

Abstract
Here, we describe the genetic pathways taken by a human immunodeficiency virus type 1 (HIV-1) isolate, D101.12, to become resistant to the small molecule CCR5 inhibitor, vicriviroc (VCV) in vitro. Resistant D101.12 variants contained at least one substitution in the gp120 V3 region (H308P), plus one of two patterns of gp41 sequence changes involving the fusion peptide (FP) and a downstream residue: G514V+V535M or M518V+F519L+V535M. Studies of Env-chimeric and point-substituted viruses in peripheral blood mononuclear cells (PBMC) and TZM-bl cells showed that resistance can arise from the cooperative action of gp120 and gp41 changes, while retaining CCR5 usage. Modeling the VCV inhibition data from the two cell types suggests that D101.12 discriminates between high- and low-VCV affinity forms of CCR5 less than D1/85.16, a resistant virus with three FP substitutions.
Keywords: HIV-1, Entry inhibitors, Vicriviroc, Escape mutants, Fusion peptide, GPCR
Introduction
Human immunodeficiency virus type 1 (HIV-1) has a well-recognized ability to acquire resistance to antiviral compounds both in vitro and in vivo, as a result of its high replication rate combined with an error-prone reverse transcriptase enzyme (Coffin, 1996; Hirsch et al., 2008). Randomly generated variants with a selective advantage over their wild type counterparts therefore expand to dominance in the presence of the selecting agent. Sometimes, the genetic pathways involved can be unexpected, particularly for resistance to compounds that affect the function of the env gene products, the gp120 and gp41 envelope glycoproteins. Both these proteins retain their abilities to mediate virus-cell entry in the face of substantial sequence variation, more so for the surface gp120 than the transmembrane gp41 subunit. When drug-resistant variants are selected for, this functional plasticity is exploited and viruses with unusual properties emerge.
We have been studying how variants resistant to the small molecule CCR5 inhibitors arise in vitro. These compounds, exemplified by the licensed drug maraviroc (MVC) and the drug candidate vicriviroc (VCV), bind to the CCR5 co-receptor that is part of the virus-cell entry pathway (Kuhmann and Hartley, 2008). By doing so, they prevent gp120 from contacting CCR5 properly, and thereby abort the sequence of conformational changes in the gp120-gp41 complex that are essential for the virus and cell membranes to fuse together (Kuhmann and Hartley, 2008). When HIV-1 is cultured for prolonged periods in the presence of the CCR5 inhibitors, fully resistant variants emerge that still use CCR5 to enter cells, by virtue of acquiring the ability to interact with the inhibitor-CCR5 complex, while still retaining the ability to use the free form of CCR5 (Pugach et al., 2007; Westby et al., 2007).
The most common genetic pathway to resistance involves the accumulation of several (usually two to four) sequence changes in the V3 region of the gp120 glycoprotein (Baba et al., 2007; Kuhmann et al., 2004; Laakso et al., 2007; Ogert et al.,; Ogert et al., 2008; Tsibris et al., 2008; Westby et al., 2007), a location that is consistent with the highly variable nature of V3 and its role in CCR5 binding (Hartley et al., 2005; Kuhmann and Hartley, 2008). The resistance phenotype of isolate CC101.19, for example, has been attributed to four substitutions in V3 (K305R, H308P, A316V and G321E), with the proline at position 308 being the most critical determinant of resistance (Kuhmann et al., 2004; Trkola et al., 2002). These V3 changes have been suggested to render the virus more dependent on the CCR5 N-terminus, to compensate for impaired interactions between the V3-crown and the second extracellular loop (ECL2) (Berro et al., 2009). Consistent with this concept are studies showing that substantial deletions in V3 from both HIV-1 and HIV-2 confer complete resistance to co-receptor antagonists, presumably by disrupting the interaction between V3 and ECL2 (Agrawal-Gamse et al., 2009; Laakso et al., 2007; Lin et al., 2007; Nolan et al., 2009; Nolan et al., 2008). Increases in the affinity of resistant viruses for CD4 and/or in the kinetics of virus entry may compensate for any adverse effect the V3 sequence changes have at the CCR5-binding stage (Agrawal-Gamse et al., 2009; Platt et al.). There is, however, considerable complexity involved in the altered virus-CCR5 interactions. Thus, a subtype D, VCV-resistant patient isolate recognizes the drug-bound form of CCR5 more efficiently but still uses both the N-terminus and ECL2 (Ogert et al., 2010). Moreover, a model has been proposed suggesting that broad cross-resistance to multiple inhibitors is associated with an increased dependence on the N-terminus, while a more specific pattern of resistance to individual compounds involves more subtle changes in how the virus interacts with both the N-terminus and ECL2 (Tilton et al.).
We have also described a CCR5 inhibitor resistant variant, D1/85.16, that contains no sequence changes in V3 (Marozsan et al., 2005), and in which resistance is conferred by three conservative substitutions in the fusion peptide (FP) region of gp41 (Anastassopoulou et al., 2009). How FP changes contribute to viral escape from VCV remains to be understood; one possibility is that they may alter the rate or location of fusion events.
Here, we describe the resistance genetics of another resistant virus, D101.12, which was generated from a weakly resistant isolate, CC101.6, under the selection pressure of VCV (Marozsan et al., 2005). The CC101.6 virus itself emerged when the parental, inhibitor-sensitive isolate CC1/85 was cultured for 6 passages in the presence of AD101, a compound in the same general family as VCV (Trkola et al., 2002). Compared to its parent, CC1/85, the CC101.6 isolate contains an amino acid change in V3, H308P, that renders it up to 4-fold less sensitive to such small molecules as AD101 and VCV (Kuhmann et al., 2004). After 12 additional passages with VCV, the resulting D101.12 isolate became fully resistant (>20,000-fold). We show here that two different gp41 lineages co-exist in the D101.12 isolate and clones derived from it, together with at least the H308P substitution in V3. Mutagenesis studies on clones derived from the D101.12 isolate show that strong resistance to VCV can be conferred when the H308P sequence change in V3 is combined with specific changes in the N-terminal region of gp41, including, but not limited to, the FP. Hence the two subunits of the Env complex can cooperate to enable a VCV-resistant virus to use the VCV-CCR5 complex for entry.
Results
D101.12 is a VCV-resistant virus with a stable phenotype
Two fully resistant isolates were generated, from different input viruses, under the selection pressure of VCV in vitro (for virus nomenclature see Table 1 and Fig. 1). Isolate D1/85.16 emerged after 16 passages of the parental, inhibitor-sensitive CC1/85 isolate, while D101.12 arose from the weakly resistant CC101.6 isolate after 12 passages (Marozsan et al., 2005). An infection-inhibition assay using PBMC confirmed the VCV sensitivities of the study viruses; the CC101.6 isolate was weakly resistant, with a low (< 5%), but consistent, level of replication occurring even at very high inhibitor concentrations (up to 5 μM). In contrast, D101.12, like D1/85.16, was completely resistant to VCV (Fig. 2A).
Table 1.
Nomenclature and properties of viruses used in this study
| Virus | Type | Origin of env subunits in clones |
Engineered gp41 mutations (*) | gp41 sequence pattern | |
|---|---|---|---|---|---|
| gp120 | gp41 | ||||
| CC1/85 | Parental isolate | NA | NA | NA | NA |
| CC101.6 | Input isolate | NA | NA | NA | NA |
| D101.12 (R#) | VCV-resistant isolate (Reversion isolate#) | NA | NA | NA | Patterns I and II (Pattern II ) |
| S | Parental clone | CC1/85 cl.6 | CC1/85 cl.6 | NA | NA |
| S+(G514V) | Mutant parental clone | CC1/85 cl.6 | CC1/85 cl.6 | G514V | Pattern I |
| S+(G514V, V535M) | Mutant parental clone | CC1/85 cl.6 | CC1/85 cl.6 | G514V, V535M | Pattern I |
| S+(M518V, F519L) | Mutant parental clone | CC1/85 cl.6 | CC1/85 cl.6 | M518V, F519L | Pattern II |
| S+(M518V, F519L, V535M) | Mutant parental clone | CC1/85 cl.6 | CC1/85 cl.6 | M518V, F519L, V535M | Pattern II |
| S+(V535M) | Mutant parental clone | CC1/85 cl.6 | CC1/85 cl.6 | V535M | Patterns I and II |
| R14 | VCV-resistant clone | D101.12 cl.14 | D101.12 cl.14 | NA | Pattern I |
| R14/S | Chimera | D101.12 cl.14 | CC1/85 cl.6 | NA | NA |
| R14/S+(G514V) | Mutant chimera | D101.12 cl.14 | CC1/85 cl.6 | G514V | Pattern I |
| R14/S+(G514V, V535M) | Mutant chimera | D101.12 cl.14 | CC1/85 cl.6 | G514V, V535M | Pattern I |
| R14/S+(M518V, F519L) | Mutant chimera | D101.12 cl.14 | CC1/85 cl.6 | M518V, F519L | Pattern II |
| R14/S+(M518V, F519L, V535M) | Mutant chimera | D101.12 cl.14 | CC1/85 cl.6 | M518V, F519L, V535M | Pattern II |
| R14/S+(V535M) | Mutant chimera | D101.12 cl.14 | CC1/85 cl.6 | V535M | Patterns I and II |
Isolates designated as D101.12R followed by a number were derived from the PBMC culture in which D101.12 was cultured for a prolonged period in the absence of VCV; the number indicates the number of weekly passages (see Fig. 2B).
