

Abstract
Background
Acrolein is a dietary aldehyde that is present in high concentrations in alcoholic beverages and foods including cheese, donuts and coffee. It is also abundant in tobacco smoke, automobile exhaust and industrial waste and is generated in vivo during inflammation and oxidative stress.
Objectives
The goal of this study was to examine the effects of dietary acrolein on atherosclerosis.
Methods
Eight-week old male apoE-null mice were gavage-fed acrolein (2.5 mg/kg/day) for 8 weeks. Atherosclerotic lesion formation and composition and plasma lipids and platelet factor 4 (PF4) levels were measured. Effects of acrolein and PF4 on endothelial cell function was measured in vitro.
Results
Acrolein feeding increased the concentration of cholesterol in the plasma. NMR analysis of the lipoproteins showed that acrolein feeding increased the abundance of small and medium VLDL particles. Acrolein feeding also increased atherosclerotic lesion formation in the aortic valve and the aortic arch. Immunohistochemical analysis showed increased macrophage accumulation in the lesions of acrolein-fed mice. Plasma PF4 levels and accumulation of PF4 in atherosclerotic lesions was increased in the acrolein-fed mice. Incubation of endothelial cells with the plasma of acrolein-fed mice augmented transmigration of monocytic cells, which was abolished by anti-PF4 antibody treatment.
Conclusions
Dietary exposure to acrolein exacerbates atherosclerosis in apoE-null mice. Consumption of foods and beverages rich in unsaturated aldehydes such as acrolein may be a contributing factor to the progression of atherosclerotic lesions.
Keywords: Acrolein, atherosclerosis, endothelial cells, lipoproteins, platelets, PF4
1. Introduction
The formation and progression of atherosclerotic lesions is affected by multiple genetic and environmental factors. Extensive work has shown that a high caloric intake and consumption of diets rich in saturated fatty acids accelerate atherogenesis; however, the effects of individual food components are less well understood. Human diet is a complex mixture of several constituents and contains reactive chemicals that have the potential to affect cardiovascular health and function. Of these, the consumption of aldehydes may be particularly significant. Aldehydes are highly reactive chemicals that are present in high abundance in most sources of food and water. More than 300 different aldehydes (e.g., crotonaldehyde, furfural, formaldehyde, acetaldehyde etc.) have been identified in different food substances1, 2. High concentrations of the unsaturated aldehydes such as acrolein are present in alcoholic beverages and foods including cheese, donuts and coffee1. The unsaturated aldehyde hexenal is used as a common flavoring agent. Heating and cooking of fats, oils and sugar also generate high concentrations of aldehydes3. Heating decreases the cis-double bond content of triglycerides and increases the formation of trans-unsaturated aldehydes such as acrolein3. Aldehydes are also present in drinking water. Storage of carbonated or non-carbonated water in plastic bottles also generates a variety of aldehydes4. At least 36 different aldehydes have been identified in water of which acrolein and endrin aldehyde are the two highest priority pollutants5. The recommended maximum concentration of acrolein in water is 65 μg/l, however these limits are often exceeded5. The overall daily human are consumption of total aldehydes is estimated to be approximately 7 mg/kg of which about 5 mg/kg is unsaturated aldehydes5, 6. Nevertheless, the cardiovascular toxicity of dietary aldehydes remains poorly understood.
We have shown that in vitro acrolein evokes delayed vasorelaxation of rat aortic rings, whereas acrolein exposure in vivo compromises vessel contractility7. We also observed that acute exposure to acrolein induces vasodilation of mesenteric bed in rats and mice8. Others have shown that in vitro acrolein is cytotoxic to endothelial cells9. In addition, our studies show that oral exposure to acrolein in mice increases the sensitivity of the heart to ischemia-reperfusion injury and abolishes the protective effects of the late phase of ischemic preconditioning6. Moreover, recent studies have also shown that oral exposure to acrolein induces hyperlipidemia in mice5 and activates matrix metalloproteinases (MMPs) by increasing the formation of free radicals in macrophages10. However, the effects of dietary acrolein on atherogenesis have not been studied. Accordingly, the current study was designed to investigate whether oral exposure to acrolein, as a model of dietary ingestion of unsaturated aldehydes, affects atherosclerotic lesion formation in mice. Results of this study show that chronic oral exposure to acrolein increases the plasma concentration of cholesterol and the chemokine platelet factor 4 (PF4), and exacerbates the formation of atherosclerotic lesions.
2. Materials and Methods
2.1 Housing and Treatment of Mice
Male apoE−/− mice (B6.129P2-Apoetm1Unc/J) were obtained from Jax Labs, Bar Harbor, ME. Mice were housed under pathogen-free conditions in the University of Louisville vivarium under controlled temperature and 12 h light/12 h dark cycle. Mice were maintained on a standard chow diet (PicoLab Rodent Chow 20 containing 4.5 % fat by weight and 0.02 % cholesterol). Starting at eight weeks of age mice were fed acrolein (2.5 mg/kg/day; n=11) or tap water (vehicle; controls; n=14) daily by gavage for 8 weeks. At 16 weeks of age, mice were euthanized and their plasma, spleen, liver, heart, and aorta were harvested.
2.2 Plasma Lipoprotein Profile
Total cholesterol and triglycerides in the plasma were measured using commercial kits as described before5, 11. Lipoprotein subclasses in the plasma were analyzed by NMR spectroscopy as described before5.
2.3 Atherosclerotic Lesion Analysis
Lesions in the aortic arch and the aortic valve and macrophages in the aortic valve were quantified as described before11.
