important National Science and Technology Specific Projects (2009ZX09301-014)
Peer reviewers: Dr. BS Anand, Professor, Digestive Diseases Section (111D), VA Medical Center, 2002 Holcombe Blvd., Houston, TX 77030, United States; Dr. Jin-Town Wang, Professor, Department of Microbiology and Internal Medicine, National Taiwan University, College of Medicine, 1, Sec 1, Jen-Ai Rd, Taipei 10016, Taiwan, China
S- Editor Tian L L- Editor Webster JR E- Editor Zheng XM
References
- 1.Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004;350:1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 2.Baumert TF, Thimme R, von Weizsäcker F. Pathogenesis of hepatitis B virus infection. World J Gastroenterol. 2007;13:82–90. doi: 10.3748/wjg.v13.i1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Summers J, O'Connell A, Millman I. Genome of hepatitis B virus: restriction enzyme cleavage and structure of DNA extracted from Dane particles. Proc Natl Acad Sci USA. 1975;72:4597–4601. doi: 10.1073/pnas.72.11.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck J, Nassal M. Hepatitis B virus replication. World J Gastroenterol. 2007;13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tillmann HL. Antiviral therapy and resistance with hepatitis B virus infection. World J Gastroenterol. 2007;13:125–140. doi: 10.3748/wjg.v13.i1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu WS, Zhao KK, Miao XH, Ni W, Cai X, Zhang RQ, Wang JX. Effect of oxymatrine on the replication cycle of hepatitis B virus in vitro. World J Gastroenterol. 2010;16:2028–2037. doi: 10.3748/wjg.v16.i16.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ying C, Li Y, Leung CH, Robek MD, Cheng YC. Unique antiviral mechanism discovered in anti-hepatitis B virus research with a natural product analogue. Proc Natl Acad Sci USA. 2007;104:8526–8531. doi: 10.1073/pnas.0609883104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tseng YP, Kuo YH, Hu CP, Jeng KS, Janmanchi D, Lin CH, Chou CK, Yeh SF. The role of helioxanthin in inhibiting human hepatitis B viral replication and gene expression by interfering with the host transcriptional machinery of viral promoters. Antiviral Res. 2008;77:206–214. doi: 10.1016/j.antiviral.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 9.Shankar S, Ganapathy S, Srivastava RK. Green tea polyphenols: biology and therapeutic implications in cancer. Front Biosci. 2007;12:4881–4899. doi: 10.2741/2435. [DOI] [PubMed] [Google Scholar]
- 10.Mukhtar H, Ahmad N. Tea polyphenols: prevention of cancer and optimizing health. Am J Clin Nutr. 2000;71:1698S–1702S; discussion 1703S-1704S. doi: 10.1093/ajcn/71.6.1698S. [DOI] [PubMed] [Google Scholar]
- 11.Hsieh TC, Wu JM. Targeting CWR22Rv1 prostate cancer cell proliferation and gene expression by combinations of the phytochemicals EGCG, genistein and quercetin. Anticancer Res. 2009;29:4025–4032. [PMC free article] [PubMed] [Google Scholar]
- 12.Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer Res. 2006;66:1234–1240. doi: 10.1158/0008-5472.CAN-05-1145. [DOI] [PubMed] [Google Scholar]
- 13.Qiao Y, Cao J, Xie L, Shi X. Cell growth inhibition and gene expression regulation by (-)-epigallocatechin-3-gallate in human cervical cancer cells. Arch Pharm Res. 2009;32:1309–1315. doi: 10.1007/s12272-009-1917-3. [DOI] [PubMed] [Google Scholar]
- 14.Philips BJ, Coyle CH, Morrisroe SN, Chancellor MB, Yoshimura N. Induction of apoptosis in human bladder cancer cells by green tea catechins. Biomed Res. 2009;30:207–215. doi: 10.2220/biomedres.30.207. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Wang J, Deng F, Hu Z, Wang H. Green tea extract and its major component epigallocatechin gallate inhibits hepatitis B virus in vitro. Antiviral Res. 2008;78:242–249. doi: 10.1016/j.antiviral.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Zhou T, Guo H, Guo JT, Cuconati A, Mehta A, Block TM. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res. 2006;72:116–124. doi: 10.1016/j.antiviral.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Sun D, Nassal M. Stable HepG2- and Huh7-based human hepatoma cell lines for efficient regulated expression of infectious hepatitis B virus. J Hepatol. 2006;45:636–645. doi: 10.1016/j.jhep.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Nassal M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol. 1992;66:4107–4116. doi: 10.1128/jvi.66.7.4107-4116.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Hussain M, Wong S, Fung SK, Yim HJ, Lok AS. A genotype-independent real-time PCR assay for quantification of hepatitis B virus DNA. J Clin Microbiol. 2007;45:553–558. doi: 10.1128/JCM.00709-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moolla N, Kew M, Arbuthnot P. Regulatory elements of hepatitis B virus transcription. J Viral Hepat. 2002;9:323–331. doi: 10.1046/j.1365-2893.2002.00381.x. [DOI] [PubMed] [Google Scholar]
- 23.Zoulim F. New insight on hepatitis B virus persistence from the study of intrahepatic viral cccDNA. J Hepatol. 2005;42:302–308. doi: 10.1016/j.jhep.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 24.Levrero M, Pollicino T, Petersen J, Belloni L, Raimondo G, Dandri M. Control of cccDNA function in hepatitis B virus infection. J Hepatol. 2009;51:581–592. doi: 10.1016/j.jhep.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 25.Gerlich WH, Heermann KH, Lu X. Functions of hepatitis B surface proteins. Arch Virol Suppl. 1992;4:129–132. doi: 10.1007/978-3-7091-5633-9_28. [DOI] [PubMed] [Google Scholar]
- 26.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol. Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acs G, Sells MA, Purcell RH, Price P, Engle R, Shapiro M, Popper H. Hepatitis B virus produced by transfected Hep G2 cells causes hepatitis in chimpanzees. Proc Natl Acad Sci USA. 1987;84:4641–4644. doi: 10.1073/pnas.84.13.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu L, Cheng YC. Characterization of novel human hepatoma cell lines with stable hepatitis B virus secretion for evaluating new compounds against lamivudine- and penciclovir-resistant virus. Antimicrob Agents Chemother. 2000;44:3402–3407. doi: 10.1128/aac.44.12.3402-3407.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doong SL, Tsai CH, Schinazi RF, Liotta DC, Cheng YC. Inhibition of the replication of hepatitis B virus in vitro by 2',3'-dideoxy-3'-thiacytidine and related analogues. Proc Natl Acad Sci USA. 1991;88:8495–8499. doi: 10.1073/pnas.88.19.8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gehring AJ, Sun D, Kennedy PT, Nolte-'t Hoen E, Lim SG, Wasser S, Selden C, Maini MK, Davis DM, Nassal M, et al. The level of viral antigen presented by hepatocytes influences CD8 T-cell function. J Virol. 2007;81:2940–2949. doi: 10.1128/JVI.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tachibana H. Molecular basis for cancer chemoprevention by green tea polyphenol EGCG. Forum Nutr. 2009;61:156–169. doi: 10.1159/000212748. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Zhang HY. Cancer preventive mechanisms of the green tea polyphenol (-)-epigallocatechin-3-gallate. Molecules. 2007;12:946–957. doi: 10.3390/12050946. [DOI] [PMC free article] [PubMed] [Google Scholar]