Amino acid numbering is based on HXB2 Env. The columns list the origins of gp120 and gp41 subunits in chimeric viruses based on the reference clones of CC1/85 (CC1/85 cl.6 = S) and D101.12 (D101.12 cl.14 = R14). Isolate CC101.6 and clone R14 both contain the critical H308P change in the gp120 V3 region.
NA, Not applicable.
Fig. 1. Passage history and associated genetics of CCR5 inhibitor-resistant viruses.

The passage history, including passage numbers and culture conditions, of the escape mutant viruses is depicted schematically, together with a summary of the specific genetic changes associated with resistance and relevant citations.
Fig. 2. The D101.12 isolate is resistant to VCV and has a stable phenotype.

(A) D101.12 and D1/85.16 were first tested for VCV sensitivity alongside their respective parental viruses, CC101.6 and CC1/85, in a PBMC-based HIV-1 replication assay. The cultures were infected with each isolate in the presence of the indicated VCV concentration, and p24 production was measured after 7 days. The data points represent the means of two independent experiments ± SEM. (B) The VCV-resistant isolate D101.12 was then cultured for 19 weeks in PBMC from two or three random blood donors, in the absence of VCV. Viruses from this reversion culture were tested for VCV sensitivity. The sensitivity profiles of representative viruses D101.12R7, D101.12R8, D101.12R10, and D101.12R19 (from weekly passages 7, 8, 10, and 19, respectively) after 7 days are shown in comparison to D101.12 and CC101.6. The values shown are the means of at least three independent experiments ± SEM.
To determine whether its VCV-resistance phenotype was stable, we returned D101.12 to culture in PBMC for 19 weekly passages without VCV, assessing its inhibitor sensitivity periodically. Each virus from this reversion culture, like the input D101.12 isolate, was completely resistant to VCV concentrations as high as 5 μM, whereas the input CC101.6 virus was again only weakly resistant (Fig. 2B). Hence, the D101.12 phenotype is highly stable in a PBMC culture, with no associated fitness cost. In this respect, D101.12 resembles the fully AD101- and VCV-resistant CC101.19 and D1/85.16 isolates (Anastassopoulou et al., 2009; Anastassopoulou et al., 2007).
Analysis of gp120 sequences
An infectious clone derived from the fully resistant D101.12 isolate, D101.12 cl.14 (= R14), retained the H308P change in V3 that was also present in the CC101.6 input virus but had no other V3 changes compared to the parental isolate, CC1/85 (Kuhmann et al., 2004; Marozsan et al., 2005). The introduction of the H308P single residue change into a parental, inhibitor-sensitive clone is known to confer low-level (~3-fold) resistance to AD101 and VCV (Kuhmann et al., 2004). However, although the H308P change itself only modestly affects VCV sensitivity, it has a substantial influence on the outcome of additional V3 substitutions in this genetic context. Thus, the three later-arising V3 changes (K305R, A316V and G321E) confer complete resistance when introduced into a weakly resistant clone that contains proline at residue 308, but they have no effect on VCV sensitivity when the residue at this position is the predominant histidine (Kuhmann et al., 2004). Clone R14 does harbor the critical proline at position 308, but it lacks the three later-arising resistance-associated changes in V3 (Kuhmann et al., 2004; Marozsan et al., 2005). Hence, additional mutations elsewhere in env must be critical determinants of the full resistance of the D101.12 virus.
To map the responsible changes, we first sequenced the env genes from the D101.12 isolate and also from the VCV-resistant D101.12R7, D101.12R8, D101.12R10 and D101.12R19 viruses that were derived from the D101.12 reversion cultures. The predicted gp160 sequences from these resistant isolates were compared to sequences from the resistant R14 clone and from sensitive viruses. No consistent pattern was discernible among the gp120 and, specifically, the V3 sequences of these various viruses (Fig. 3A,B and data not shown). Thus, the uncloned D101.12 isolate had all four resistance-associated V3 substitutions (K305R, H308P, A316V and G321E), but while the first two changes were fixed, the other two were unstable. And, as noted above, the R14 clone contained only the H308P change. Finally, the reversion-culture isolates consistently retained two of the four resistance-associated V3 changes (K305R and A316V), but the G321E change was consistently absent (i.e., it had reverted to the cognate parental sequence, 321G). Of note is that the resistance-relevant proline at residue 308 was absent from the reversion isolates, and although a change arose at this position, it was not to the dominant parental histidine, but to a new amino acid, threonine (i.e., P308T). Other changes were also evident at V3 positions adjacent to the first two established resistance-associated substitutions, although only the second of those changes (I309R) had become stabilized by passage 19. There was also a new substitution (F317I) in the D101.12R19 virus, located adjacent to the third of the resistance-associated V3 changes, A316V (Fig. 3B).
Fig. 3. V3 and N-terminal gp41 sequence alignments from VCV-sensitive and VCV-resistant viruses.
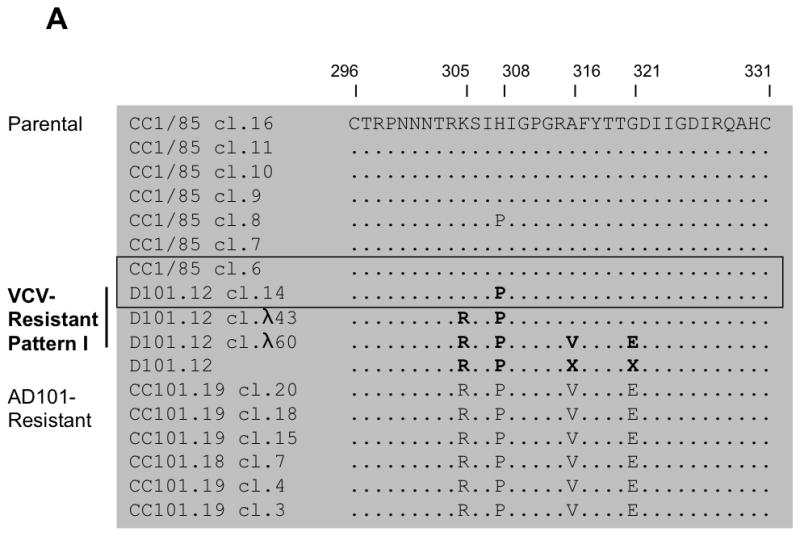
(A, B) The 35 residues from the gp120 V3 region and (C, D) the first 29 amino acid residues from the gp41 N-terminus are shown for seven clones from the parental, VCV-sensitive virus CC1/85, 13 clones (3 and 10 of gp41 Patterns I and II, respectively) and four isolates based on the VCV-resistant isolate D101.12 as well as six clones from the AD101- and VCV-resistant isolate CC101.19. Ten clones (one of gp41 Pattern-I and nine of gp41 Pattern-II, two of which contained only the H308P substitution) were excluded from the analysis since they contained premature stop codons and were presumably non-infectious (data not shown). The sequences are recorded relative to that of CC1/85 cl.16 (top line), with dots indicating amino acid sequence identity. Substitutions that influence resistance, as discussed in the main text, are highlighted in bold. Among the seven parental clones, CC1/85 cl.6 (= S, GenBank accession no. AY 357338) is the most closely related to D101.12 cl.14 (= R14); these two clones, which were used for subsequent genetic studies, are boxed. Amino acid numbering is based on HXB2 Env, with the first residue of V3 at position 296 and of gp41 at position 512.
Two genetic patterns are present in the D101.12 gp41 subunit
The above sequence analyses imply that the determinants for the full resistance of D101.12 and related viruses do not lie solely within gp120. We therefore assessed whether a common genetic determinant of resistance could be found in the gp41 subunit, by sequencing and examining 13 env genes cloned from D101.12. Approximately half (5/13) of the D101.12 clones contained all four of the resistance-associated V3 changes, but others possessed only three (3/13), two (4/13), or one of them (1/13); the H308P change was, however, invariably present in all the D101.12 clones (Fig. 3A,B). Two clearly distinct patterns of gp41 sequences were apparent: A valine to methionine substitution at position 535 of the gp41 ectodomain (V535M) was present in all resistant isolates and clones, but it was accompanied by either a single G514V change in the FP (Pattern I, 3/13 clones, Fig. 3C) or two changes, M518V and F519L (Pattern II, 10/13 clones, Fig. 3D). The Pattern-I change adds a bulky hydrophobic side-chain (G514V), whereas the other FP changes involve semi-conservative substitutions. Of note is that, in each case, one of the changes results in the introduction of a C-β branched amino acid (Val), just as arises in the resistant D1/85.16 virus (Anastassopoulou et al., 2009). A valine residue is also present at position 518 in 6 of 6 CC101.19 clones (Fig. 3C), although resistance is conferred to this virus by the four changes in V3 (Kuhmann et al., 2004).