2.4 Platelet Factor 4 Assay
Plasma PF4 levels were assayed by a sandwich ELISA using DeoSet Mouse PF4/CXCL4 ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
2.5 Immunofluorescence Staining and Confocal Microscopy
Sections of the aortic valve were stained with Alexa 647-conjugated rat anti-mouse CD68 as described before11. Nuclei were stained with DAPI. For the PF4 and protein-acrolein adduct staining, samples were incubated with the polyclonal rabbit anti-PF4 antibody (Santa Cruz Biotech, Santa Cruz, CA) or anti-KLH-acrolein antibody12 for 16 h. Samples were then incubated with Alexa-488-labeled appropriate secondary antibodies. Confocal images were acquired on a Zeiss LSM 510 microscope (Zeiss, Oberkochen, Germany) as described before11.
2.6 Protein-Acrolein Adducts
Protein-acrolein adducts in the plasma of control and acrolein-fed mice were measured by Western blotting, using the anti-KLH-acrolein antibody13.
2.7 Measurement of Markers of Endothelial Activation, Inflammation and Oxidative Stress in the Plasma
Concentrations of E-selectin and PAI-1 in the plasma were measured as markers of endothelial activation by ELISA using the kits from R&D Systems Inc., Minneapolis, MN and Innovative Research, Novi, MI, respectively, as per manufacturer's instructions. Plasma serum amyloid A (SAA; Caymen Chemical Co, Ann Arbor, MI) and cytokines and chemokines levels were measured as markers of systemic inflammation. SAA and monocyte chemotactic protein-1 (MCP-1; BD Biosciences, San Diego, CA) were measured by ELISA. Plasma TNF-α, IL-6 and GMCSF levels were measured by Millipore's Milliplex MAP kit on Luminex 100 IS, as per manufacturer's instructions. Systemic oxidative stress was measured by measuring the concentration of 8-iso prostaglandin F2αby ELISA using the kit from Caymen Chemical Co, Ann Arbor, MI.
2.8 Cell Culture
Human umbilical vein endothelial cells (HUVEC) and human monocytic cells (THP-1) were obtained from Clonetics (San Diego, CA). Cells were cultured as described before14. Experiments were performed on passages 4–8 cells.
2.9 Adhesion Assay
Stock solutions of acrolein (10 mM) and PF4 (50 μg/ml) were prepared fresh in Hank's balanced salt solution (HBSS). Working solution of acrolein and PF4 were made by successive dilutions in DMEM without serum. HUVEC (5 × 104) cultured for 24h in 96-well plates and incubated with acrolein (0, 250, 500 and 1000 nM) or PF4 (0, 250, 100 nM) in DMEM (100μl) without serum for 1h at 37°C. Appropriate amount of HBSS was used as vehicle control. Endothelial cells were then incubated with Calcein-labeled THP-1 cells (2 × 105) for 60 min at 37°C and adhesion of THP-1 cells to endothelial cells was measured as described before15.
2.10 Transmigration Assay
HUVEC (4 × 104 cells) were incubated with PF4 (0, 250, 1000 nM), acrolein (0, 250, 500, 1000 nM) or PF4 + acrolein (250 nM each) in DMEM (100μl) without serum for 1h at 37°C in Transwell chambers. THP-1 (2 × 105 cells) were added to the transwells and transmigration was measured as described before16. To examine the effect of acrolein feeding on the transmigration of THP-1 cells, HUVEC were incubated with the plasma of control (pooled from 8 mice) or acrolein (pooled from 8 mice)-fed mice for 1 h and transmigration of THP-1 cells was measured as described above. To examine the effect of anti-PF4 antibody on monocyte transmigration, HUVEC were incubated without or with anti- PF4 antibody (20 μg/ml) for 30 min in DMEM, prior to the incubation with acrolein, PF4 or plasma.
2.11 Statistical Analysis
Statistical analyses were performed using Sigma Stat (version 3) software. Values are reported as mean ± standard error (SEM). Student's t-test was used for statistical comparison between two groups. For comparing more than two groups, one-way ANOVA and Student-Newman-Keuls post-hoc test was used. P <0.05 was considered statistically significant.
3. Experimental Results
3.1 Exposure to Acrolein Causes Hypercholesterolemia
To examine whether chronic exposure to acrolein affects the concentrations of plasma lipoproteins, we measured total cholesterol and triglycerides concentration in the plasma of acrolein-fed apoE-null mice. As shown in Table 1, exposure to acrolein significantly increased plasma cholesterol levels (P<0.05). A modest increase was also observed in the plasma triglyceride concentration of acrolein-fed mice, but it was not statistically significant. NMR analysis of plasma lipoproteins showed that exposure to acrolein did not affect the size of VLDL or LDL particle (Table 1). However, a significant increase in the abundance of total VLDL and LDL particles was observed in acrolein-fed mice. Further analysis showed a significant (P<0.01) increase in the concentration of small and medium VLDL particles in acrolein-fed mice. Abundance of large LDL particle was also increased by 25% in acrolein-fed mice, but this change was not statistically significant. Exposure to acrolein had no effect on either the abundance or the size of HDL particles.
Table 1.
Parameters measured in the plasma of apoE-null mice exposed to water or acrolein.