The corresponding V3 sequence pattern of the D101.12 clones was complex. Among the three clones with gp41 sequence Pattern I, one (R14) possessed only the H308P change, one had the K305R and H308P substitutions, while the third, λ60, harbored all four V3 changes (Fig. 3A). Outside of V3, all three Pattern-I clones had lost a potential N-linked glycosylation site in the V4 region as a result of an N406K substitution (data not shown). However, the majority (10/13) of the D101.12 clones were of gp41 Pattern II (Fig. 3D), with the H308P change being the only common feature among their V3 and gp120 sequences (Fig. 3B and data not shown). Most Pattern II clones (4/13) contained all four V3 substitutions, three had three changes (although not consistently in the same combination), while another three possessed the K305R and H308P changes (Fig. 3B). Among the Pattern-II clones, 5/13 contained the N406K change in V4 (data not shown).
Clearly, the genetics of VCV resistance are complex for the D101.12 isolate. Two distinct genetic lineages are apparent in gp41, together with a minimum of one (H308P), and up to all four, of the resistance-associated mutations in the gp120 V3 region.
VCV-resistance can be conferred by the cooperative action of changes in both gp41 and gp120
The fully resistant R14 clone contains the H308P change in V3, but no other changes in this region, together with the Pattern-I gp41 substitutions, V535M + G514V (Fig. 3). To assess the relative contributions of the changes in the two Env subunits, we first interchanged the gp120 and gp41 components from the R14 clone and the parental clone CC1/85 cl.6 (= S). Hence the gp120 component of the resulting R14/S chimera is derived from R14, the gp41 component from S (Table 1). The R14/S virus was replication-competent in PBMC with titers of 105 TCID50/mL on day 7 post-infection (p24 production, 1–4 ng/mL). When VCV resistance was determined in a PBMC infection-inhibition assay, it was found not to track with the source of gp120. Thus, the R14/S chimera was only weakly resistant to VCV, which is consistent with its possession of the H308P change in V3 (Fig. 4A). Overall, R14/S behaved more like the sensitive clone, S, than the resistant clone, R14. The weak resistance of the R14/S chimera, which is attributable to the H308P change, is mainly manifested by the small (~6%), but consistent, level of replication that occurs at VCV concentrations as high as 5 μM (MPI = 94 for R14/S and 100 for S; Fig. 4A, Table 2). We can infer from these results that the full resistance of the R14 clone must, therefore, be attributable to the influence of the Pattern-I gp41 changes, acting in concert with the H308P change in V3.
Fig. 4. VCV resistance maps to both gp41 and gp120.

The engineered R14/S chimera and mutant viruses were tested for VCV sensitivity in a PBMC-based, multi-cycle replication assay with p24 production measured after 7 days. For comparison, the parental inhibitor-sensitive clone S and the VCV-resistant clone R14 were also tested in the same experiments. For clarity, the dose-response curves are shown in separate panels for the R14/S chimera (A), and the mutants harboring either the V535M change only (B), or the complete set of gp41 changes of Pattern I (C), or Pattern II (D). VCV-inhibition curves were created by fitting the model function, Q=(1−(1−(C/(Kdi+C))+w *(C/(4.3*Kdi+C))))*100%, to the average PBMC data, since sigmoid dose-response curves did not fit the data for viruses that were resistant to various degrees in these cells. The results shown are the average of 3–11 independent experiments, with the error bars indicating the SEM.
Table 2.
VCV inhibition of HIV-1 replication in PBMC and TZM-bl cellsa
| Virus | PBMC | TZM-bl cells | VCV phenotypeb | ||
|---|---|---|---|---|---|
| MPI | IC50 (nM) | MPI | IC50 (nM) | ||
| S | 100 | 0.73 | 99 | 21 | Sensitive |
| S+(G514V) | 100 | 0.13 | 97 | 4.4 | Sensitive |
| S+(G514V, V535M) | 73 | 0.84 | 96 | 5.2 | Partially resistant |
| S+(M518V, F519L) | 99 | 0.13 | 100 | 2.2 | Sensitive |
| S+(M518V, F519L, V535M) | 92 | 0.71 | 98 | 4.1 | Weakly resistant |
| S+(V535M) | 100 | 0.69 | 99 | 25 | Sensitive |
| R14 | 42 | NA | 79 | 3.7 | Resistant |
| R14/S | 94 | 0.21 | 95 | 9.6 | Weakly resistant |
| R14/S+(G514V) | 72 | 0.25 | 85 | 3.4 | Partially resistant |
| R14/S+(G514V, V535M) | 41 | NA | 83 | 2.4 | Resistant |
| R14/S+(M518V, F519L) | 53 | 0.82 | 93 | 1.0 | Partially resistant |
| R14/S+(M518V, F519L, V535M) | 82 | 0.19 | 91 | 1.9 | Partially resistant |
| R14/S+(V535M) | 93 | 0.45 | 96 | 30 | Weakly resistant |
The model function for inhibition, Q=(1−(1−(C/(Kdi+C))+w *(C/(4.3*Kdi+C))))*100%, was fitted to the average PBMC and TZM-bl data (see Figs. 4 and 5) to determine MPI and IC50 values (see Materials and Methods and Results; R2 values are given in Table 3). The IC50 is the VCV concentration (C) that yields 50% inhibition (Q). The IC50 therefore applies only when MPI > 50%, and it can be substantially larger than the VCV concentration that would inhibit to an extent equal to 50% of the MPI value. MPI is defined as the upper plateau for sigmoid curves, or as the maximum inhibition value for peaked curves.
Viruses with MPI values in the range 90–95 for VCV inhibition in PBMC are considered to be weakly resistant, 50–90 partially resistant, <50 fully resistant. The fully resistant viruses, according to this criterion, are highlighted in bold.
MPI, maximum percent inhibition; NA, Not applicable
Site-directed mutagenesis of gp41 residues
To further examine the genetics of resistance of the D101.12 isolate, we introduced the gp41 substitutions highlighted by the sequence analysis into both the parental, VCV-sensitive clone S and the weakly resistant R14/S chimera that contains the H308P change in V3. Both single residue and combination changes corresponding to gp41 Patterns-I and -II were made in these viruses, which were tested for VCV sensitivity in PBMC in comparison to clones S, R14 and R14/S (Fig. 4). Similar results were obtained when AD101 was used instead of VCV (data not shown).
The V535M substitution is common to all D101.12 isolates and clones, irrespective of which gp41 sequence pattern was present (Fig. 3C,D). Introducing this change alone into the parental clone S had no notable effect on the VCV infection-inhibition curve and the resulting MPI and IC50 values (Fig. 4B, Table 2). The same V535M change in the R14/S chimera created a virus, R14/S+(V535M), with a slightly higher IC50 value (0.45 nM vs. 0.21 nM) but with an unchanged MPI (93 vs. 94) (Fig. 4B, Table 2). Having a methionine at position 535 of the gp41 ectodomain when V3-residue 308 is a proline therefore modestly increases the IC50, but without affecting the incomplete inhibition plateau that stems from the presence of the proline.
Introducing the two Pattern-I gp41 changes (G514V + V535M) into the R14/S chimera that also harbors the H308P substitution mimicked the fully resistant phenotype of clone R14 (Fig. 4C). These two gp41 mutations are, however, necessary, but not sufficient, to confer resistance, since their insertion into the sensitive clone S resulted in only a partially resistant virus; hence, the MPI value of the engineered S+(G514V, V535M) mutant was reduced from 100 to 73 (Fig. 4C, Table 2). When only the G514V change was made in the S clone, the VCV dose-response curve was shifted to the left, resulting in a 6-fold lower IC50 value (0.13 nM vs. 0.73 nM, Table 2). Hence, the S+(G514V) virus, like S+(V535M), remained VCV sensitive (Fig. 4B,C). When the same G514V mutation was engineered in the context of the H308P change, the resulting R14/S+(G514V) virus was difficult to grow; it replicated sufficiently for its response to VCV to be properly quantifiable in only 3/11 of PBMC-based infection-inhibition assays. However, this virus also was partially VCV resistant, with an MPI value of 72 (Fig. 4C, Table 2). The further addition of the V535M change to R14/S, creating the R14/S+(G514V, V535M) mutant made the virus replication-competent and rendered it fully resistant (Fig. 4C, Table 2). Thus, the VCV dose-response curve for R14/S+(G514V, V535M) was almost completely coincident with that of the reference, VCV-resistant clone R14. Of note is that both curves had complex shapes. There was an inhibition peak of ~30–40% at low VCV concentrations (~0.3–2 nM) but a lesser effect at higher concentrations with the curves leveling off at ~18–20% inhibition at 5 μM of VCV (Fig. 4C). We have previously described, and modeled, dose-response curves with this unusual configuration for viruses for which VCV-resistance is due to FP changes (Anastassopoulou et al., 2009).
As noted above, most D101.12 clones were of gp41 sequence Pattern II, for which the important changes were M518V and F519L in the FP, together with the downstream V535M substitution (Fig. 3D). The introduction of the two FP changes into the parental S clone created a VCV-sensitive virus, S+(M518V, F519L) (Fig. 4D). The dose-response curve for this virus was shifted to the left, similarly to the effect of the Pattern-I FP change creating the S+(G514V) virus (IC50 = 0.13 nM for both viruses) (Fig. 4C,D, Table 2). The further addition of V535M to make the triple mutant, S+(M518V, F519L, V535M) created a virus that was weakly resistant; its dose response curve was shifted rightward (IC50 = 0.71 nM), but with an MPI of 92 compared to 73 for S+(G514V, V535M) (Fig. 4C,D, Table 2). Thus, when the gp41 Pattern-I changes (G514V + V535M) are combined in the context of the parental clone S (i.e. in the absence of H308P), they have a more pronounced effect than the combined Pattern-II changes (M518V, F519L, V535M), as manifested by the lower MPI value (Fig. 4C,D, Table 2).
Introducing the Pattern II gp41 changes into the R14/S chimera, and hence in the context of the H308P change in V3, created a virus, R14/S+(M518V, F519L, V535M), that was only partially resistant. The inhibition plateau of this triple mutant was ~1.5-fold higher than that of the double mutant R14/S+(M518V, F519L) virus (MPI = 82 vs. 53, respectively) (Fig. 4D, Table 2). Hence when a proline is present at position 308, the V535M substitution reinforces the effects of the Pattern-I change in the FP (G514V), but not the Pattern-II changes (M518V, F519L) (Fig. 4C,D, Table 2).
Manifestation of VCV-resistance is cell type-dependent
Resistance to small molecule CCR5 inhibitors is known to vary with the cell type (Anastassopoulou et al., 2009; Ogert et al., 2008; Pugach et al., 2007; Westby et al., 2007). To further characterize the resistance profiles of the engineered infectious clones, we tested them for their VCV sensitivity in TZM-bl cells (Fig. 5). The MPI reflects the relative ability of the virus to use the inhibitor-bound and the inhibitor-free forms of the co-receptor for entry; the higher the MPI value, the less efficiently a resistant virus uses the inhibitor-CCR5 complex (Pugach et al., 2007; Westby et al., 2007). As in the PBMC assay, the replication-competent mutants were tested for VCV sensitivity in comparison to the reference parental clone S, the weakly resistant R14/S chimera, and the VCV-resistant clone R14 (Fig. 5). Similar results were obtained when AD101 was used instead of VCV (data not shown).
Fig. 5. Determinants of VCV resistance in a TZM-bl cell-based assay.

The same clonal viruses studied in PBMC were assessed for VCV sensitivity in TZM-bl cells with a luciferase reporter gene endpoint. For comparison, the parental inhibitor-sensitive clone S, the engineered R14/S chimera, and the VCV-resistant clone R14 were also tested. For clarity, the dose-response curves are shown in separate panels for the R14/S chimera (A), and for the mutants containing either the V535M change only (B), or the complete set of gp41 changes of Pattern-I (C) or Pattern-II (D). The uncloned isolates were also studied (E). For consistency, VCV-inhibition curves were again created by fitting the model function, Q=(1−(1−(C/(Kdi+C))+w *(C/(4.3*Kdi+C))))*100%, to the average TZM-bl data. The results shown are the average of 3 independent experiments for the mutant clones (Panels A–D) and 2–6 experiments for the isolates (Panel E), with the error bars indicating the SEM.
The model function was fitted to the average TZM-bl cell inhibition data to determine MPI and IC50 values (Fig. 5, Table 2). The VCV sensitivity profiles of the parental clone S and the R14/S chimera were similar to those seen for each virus in the PBMC assay (Fig. 4A). The MPI values were 99 and 95, respectively, but the IC50 value (9.6 nM) for R14/S was about half that for S (21 nM) (Fig. 5A, Table 2). The VCV-resistant R14 virus, in contrast, had a reduced MPI value of 79, and a leftward shift in the dose response curve that lowered the IC50 to 3.7 nM (Fig. 5A, Table 2).
Introducing the V535M substitution, which is common to both gp41 patterns, into the parental clone S had no effect on the VCV infection-inhibition curve and the resulting MPI and IC50 values (Fig. 5B, Table 2). In contrast, making the same V535M change in R14/S (i.e., in the context of the H308P change in V3) caused a rightward shift of the dose-response curve, just as it did in PBMC (Fig. 4B), and an ~3-fold increased IC50 (30 nM vs. 9.6 nM for R14/S) (Fig. 5B, Table 2).
The fully resistant phenotype of clone R14 was again mimicked by the introduction of the two Pattern-I gp41 changes (G514V + V535M) into the R14/S chimera (Fig. 5C). The resulting, leftward-shifted dose-response curve yielded MPI and IC50 values that were both reduced compared to the R14/S chimera (Table 2). When the same two changes were made in the parental clone S, the dose-response curve was again shifted leftward, but not downward, yielding an IC50 value ~4-fold lower than for S (5.2 nM vs. 21 nM, respectively) (Fig. 5C, Table 2).
The R14/S+(G514V) single mutant was consistently able to replicate in TZM-bl cells, which contrasts with its replication-incompetence in PBMC (data not shown). The VCV dose-response curve for R14/S+(G514V) was very similar to those of R14 and R14/S+(G514V, V535M), as were the corresponding MPI (85 vs. 79 and 83, respectively) and IC50 values (3.4 nM vs. 3.7 nM and 2.4 nM, respectively) (Fig. 5C, Table 2). The S+(G514V) single mutant and the S+(G514V, V535M) double mutant had similar MPI and IC50 values (Fig. 5C and Table 2). In both cases, the virus was almost fully inhibited (MPI values >96), but with IC50 values ~4-fold lower than for S (Table 2).
Introducing the Pattern-II gp41 changes into both S and the R14/S chimera also caused leftward shifts in dose-response curves (Fig. 5D). The S+(M518V, F519L) double mutant and the S+(M518V, F519L+V535M) triple mutant had similar MPI (>98) and IC50 (2.2 nM and 4.1 nM, respectively) values, the latter being ~7-fold lower than for S (Fig. 5D and Table 2). When the same changes were made in the R14/S chimera, the leftward shifts were now accompanied by lower inhibition plateaus (Fig. 5D). The resulting MPI values of 93 and 91 for R14/S+(M518V, F519L) and R14/S+(M518V, F519L, V535M) were comparable to that of R14/S, but the corresponding IC50 values of 1.0 nM and 1.9 nM were lower (Table 2). The reductions in the IC50 values for the R14/S mutants are therefore attributable to the combination of the Pattern-II gp41 changes, while the reduced MPI values arise from the presence of Pro-308 in the V3 region.
The engineered mutant viruses that are VCV-resistant in PBMC therefore display the non-competitive resistance indicator that is a feature of the TZM-bl cell assay; i.e. MPI values significantly below 100%. However, the mutants also have lower IC50 values than the parental virus. This property is not just atypical for a resistant virus, it is apparently paradoxical, as discussed previously for the D1/85.16 isolate (Anastassopoulou et al., 2009).
To gain a better understanding of how VCV-resistance is influenced by sequence variation in only the FP (e.g., as in isolate D1/85.16), compared to changes in both gp120 and gp41 (e.g., as in isolate D101.12), we tested uncloned isolates for VCV sensitivity in the same TZM-bl cell assay (Fig. 5E). Similar results were obtained when AD101 was used instead of VCV (data not shown). The input CC1/85 and CC101.6 isolates had comparable MPI values of 91 and 90, respectively, although CC101.6, which is weakly resistant in PBMC (Fig. 2A), had a slightly lower IC50 value (11 nM, compared to 18 nM). The two isolates that are fully resistant in PBMC (Fig. 2A) had unusual VCV dose-response curves in TZM-bl cells. Thus, D101.12 was only minimally inhibited (0–5%) by VCV concentrations up to ~200 nM, while infection by the D1/85.16 isolate was modestly but consistently enhanced (~25%) at very low concentrations (up to ~0.3 nM). The lower MPI value for D101.12 compared to D1/85.16 (24 vs. 51, respectively) implies that it uses the inhibitor-CCR5 complex more efficiently. For D101.12, this outcome arises from the cooperative action of sequence change in both gp120 and gp41, whereas for D1/85.16 the three substitutions in the FP have the same effect, although to a lesser extent.
Sensitivity to other inhibitors
All the test viruses had been carefully titrated in each cell system, to allow nominally equivalent titers (100 TCID50 per well of a 96-well plate) to be used in subsequent inhibitor-sensitivity experiments. Despite these precautions, we considered it possible that titer variations could influence the resulting IC50 values for the different viruses, and hence perhaps the diagnosis of resistance. To gauge the magnitude of any such variations, we tested zidovudine (AZT), an antiviral drug that does not target Env, under the same conditions. The similar AZT-sensitivity profiles of the test viruses contrasts markedly with the differences seen using VCV in the same experiment (SI Fig. 1A,B). We observed similar results using the broadly neutralizing monoclonal antibody 2F5, which inhibits virus entry at a later stage than VCV (SI Fig. 1C). The observed differences in VCV IC50 values are, therefore, not merely due to virus titer variations. Of note, however, is that viruses harboring the gp41 Pattern-I changes, G514V and V535M, were more sensitive to the fusion inhibitor enfuvirtide (T20) than other viruses from this lineage, irrespective whether the H308P change in gp120 was present or not (SI Fig. 1D). Future studies will focus on understanding the latter observation.