| Control | Acrolein | |
|---|---|---|
| Lipids | ||
| Cholesterol (mg/dL) | 473 ± 18 | 564 ± 17* |
| Triglycerides (mg/dL) | 105 ± 5 | 118 ± 8 |
| Total Lipoprotein abundance (nmoles/L) | ||
| Total VLDL and chylomicron particles | 183 ± 5 | 284 ± 14* |
| Total LDL Particles | 677 ± 58 | 924 ± 10* |
| Total IDL Particles | 378 ± 38 | 552 ± 27* |
| Total HDL Particles | 11040 ± 697 | 11222 ± 2000 |
| VLDL Particle abundance (nmoles/L) | ||
| Large VLDL Particles | 14 ± 2 | 20 ± 3 |
| Medium VLDL Particles | 84 ± 5 | 126 ± 6* |
| Small VLDL Particles | 85 ± 3 | 138 ± 9* |
| LDL Particle abundance (nmoles/L) | ||
| Large LDL Particles | 296 ± 41 | 372 ± 25 |
| Medium LDL Particles | N.D. | N.D. |
| Small LDL Particles | N.D. | N.D. |
| HDL Particle abundance (nmoles/L) | ||
| Large HDL Particles | N.D. | N.D. |
| Medium HDL Particles | 1104 ± 80 | 1303 ± 227 |
| Small HDL Particles | 8732 ± 167 | 9771 ± 1716 |
| Mean particle size (nm) | ||
| VLDL | 62.8 ± 1.4 | 61.5± 1.1 |
| LDL | 22.6 ± 0 | 22 ± 0 |
| HDL | 8.2 ± 0.1 | 8±0 |
| Markers of Endothelial Activation | ||
| E-Selectin (ng/ml) | 57.0 ± 2.8 | 81 ± 5.9* |
| PAI-1 (ng/ml) | 27.4 ± 2.0 | 35.3 ± 2.9# |
| Markers of Inflammation | ||
| TNFα (pg/ml) | 1.7 ± 0.3 | 1.25 ± 0.3 |
| IL-6 (pg/ml) | 5.8 ± 1.8 | 6.1 ± 1.2 |
| MCP-1(pg/ml) | 19.3 ± 7.5 | 31.6 ± 6.4 |
| GMCSF(pg/ml) | 3.5 ± 0.9 | 2.4± 0.6 |
| Serum Amyloid A (μg/ml) | 11.4 ± 2.2 | 12.1 ±3 |
| Marker of Oxidative Stress | ||
| 8-iso Prostaglandin F2α (pg/ml) | 817 ± 123 | 874 ± 147 |
Value are mean ± SE.
P<0.05
P<0.01
N.D.- Not determined.
3.2 Exposure to Acrolein Causes Endothelial activation
To examine the effect of acrolein exposure on endothelial activation, we measured the concentrations of E-selectin and PAI-1in the plasma of acroleinfed mice. Expressed selectively on endothelial cells, E-selectin plays a significant role on the recruitment of leukocytes on activated endothelium. PAI-1 is a serine protease inhibitor and is formed mainly by activated endothelial cells. However, it can also be formed by adipose tissue. As shown in the Table 1, acrolein feeding significantly increased E-selectin and PAI-1 levels. These data suggest that exposure to acrolein causes endothelial activation in vivo, which would promote monocyte recruitment.
3.3 Exposure to Acrolein Exacerbates Atherosclerotic Lesion Formation in ApoE-null Mice
Treatment with acrolein did not affect the body weight of the animals or their general health (data not shown). To examine the effect of acrolein-feeding on atherosclerosis, we examined lesion formation in the aortic valve (Fig. 1A). Staining of the aortic valve with Oil red O showed small lipid-laden foam cells in the cusps of the aortic valves of vehicle-fed controls (Fig. 1Ai and Fig. 1Aii). Accumulation of cholesterol-rich foam cells was markedly increased in aortic valve of acrolein-fed mice (Fig. 1Aiii and Fig. 1Aiv). Morphometric analysis of the aortic valve showed that lesion area was increased by > 2-fold in acrolein-fed mice (Fig. 1Av; P<0.01). Similarly, quantification of the lesions in the aortic arch showed 1.9-fold increase in lesion formation in acrolein-fed mice as compared with controls (Fig. 1B; P<0.05). No lesions were detected in the abdominal aorta of acrolein-fed or control mice (data not shown). To examine lesion composition, sections of the aortic valve of control and acrolein-fed mice were stained with MOMA-2 for macrophages and α-smooth muscle cell actin for smooth muscle cells. Staining for the macrophages in acrolein-fed mice was 2.5-fold greater than the lesions of control mice (Fig. 1C; P<0.01). Only a few cells (<2 %) showed positive staining for smooth muscle cells. The extent of staining for smooth muscle cells was similar in control and acrolein-fed mice (data not shown). These observations suggest that oral exposure to acrolein accelerates early lesion formation and macrophage accumulation in apoE-null mice.
Figure 1. Acrolein-feeding exacerbates early phase of atherogenesis and enhances accumulation of macrophages in lesions.
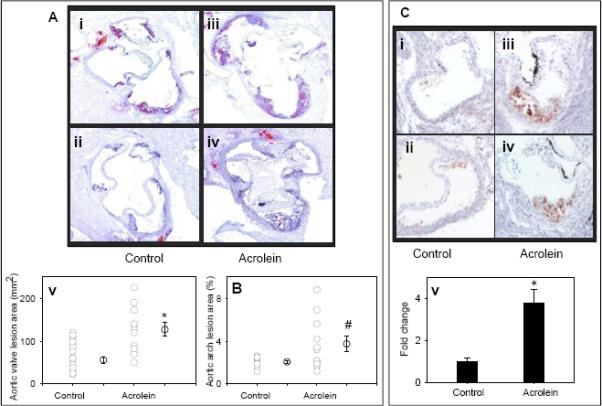
Eight week old male apoE-null mice were fed acrolein (2.5 mg/kg in drinking water) by gavage for 8 weeks. Mice gavage-fed with drinking water served as controls. A) Lesions in the aortic valve. Lipids were visualized by Oil red O staining. Panels i and ii show representative photomicrographs of aortic valve of control and iii and iv of acrolein-fed mice. Panel v shows the group data. Panel B shows the group data of lesions in the aortic arch of acrolein-fed and control mice. C) Macrophage accumulation in the aortic valve was examined by staining with MOMA-2. Panels i and ii show representative photomicrographs of aortic valve of control and iii and iv of acrolein-fed mice. Panel v shows the group data. Values are means ± SEM. #P<0.05 and *P<0.01 vs controls.