VCV resistance is not due to a switch in co-receptor usage
To determine whether any of the engineered resistant variants of D101.12 had acquired the ability to use CXCR4 for entry, we assessed their replication in PBMC from a CCR5-Δ32/Δ32 homozygous donor, and their sensitivity to the CXCR4-specific small molecule inhibitor AMD3465. In contrast to the reference X4 and R5X4 viruses (NL4-3 and CC2/86, respectively), the fully resistant clone R14 and the engineered resistant virus R14/S+(G514V, V535M) did not produce detectable amounts of p24 in CCR5-Δ32/Δ32 PBMC. Moreover, their replication in normal donor PBMC was insensitive to 500 nM of AMD3465 (Fig. 6). The extent of replication with AMD3465, relative to its absence, did not differ between the VCV-sensitive viruses (S and R14/S) and -resistant viruses (R14 and R14/S+(G514V, V535M)), Mann-Whitney U two-tailed test for pooled replicates of each category, p=1.0. Similar results were obtained using the other D101.12 variants described above (data not shown).
Fig. 6. VCV resistance does not involve switching to CXCR4 use.

Replication of the VCV-sensitive clone S, the weakly resistant R14/S chimera, and the VCV-resistant viruses R14 and R14/S+(G514V, V535M) was determined in PBMC from CCR5 wild type and CCR5-Δ32/Δ32 homozygous donors in the presence and absence of the CXCR4-specific inhibitor AMD3465 (500 nM). The reference viruses NL4-3 (X4) and CC2/86 (R5X4) were included for comparison. HIV-1 replication was quantified by measuring p24 production on day 14 post-infection.
The VCV-resistant viruses were also tested in TZM-bl cells in the presence and absence of AMD3465 (500 nM). The incomplete inhibition plateaus described above (Fig. 5 and Table 2) were unaffected by the presence of the CXCR4-specific inhibitor (data not shown). We conclude that the VCV resistance of D101.12 and the engineered mutants is not due to a switch to CXCR4 usage in PBMC or TZM-bl cells.
Modeling VCV inhibition and resistance
We applied a previously developed model to analyze the inhibitory effect of VCV on HIV-1 infection of PBMC and TZM-bl cells (Anastassopoulou et al., 2009). The model is a quantitative formulation of the hypothesis that two distinct forms of CCR5 with high or low affinity for VCV exist on the surfaces of different cell types in varying proportions. The inhibition of viral infectivity, Q, is a function of the concentration, C, of VCV according to the equation
where Kdi is the postulated dissociation constant for the binding of VCV that blocks viral usage of CCR5 and Kde, correspondingly, for any such binding that allows it. The w term reflects the relative abundance of the CCR5 form that can bind VCV with low affinity, and also the efficacy with which a resistant virus can use that CCR5 form as a VCV-complex. According to the model, a VCV-sensitive virus may be able to use both the high- and low-affinity forms of inhibitor-free CCR5 to various extents, whereas a resistant virus has a preference for the unoccupied high-affinity form of the receptor and for the occupied low-affinity receptor (Anastassopoulou et al., 2009).
The resistant virus with three FP changes (D1/85.16 cl.23; i.e., clone R) that we previously described was characterized by w > 0 (large values on PBMC and smaller on TZM-bl cells), and its Kdi value on TZM-bl cells was lower than those for VCV-sensitive viruses (Anastassopoulou et al., 2009). To assess whether the various engineered viruses described above behaved similarly to clone R, and to explore which amino acid substitutions segregated with the various aspects of the resistance phenotype, we modeled data for VCV inhibition of infection of PBMC and TZM-bl cells.
We lacked a sufficient number of data points to determine all three parameters (w, Kdi and Kde) simultaneously. We therefore optimized the ratio of Kde/Kdi to 4.3 based on data for the reference resistant virus, the R14 clone. In our previous study of other mutants, the optimal Kde/Kdi ratio was 22 (Anastassopoulou et al., 2009). Here, the lower ratio of 4.3 gave at least as good fits to the experimental data for all the resistant and sensitive viruses as it did for R14. The implication is that the resistant viruses being analyzed here discriminate less than the previously studied ones between the high- and low-VCV affinity forms of CCR5. However, they do not use the VCV-CCR5 complexes less efficiently since their w values were, generally, not lower. The resulting non-linear regression fits all yielded R2 values > 0.75 and in all but three cases > 0.96 (Table 3). The modeling results for the viruses S and R14 resembled what we found previously for S and the resistant virus S+3FP (Anastassopoulou et al., 2009). Thus for S, w = 0 while for R14, w was substantial for both cell types but larger for PBMC than TZM-bl cells.
Table 3.
Modeling how VCV inhibits HIV-1 usage of two hypothetical forms of CCR5
| Virus: | S | R14 | R14/S | S+(V535M) | R14/S+(V535M) | S+(G514V) | S+(G514V, V535M) | R14/S+(G514V) | R14/S+(G514V, V535M) | S+(M518V, F519L) | R14/S+(M518V, F519L) | S+(M518V, F519L, V535M) | R14/S+(M518V, F519L, V535M) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PBMC | Kdia | 0.73 ±0.065 | 0.39 ±0.13 | 0.20 ±0.037 | 0.68 ±0.074 | 0.42 ±0.036 | 0.13 ±0.012 | 0.65 ±0.095 | 0.19 ±0.12 | 0.38 ±0.14 | 0.13 ±0.022 | 0.35 ±0.11 | 0.67 ±0.084 | 0.16 ±0.026 |
| wb | 0 ±0.014 | 0.80 ±0.028 | 0.060 ±0.025 | 0.0017 ±0.016 | 0.066 ±0.012 | 0.0011 ±0.012 | 0.28 ±0.017 | 0.29 ±0.067 | 0.82 ±0.032 | 0.011 ±0.021 | 0.57 ±0.028 | 0.081 ±0.018 | 0.18 ±0.019 | |
| R2 | 1.0 | 0.77 | 0.98 | 0.99 | 1.0 | 1.0 | 0.99 | 0.80 | 0.76 | 0.98 | 0.90 | 0.99 | 0.98 | |
| TZM-bl | Kdi | 21±4.3 | 3.0±0.71 | 9.2±1.9 | 25±6.1 | 30±3.9 | 4.3±0.21 | 5.1±1.0 | 3.0±0.20 | 2.1±0.0.39 | 2.2±0.10 | 0.97±0.093 | 4.1±0.69 | 1.8±0.15 |
| wb | 0.0019 ±0.042 | 0.21 ±0.033 | 0.049 ±0.038 | 0.0049 ±0.051 | 0.039 ±0.028 | 0.026 ±0.0081 | 0.043 ±0.035 | 0.15 ±0.0098 | 0.17 ±0.027 | 0.0015 ±0.0077 | 0.073 ±0.014 | 0.018 ±0.029 | 0.089 ±0.013 | |
| R2 | 0.98 | 0.97 | 0.98 | 0.97 | 0.99 | 1.0 | 0.98 | 1.0 | 0.98 | 0.98 | 1.0 | 0.99 | 1.0 | |
Modeled inhibitory dissociation constants, Kdi, have the dimension of concentration [nM].
Values of w ≥ 20% of that for clone R14 on the respective cells are shown in italics; values of w <20% of that for clone R14 are in bold.
The high w value for R14 on TZM-bl cells, which indicates efficient use of the VCV-CCR5 complex, is largely reproduced for the R14/S+(G514V) and R14/S+(G514V, V535M) mutants and partly so for R14/S+(M518V, F519L, V535M) and R14/S+(M518V, F519L). The paradoxical reduction in the inhibitory Kd value for resistant viruses on TZM-bl cells (i.e., the leftward shift in the VCV dose-response curve, Fig. 5C,D) could be partly attributable to mutations other than those responsible for raising w. Thus, one or more of the three FP changes (G514V, M518V, F519L), but not V535M (Fig. 5B), may contribute to this particular phenotype of the resistant viruses. We will address this point further in a future report on the contribution of the individual FP changes to various aspects of VCV resistance.
Overall, the VCV resistance profile for the R14 clone is complex. Thus, reasonable data fits for VCV inhibition of R14 infection of PBMC can only be obtained if we factor in some affinity heterogeneity among the various forms of CCR5. The mutants that apparently use VCV-CCR5 complexes on PBMC most efficiently (i.e. with a high w value) were R14/S+(G514V, V535M) and R14/S+(M518V, F519L). The G514V and V535M changes generate robust resistance on both cell types when they are introduced together into the S clone, as does the single G514V change when it is made in the context of R14/S but not S (Table 3). The V535M change enhances the usage of VCV-CCR5 complexes for viruses with gp41-Pattern I sequences, but when it was added to the Pattern II-derived virus R14/S+(M518V, F519L) it reduced the w-value from 0.57 to 0.18. The various gp41 mutations may therefore interact to enable the virus to take different escape routes.