3.4 Effect of Acrolein of Systemic Inflammation, Endoplasmic Reticulum Stress and Oxidative Stress
We have recently shown that incubation of endothelial cells with acrolein increases the expression of pro-inflammatory cytokine IL-814. Therefore, next we examined the effect of acrolein-feeding on systemic inflammation. For this, we measured the concentration of pro-inflammatory cytokines and chemokines (TNF-α, IL-6, MCP-1 and GMCSF) in the plasma of control and acrolein-fed apoE-null mice. As shown in Table 1 acrolein feeding did not increase the abundance of any of these pro-inflammatory proteins in the plasma. Similarly, acrolein feeding did not increase the expression of TNF-α, IL-1βand IL-6 in the spleen (data not shown). Our observation are in agreement with a recent report by Kasahara et al.,17 showing that intratracheal exposure to acrolein (5ppm; 6h/day for 1–3 days) did not affect the cytokine expression in the lungs, however acrolein also inhibited the lipopolysaccharide (LPS)-induced formation of cytokines. To further examine the effect of acrolein feeding on systemic inflammation, we measured the concentration of serum amyloid A (SAA) in the plasma. As shown in Table 1, acrolein feeding did not affect the plasma levels of SAA in apoE-null mice. Together, these observations suggest that exposure to acrolein does not increase systemic inflammation.
Recently, we have also shown that acrolein increases oxidative stress and endoplasmic reticulum (ER) stress in endothelial cells10, 14. To examine the effect of acrolein feeding on systemic oxidative stress and ER-stress, we measured the expression of oxidative defense enzymes (SOD2, catalase, Nrf2, NQO1), phase 2 detoxification enzymes (GSTA1) and markers of ER-stress (Grp78 and ATF3) in the liver. Expression of all of these genes in acrolein-fed mice was comparable with controls (data not shown). To further examine the effect of acrolein feeding on systemic oxidative stress we measured the concentration of isoprostane, 8-iso prostaglandin F22α, in the plasma of control and acrolein-fed apoE-null mice. As shown in Table 1, acrolein feeding did not affect the 8-iso Prostaglandin F2α levels in the plasma. Together, these data suggest that acrolein feeding did not cause systemic oxidative stress or ER-stress under the experimental conditions.
3.5 Exposure to Acrolein Increases the Accumulation of Protein-acrolein Adducts in the Plasma and Atherosclerotic Lesions of ApoE-null Mice
Acrolein feeding increased the abundance of protein-acrolein adducts in the plasma of apoE-null mice (Fig. 2A). Specifically, we observed a marked increase in the accumulation of a 150 kDa - acrolein-modified protein in the plasma of acrolein-fed mice. Recently, we have shown that exposure to acrolein in C57BL/6 mice causes the modification of a 150 kDA protein in the platelets13. Further studies are required to examine whether the acrolein modified protein detected in the plasma of acrolein-fed apoE-null mice is due to direct modification of a plasma protein or the acrolein-modified protein in the platelet is secreted into the plasma. Nonetheless, these observation clearly demonstrate that ingested acrolein is present in circulating blood.
Figure 2. Accumulation of protein-acrolein adducts and PF4 in the plasma and atherosclerotic lesions of acrolein-fed apoE-null mice.
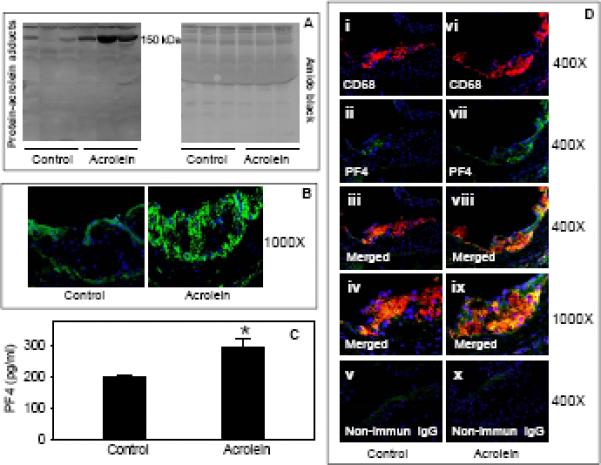
Eight week old male apoE-null mice were fed acrolein (2.5 mg/kg) or water (controls) by gavage for 8 weeks. A) Western blot analysis of protein-acrolein adducts in the plasma. Plasma obtained from control and acrolein-fed apoE-null mice, was probed with anti-acrolein-KLH antibody. B) Expression of protein-acrolein adducts in the aortic valve. Frozen sections of control and acrolein-fed apoE-null mice were stained with anti KLH-acrolein antibody, followed by Alexa488-conjugated goat-anti-rabbit secondary antibody. C) Plasma levels of PF4. Concentration of PF4 in the plasma was measured by sandwich ELISA. Values are means ± SEM. *P<0.01 vs controls. D) Expression and co-localization of PF4 with macrophages in the aortic valve. OCT-fixed frozen sections of control (i–v) and acrolein-fed (vi–x) apoE-null mice were stained with Alexa 647-conjugated anti-CD68 (i and vi; red) and PF4 (PF4; ii and vii; green, Alexa 488). Sections incubated with non-immune rabbit IgG (v and x) served as negative controls. The yellow fluorescence in the merged image (iii and viii) indicates PF4 co-localization with macrophages. Nuclei are identified in blue (DAPI). Panels iv and ix show higher magnification images of the colocalization of PF4 with macrophages.
We also observed some immunopositive staining with the anti-KLH antibody in the aortic valve of control apoE-null mice. These observations are in agreement with a previous report illustrating that protein-acrolein adducts are present in human atherosclerotic lesions18. Acrolein feeding markedly increased the accumulation of protein-acrolein adducts in the aortic valve (Fig. 2B). Together, these observations suggest that exposure to acrolein results in the accumulation of acrolein-modified proteins in the plasma and in atherosclerotic lesions.