The data we have modeled here represent averages derived from 3–11 experiments for 13 different viruses (1690 data points), each experiment using a mixture of PBMC from two individuals. VCV inhibition curves sometimes differed from experiment to experiment, in that the curves were sometimes fairly flat, but in other cases with an inhibition maximum in the middle or a plateau to the right (SI Fig. 2A,B). Indeed, on occasion, we even saw strong enhancement of infection at the highest VCV concentrations (SI Fig. 2C). These types of profile were reproduced when the two fully resistant viruses, R14 and R14/S+(G514V, V535M), were included in the same eight experiments, each performed using PBMC from the same donor pair. When we modeled the data set, derived from a single pair of PBMC donors, that displayed the most pronounced VCV-mediated infectivity enhancement, we obtained w values of 1.5 ± 0.11 for R14 and 1.6 ± 0.051 for R14/S+(G514V, V535M) (R2=0.81 and 0.87, respectively). These w values are higher than ones based on the average inhibition seen in all the experiments taken together. They indicate the presence of particularly prevalent VCV-CCR5 complexes that could be used by these viruses. Taken together, these observations suggest that the forms of CCR5 on the cell surface vary from donor to donor.
Discussion
In this study, we investigated the genetic pathway involved in the resistance of the HIV-1 isolate D101.12 to VCV and other small molecule CCR5 inhibitors. Resistance to this class of compound is typically caused by changes in the gp120 V3 region (Baba et al., 2007; Kuhmann et al., 2004; Laakso et al., 2007; Ogert et al.,; Ogert et al., 2008; Tsibris et al., 2008; Westby et al., 2007). Although derived from a common parent, CC1/85, two resistant viruses that we previously described, CC101.19 and D1/85.16, followed different genetic pathways to reach the same phenotypic endpoint: CC101.19 acquired four substitutions (K305R, H308P, A316V and G321E) in the V3 region (Kuhmann et al., 2004), while the key determinants of resistance in D1/85.16 were three changes in the gp41 FP (G516V, M518V and F519I) (Anastassopoulou et al., 2009). Both resistant variants acquired the ability to use the VCV-CCR5 complex for entry (Anastassopoulou et al., 2009; Kuhmann et al., 2004; Pugach et al., 2007; Trkola et al., 2002); they do, however, differ in their dependence on the CCR5 N-terminus and their exposure of regions of gp120 associated with the CCR5-binding site, with the FP changes having a much lesser effect on the overall topology of the Env complex (Berro et al., 2009).
The D101.12 isolate arose from the weakly resistant CC101.6 virus that harbors the critical H308P change and, like D1/85.16, it is completely resistant to VCV in PBMC assays. Both D1/85.16 and D101.12 have highly stable phenotypes, in that they retain their resistance when cultured in PBMC for multiple passages in the absence of the inhibitor (Anastassopoulou et al., 2009). Hence these viruses resemble CC101.19 in being fit, well adapted to PBMC and not requiring the selecting compound to replicate efficiently (Anastassopoulou et al., 2007).
The fully resistant D101.12 clone R14 contains an H308P substitution in V3 that confers modest VCV resistance, but lacks the three later-arising, resistance-associated changes in V3 that are often needed for full resistance (Kuhmann et al., 2004; Marozsan et al., 2005). Clones derived from the D101.12 isolate and from its reversion culture also did not consistently possess all four of the V3 resistance-associated changes. Approximately half of the D101.12 clones (5/13) contained all four V3 changes, the rest possessing fewer, with only H308P being invariably present. We therefore considered it possible that additional mutations elsewhere in env could be contributing to the full VCV resistance of the D101.12 isolate. The R14 clone is not representative of the D101.12 isolate from which it was derived, but it provided an opportunity to study the interactions between V3 and other Env sequences that arise during resistance development. Our hypothesis was that gp41 changes contributed to the resistance of the D101.12 isolate, but we could not easily test this idea using a clone that harbored additional V3 changes. Hence we used the R14 clone, which contains only the H308P change in V3 and yet is fully VCV-resistant, in our domain swapping and mutagenesis strategies. We did this both to study how R14 had become resistant and to gain more general insights into the interplay between gp120 and gp41.
An N406K substitution that results in the deletion of a potential N-linked glycosylation site was present in the gp120 V4 region in more than half of the D101.12 clones (8/13), including clone R14 as previously described (Marozsan et al., 2005). The loss of a glycosylation site in the C-terminal part of V4 from the JR-CSF isolate has been associated with the more efficient use of an N-terminally-deleted CCR5 mutant, suggesting that sequence changes in this area can influence Env-CCR5 interactions (Platt et al., 2005). Other changes in V4, D407G and the loss of a glycosylation site at residue 386, have been reported to modulate the magnitude of clinical resistance to MVC that is conferred principally by V3 changes (Tilton et al.). We did not, however, further study the N406K change here, because it was not consistently present in all the D101.12 clones and it was not associated with the number or identity of the changes in V3 (data not shown). Inspection of the location of the N406K change on the gp120 core structure does not reveal any clues as to how it might affect the co-receptor interactions of the native Env trimer (data not shown).
We used the R14 clone to study the interplay between the sequence changes in V3 and gp41 that conferred full resistance on the D101.12 isolate. The two separate gp41 sequence patterns both involved the substitution of hydrophobic amino acids in the FP (G514V, M518V, F519L), together with an additional V535M change. The gp41 sequence variations were accompanied by a minimum of one (H308P), but up to all four, of the resistance-associated substitutions in V3. The gp41 Pattern I changes were G514V + V535M, while Pattern II was M518V + F519L + V535M. Studies with chimeric viruses in which the gp120 and gp41 subunits of VCV-sensitive and -resistant viruses were exchanged showed that resistance did not track with the source of gp120, but was, instead, conferred by the cooperative action of changes in both gp120 and gp41. Of note is that the majority of D101.12 clones of both gp41 patterns simultaneously harbored three, if not all four, V3 resistance-associated changes; changes in both Env subunits appear to contribute to the survival of these viruses under the selection pressure of VCV. These changes persist even when VCV is absent, presumably because they do not carry an associated fitness cost. A simian-human immunodeficiency virus selected in macaques for resistance to PSC-RANTES, a CCR5 inhibitor based on one of its chemokine ligands, also contains a combination of V3 (K315R) and gp41 (N640D) changes (Dudley et al., 2009). Clinical resistance to the small molecule CCR5 inhibitor, aplaviroc, is conferred by V3 changes although changes elsewhere in gp120 and gp41 modulate their influence (Pfaff et al.).
Mutagenesis studies followed by PBMC infection-inhibition assays showed that individual gp41 amino acid changes generally had a minor and often context-dependent effect on the resistance phenotype. The V535M substitution appears to compensate for the otherwise replication impairing effect that is created when the G514V and H308P changes are combined, rather than having a direct impact on resistance. The two Pattern-I gp41 changes conferred full resistance when they were both present in a virus that also contained the H308P substitution in V3 (e.g., as in clone R14), but when H308P was absent they created only a partially resistant virus. Hence the V535M + G514V substitutions are necessary, but not sufficient, to cause VCV resistance, while neither change has much, if any effect, when made in isolation. The increased T20 sensitivity of viruses bearing the gp41 Pattern-I sequence combination compared to other viruses from this lineage, irrespective of the presence or absence of the H308P change in gp120, provides clues to how these gp41 changes may be acting. Follow-up studies will be designed accordingly.
Resistance to a novel entry inhibitor, PF-68742, is associated with a G514R change in the FP, together with additional changes in gp41 that probably affect how this subunit interacts with gp120 (Murray et al.). The FP residue involved is the same one that changes in the VCV escape mutants of gp41 Pattern-I, although the substitution associated with PF-68742 resistance involves the introduction of a positively charged residue (G514R vs. G514V). Although PF-68742 and VCV have different targets, the Env proteins and CCR5 respectively, there seem to be similarities in the process by which HIV-1 escapes from the selection pressure they impose.
Among D101.12 clones, Pattern-II gp41 changes were much more common than Pattern-I. The two Pattern-II changes, M518V + F519L in the FP, did not confer resistance when introduced into a virus lacking the H308P change, and the further introduction of the V535M change to create the triple mutant produced only a weakly resistant virus. Indeed, in a virus lacking the H308P change, the Pattern-II changes had less of an effect than Pattern I. As with the Pattern-I changes, the resistance-conferring effect of the Pattern-II substitutions was only manifested in the context of a virus containing the H308P substitution in V3, although the overall effect was to confer partial and not full resistance.
We, and others, have reported previously that the manifestation of CCR5 inhibitor resistance is dependent on the cell type in which the assays are performed. In engineered cell lines, resistance is usually detected not by a rightward shift in a dose-response curve and a concomitant rise in the IC50 value, but by a lower plateau that is quantified by a reduction in the MPI value (Anastassopoulou et al., 2009; Ogert et al., 2008; Pugach et al., 2007; Westby et al., 2007). We therefore repeated the infection-inhibition assays for the various chimeric and mutant clones, using TZM-bl cells. In general, the relationships between genetic changes and the manifestations of resistance were similar in TZM-bl cells and PBMC, although some differences were apparent. Thus, unlike in PBMC, the V535M change is not required to compensate for the presence of the H308P and G514V changes when the relevant viruses replicate in TZM-bl cells. Why this is the case remains to be determined. Various viruses that are VCV-resistant in PBMC had MPI values significantly below 100% in the TZM-bl cell assay, an expected outcome of experiments in these cells. However, in many cases, the mutants also have lower IC50 values than the parental virus. This apparently paradoxical property of a resistant virus is something we have observed and discussed previously (Anastassopoulou et al., 2009). By comparing the properties of various gp41 mutant clones of both sequence patterns that either contained or lacked the H308P change in V3, we found that the IC50 reductions are attributable to FP changes while the reduced MPI values are driven by the H308P substitution.