3.6 Exposure to Acrolein Increases PF4 Accumulation in the Plasma and Atherosclerotic Lesions
Our recent studies13 suggest that acrolein feeding enhances platelet activation in C57BL/6 mice. Upon activation, platelets release their granular contents. PF4 is a chemokine and is released from the α-granules of activated platelets. PF4 has been shown to be atherogenic and genetic ablation of PF4 has been shown to diminish lesion formation in atherogenic mice19. Therefore, we measured the concentration of PF4 in the plasma of acrolein-fed mice. As shown in Fig. 2C, the concentration of PF4 in the plasma of acrolein-fed mice was significantly higher (P<0.01) than in the water-fed controls. Next, we examined the expression of PF4 in atherosclerotic lesions of controls and acrolein-fed mice. As shown in Fig. 2D, anti-PF4 antibody showed sparse immunopositive reactivity within the aortic root lesions of control mice. Confocal imaging showed weak staining with anti-PF4 antibody that was co-localized with the CD68+ macrophage staining. The accumulation of PF4 was markedly greater in the aortic valve of acrolein-fed mice and most of the PF4 staining co-localized with the CD68+ macrophage staining. Together, these observations suggest that exposure to acrolein enhances plasma PF4 levels and its accumulation in atherosclerotic lesions. To examine whether increase in the plasma PF4 levels in acrolein-fed apoE-null mice are due to an increase in lesional PF4 or they could be causally involved in atherogenesis, we measured plasma PF4 levels in C57BL/6 mice fed with acrolein for 8 weeks by gavage. Similar to apoE-null mice, we observed a significant increase in plasma PF4 levels in acroleinfed mice (data not shown), suggesting that the increase in plasma PF4 levels in acrolein-fed mice are not due to the release of PF4 from the lesions but that PF4 could be directly involved in promoting lesion progression.
3.7 Platelet Factor 4 Enhances Acrolein-Induced Endothelial Activation
To further examine the atherogenic mechanisms of acrolein, we measured the effect of acrolein on endothelial function. As shown in Fig. 3Ai, incubation of HUVEC with acrolein enhanced the adhesion of THP-1 cells to endothelial cells in a concentration-dependent manner. Moreover, incubation of HUVEC with acrolein also increased the transmigration of THP-1 cells through the endothelial monolayer in a concentration-dependent manner (Fig. 3Aii). Similar to acrolein, PF4 also increased THP-1 cell adhesion (Fig. 3Bi) and transmigration (Fig. 3Bii) in a concentration-dependent manner. Next, we examined whether PF4 potentiates acrolein-induced transmigration of the monocytic cells. For these experiments, we used the concentrations at which PF4 (250 ng/ml) and acrolein (250 nM) by themselves, did not increase the transmigration of monocytic cells. However, treatment of endothelial HUVEC with PF4 (250 ng/ml) + acrolein (250 nM) increased the transmigration of THP-1 cells by > 2-fold (Fig. 3C). Anti-PF4 antibody (20 μg/ml) inhibited the transmigration of THP-1 cells by >80% (Fig. 3C). Non-immunized rabbit IgG (used as an isotype control for polyclonal anti-PF4 antibody raised in rabbit) did not affect the transmigration of control or acrolein-treated HUVEC (Fig. 3C). Similar to reagent acrolein and PF4, incubation of HUVEC with the plasma of acrolein-fed mice (pooled from 8 mice) increased the transmigration of THP-1 cells by 2-fold as compared with the plasma of water-fed controls (pooled from 8 mice; Fig. 3D). Anti-PF4 (20 μg/ml) antibody treatment did not inhibit the transmigration of THP-1 cells by the plasma of control mice but completely abolished the increase in the transmigration of THP-1 cells by the plasma of acrolein-fed mice (Fig. 3D). We did not have sufficient plasma from these mice to examine the effect of non-immune rabbit IgG on the transmigration of THP-1 cells. However, in an independent experiment, we incubated plasma of the naive C57BL/6 mice (in triplicate from three different mice) without or with non-immunized rabbit IgG under the incubation conditions identical to those described in Fig. 3D. Our data showed that non-immunized rabbit IgG does not affect transmigration of THP-1 cells (data not shown). Together, these data suggest that acrolein enhances endothelial activation and PF4 potentiates acrolein-induced endothelial activation.
Figure 3. Acrolein and platelet factor 4 enhance endothelial activation.

A) Acrolein causes endothelial activation. HUVEC were incubated with acrolein in DMEM without serum and THP-1 cells adhesion (i) and transmigration (ii) were measured as described under Materials and Methods. B) PF4 causes endothelial activation. HUVEC were incubated with PF4 in DMEM without serum and THP-1 cells adhesion (i) and transmigration (ii) were measured. C) Anti-PF4 antibody diminishes acrolein-induced endothelial activation. HUVEC were pre-incubated with anti-PF4 antibody (20 μg/ml) or non-immunized rabbit IgG (iso type control for anti-PF4 antibody; 20 μg/ml) for 30 min in DMEM without serum. Sub-threshold concentrations of acrolein (250 nM), PF4 (250 ng/ml) or acrolein (250 nM) + PF4 (250 ng/ml) was added to the medium and transmigration of THP-1 cells was measured as described under Materials and Methods. D) Anti-PF4 antibody diminishes the transmigration of THP-1 induced by the plasma acrolein-fed mice. Plasma of control (pooled from 8 mice) and acrolein-fed (pooled from 8 mice) mice was incubated with anti-PF4 antibody (20 μg/ml) for 30 min and transmigration of THP-1 cells was measured. Values are means ± SEM. *P<0.01 vs control, §P<0.01 vs acrolein + PF4.
4. Discussion
Atherosclerosis is a complex and multi-factorial disease. Interplay of multiple factors including hyperlipidemia, endothelial activation, monocyte recruitment and their differentiation into macrophages, foam cell formation and activation of platelets, is well documented in the pathogenesis of atherosclerosis. In the present study, we show that exposure to a dietary and environmental aldehyde, acrolein, exacerbates atherosclerotic lesion formation in mice. This outcome is likely because exposure to acrolein increases plasma cholesterol concentration, augments endothelial activation and enhances the circulating and lesional concentration of platelet-derived chemokine PF4. These changes can accelerate atherogenesis without causing an increase in systemic inflammation or oxidative stress.
Hypercholesterolemia is also well known risk factor for atherosclerosis. Recently, we have shown that a single dose of acrolein by gavage increases plasma cholesterol and triglycerides in C57BL/6 mice5. The effect is most pronounced in VLDL particles, primarily because of decreased clearance of VLDL due to hepatic lipase activity and the down regulation of the LDL receptor5. In agreement with these data, in the present study we observed that chronic feeding of acrolein to apoE-null mice significantly increases the concentration of total cholesterol in the plasma (Table 1). We observed only a modest increase in the plasma triglyceride levels in acrolein-fed apoE-null mice (Table 1) which was further confirmed by the calculated plasma triglyceride concentration by the NMR analysis of lipoproteins. However, this increase in plasma triglyceride in acrolein-fed mice did not reach statistical significance perhaps in part, due to large variations in acrolein-exposed mice. It is also possible that acrolein affects triglyceride clearance. Further studies are required to examine why chronic acrolein feeding does not significantly increase plasma triglycerides. Nonetheless, despite only a modest increase in plasma triglyceride levels, NMR analysis of the lipoproteins showed a significant increase in the levels of small and medium VLDL particles in acrolein-fed mice. Increase in plasma E-selectin and PAI-1 suggests that exposure to acrolein causes endothelial activation. Therefore, these smaller lipoproteins particles can readily accumulate in the sub-endothelial space and can further activate the endothelium to bind and facilitates the transmigration of more monocytes. Moreover, it is also possible that monocytes can home to the perivascular adipose and influence aortic remodeling from there20. These monocytes, upon differentiation into macrophages can engulf more VLDL and LDL particles and enhance foam cell formation. Indeed we observed that in vitro, low concentrations of acrolein augments monocyte adhesion and transmigration. These observations are in agreement with our earlier studies showing that acrolein and structurally-related lipid peroxidation-derived aldehyde, 4-hydroxynonenal, enhances the TNF-α-induced expression of ICAM-121. Alternatively, incubation of endothelial cells with acrolein can result in the cross-linking of acrolein with extracellular matrix proteins in endothelial cells and this may facilitate monocyte adhesion. Studies by Kirkham et al., showed that exposure of collagen IV to acrolein or HNE causes the modification of collagen and enhances the adhesion of macrophages22. In the present study we observed that chronic acrolein-feeding to apoE-null mice exacerbated early atherosclerotic lesion formation and the lesions in acrolein-fed mice are rich in macrophages. Together, these data suggest that acrolein-induced hypercholesterolemia and endothelial activation could augment atherosclerosis.
Emerging data suggest that platelet activation could be involved in the development of atherogenesis. In humans, adherence of platelets to the damaged endothelium increases thrombus formation, accelerates atherogenesis and increases the risk of ischemic heart disease. Deficiency of P-selectin, which enhances platelet-endothelial interaction, diminishes atherosclerotic lesion formation in atherogenic mice23. Massberg et al., have shown that in apoE-null mice maintained on Western diet, platelet adhesion in carotid artery precedes leukocyte adhesion, suggesting that platelets play a pivotal role in early atheosclerotic lesion formation24. Recently, we have shown that acute exposure to acrolein causes the accumulation of protein-acrolein adducts in platelets and enhances platelet activation in C57BL/6 mice and releases its granular contents including PF4 and serotonin13. PF4 is synthesized in megakaryocytes and stored in α-granules. PF4 in released from activated platelets binds to heparin-like molecules and facilitate thrombus formation at the site of injury13. In addition, PF4 has been shown to enhance the accumulation of lipoproteins in vascular cells25 and augment the binding of oxLDL to endothelial cells, smooth muscle cells and macrophages26. Concentration of PF4 closer to the vessel wall has been estimated to be 25 μg/ml27. Moreover, apoE-null mice lacking PF4 have diminished atherosclerotic lesion formation as compared with apoE-null mice with PF419. In the present study, we observed that chronic acrolein-feeding in apoE-null mice increased the concentration of PF4 in the plasma and accumulation of PF4 in atherosclerotic lesions. It is conceivable that chronic hypercholesterolemia in these mice could enhance platelet activation, resulting in the release of pro-atherogenic PF4 from the α-granules of platelets. Increased PF4 in acrolein-fed mice could then enhance the accumulation of lipoprotein particles in the sub-endothelial space and propagate atherogenesis.
Because PF4 is also a chemokine, we also examined the effect of PF4 on endothelial functions. Incubation of endothelial cells with PF4 (250–1000 ng/ml) enhanced monocyte adhesion and transmigration. More importantly, incubation of endothelial cells with the sub-threshold concentrations of PF4 augmented the acrolein-induced monocyte adhesion and transmigration. This is quite significant, considering the fact that the concentration of PF4 closer to the vessel wall has been estimated to be 25 μg/ml26. Our observations are consistent with previous studies showing that PF4 induces the expression of adhesion molecule, E-selectin in endothelial cells25 and activated platelets enhance the recruitment of monocytes to the vessel wall by increasing the expression of MCP-1 and ICAM-1 in endothelial cells28. In early human atherosclerotic lesions PF4 co-localizes with oxLDL in macrophages26. Significantly, our data shows that chronic feeding of acrolein to apoE-null mice increases PF4 concentration in the plasma and accumulation of PF4 in early atherosclerotic lesions. Immunostaining for PF4 in the lesions co-localized with macrophages. Ex-vivo, we observed that incubation of endothelial cells with the plasma of acrolein-fed mice, significantly augmented transmigration of monocytic cells, and anti-PF4 antibody abolished the increase in transmigration of monocytes by the plasma of acrolein-fed mice. Together, these data suggest that exposure to acrolein or related aldehydes could enhance platelet activation and atherosclerosis in humans and may thereby elevate the risk of developing cardiovascular disease.