The resistance pattern for the R14 virus is more complex than can be explained by the equal and interchangeable use of all the VCV-bound and free forms of CCR5. Our modeling has indicated that multiple forms of CCR5 with heterogeneous affinities for VCV are present on the cell surface (Anastassopoulou et al., 2009). The model shows that R14 appears to use VCV-CCR5 complexes strongly (i.e., it has a high w). The R14/S+(G514V, V535M) mutant that recapitulates the R14 phenotype also has a comparable w value. However, no mutations in either gp120 or gp41 alone could entirely reproduce the R14 phenotype, although some of the FP sequence changes were sufficient to have substantial effects on w. The other phenotypic trait possessed by the resistant virus is the paradoxical reduction in the inhibitory concentration of VCV that is primarily seen with TZM-bl cells but is also discernable by modeling the PBMC data. This property could be recreated by introducing several different permutations of single, double and triple FP mutations into the VCV-sensitive, parental virus, S. We suggest that the paradoxical shift to a lower IC50 is a byproduct of a subset of escape routes that involve changes in the FP; as noted, this aspect of the phenotype is most pronounced in TZM-bl and other cell lines that were neither used in the original selection nor are physiologically relevant targets for infection. We have proposed that the IC50 reductions are explained by the mutant virus switching to use unoccupied forms of CCR5 for which VCV has a higher affinity, compared to the forms that the wild type virus can use (Anastassopoulou et al., 2009). This side effect of the escape phenotype illustrates that, to the virus, drug efficacy is matter of survival, whereas drug potency is a matter of convenience.
Cooperative interactions between the subunits are a general feature of the functional Env trimer (Salzwedel and Berger, 2009). For some resistant viruses from the D101.12 lineage, the use of VCV-CCR5 complexes may be facilitated by substitutions in gp41 that work in concert with at least the H308P change in V3. In the alternative, more frequently seen and less complex, pathway, multiple changes in V3 achieve the same effect (i.e., for the fully resistant CC101.19 virus, H308P is supplemented by the three additional V3 changes) (Kuhmann et al., 2004). But changes only in the FP can also be both necessary and sufficient for full resistance, as occurs in D1/85.16 (Anastassopoulou et al., 2009). It remains to be understood how these different, but inter-related, genetic pathways affect the overall structure and function of Env in a way that enables it to use VCV-CCR5 complexes for entry. The impact the resistance-associated changes have on how the FP functions also remains to be defined, taking into account current models of its structure (Buzon et al.; Jaroniec et al., 2005; Qiang et al., 2008; Qiang and Weliky, 2009).
Materials and Methods
Reagents
The small molecule CCR5 inhibitors VCV (SCH-D, SCH-417690) and AD101 (SCH-350581) were provided by Dr. Julie Strizki (Schering-Plough Research Institute), while the small molecule CXCR4 inhibitor AMD3465 was obtained from Gary Bridger of AnorMED Inc (now Genzyme Inc).
Viruses
The study viruses are listed in Table 1, with their passage history portrayed graphically in Fig. 1. In brief, D101.12 arose from the weakly resistant CC101.6 isolate after 12 passages with VCV (Marozsan et al., 2005). Samples from intermediate stages of this escape culture were unavailable, so the evolution of env sequence changes in D101.12 during the generation of resistance could not be studied. CC101.6 had been isolated from the passage 6 culture of the parental CC1/85 isolate with AD101 (Trkola et al., 2002). The ~4-fold resistance of CC101.6 compared to its parent virus stems largely from an H308P polymorphism in the V3 region that presumably allows for improved exploitation of lower levels of free CCR5 (Kuhmann et al., 2004; Trkola et al., 2002). Infectious stocks were prepared by transient transfection of 293T cells with pNL4-3/env proviral plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and titrated before use as described (Anastassopoulou et al., 2009; Kuhmann et al., 2004). Env genes were sequenced as described (Marozsan et al., 2005; Trkola et al., 2002), and aligned with MacVector 10.5.1. The sequences have been submitted to GenBank (accession numbers HQ591389-HQ591416).
Construction of chimeric NL4-3/env proviruses
The env gene in the R14/S chimera, which contains gp120 from VCV-resistant D101.12 cl.14 (= R14) (Marozsan et al., 2005) and gp41 from parental CC1/85 cl.6 (= S) (Kuhmann et al., 2004), was constructed using the unique MfeI sites, sequenced, then subcloned into pNL4-3 to produce chimeric infectious viruses, as previously described (Anastassopoulou et al., 2009). To introduce mutations into the gp41 subunit of the S and the R14/S viruses, site-directed mutagenesis was performed with QuickChange II (Stratagene), using pBluescript KS(+) plasmids containing EcoRI/XhoI fragments that were then subcloned into pNL4-3.
Infection-inhibition assays
HIV-1 sensitivity to VCV or AD101 was assessed using PBMC from two or three random blood donors or TZM-bl cells, as previously described (Anastassopoulou et al., 2009; Ketas et al., 2007; Kuhmann et al., 2004; Marozsan et al., 2005); the PBMC assay endpoint was p24 production after seven days of infection, whereas with TZM-bl cells, luciferase expression in relative light units (RLU) was measured after two days. The model function for inhibition, Q=(1−(1−(C/(Kdi+C))+w*(C/(4.3*Kdi+C))))*100% (Anastassopoulou et al., 2009; Klasse, 2007), was fitted to the average PBMC and TZM-bl data by nonlinear regression (Prism, Graphpad) (Anastassopoulou et al., 2009; Klasse, 2007). The model function was fitted by nonlinear regression (Prism, Graphpad) to determine MPI and IC50 (as defined in Table 2) as well as Kdi and w (Table 3).
Supplementary Material
The following viruses were tested for their sensitivity to (A) VCV, (B) Zidovudine (AZT), (C) 2F5, and (D) Enfuvirtide (T20) in TZM-bl cells: VCV-resistant reference clone R14 (black squares); engineered resistant mutant R14/S+(G514V, V535M) (dark gray squares); parental, inhibitor-sensitive clone S (black circles); weakly resistant chimera R14/S (gray diamonds). The data are the average of five independent experiments, each performed using duplicate wells, with the error bars indicating the standard deviation.
Dose-response curves derived in PBMC from three different donor pools for VCV inhibition of the following viruses are shown: VCV-resistant reference clone R14 (black squares); engineered resistant mutant R14/S+(G514V, V535M) (gray diamonds); parental, inhibitor-sensitive clone S (dark gray triangles). The data are the average of three independent experiments, each performed using triplicate wells, with the error bars indicating the SEM. Depending on the donor, the curves for the resistant viruses can (A) peak at mid-range VCV concentrations, (B) be flat or (C) indicate that high VCV concentrations enhance infection.