Apart from dietary exposures, humans are also exposed to acrolein via environmental and occupational exposures. Aldehydes such as acrolein are ubiquitous pollutants in the air. Acrolein is present in high concentration in automobile exhaust, cigarette and wood smoke29. Due to incomplete combustion, side-stream smoke contains 12-fold more acrolein than mainstream smoke; consistent with the high cardiovascular risk of second-hand tobacco smoke30. In apoE-null mice side-stream cigarette smoke has been shown to accelerate atherosclerosis30. Acrolein is also generated during the metabolism of several drugs (e.g., cyclophosphamide), toxins (e.g. allylamine), or atmospheric transformation of pollutants such as 1,3-butadiene29. Large amounts of acrolein are synthesized for industrial use in the production of acrylic acid, polymers and herbicides and it is used widely as a biocide against aqueous organisms and as a slimicide in the manufacture of paper29. Hence exposure to acrolein from these environmental sources could also increase atherogenesis.
Furthermore, acrolein is also generated endogenously by myeloperoxidase-catalyzed oxidation and is present in high amounts at the sites of inflammation29. Aldehydes such as acrolein, malonaldialdehyde, and 4-hydroxy trans-2-nonenal (HNE) are also generated endogenously during lipoprotein oxidation29. These aldehydes combine readily with nucleophilic sites in proteins, phospholipids and DNA to form covalent adducts that can induce structural damage or cause functional changes. An increase in endogenous production of aldehydes by oxidized lipids has been linked to the pathogenesis of several diseases including cardiovascular diseases29 and aldehydes-modified proteins have been detected in vascular lesions11_ENREF_11_ENREF_11. Thus, our finding that exposure to acrolein increases atherogenesis may be significant also for understanding the atherogenic effects of inflammation and oxidative stress.
In summary, our studies show for the first time that acrolein induces endothelial and platelet activation, cause hypercholesterolemia and exacerbates atherosclerosis (Scheme I). Based on these observations, we speculate that human exposure to reactive dietary aldehydes such as acrolein might have pro-atherogenic effects similar to those observed in mice. Moreover, our data support the idea that acrolein may be responsible for the well-documented pro-atherogenic effects of direct or second-hand smoke exposure or exposure to polluted environments rich in combustion products.
Scheme 1.

Atherogenic mechanisms of acrolein.
Acknowledgements
The authors thank Mr. David Young and Ms. Barbara Bishop for expert technical assistance. This work was supported in part by NIH grants ES17260, ES11594, ES11860, HL55477, HL59378, HL89380 and RR 24489.
Abbreviations
- (PF4)
Platelet factor 4
- (HUVEC)
Human Umbilical Vein Endothelial Cells
- (LDL)
low density lipoproteins
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests Declaration: None
References
- 1.Feron VJ, Til HP, de Vrijer F, Woutersen RA, Cassee FR, van Bladeren PJ. Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutat Res. 1991;259:363–385. doi: 10.1016/0165-1218(91)90128-9. [DOI] [PubMed] [Google Scholar]
- 2.Luo J, Hill BG, Gu Y, Cai J, Srivastava S, Bhatnagar A, Prabhu SD. Mechanisms of acrolein-induced myocardial dysfunction: implications for environmental and endogenous aldehyde exposure. Am J Physiol Heart Circ Physiol. 2007;293:H3673–3684. doi: 10.1152/ajpheart.00284.2007. [DOI] [PubMed] [Google Scholar]
- 3.Fullana A, Carbonell-Barrachina AA, Sidhu S. Comparison of volatile aldehydes present in the cooking fumes of extra virgin olive, olive, and canola oils. J Agric Food Chem. 2004;52:5207–5214. doi: 10.1021/jf035241f. [DOI] [PubMed] [Google Scholar]
- 4.Nawrocki J, Dabrowska A, Borcz A. Investigation of carbonyl compounds in bottled waters from Poland. Water Res. 2002;36:4893–4901. doi: 10.1016/s0043-1354(02)00201-4. [DOI] [PubMed] [Google Scholar]
- 5.Conklin DJ, Barski OA, Lesgards JF, Juvan P, Rezen T, Rozman D, Prough RA, Vladykovskaya E, Liu S, Srivastava S, Bhatnagar A. Acrolein consumption induces systemic dyslipidemia and lipoprotein modification. Toxicol Appl Pharmacol. 2010;243:1–12. doi: 10.1016/j.taap.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang GW, Guo Y, Vondriska TM, Zhang J, Zhang S, Tsai LL, Zong NC, Bolli R, Bhatnagar A, Prabhu SD. Acrolein consumption exacerbates myocardial ischemic injury and blocks nitric oxide-induced PKCepsilon signaling and cardioprotection. J Mol Cell Cardiol. 2008;44:1016–1022. doi: 10.1016/j.yjmcc.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Tsakadze NL, Srivastava S, Awe SO, Adeagbo AS, Bhatnagar A, D'Souza SE. Acrolein-induced vasomotor responses of rat aorta. Am J Physiol Heart Circ Physiol. 2003;285:H727–734. doi: 10.1152/ajpheart.00269.2003. [DOI] [PubMed] [Google Scholar]
- 8.Awe SO, Adeagbo AS, D'Souza SE, Bhatnagar A, Conklin DJ. Acrolein induces vasodilatation of rodent mesenteric bed via an EDHF-dependent mechanism. Toxicol Appl Pharmacol. 2006;217:266–276. doi: 10.1016/j.taap.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kachel DL, Martin WJ., 2nd Cyclophosphamide-induced lung toxicity: mechanism of endothelial cell injury. J Pharmacol Exp Ther. 1994;268:42–46. [PubMed] [Google Scholar]