Acknowledgments
We thank Samson Jacob and Nicole Labutong for technical support, Min Lu, Rogier Sanders, Reem Berro and Antu Dey for helpful discussions, and Julie Strizki for supplying CCR5 inhibitors. This work was supported by NIH grant R01 AI41420.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal-Gamse C, Lee FH, Haggarty B, Jordan AP, Yi Y, Lee B, Collman RG, Hoxie JA, Doms RW, Laakso MM. Adaptive mutations in a human immunodeficiency virus type 1 envelope protein with a truncated V3 loop restore function by improving interactions with CD4. J Virol. 2009;83 (21):11005–11015. doi: 10.1128/JVI.01238-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassopoulou CG, Ketas TJ, Klasse PJ, Moore JP. Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41. Proc Natl Acad Sci USA. 2009;106 (13):5318–5323. doi: 10.1073/pnas.0811713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastassopoulou CG, Marozsan AJ, Matet A, Snyder AD, Arts EJ, Kuhmann SE, Moore JP. Escape of HIV-1 from a small molecule CCR5 inhibitor is not associated with a fitness loss. PLoS Pathog. 2007;3 (6):e79. doi: 10.1371/journal.ppat.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Miyake H, Wang X, Okamoto M, Takashima K. Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652. Antimicrob Agents Chemother. 2007;51 (2):707–715. doi: 10.1128/AAC.01079-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berro R, Sanders RW, Lu M, Klasse PJ, Moore JP. Two HIV-1 variants resistant to small molecule CCR5 inhibitors differ in how they use CCR5 for entry. PLoS Pathog. 2009;5 (8):e1000548. doi: 10.1371/journal.ppat.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzon V, Natrajan G, Schibli D, Campelo F, Kozlov MM, Weissenhorn W. Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 2010;6 (5):e1000880. doi: 10.1371/journal.ppat.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin JM. HIV viral dynamics. Aids. 1996;10(Suppl 3):S75–84. [PubMed] [Google Scholar]
- Dudley DM, Wentzel JL, Lalonde MS, Veazey RS, Arts EJ. Selection of a simian-human immunodeficiency virus strain resistant to a vaginal microbicide in macaques. J Virol. 2009;83 (10):5067–5076. doi: 10.1128/JVI.00055-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley O, Klasse PJ, Sattentau QJ, Moore JP. V3: HIV’s switch-hitter. AIDS Res Hum Retroviruses. 2005;21 (2):171–189. doi: 10.1089/aid.2005.21.171. [DOI] [PubMed] [Google Scholar]
- Hirsch MS, Gunthard HF, Schapiro JM, Brun-Vezinet F, Clotet B, Hammer SM, Johnson VA, Kuritzkes DR, Mellors JW, Pillay D, Yeni PG, Jacobsen DM, Richman DD. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society-USA panel. Top HIV Med. 2008;16 (3):266–285. [PubMed] [Google Scholar]
- Jaroniec CP, Kaufman JD, Stahl SJ, Viard M, Blumenthal R, Wingfield PT, Bax A. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005;44 (49):16167–16180. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- Ketas TJ, Kuhmann SE, Palmer A, Zurita J, He W, Ahuja SK, Klasse PJ, Moore JP. Cell surface expression of CCR5 and other host factors influence the inhibition of HIV-1 infection of human lymphocytes by CCR5 ligands. Virology. 2007;364 (2):281–290. doi: 10.1016/j.virol.2007.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse PJ. Modeling how many envelope glycoprotein trimers per virion participate in human immunodeficiency virus infectivity and its neutralization by antibody. Virology. 2007;369 (2):245–262. doi: 10.1016/j.virol.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhmann SE, Hartley O. Targeting chemokine receptors in HIV: a status report. Annu Rev Pharmacol Toxicol. 2008;48:425–461. doi: 10.1146/annurev.pharmtox.48.113006.094847. [DOI] [PubMed] [Google Scholar]
- Kuhmann SE, Pugach P, Kunstman KJ, Taylor J, Stanfield RL, Snyder A, Strizki JM, Riley J, Baroudy BM, Wilson IA, Korber BT, Wolinsky SM, Moore JP. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J Virol. 2004;78 (6):2790–2807. doi: 10.1128/JVI.78.6.2790-2807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakso MM, Lee FH, Haggarty B, Agrawal C, Nolan KM, Biscone M, Romano J, Jordan AP, Leslie GJ, Meissner EG, Su L, Hoxie JA, Doms RW. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies. PLoS Pathog. 2007;3 (8):e117. doi: 10.1371/journal.ppat.0030117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Bertolotti-Ciarlet A, Haggarty B, Romano J, Nolan KM, Leslie GJ, Jordan AP, Huang CC, Kwong PD, Doms RW, Hoxie JA. Replication-competent variants of human immunodeficiency virus type 2 lacking the V3 loop exhibit resistance to chemokine receptor antagonists. J Virol. 2007;81 (18):9956–9966. doi: 10.1128/JVI.00385-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marozsan AJ, Kuhmann SE, Morgan T, Herrera C, Rivera-Troche E, Xu S, Baroudy BM, Strizki J, Moore JP. Generation and properties of a human immunodeficiency virus type 1 isolate resistant to the small molecule CCR5 inhibitor, SCH-417690 (SCH-D) Virology. 2005;338 (1):182–199. doi: 10.1016/j.virol.2005.04.035. [DOI] [PubMed] [Google Scholar]
- Murray EJ, Leaman DP, Pawa N, Perkins H, Pickford C, Perros M, Zwick MB, Butler SL. A low-molecular-weight entry inhibitor of both CCR5- and CXCR4-tropic strains of human immunodeficiency virus type 1 targets a novel site on gp41. J Virol. 2010;84 (14):7288–7299. doi: 10.1128/JVI.00535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan KM, Del Prete GQ, Jordan AP, Haggarty B, Romano J, Leslie GJ, Hoxie JA. Characterization of a human immunodeficiency virus type 1 V3 deletion mutation that confers resistance to CCR5 inhibitors and the ability to use aplaviroc-bound receptor. J Virol. 2009;83 (8):3798–3809. doi: 10.1128/JVI.01751-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan KM, Jordan AP, Hoxie JA. Effects of partial deletions within the human immunodeficiency virus type 1 V3 loop on coreceptor tropism and sensitivity to entry inhibitors. J Virol. 2008;82 (2):664–673. doi: 10.1128/JVI.01793-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogert RA, Hou Y, Ba L, Wojcik L, Qiu P, Murgolo N, Duca J, Dunkle LM, Ralston R, Howe JA. Clinical resistance to vicriviroc through adaptive V3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5. Virology. 2010;400 (1):145–155. doi: 10.1016/j.virol.2010.01.037. [DOI] [PubMed] [Google Scholar]
- Ogert RA, Wojcik L, Buontempo C, Ba L, Buontempo P, Ralston R, Strizki J, Howe JA. Mapping resistance to the CCR5 co-receptor antagonist vicriviroc using heterologous chimeric HIV-1 envelope genes reveals key determinants in the C2-V5 domain of gp120. Virology. 2008;373 (2):387–399. doi: 10.1016/j.virol.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Pfaff JM, Wilen CB, Harrison JE, Demarest JF, Lee B, Doms RW, Tilton JC. HIV-1 resistance to CCR5 antagonists associated with highly efficient use of CCR5 and altered tropism on primary CD4+ T cells. J Virol. 2010;84 (13):6505–6514. doi: 10.1128/JVI.00374-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Kozak SL, Durnin JP, Hope TJ, Kabat D. Rapid dissociation of HIV-1 from cultured cells severely limits infectivity assays, causes the inactivation ascribed to entry inhibitors, and masks the inherently high level of infectivity of virions. J Virol. 2010;84 (6):3106–3110. doi: 10.1128/JVI.01958-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Shea DM, Rose PP, Kabat D. Variants of human immunodeficiency virus type 1 that efficiently use CCR5 lacking the tyrosine-sulfated amino terminus have adaptive mutations in gp120, including loss of a functional N-glycan. J Virol. 2005;79 (7):4357–4368. doi: 10.1128/JVI.79.7.4357-4368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugach P, Marozsan AJ, Ketas TJ, Landes EL, Moore JP, Kuhmann SE. HIV-1 clones resistant to a small molecule CCR5 inhibitor use the inhibitor-bound form of CCR5 for entry. Virology. 2007;361 (1):212–228. doi: 10.1016/j.virol.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Bodner ML, Weliky DP. Solid-state NMR spectroscopy of human immunodeficiency virus fusion peptides associated with host-cell-like membranes: 2D correlation spectra and distance measurements support a fully extended conformation and models for specific antiparallel strand registries. J Am Chem Soc. 2008;130 (16):5459–5471. doi: 10.1021/ja077302m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Weliky DP. HIV fusion peptide and its cross-linked oligomers: efficient syntheses, significance of the trimer in fusion activity, correlation of beta strand conformation with membrane cholesterol, and proximity to lipid headgroups. Biochemistry. 2009;48 (2):289–301. doi: 10.1021/bi8015668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzwedel K, Berger EA. Complementation of diverse HIV-1 Env defects through cooperative subunit interactions: a general property of the functional trimer. Retrovirology. 2009;6:75. doi: 10.1186/1742-4690-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton JC, Wilen CB, Didigu CA, Sinha R, Harrison JE, Agrawal-Gamse C, Henning EA, Bushman FD, Martin JN, Deeks SG, Doms RW. A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5 antagonists depends on both N-terminal and extracellular loop domains of drug-bound CCR5. J Virol. 2010 doi: 10.1128/JVI.01109-10. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trkola A, Kuhmann SE, Strizki JM, Maxwell E, Ketas T, Morgan T, Pugach P, Xu S, Wojcik L, Tagat J, Palani A, Shapiro S, Clader JW, McCombie S, Reyes GR, Baroudy BM, Moore JP. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc Natl Acad Sci USA. 2002;99 (1):395–400. doi: 10.1073/pnas.012519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsibris AM, Sagar M, Gulick RM, Su Z, Hughes M, Greaves W, Subramanian M, Flexner C, Giguel F, Leopold KE, Coakley E, Kuritzkes DR. In vivo emergence of vicriviroc resistance in a human immunodeficiency virus type 1 subtype C-infected subject. J Virol. 2008;82 (16):8210–8214. doi: 10.1128/JVI.00444-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, Dorr P, Ciaramella G, Perros M. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol. 2007;81 (5):2359–2371. doi: 10.1128/JVI.02006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following viruses were tested for their sensitivity to (A) VCV, (B) Zidovudine (AZT), (C) 2F5, and (D) Enfuvirtide (T20) in TZM-bl cells: VCV-resistant reference clone R14 (black squares); engineered resistant mutant R14/S+(G514V, V535M) (dark gray squares); parental, inhibitor-sensitive clone S (black circles); weakly resistant chimera R14/S (gray diamonds). The data are the average of five independent experiments, each performed using duplicate wells, with the error bars indicating the standard deviation.
Dose-response curves derived in PBMC from three different donor pools for VCV inhibition of the following viruses are shown: VCV-resistant reference clone R14 (black squares); engineered resistant mutant R14/S+(G514V, V535M) (gray diamonds); parental, inhibitor-sensitive clone S (dark gray triangles). The data are the average of three independent experiments, each performed using triplicate wells, with the error bars indicating the SEM. Depending on the donor, the curves for the resistant viruses can (A) peak at mid-range VCV concentrations, (B) be flat or (C) indicate that high VCV concentrations enhance infection.