- 10.O'Toole TE, Zheng YT, Hellmann J, Conklin DJ, Barski O, Bhatnagar A. Acrolein activates matrix metalloproteinases by increasing reactive oxygen species in macrophages. Toxicol Appl Pharmacol. 2009;236:194–201. doi: 10.1016/j.taap.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava S, Vladykovskaya E, Barski OA, Spite M, Kaiserova K, Petrash JM, Chung SS, Hunt G, Dawn B, Bhatnagar A. Aldose reductase protects against early atherosclerotic lesion formation in apolipoprotein E-null mice. Circ Res. 2009;105:793–802. doi: 10.1161/CIRCRESAHA.109.200568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conklin DJ, Haberzettl P, Prough RA, Bhatnagar A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am J Physiol Heart Circ Physiol. 2009;296:H1586–1597. doi: 10.1152/ajpheart.00867.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sithu SD, Srivastava S, Siddiqui MA, Vladykovskaya E, Riggs DW, Conklin DJ, Haberzettl P, O'Toole TE, Bhatnagar A, D'Souza SE. Exposure to acrolein by inhalation causes platelet activation. Toxicol Appl Pharmacol. 2010;248:100–110. doi: 10.1016/j.taap.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haberzettl P, Vladykovskaya E, Srivastava S, Bhatnagar A. Role of endoplasmic reticulum stress in acrolein-induced endothelial activation. Toxicol Appl Pharmacol. 2009;234:14–24. doi: 10.1016/j.taap.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsakadze NL, Sithu SD, Sen U, English WR, Murphy G, D'Souza SE. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1) J Biol Chem. 2006;281:3157–3164. doi: 10.1074/jbc.M510797200. [DOI] [PubMed] [Google Scholar]
- 16.Sithu SD, English WR, Olson P, Krubasik D, Baker AH, Murphy G, D'Souza SE. Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J Biol Chem. 2007;282:25010–25019. doi: 10.1074/jbc.M611273200. [DOI] [PubMed] [Google Scholar]
- 17.Kasahara DI, Poynter ME, Othman Z, Hemenway D, van der Vliet A. Acrolein inhalation suppresses lipopolysaccharide-induced inflammatory cytokine production but does not affect acute airways neutrophilia. J Immunol. 2008;181:736–745. doi: 10.4049/jimmunol.181.1.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 19.Sachais BS, Turrentine T, Dawicki McKenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE–/– mice. Thromb Haemost. 2007;98:1108–1113. [PubMed] [Google Scholar]
- 20.Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29:1458–1464. doi: 10.1161/ATVBAHA.109.192658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srivastava S, Ramana KV, Srivastava SK, D'Souza SE, Bhatnagar A. Role of Aldose Reductase in the Detoxification of Oxidized Phospholipids. In: Penning TM, Petras Mark. J., editors. Aldo-Keto Reductases and Toxicant Metabolism. American Chemical Society; 2004. pp. 49–64. (ACH Symposium Series 865). [Google Scholar]
- 22.Kirkham PA, Spooner G, Ffoulkes-Jones C, Calvez R. Cigarette smoke triggers macrophage adhesion and activation: role of lipid peroxidation products and scavenger receptor. Free Radic Biol Med. 2003;35:697–710. doi: 10.1016/s0891-5849(03)00390-3. [DOI] [PubMed] [Google Scholar]
- 23.Manka D, Collins RG, Ley K, Beaudet AL, Sarembock IJ. Absence of p-selectin, but not intercellular adhesion molecule-1, attenuates neointimal growth after arterial injury in apolipoprotein e-deficient mice. Circulation. 2001;103:1000–1005. doi: 10.1161/01.cir.103.7.1000. [DOI] [PubMed] [Google Scholar]
- 24.Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, Bergmeier W, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu G, Rux AH, Ma P, Bdeir K, Sachais BS. Endothelial expression of E-selectin is induced by the platelet-specific chemokine platelet factor 4 through LRP in an NF-kappaB-dependent manner. Blood. 2005;105:3545–3551. doi: 10.1182/blood-2004-07-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nassar T, Sachais BS, Akkawi S, Kowalska MA, Bdeir K, Leitersdorf E, Hiss E, Ziporen L, Aviram M, Cines D, Poncz M, Higazi AA. Platelet factor 4 enhances the binding of oxidized low-density lipoprotein to vascular wall cells. J Biol Chem. 2003;278:6187–6193. doi: 10.1074/jbc.M208894200. [DOI] [PubMed] [Google Scholar]
- 27.Stuckey JA, St Charles R, Edwards BF. A model of the platelet factor 4 complex with heparin. Proteins. 1992;14:277–287. doi: 10.1002/prot.340140213. [DOI] [PubMed] [Google Scholar]
- 28.Gawaz M, Neumann FJ, Dickfeld T, Koch W, Laugwitz KL, Adelsberger H, Langenbrink K, Page S, Neumeier D, Schomig A, Brand K. Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation. 1998;98:1164–1171. doi: 10.1161/01.cir.98.12.1164. [DOI] [PubMed] [Google Scholar]
- 29.Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ Res. 2006;99:692–705. doi: 10.1161/01.RES.0000243586.99701.cf. [DOI] [PubMed] [Google Scholar]
- 30.Gairola CG, Drawdy ML, Block AE, Daugherty A. Sidestream cigarette smoke accelerates atherogenesis in apolipoprotein E−/− mice. Atherosclerosis. 2001;156:49–55. doi: 10.1016/s0021-9150(00)00621-3. [DOI] [PubMed] [Google Scholar]